Isoproterenol Increases RANKL Expression in a ATF4/NFATc1-Dependent Manner in Mouse Osteoblastic Cells


Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
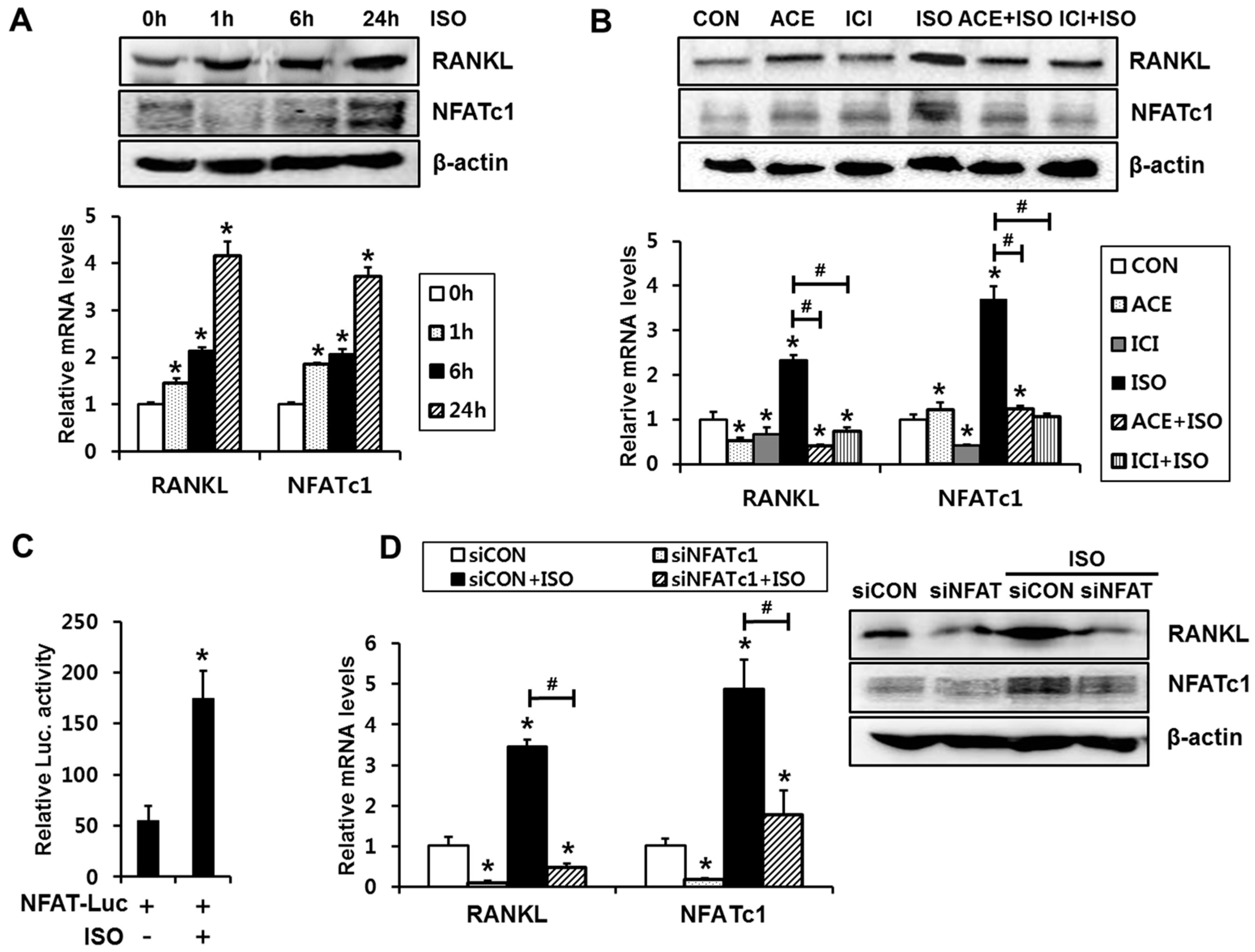
2.1. Isoproterenol Induces Expression Levels and Transcriptional Activity of NFATc1, Which Is Necessary for Isoproterenol-Induced RANKL Expression
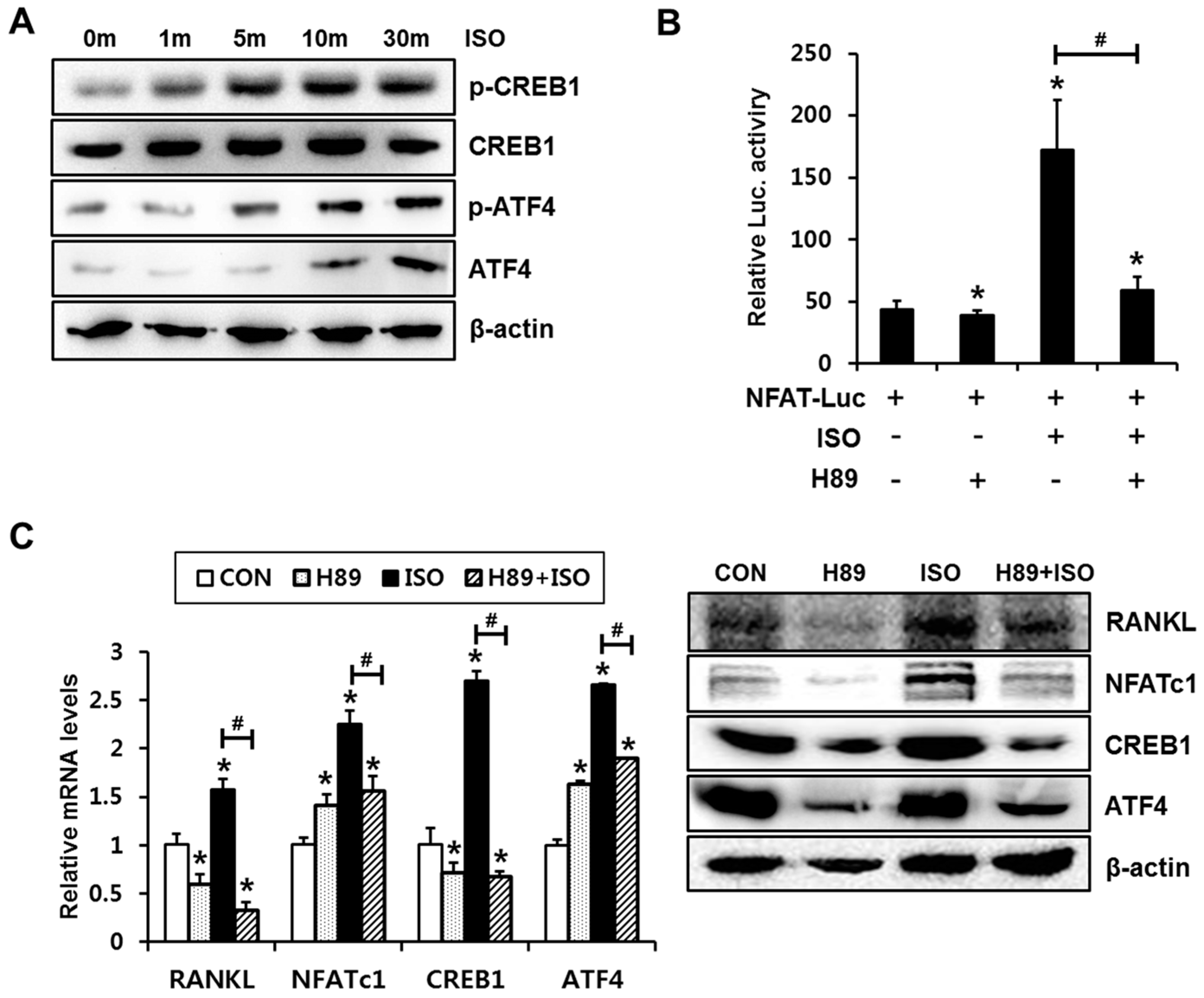
2.2. PKA Signaling Is Involved in Isoproterenol-Induced NFATc1 Activation and Expression
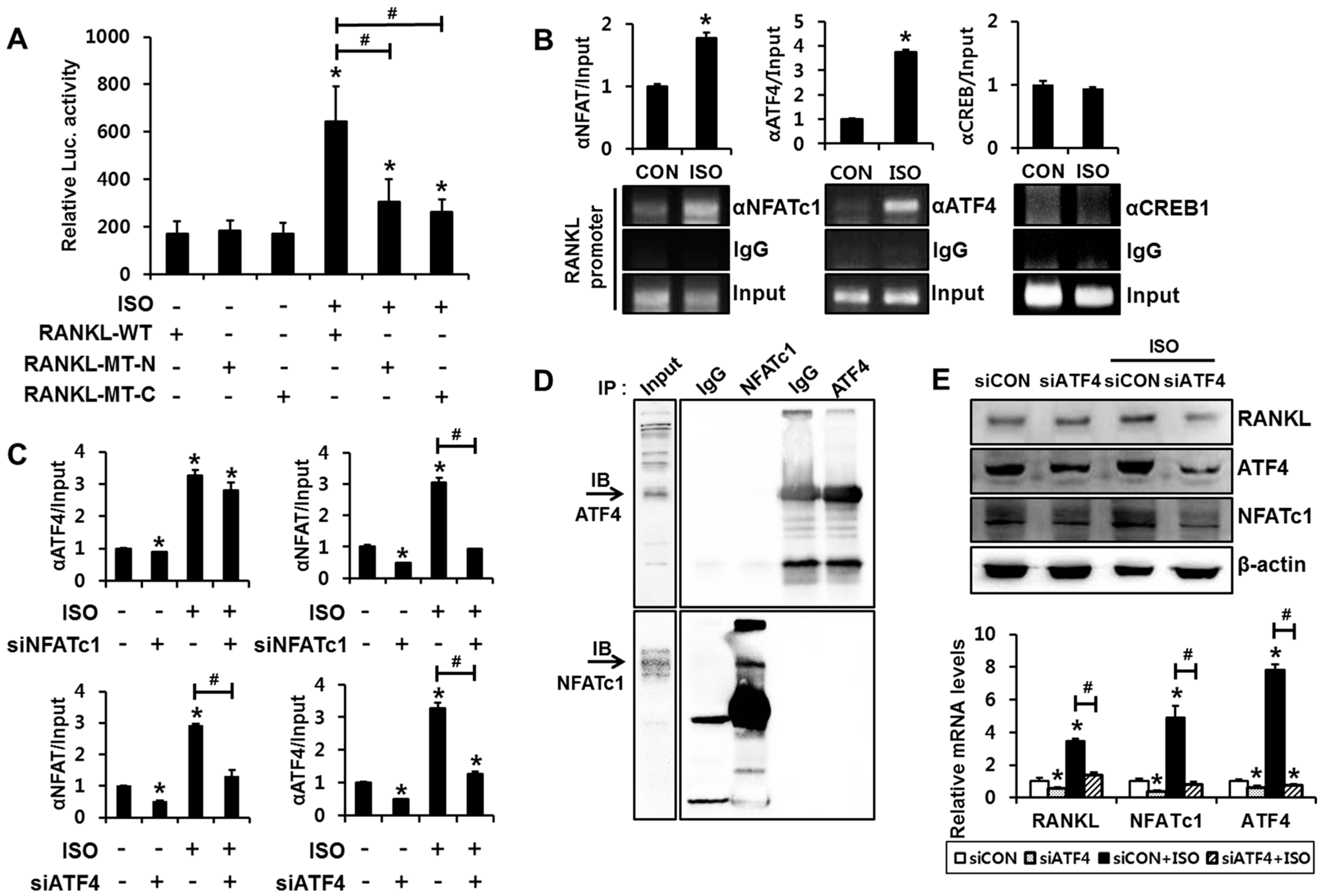
2.3. Binding of Both NFATc1 and ATF4 to the RANKL Promoter Is Necessary for Transactivation of RANKL Gene
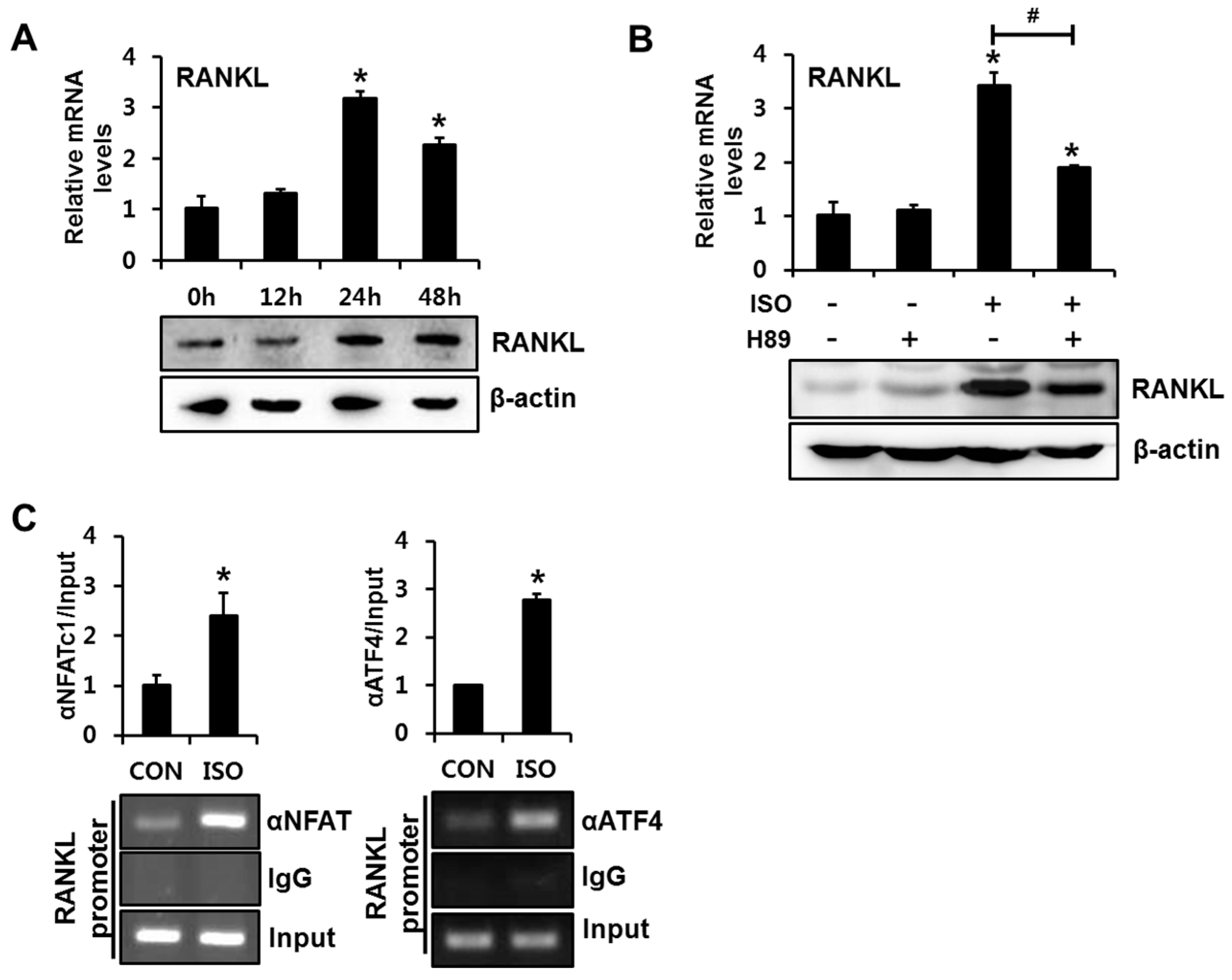
2.4. Isoproterenol Induced the Binding of NFATc1 and ATF4 to the RANKL Promoter in Primary Cultured Mouse Calvarial Cells
3. Discussion
4. Materials and Methods
4.1. Reagents and Antibodies
4.2. Cell Culture
4.3. Plasmid Construction
4.4. Reverse Transcription-Polymerase Chain Reaction
4.5. Western Blot Analysis and Co-Immunoprecipitation
4.6. Chromatin Immunoprecipitation Assay
4.7. Luciferase Reporter Assays
4.8. Statistical Analysis
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Bjurholm, A. Neuroendocrine peptides in bone. Int. Orthop. 1991, 15, 325–329. [Google Scholar] [CrossRef] [PubMed]
- Takeda, S.; Elefteriou, F.; Levasseur, R.; Liu, X.; Zhao, L.; Parker, K.L.; Armstrong, D.; Ducy, P.; Karsenty, G. Leptin regulates bone formation via the sympathetic nervous system. Cell 2002, 111, 305–317. [Google Scholar] [CrossRef]
- Elefteriou, F.; Ahn, J.D.; Takeda, S.; Starbuck, M.; Yang, X.; Liu, X.; Kondo, H.; Richards, W.G.; Bannon, T.W.; Noda, M.; et al. Leptin regulation of bone resorption by the sympathetic nervous system and CART. Nature 2005, 434, 514–520. [Google Scholar] [CrossRef] [PubMed]
- Bouxsein, M.L.; Devlin, M.J.; Glatt, V.; Dhillon, H.; Pierroz, D.D.; Ferrari, S.L. Mice lacking β-adrenergic receptors have increased bone mass but are not protected from deleterious skeletal effects of ovariectomy. Endocrinology 2009, 150, 144–152. [Google Scholar] [CrossRef] [PubMed]
- Baek, K.; Bloomfield, S.A. β-adrenergic blockade and leptin replacement effectively mitigate disuse bone loss. J. Bone Miner. Res. 2009, 24, 792–799. [Google Scholar] [CrossRef] [PubMed]
- Baek, K.; Hwang, H.R.; Park, H.J.; Kwon, A.; Qadir, A.S.; Baek, J.H. Propranolol, a β-adrenergic antagonist, attenuates the decrease in trabecular bone mass in high calorie diet fed growing mice. BMB Rep. 2014, 47, 506–511. [Google Scholar] [CrossRef] [PubMed]
- Bonnet, N.; Beaupied, H.; Vico, L.; Dolleans, E.; Laroche, N.; Courteix, D.; Benhamou, C.L. Combined effects of exercise and propranolol on bone tissue in ovariectomized rats. J. Bone Miner. Res. 2007, 22, 578–588. [Google Scholar] [CrossRef] [PubMed]
- Udagawa, N.; Takahashi, N.; Jimi, E.; Matsuzaki, K.; Tsurukai, T.; Itoh, K.; Nakagawa, N.; Yasuda, H.; Goto, M.; Tsuda, E.; et al. Osteoblasts/stromal cells stimulate osteoclast activation through expression of osteoclast differentiation factor/RANKL but not macrophage colony-stimulating factor: Receptor activator of NF-κB ligand. Bone 1999, 25, 517–523. [Google Scholar] [CrossRef]
- Wada, T.; Nakashima, T.; Hiroshi, N.; Penninger, J.M. RANKL-RANK signaling in osteoclastogenesis and bone disease. Trends Mol. Med. 2006, 12, 17–25. [Google Scholar] [CrossRef] [PubMed]
- Sitara, D.; Aliprantis, A.O. Transcriptional regulation of bone and joint remodeling by NFAT. Immunol. Rev. 2010, 233, 286–300. [Google Scholar] [CrossRef] [PubMed]
- Fu, Q.; Manolagas, S.C.; O‘Brien, C.A. Parathyroid hormone controls receptor activator of NF-κB ligand gene expression via a distant transcriptional enhancer. Mol. Cell. Biol. 2006, 26, 6453–6468. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Yamazaki, M.; Shevde, N.K.; Pike, J.W. Transcriptional control of receptor activator of nuclear factor-κB ligand by the protein kinase A activator forskolin and the transmembrane glycoprotein 130-activating cytokine, oncostatin M, is exerted through multiple distal enhancers. Mol. Endocrinol. 2007, 21, 197–214. [Google Scholar] [CrossRef] [PubMed]
- Park, H.J.; Baek, K.; Baek, J.H.; Kim, H.R. The cooperation of CREB and NFAT is required for PTHrP-induced RANKL expression in mouse osteoblastic cells. J. Cell. Physiol. 2015, 230, 667–679. [Google Scholar] [CrossRef] [PubMed]
- Jiao, K.; Niu, L.N.; Li, Q.H.; Ren, G.T.; Zhao, C.M.; Liu, Y.D.; Tay, F.R.; Wang, M.Q. β2-Adrenergic signal transduction plays a detrimental role in subchondral bone loss of temporomandibular joint in osteoarthritis. Sci. Rep. 2015, 5, 12593. [Google Scholar] [CrossRef] [PubMed]
- Chuvpilo, S.; Jankevics, E.; Tyrsin, D.; Akimzhanov, A.; Moroz, D.; Jha, M.K.; Schulze-Luehrmann, J.; Santner-Nanan, B.; Feoktistova, E.; Konig, T.; et al. Autoregulation of NFATc1/A expression facilitates effector T cells to escape from rapid apoptosis. Immunity 2002, 16, 881–895. [Google Scholar] [CrossRef]
- Gilman, A.G. G proteins: Transducers of receptor-generated signals. Annu. Rev. Biochem. 1987, 56, 615–649. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.L.; Bae, O.Y.; Baek, K.H.; Kwon, A.; Hwang, H.R.; Qadir, A.S.; Park, H.J.; Woo, K.M.; Ryoo, H.M.; Baek, J.H. High extracellular calcium-induced NFATc3 regulates the expression of receptor activator of NF-κB ligand in osteoblasts. Bone 2011, 49, 242–249. [Google Scholar] [CrossRef] [PubMed]
- Park, H.J.; Baek, K.; Baek, J.H.; Kim, H.R. TNF α Increases RANKL Expression via PGE(2)-Induced Activation of NFATc1. Int. J. Mol. Sci. 2017, 18, 495. [Google Scholar] [CrossRef] [PubMed]
- Yuvaraj, S.; Griffin, A.C.; Sundaram, K.; Kirkwood, K.L.; Norris, J.S.; Reddy, S.V. A novel function of CXCL13 to stimulate RANK ligand expression in oral squamous cell carcinoma cells. Mol. Cancer Res. 2009, 7, 1399–1407. [Google Scholar] [CrossRef] [PubMed]
- Cao, H.; Yu, S.; Yao, Z.; Galson, D.L.; Jiang, Y.; Zhang, X.; Fan, J.; Lu, B.; Guan, Y.; Luo, M.; et al. Activating transcription factor 4 regulates osteoclast differentiation in mice. J. Clin. Investig. 2010, 120, 2755–2766. [Google Scholar] [CrossRef] [PubMed]
- Kondo, H.; Nifuji, A.; Takeda, S.; Ezura, Y.; Rittling, S.R.; Denhardt, D.T.; Nakashima, K.; Karsenty, G.; Noda, M. Unloading induces osteoblastic cell suppression and osteoclastic cell activation to lead to bone loss via sympathetic nervous system. J. Biol. Chem. 2005, 280, 30192–30200. [Google Scholar] [CrossRef] [PubMed]
- Bonnet, N.; Benhamou, C.L.; Malaval, L.; Goncalves, C.; Vico, L.; Eder, V.; Pichon, C.; Courteix, D. Low dose β-blocker prevents ovariectomy-induced bone loss in rats without affecting heart functions. J. Cell. Physiol. 2008, 217, 819–827. [Google Scholar] [CrossRef] [PubMed]
- Bonnet, N.; Laroche, N.; Vico, L.; Dolleans, E.; Benhamou, C.L.; Courteix, D. Dose effects of propranolol on cancellous and cortical bone in ovariectomized adult rats. J. Pharmacol. Exp. Ther. 2006, 318, 1118–1127. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.H.; Kim, J.M.; Kim, S.N.; Kim, G.S.; Baek, J.H. p44/42 MAPK activation is necessary for receptor activator of nuclear factor-κB ligand induction by high extracellular calcium. Biochem. Biophys. Res. Commun. 2003, 304, 729–735. [Google Scholar] [CrossRef]
- Takayanagi, H.; Kim, S.; Koga, T.; Nishina, H.; Isshiki, M.; Yoshida, H.; Saiura, A.; Isobe, M.; Yokochi, T.; Inoue, J.; et al. Induction and activation of the transcription factor NFATc1 (NFAT2) integrate RANKL signaling in terminal differentiation of osteoclasts. Dev. Cell 2002, 3, 889–901. [Google Scholar] [CrossRef]
- Mori, K.; Kitazawa, R.; Kondo, T.; Maeda, S.; Yamaguchi, A.; Kitazawa, S. Modulation of mouse RANKL gene expression by Runx2 and PKA pathway. J. Cell. Biochem. 2006, 98, 1629–1644. [Google Scholar] [CrossRef] [PubMed]




© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Baek, K.; Park, H.-J.; Baek, J.-H.; Kim, H.-R. Isoproterenol Increases RANKL Expression in a ATF4/NFATc1-Dependent Manner in Mouse Osteoblastic Cells. Int. J. Mol. Sci. 2017, 18, 2204. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms18102204
Baek K, Park H-J, Baek J-H, Kim H-R. Isoproterenol Increases RANKL Expression in a ATF4/NFATc1-Dependent Manner in Mouse Osteoblastic Cells. International Journal of Molecular Sciences. 2017; 18(10):2204. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms18102204
Chicago/Turabian StyleBaek, Kyunghwa, Hyun-Jung Park, Jeong-Hwa Baek, and Hyung-Ryong Kim. 2017. "Isoproterenol Increases RANKL Expression in a ATF4/NFATc1-Dependent Manner in Mouse Osteoblastic Cells" International Journal of Molecular Sciences 18, no. 10: 2204. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms18102204