Hypothalamic-Pituitary-Adrenal Axis Modulation of Glucocorticoids in the Cardiovascular System

Abstract
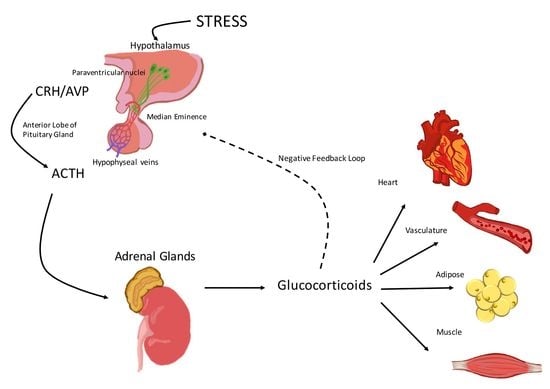
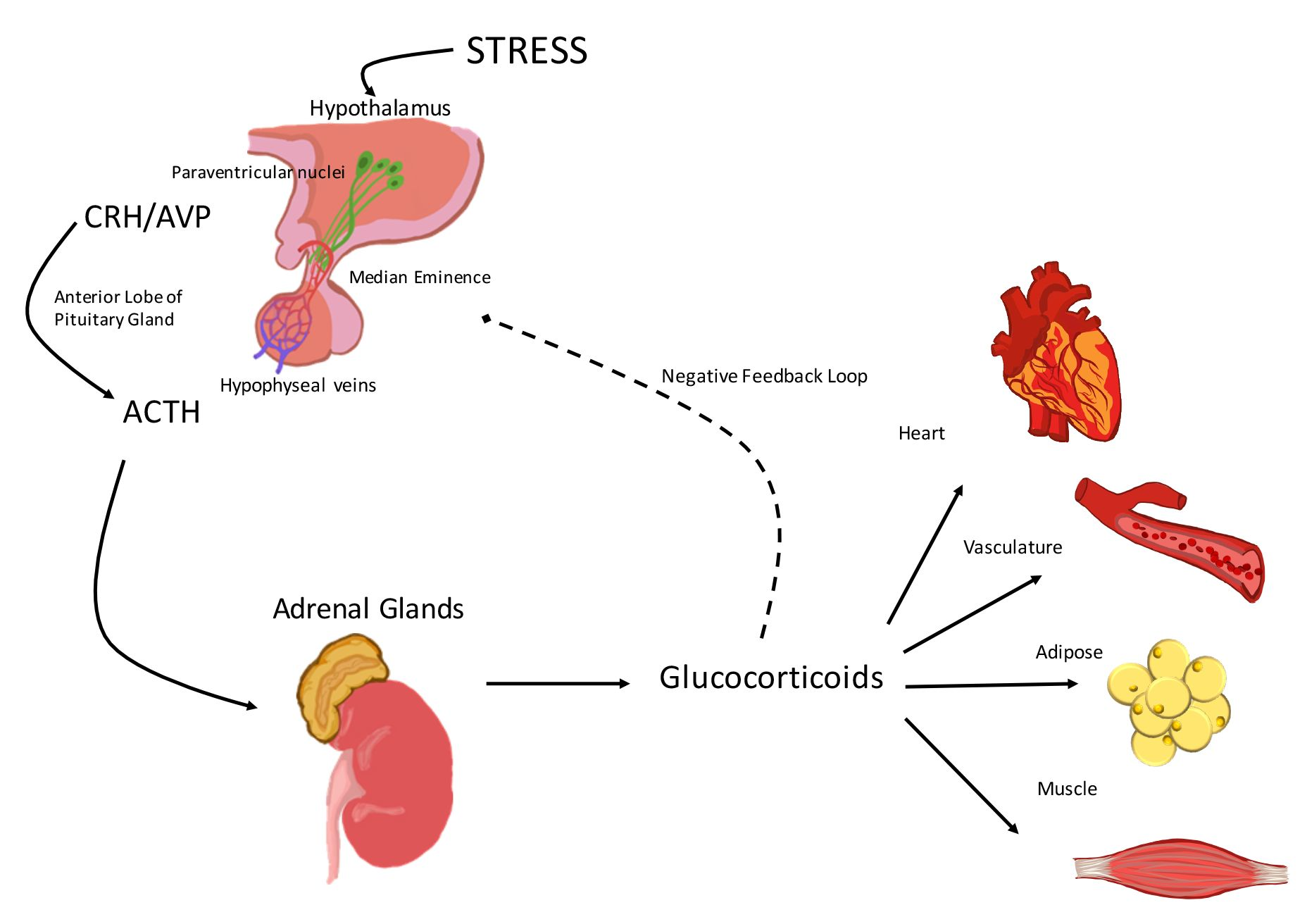
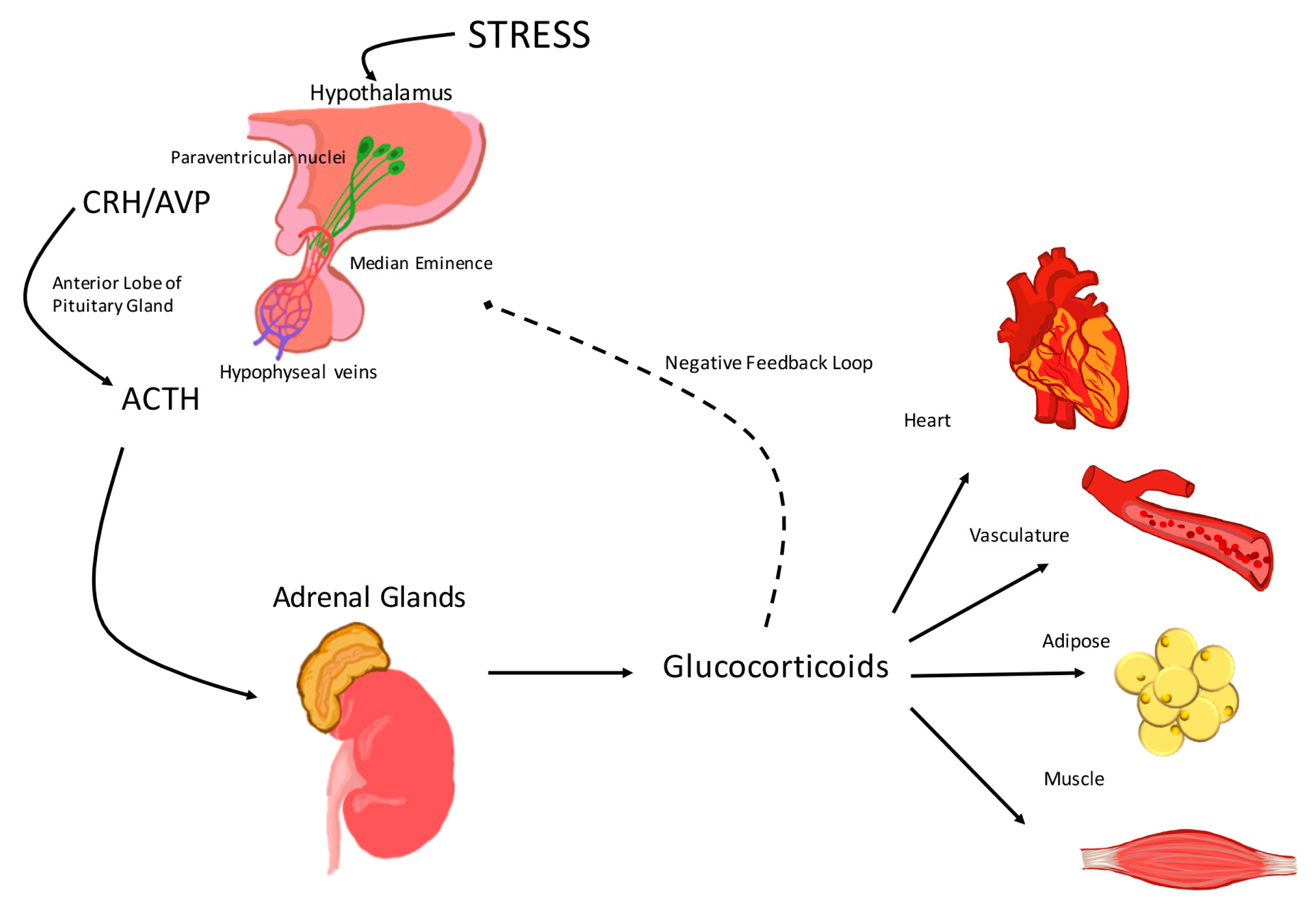
:
{kind=link}
{kind=link}
{kind=link}
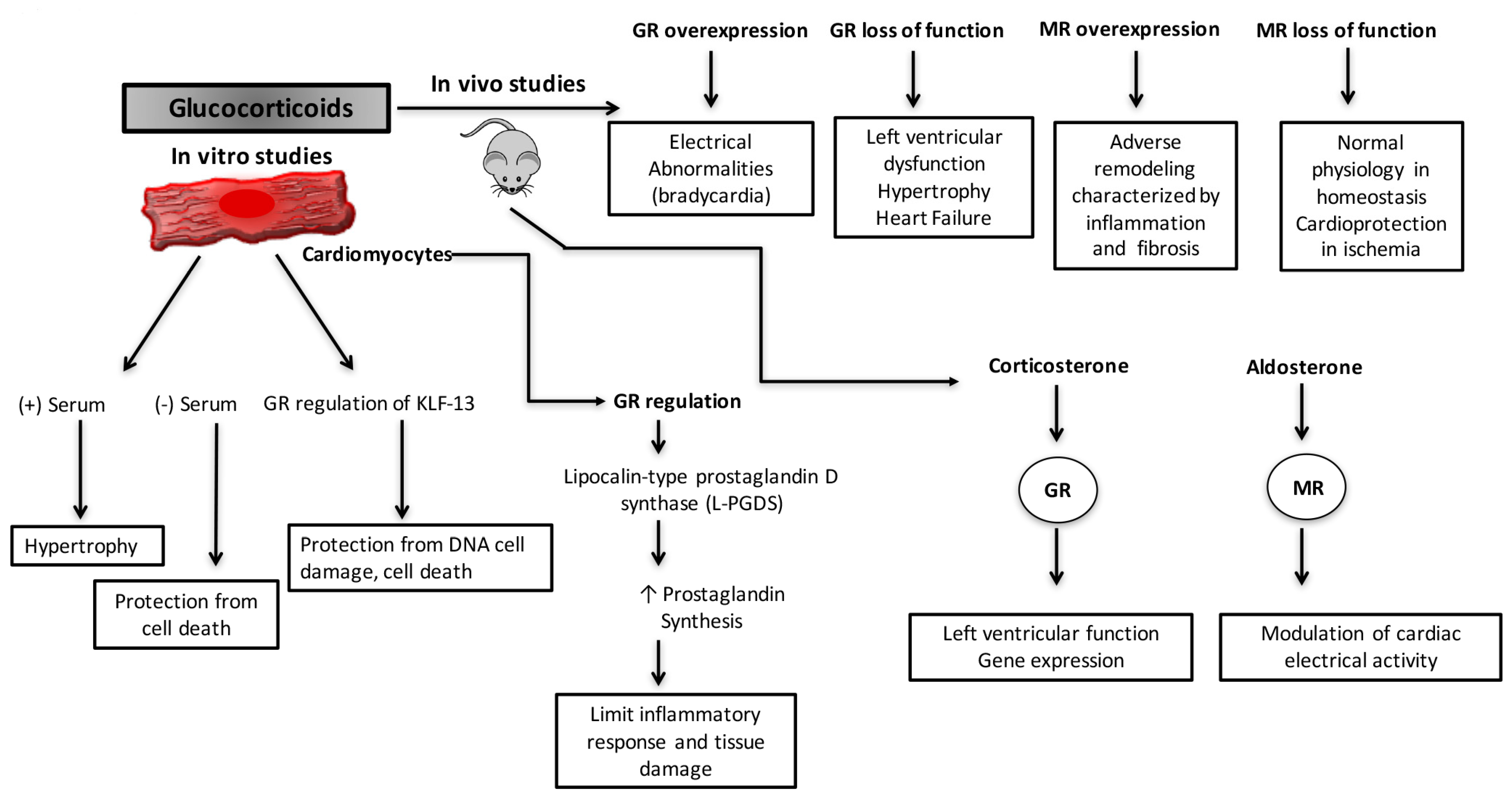{kind=link}
1. Activation of the Hypothalamic-Pituitary-Adrenal (HPA) Axis
2. The Hypothalamic-Pituitary-Adrenal (HPA) Axis and the Cardiovascular System
2.1. Corticotropin-Releasing Hormone (CRH)
2.2. Arginine Vasopressin (AVP)
2.3. Adrenocorticotropic Hormone (ACTH)
2.4. Glucocorticoids
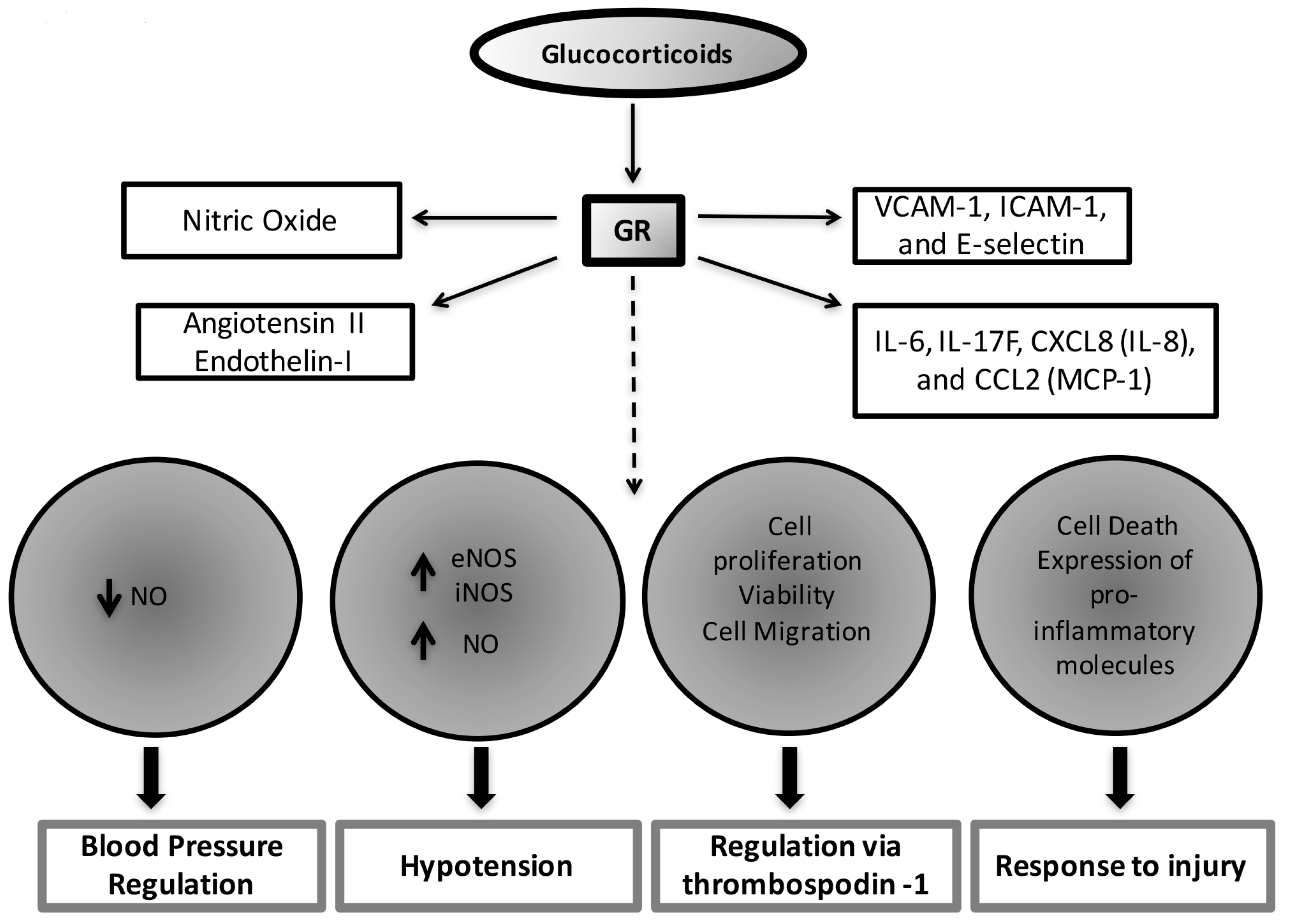
3. Glucocorticoid Effects on the Vasculature and the Heart
3.1. Glucocorticoid Effects on the Vascular Endothelium
3.2. Glucocorticoid Effects on the Heart
4. Concluding Remarks
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Selye, H. Endocrine reactions during stress. Curr. Res. Anesth. Analg. 1956, 35, 182–193. [Google Scholar] [CrossRef] [PubMed]
- Smith, S.M.; Vale, W.W. The role of the hypothalamic-pituitary-adrenal axis in neuroendocrine responses to stress. Dialogues Clin. Neurosci. 2006, 8, 383–395. [Google Scholar] [PubMed]
- Vale, W.; Spiess, J.; Rivier, C.; Rivier, J. Characterization of a 41-residue ovine hypothalamic peptide that stimulates secretion of corticotropin and β-endorphin. Science 1981, 213, 1394–1397. [Google Scholar] [CrossRef] [PubMed]
- Rivier, C.; Vale, W. Modulation of stress-induced ACTH release by corticotropin-releasing factor, catecholamines and vasopressin. Nature 1983, 305, 325–327. [Google Scholar] [CrossRef] [PubMed]
- Kadmiel, M.; Cidlowski, J.A. Glucocorticoid receptor signaling in health and disease. Trends Pharmacol. Sci. 2013, 34, 518–530. [Google Scholar] [CrossRef] [PubMed]
- Reyes, T.M.; Lewis, K.; Perrin, M.H.; Kunitake, K.S.; Vaughan, J.; Arias, C.A.; Hogenesch, J.B.; Gulyas, J.; Rivier, J.; Vale, W.W.; et al. Urocortin II: A member of the corticotropin-releasing factor (CRF) neuropeptide family that is selectively bound by type 2 CRF receptors. Proc. Natl. Acad. Sci. USA 2001, 98, 2843–2848. [Google Scholar] [CrossRef] [PubMed]
- Lewis, K.; Li, C.; Perrin, M.H.; Blount, A.; Kunitake, K.; Donaldson, C.; Vaughan, J.; Reyes, T.M.; Gulyas, J.; Fischer, W.; et al. Identification of urocortin III, an additional member of the corticotropin-releasing factor (CRF) family with high affinity for the CRF2 receptor. Proc. Natl. Acad. Sci. USA 2001, 98, 7570–7575. [Google Scholar] [CrossRef] [PubMed]
- Vaughan, J.; Donaldson, C.; Bittencourt, J.; Perrin, M.H.; Lewis, K.; Sutton, S.; Chan, R.; Turnbull, A.V.; Lovejoy, D.; Rivier, C.; et al. Urocortin, a mammalian neuropeptide related to fish urotensin I and to corticotropin-releasing factor. Nature 1995, 378, 287–292. [Google Scholar] [CrossRef] [PubMed]
- Rivier, C.; Rivier, J.; Lederis, K.; Vale, W. In vitro and in vivo ACTH-releasing activity of ovine CRF, sauvagine and urotensin I. Regul. Pept. 1983, 5, 139–143. [Google Scholar] [CrossRef]
- Rivier, C.; Brownstein, M.; Spiess, J.; Rivier, J.; Vale, W. In vivo corticotropin-releasing factor-induced secretion of adrenocorticotropin, β-endorphin, and corticosterone. Endocrinology 1982, 110, 272–278. [Google Scholar] [CrossRef] [PubMed]
- Perrin, M.H.; Vale, W.W. Corticotropin releasing factor receptors and their ligand family. Ann. N. Y. Acad. Sci. 1999, 885, 312–328. [Google Scholar] [CrossRef] [PubMed]
- Perrin, M.; Donaldson, C.; Chen, R.; Blount, A.; Berggren, T.; Bilezikjian, L.; Sawchenko, P.; Vale, W. Identification of a second corticotropin-releasing factor receptor gene and characterization of a cDNA expressed in heart. Proc. Natl. Acad. Sci. USA 1995, 92, 2969–2973. [Google Scholar] [CrossRef] [PubMed]
- Stenzel, P.; Kesterson, R.; Yeung, W.; Cone, R.D.; Rittenberg, M.B.; Stenzel-Poore, M.P. Identification of a novel murine receptor for corticotropin-releasing hormone expressed in the heart. Mol. Endocrinol. 1995, 9, 637–645. [Google Scholar] [PubMed]
- Smith, G.W.; Aubry, J.M.; Dellu, F.; Contarino, A.; Bilezikjian, L.M.; Gold, L.H.; Chen, R.; Marchuk, Y.; Hauser, C.; Bentley, C.A.; et al. Corticotropin releasing factor receptor 1-deficient mice display decreased anxiety, impaired stress response, and aberrant neuroendocrine development. Neuron 1998, 20, 1093–1102. [Google Scholar] [CrossRef]
- Timpl, P.; Spanagel, R.; Sillaber, I.; Kresse, A.; Reul, J.M.; Stalla, G.K.; Blanquet, V.; Steckler, T.; Holsboer, F.; Wurst, W. Impaired stress response and reduced anxiety in mice lacking a functional corticotropin-releasing hormone receptor 1. Nat. Genet. 1998, 19, 162–166. [Google Scholar] [CrossRef] [PubMed]
- Jeong, K.H.; Jacobson, L.; Widmaier, E.P.; Majzoub, J.A. Normal suppression of the reproductive axis following stress in corticotropin-releasing hormone-deficient mice. Endocrinology 1999, 140, 1702–1708. [Google Scholar] [CrossRef] [PubMed]
- Fisher, L.A.; Jessen, G.; Brown, M.R. Corticotropin-releasing factor (CRF): Mechanism to elevate mean arterial pressure and heart rate. Regul. Pept. 1983, 5, 153–161. [Google Scholar] [CrossRef]
- Nijsen, M.J.; Croiset, G.; Stam, R.; Bruijnzeel, A.; Diamant, M.; de Wied, D.; Wiegant, V.M. The role of the CRH type 1 receptor in autonomic responses to corticotropin-releasing hormone in the rat. Neuropsychopharmacology 2000, 22, 388–399. [Google Scholar] [CrossRef]
- Hillhouse, E.W.; Grammatopoulos, D.K. The molecular mechanisms underlying the regulation of the biological activity of corticotropin-releasing hormone receptors: Implications for physiology and pathophysiology. Endocr. Rev. 2006, 27, 260–286. [Google Scholar] [CrossRef] [PubMed]
- Tojo, K.; Sato, S.; Tokudome, G.; Ohta, M.; Kawaguchi, Y.; Sakai, O.; Nakagawa, O.; Nakao, K. Stimulation by corticotropin-releasing factor of atrial natriuretic peptide and brain natriuretic peptide secretions from cultured neonatal rat cardiomyocytes. Biochem. Biophys. Res. Commun. 1996, 225, 340–346. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, K.; Tojo, K.; Sato, S.; Ebisawa, T.; Tokudome, G.; Hosoya, T.; Harada, M.; Nakagawa, O.; Nakao, K. Urocortin, a newly identified corticotropin-releasing factor-related mammalian peptide, stimulates atrial natriuretic peptide and brain natriuretic peptide secretions from neonatal rat cardiomyocytes. Biochem. Biophys. Res. Commun. 1998, 250, 298–304. [Google Scholar] [CrossRef] [PubMed]
- Holmes, C.L.; Landry, D.W.; Granton, J.T. Science review: Vasopressin and the cardiovascular system part 1—Receptor physiology. Crit. Care 2003, 7, 427–434. [Google Scholar] [CrossRef] [PubMed]
- Bankir, L.; Bouby, N.; Ritz, E. Vasopressin: A novel target for the prevention and retardation of kidney disease? Nat. Rev. Nephrol. 2013, 9, 223–239. [Google Scholar] [CrossRef] [PubMed]
- Verney, E.B. The antidiuretic hormone and the factors which determine its release. Proc. R. Soc. Lond. B 1947, 135, 25–106. [Google Scholar] [CrossRef] [PubMed]
- Arnauld, E.; Czernichow, P.; Fumoux, F.; Vincent, J.D. The effects of hypotension and hypovolaemia on the liberation of vasopressin during haemorrhage in the unanaesthetized monkey (Macaca mulatta). Pflugers Arch. 1977, 371, 193–200. [Google Scholar] [CrossRef] [PubMed]
- Holmes, C.L.; Landry, D.W.; Granton, J.T. Science Review: Vasopressin and the cardiovascular system part 2—Clinical physiology. Crit. Care 2004, 8, 15–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okamura, T.; Toda, M.; Ayajiki, K.; Toda, N. Receptor subtypes involved in relaxation and contraction by arginine vasopressin in canine isolated short posterior ciliary arteries. J. Vasc. Res. 1997, 34, 464–472. [Google Scholar] [CrossRef] [PubMed]
- Evora, P.R.; Pearson, P.J.; Schaff, H.V. Arginine vasopressin induces endothelium-dependent vasodilatation of the pulmonary artery. V1-receptor-mediated production of nitric oxide. Chest 1993, 103, 1241–1245. [Google Scholar] [CrossRef] [PubMed]
- Tamaki, T.; Kiyomoto, K.; He, H.; Tomohiro, A.; Nishiyama, A.; Aki, Y.; Kimura, S.; Abe, Y. Vasodilation induced by vasopressin V2 receptor stimulation in afferent arterioles. Kidney Int. 1996, 49, 722–729. [Google Scholar] [CrossRef] [PubMed]
- Vanhoutte, P.M.; Katusic, Z.S.; Shepherd, J.T. Vasopressin induces endothelium-dependent relaxations of cerebral and coronary, but not of systemic arteries. J. Hypertens Suppl. 1984, 2, S421–S422. [Google Scholar] [PubMed]
- Katusic, Z.S.; Shepherd, J.T.; Vanhoutte, P.M. Vasopressin causes endothelium-dependent relaxations of the canine basilar artery. Circ. Res. 1984, 55, 575–579. [Google Scholar] [CrossRef] [PubMed]
- Maturi, M.F.; Martin, S.E.; Markle, D.; Maxwell, M.; Burruss, C.R.; Speir, E.; Greene, R.; Ro, Y.M.; Vitale, D.; Green, M.V.; et al. Coronary vasoconstriction induced by vasopressin. Production of myocardial ischemia in dogs by constriction of nondiseased small vessels. Circulation 1991, 83, 2111–2121. [Google Scholar] [CrossRef] [PubMed]
- Bax, W.A.; van der Graaf, P.H.; Stam, W.B.; Bos, E.; Nisato, D.; Saxena, P.R. [Arg8]vasopressin-induced responses of the human isolated coronary artery: Effects of non-peptide receptor antagonists. Eur. J. Pharmacol. 1995, 285, 199–202. [Google Scholar] [CrossRef]
- Boyle, W.A., 3rd; Segel, L.D. Attenuation of vasopressin-mediated coronary constriction and myocardial depression in the hypoxic heart. Circ. Res. 1990, 66, 710–721. [Google Scholar] [CrossRef] [PubMed]
- Gillies, G.E.; Linton, E.A.; Lowry, P.J. Corticotropin releasing activity of the new CRF is potentiated several times by vasopressin. Nature 1982, 299, 355–357. [Google Scholar] [CrossRef] [PubMed]
- Herman, J.P.; McKlveen, J.M.; Ghosal, S.; Kopp, B.; Wulsin, A.; Makinson, R.; Scheimann, J.; Myers, B. Regulation of the Hypothalamic-Pituitary-Adrenocortical Stress Response. Compr. Physiol. 2016, 6, 603–621. [Google Scholar] [PubMed]
- Aguilera, G.; Rabadan-Diehl, C. Vasopressinergic regulation of the hypothalamic-pituitary-adrenal axis: Implications for stress adaptation. Regul. Pept. 2000, 96, 23–29. [Google Scholar] [CrossRef]
- Bilezikjian, L.M.; Woodgett, J.R.; Hunter, T.; Vale, W.W. Phorbol ester-induced down-regulation of protein kinase C abolishes vasopressin-mediated responses in rat anterior pituitary cells. Mol. Endocrinol. 1987, 1, 555–560. [Google Scholar] [CrossRef] [PubMed]
- Carvallo, P.; Aguilera, G. Protein kinase C mediates the effect of vasopressin in pituitary corticotrophs. Mol. Endocrinol. 1989, 3, 1935–1943. [Google Scholar] [CrossRef] [PubMed]
- Stalla, G.K.; Stalla, J.; Loeffler, J.P.; von Werder, K.; Muller, O.A. Pharmacological modulation of CRH-stimulated ACTH secretion by ketoconazole. Horm. Metab. Res. Suppl. 1987, 16, 31–36. [Google Scholar] [PubMed]
- Grammatopoulos, D.K. Insights into mechanisms of corticotropin-releasing hormone receptor signal transduction. Br. J. Pharmacol. 2012, 166, 85–97. [Google Scholar] [CrossRef] [PubMed]
- Aguilera, G. Regulation of pituitary ACTH secretion during chronic stress. Front. Neuroendocrinol. 1994, 15, 321–350. [Google Scholar] [CrossRef] [PubMed]
- Jia, L.G.; Canny, B.J.; Leong, D.A. Paracrine communication regulates adrenocorticotropin secretion. Endocrinology 1992, 130, 534–539. [Google Scholar] [CrossRef] [PubMed]
- Agulleiro, M.J.; Sanchez, E.; Leal, E.; Cortes, R.; Fernandez-Duran, B.; Guillot, R.; Davis, P.; Dores, R.M.; Gallo-Payet, N.; Cerda-Reverter, J.M. Molecular characterization and functional regulation of melanocortin 2 receptor (MC2R) in the sea bass. A putative role in the adaptation to stress. PLoS ONE 2013, 8, e65450. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bicknell, A.B. The tissue-specific processing of pro-opiomelanocortin. J. Neuroendocrinol. 2008, 20, 692–699. [Google Scholar] [CrossRef] [PubMed]
- Lim, C.T.; Grossman, A.; Khoo, B. Normal Physiology of ACTH and GH Release in the Hypothalamus and Anterior Pituitary in Man. In Endotext; De Groot, L.J., Chrousos, G., Dungan, K., Feingold, K.R., Grossman, A., Hershman, J.M., Koch, C., Korbonits, M., McLachlan, R., New, M., et al., Eds.; MDtext.Com, Inc.: South Dartmouth, MA, USA, 2000. [Google Scholar]
- Spiga, F.; Waite, E.J.; Liu, Y.; Kershaw, Y.M.; Aguilera, G.; Lightman, S.L. ACTH-dependent ultradian rhythm of corticosterone secretion. Endocrinology 2011, 152, 1448–1457. [Google Scholar] [CrossRef] [PubMed]
- Lefebvre, H.; Thomas, M.; Duparc, C.; Bertherat, J.; Louiset, E. Role of ACTH in the Interactive/Paracrine Regulation of Adrenal Steroid Secretion in Physiological and Pathophysiological Conditions. Front. Endocrinol. 2016, 7, 98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, W.L.; Auchus, R.J. The molecular biology, biochemistry, and physiology of human steroidogenesis and its disorders. Endocr. Rev. 2011, 32, 81–151. [Google Scholar] [CrossRef] [PubMed]
- Rhee, S.S.; Pearce, E.N. Update: Systemic Diseases and the Cardiovascular System (II). The endocrine system and the heart: A review. Rev. Esp. Cardiol. 2011, 64, 220–231. [Google Scholar] [CrossRef] [PubMed]
- De Leo, M.; Pivonello, R.; Auriemma, R.S.; Cozzolino, A.; Vitale, P.; Simeoli, C.; de Martino, M.C.; Lombardi, G.; Colao, A. Cardiovascular disease in Cushing’s syndrome: Heart versus vasculature. Neuroendocrinology 2010, 92, 50–54. [Google Scholar] [CrossRef] [PubMed]
- Whitworth, J.A.; Williamson, P.M.; Mangos, G.; Kelly, J.J. Cardiovascular consequences of cortisol excess. Vasc. Health Risk Manag. 2005, 1, 291–299. [Google Scholar] [CrossRef] [PubMed]
- Hatakeyama, H.; Inaba, S.; Taniguchi, N.; Miyamori, I. Functional adrenocorticotropic hormone receptor in cultured human vascular endothelial cells: Possible role in control of blood pressure. Hypertension 2000, 36, 862–865. [Google Scholar] [CrossRef] [PubMed]
- Boston, B.A.; Cone, R.D. Characterization of melanocortin receptor subtype expression in murine adipose tissues and in the 3T3-L1 cell line. Endocrinology 1996, 137, 2043–2050. [Google Scholar] [CrossRef] [PubMed]
- Chida, D.; Nakagawa, S.; Nagai, S.; Sagara, H.; Katsumata, H.; Imaki, T.; Suzuki, H.; Mitani, F.; Ogishima, T.; Shimizu, C.; et al. Melanocortin 2 receptor is required for adrenal gland development, steroidogenesis, and neonatal gluconeogenesis. Proc. Natl. Acad. Sci. USA 2007, 104, 18205–18210. [Google Scholar] [CrossRef] [PubMed]
- Walker, B.R. Glucocorticoids and cardiovascular disease. Eur. J. Endocrinol. 2007, 157, 545–559. [Google Scholar] [CrossRef] [PubMed]
- Oakley, R.H.; Cidlowski, J.A. Glucocorticoid signaling in the heart: A cardiomyocyte perspective. J. Steroid Biochem. Mol. Biol. 2015, 153, 27–34. [Google Scholar] [CrossRef] [PubMed]
- Chapman, K.; Holmes, M.; Seckl, J. 11β-hydroxysteroid dehydrogenases: Intracellular gate-keepers of tissue glucocorticoid action. Physiol. Rev. 2013, 93, 1139–1206. [Google Scholar] [CrossRef] [PubMed]
- Funder, J.W. Mineralocorticoid receptors: Distribution and activation. Heart Fail. Rev. 2005, 10, 15–22. [Google Scholar] [CrossRef] [PubMed]
- Souverein, P.C.; Berard, A.; van Staa, T.P.; Cooper, C.; Egberts, A.C.; Leufkens, H.G.; Walker, B.R. Use of oral glucocorticoids and risk of cardiovascular and cerebrovascular disease in a population based case-control study. Heart 2004, 90, 859–865. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, L.; MacDonald, T.M.; Walker, B.R. Taking glucocorticoids by prescription is associated with subsequent cardiovascular disease. Ann. Int. Med. 2004, 141, 764–770. [Google Scholar] [CrossRef] [PubMed]
- Krug, J.J. Cardiac arrest secondary to Addison’s disease. Ann. Emerg. Med. 1986, 15, 735–737. [Google Scholar] [CrossRef]
- Van den Akker, E.L.; Koper, J.W.; van Rossum, E.F.; Dekker, M.J.; Russcher, H.; de Jong, F.H.; Uitterlinden, A.G.; Hofman, A.; Pols, H.A.; Witteman, J.C.; et al. Glucocorticoid receptor gene and risk of cardiovascular disease. Arch. Int. Med. 2008, 168, 33–39. [Google Scholar] [CrossRef] [PubMed]
- Lefer, A.M.; Verrier, R.L.; Carson, W.W. Cardiac performance in experimental adrenal insufficiency in cats. Circ. Res. 1968, 22, 817–827. [Google Scholar] [CrossRef] [PubMed]
- Lefer, A.M. Influence of corticosteroids on mechanical performance of isolated rat papillary muscles. Am. J. Physiol. 1968, 214, 518–524. [Google Scholar] [PubMed]
- Cruz-Topete, D.; Myers, P.H.; Foley, J.F.; Willis, M.S.; Cidlowski, J.A. Corticosteroids Are Essential for Maintaining Cardiovascular Function in Male Mice. Endocrinology 2016, 157, 2759–2771. [Google Scholar] [CrossRef] [PubMed]
- Penefsky, Z.J.; Kahn, M. Inotropic effects of dexamethasone in mammalian heart muscle. Eur. J. Pharmacol. 1971, 15, 259–266. [Google Scholar] [CrossRef]
- Hadoke, P.W.; Iqbal, J.; Walker, B.R. Therapeutic manipulation of glucocorticoid metabolism in cardiovascular disease. Br. J. Pharmacol. 2009, 156, 689–712. [Google Scholar] [CrossRef] [PubMed]
- Xu, B.; Strom, J.; Chen, Q.M. Dexamethasone induces transcriptional activation of Bcl-xL gene and inhibits cardiac injury by myocardial ischemia. Eur. J. Pharmacol. 2011, 668, 194–200. [Google Scholar] [CrossRef] [PubMed]
- Sudhir, K.; Jennings, G.L.; Esler, M.D.; Korner, P.I.; Blombery, P.A.; Lambert, G.W.; Scoggins, B.; Whitworth, J.A. Hydrocortisone-induced hypertension in humans: Pressor responsiveness and sympathetic function. Hypertension 1989, 13, 416–421. [Google Scholar] [CrossRef] [PubMed]
- Maca, R.D.; Fry, G.L.; Hoak, J.C. The effects of glucocorticoids on cultured human endothelial cells. Br. J. Haematol. 1978, 38, 501–509. [Google Scholar] [CrossRef] [PubMed]
- Longenecker, J.P.; Kilty, L.A.; Johnson, L.K. Glucocorticoid influence on growth of vascular wall cells in culture. J. Cell. Physiol. 1982, 113, 197–202. [Google Scholar] [CrossRef] [PubMed]
- Longenecker, J.P.; Kilty, L.A.; Johnson, L.K. Glucocorticoid inhibition of vascular smooth muscle cell proliferation: Influence of homologous extracellular matrix and serum mitogens. J. Cell. Biol. 1984, 98, 534–540. [Google Scholar] [CrossRef] [PubMed]
- Berk, B.C.; Vallega, G.; Griendling, K.K.; Gordon, J.B.; Cragoe, E.J., Jr.; Canessa, M.; Alexander, R.W. Effects of glucocorticoids on Na+/H+ exchange and growth in cultured vascular smooth muscle cells. J. Cell. Physiol. 1988, 137, 391–401. [Google Scholar] [CrossRef] [PubMed]
- Zielinska, K.A.; van Moortel, L.; Opdenakker, G.; de Bosscher, K.; van den Steen, P.E. Endothelial Response to Glucocorticoids in Inflammatory Diseases. Front. Immunol. 2016, 7, 592. [Google Scholar] [CrossRef] [PubMed]
- Limbourg, F.P.; Liao, J.K. Nontranscriptional actions of the glucocorticoid receptor. J. Mol. Med. 2003, 81, 168–174. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Zhang, L. Glucocorticoids and vascular reactivity. Curr. Vasc. Pharmacol. 2004, 2, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Baum, M.; Moe, O.W. Glucocorticoid-mediated hypertension: Does the vascular smooth muscle hold all the answers? J. Am. Soc. Nephrol. 2008, 19, 1251–1253. [Google Scholar] [CrossRef] [PubMed]
- Goodwin, J.E.; Zhang, J.; Geller, D.S. A critical role for vascular smooth muscle in acute glucocorticoid-induced hypertension. J. Am. Soc. Nephrol. 2008, 19, 1291–1299. [Google Scholar] [CrossRef] [PubMed]
- Goodwin, J.E.; Geller, D.S. Glucocorticoid-induced hypertension. Pediatr. Nephrol. 2012, 27, 1059–1066. [Google Scholar] [CrossRef] [PubMed]
- Goodwin, J.E.; Feng, Y.; Velazquez, H.; Sessa, W.C. Endothelial glucocorticoid receptor is required for protection against sepsis. Proc. Natl. Acad. Sci. USA 2013, 110, 306–311. [Google Scholar] [CrossRef] [PubMed]
- Logie, J.J.; Ali, S.; Marshall, K.M.; Heck, M.M.; Walker, B.R.; Hadoke, P.W. Glucocorticoid-mediated inhibition of angiogenic changes in human endothelial cells is not caused by reductions in cell proliferation or migration. PLoS ONE 2010, 5, e14476. [Google Scholar] [CrossRef] [PubMed]
- Kadl, A.; Leitinger, N. The role of endothelial cells in the resolution of acute inflammation. Antioxid. Redox Signal. 2005, 7, 1744–1754. [Google Scholar] [CrossRef] [PubMed]
- Tokudome, S.; Sano, M.; Shinmura, K.; Matsuhashi, T.; Morizane, S.; Moriyama, H.; Tamaki, K.; Hayashida, K.; Nakanishi, H.; Yoshikawa, N.; et al. Glucocorticoid protects rodent hearts from ischemia/reperfusion injury by activating lipocalin-type prostaglandin D synthase-derived PGD2 biosynthesis. J. Clin. Investig. 2009, 119, 1477–1488. [Google Scholar] [CrossRef] [PubMed]
- Ren, R.; Oakley, R.H.; Cruz-Topete, D.; Cidlowski, J.A. Dual role for glucocorticoids in cardiomyocyte hypertrophy and apoptosis. Endocrinology 2012, 153, 5346–5360. [Google Scholar] [CrossRef] [PubMed]
- Lavallee, G.; Andelfinger, G.; Nadeau, M.; Lefebvre, C.; Nemer, G.; Horb, M.E.; Nemer, M. The Kruppel-like transcription factor KLF13 is a novel regulator of heart development. EMBO J. 2006, 25, 5201–5213. [Google Scholar] [CrossRef] [PubMed]
- Cruz-Topete, D.; He, B.; Xu, X.; Cidlowski, J.A. Kruppel-like Factor 13 Is a Major Mediator of Glucocorticoid Receptor Signaling in Cardiomyocytes and Protects These Cells from DNA Damage and Death. J. Biol. Chem. 2016, 291, 19374–19386. [Google Scholar] [CrossRef] [PubMed]
- Sainte-Marie, Y.; Nguyen Dinh Cat, A.; Perrier, R.; Mangin, L.; Soukaseum, C.; Peuchmaur, M.; Tronche, F.; Farman, N.; Escoubet, B.; Benitah, J.P.; et al. Conditional glucocorticoid receptor expression in the heart induces atrio-ventricular block. FASEB J. 2007, 21, 3133–3141. [Google Scholar] [CrossRef] [PubMed]
- Young, M.J.; Rickard, A.J. Mineralocorticoid receptors in the heart: Lessons from cell-selective transgenic animals. J. Endocrinol. 2015, 224, R1–R13. [Google Scholar] [CrossRef] [PubMed]
- Muller, O.; Pradervand, S.; Berger, S.; Centeno, G.; Milet, A.; Nicod, P.; Pedrazzini, T.; Tronche, F.; Schutz, G.; Chien, K.; et al. Identification of corticosteroid-regulated genes in cardiomyocytes by serial analysis of gene expression. Genomics 2007, 89, 370–377. [Google Scholar] [CrossRef] [PubMed]
- Ouvrard-Pascaud, A.; Sainte-Marie, Y.; Benitah, J.P.; Perrier, R.; Soukaseum, C.; Nguyen Dinh Cat, A.; Royer, A.; le Quang, K.; Charpentier, F.; Demolombe, S.; et al. Conditional mineralocorticoid receptor expression in the heart leads to life-threatening arrhythmias. Circulation 2005, 111, 3025–3033. [Google Scholar] [CrossRef] [PubMed]
- Gomez, A.M.; Rueda, A.; Sainte-Marie, Y.; Pereira, L.; Zissimopoulos, S.; Zhu, X.; Schaub, R.; Perrier, E.; Perrier, R.; Latouche, C.; et al. Mineralocorticoid modulation of cardiac ryanodine receptor activity is associated with downregulation of FK506-binding proteins. Circulation 2009, 119, 2179–2187. [Google Scholar] [CrossRef] [PubMed]
- Messaoudi, S.; Gravez, B.; Tarjus, A.; Pelloux, V.; Ouvrard-Pascaud, A.; Delcayre, C.; Samuel, J.; Launay, J.M.; Sierra-Ramos, C.; Alvarez de la Rosa, D.; et al. Aldosterone-specific activation of cardiomyocyte mineralocorticoid receptor in vivo. Hypertension 2013, 61, 361–367. [Google Scholar] [CrossRef] [PubMed]
- Oakley, R.H.; Ren, R.; Cruz-Topete, D.; Bird, G.S.; Myers, P.H.; Boyle, M.C.; Schneider, M.D.; Willis, M.S.; Cidlowski, J.A. Essential role of stress hormone signaling in cardiomyocytes for the prevention of heart disease. Proc. Natl. Acad. Sci. USA 2013, 110, 17035–17040. [Google Scholar] [CrossRef] [PubMed]
- Fraccarollo, D.; Berger, S.; Galuppo, P.; Kneitz, S.; Hein, L.; Schutz, G.; Frantz, S.; Ertl, G.; Bauersachs, J. Deletion of cardiomyocyte mineralocorticoid receptor ameliorates adverse remodeling after myocardial infarction. Circulation 2011, 123, 400–408. [Google Scholar] [CrossRef] [PubMed]
- Fraccarollo, D.; Bauersachs, J. Cardiomyocyte mineralocorticoid receptor function post myocardial infarction. Trends Cardiovasc. Med. 2011, 21, 42–47. [Google Scholar] [CrossRef] [PubMed]
- Lother, A.; Berger, S.; Gilsbach, R.; Rosner, S.; Ecke, A.; Barreto, F.; Bauersachs, J.; Schutz, G.; Hein, L. Ablation of mineralocorticoid receptors in myocytes but not in fibroblasts preserves cardiac function. Hypertension 2011, 57, 746–754. [Google Scholar] [CrossRef] [PubMed]
- Rickard, A.J.; Morgan, J.; Bienvenu, L.A.; Fletcher, E.K.; Cranston, G.A.; Shen, J.Z.; Reichelt, M.E.; Delbridge, L.M.; Young, M.J. Cardiomyocyte mineralocorticoid receptors are essential for deoxycorticosterone/salt-mediated inflammation and cardiac fibrosis. Hypertension 2012, 60, 1443–1450. [Google Scholar] [CrossRef] [PubMed]



© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Burford, N.G.; Webster, N.A.; Cruz-Topete, D. Hypothalamic-Pituitary-Adrenal Axis Modulation of Glucocorticoids in the Cardiovascular System. Int. J. Mol. Sci. 2017, 18, 2150. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms18102150
Burford NG, Webster NA, Cruz-Topete D. Hypothalamic-Pituitary-Adrenal Axis Modulation of Glucocorticoids in the Cardiovascular System. International Journal of Molecular Sciences. 2017; 18(10):2150. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms18102150
Chicago/Turabian StyleBurford, Natalie G., Natalia A. Webster, and Diana Cruz-Topete. 2017. "Hypothalamic-Pituitary-Adrenal Axis Modulation of Glucocorticoids in the Cardiovascular System" International Journal of Molecular Sciences 18, no. 10: 2150. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms18102150