The Interactive Roles of Lipopolysaccharides and dsRNA/Viruses on Respiratory Epithelial Cells and Dendritic Cells in Allergic Respiratory Disorders: The Hygiene Hypothesis


Abstract
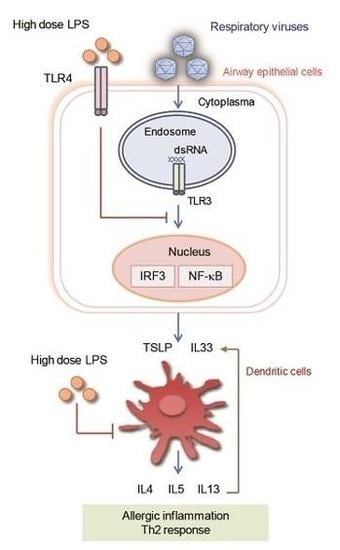
:
1. Introduction
2. Four Major Players in the Proposed Simplified Model of Hygiene Hypothesis: Epithelial Cells (ECs), Dendritic Cells (DCs), dsRNA and LPS
2.1. Epithelial Cells Play Key Roles in Bridging the Innate and Adaptive Immune System
2.2. DCs Interact Closely with ECs to Orchestrate the Immune Responses
2.3. Most Bridging Effects Start from Activation via TLRs and Other Receptors of Epithelial Cells and DCs
2.4. dsRNA or Viruses Can Activate TLR3 of Respiratory Epithelial Cells and Stimulate the Production of Various Proallergic Cytokines
2.5. LPS Activate TLR4 of Respiratory Epithelial Cell, Using Bidirectional Capacity to Modulate Allergic Disorders through Multiple Pathways
2.5.1. Protective Role of LPS against Allergic Disorders
Focusing on the Role of LPS
Focusing on the Role of LPS-Related Actions in Farming Households
2.5.2. The Proallergic Role of LPS
2.5.3. The Pro-Inflammatory Non-Allergic Role of LPS
2.6. dsRNA or Many Viruses Activate TLR3 of Dendritic Cells, Thus, Induce DCs with Th1-Promoting Capacity with Some Exceptions
2.6.1. dsRNA Activates the TLR3 of DCs, and Cause Them to Become DCs with Th1-Promoting Capacity
2.6.2. Respiratory Syncytial Virus (RSV) Is Probably an Exception, Which Likely Skew DC towards DC with Th2-Promoting Capacity
2.6.3. Is Rhinovirus (RV) Another Exception?
Related Mechanisms of Rhinovirus Infection
Question: Under Normal Circumstances, Will Rhinoviruses Easily Approach DCs, as Shown in the In Vitro Study Above?
In Transgenic Mouse Model
Conclusion on the Protective or Pro-Inflammatory Role of RV
2.7. LPS Has the Potential to Activate Immature Dendritic Cells (DC) into Mature DCs with Th1- or Th2-Promoting Capacity
3. The Protective Role and Mechanism of LPS: Pre-Exposure to LPS Protects the Respiratory Epithelial Cells and Downregulates the Effect of dsRNA or Allergen in Producing Proallergic Cytokines, Indicating a Delicate Cross-Regulation Mechanism Exists between dsRNA (TLR3 Pathway) or Allergen and LPS (TLR4 Pathway), at Least at Epithelial Level
3.1. Pretreatment with Lps Attenuates Induction of Proallergic Cytokines, TSLP and IL33 in Respiratory Epithelial Cells Stimulated with polyI:C and Human Parechovirus.
3.2. Pretreatment with LPS Protects against Allergy through A20 Induction in Lung Epithelial Cells
3.3. Pretreatment with E. coli in Mice Models Protects against Allergy via Two Pathways
3.4. Pretreatment with Salmonella enterica Serovar Typhimurium Protects against Allergic Airway Inflammation in Mice
3.5. LPS Suppresses Asthma-Like Responses via Nitric Oxide Synthase (NOS2) Activity
4. The Pro-Inflammatory Role of LPS: Why Does LPS Induce Inflammation, Instead of Protecting against Inflammation on Many Occasions?
4.1. First, the Timing of Delivering LPS
4.2. Second, the Dose of LPS Delivered
4.3. Third, the Monocytes/Macrophage or Dendritic Cells Which Are Also Activated by LPS
4.4. Fourth, the Synergistic Effect between LPS and Environmental Cofactors
4.5. Fifth, the Resource of LPS Delivered
4.6. Sixth, the Presence of Serum or Whole Blood on Lung Alveolar Cells and Bronchial Epithelial Cells
4.7. Seventh, the Type of Epithelial Cells Tested
5. Proposed Mechanism Supporting Hygiene Hypothesis
5.1. First, Why Early Exposure to Environmental LPS, Such as Farm Dust, Would Protect against the Development of Allergic Disorder in Later Life? Two Mechanisms May Possibly Explain the Observed Phenomenon
5.1.1. Pre-Exposure to LPS Attenuates the Signaling Pathway Necessary for Allergic Cytokines Production, but Spares the dsRNA/DCs Route
5.1.2. Pre-Exposure to LPS Suppresses Responsiveness of Airway Epithelial Cells via Increased Synthesis of A20
5.2. Second, Why Is More Common Cold in Early Life Associated with Less Allergy in Later Life?
5.3. Summary of the Four Major Players in Hygiene Hypothesis
6. Why Does the Hygiene Hypothesis Work Only before One Critical Time Point in Early life? After Allergy Is Established, Why Do the dsRNA and LPS No More Play Protective Roles?
6.1. Why, in Established Atopic Patients, Does Exposure to More Environmental LPS No Longer Protect Them against Allergy?
6.2. Why in Established Atopic Patients, Does Exposure to Viral Infections No Longer Protect Them against Allergies, but, Instead, Worsen the Allergic Disorders
7. What Factors Initiate the Disruption of Immune Balance towards the Allergic Predisposition?
7.1. Genetic Deficiency
7.2. Microbial Dysbiosis
7.2.1. Airway Microbial Dysbiosis
7.2.2. Gut Microbial Dysbiosis
7.3. Environmental Hazard Factors
8. Conclusions
8.1. LPS Has a High Potential for Prevention Modality; However, Application of LPS as Treatment Modality Should Be Considered Cautiously
8.2. Limitations of the Proposed Model
8.3. The Content of “Hygiene Hypothesis” Could Be Modified
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Strachan, D.P. Hay fever, hygiene, and household size. BMJ 1989, 299, 1259–1260. [Google Scholar] [CrossRef] [PubMed]
- Strachan, D.P. Family size, infection and atopy: The first decade of the “hygiene hypothesis”. Thorax 2000, 55 (Suppl. 1), S2–S10. [Google Scholar] [CrossRef] [PubMed]
- Braun-Fahrlander, C.; Riedler, J.; Herz, U.; Eder, W.; Waser, M.; Grize, L.; Maisch, S.; Carr, D.; Gerlach, F.; Bufe, A.; et al. Environmental exposure to endotoxin and its relation to asthma in school-age children. N. Engl. J. Med. 2002, 347, 869–877. [Google Scholar] [CrossRef] [PubMed]
- Braun-Fahrlander, C. Environmental exposure to endotoxin and other microbial products and the decreased risk of childhood atopy: Evaluating developments since April 2002. Curr. Opin. Allergy Clin. Immunol. 2003, 3, 325–329. [Google Scholar] [CrossRef] [PubMed]
- Riedler, J.; Braun-Fahrlander, C.; Eder, W.; Schreuer, M.; Waser, M.; Maisch, S.; Carr, D.; Schierl, R.; Nowak, D.; von Mutius, E. Exposure to farming in early life and development of asthma and allergy: A cross-sectional survey. Lancet 2001, 358, 1129–1133. [Google Scholar] [CrossRef]
- Lynch, S.V.; Wood, R.A.; Boushey, H.; Bacharier, L.B.; Bloomberg, G.R.; Kattan, M.; O’Connor, G.T.; Sandel, M.T.; Calatroni, A.; Matsui, E.; et al. Effects of early-life exposure to allergens and bacteria on recurrent wheeze and atopy in urban children. J. Allergy Clin. Immunol. 2014, 134, 593–601. [Google Scholar] [CrossRef] [PubMed]
- Illi, S.; von Mutius, E.; Lau, S.; Bergmann, R.; Niggemann, B.; Sommerfeld, C.; Wahn, U. Early childhood infectious diseases and the development of asthma up to school age: A birth cohort study. BMJ 2001, 322, 390–395. [Google Scholar] [CrossRef] [PubMed]
- Bach, J.F. Six questions about the hygiene hypothesis. Cell. Immunol. 2005, 233, 158–161. [Google Scholar] [CrossRef] [PubMed]
- Lin, T.-H.; Cheng, C.-C.; Su, H.-H.; Huang, N.-C.; Chen, J.-J.; Kang, H.-Y.; Chang, T.-H. Lipopolysaccharide Attenuates Induction of Proallergic Cytokines, Thymic Stromal Lymphopoietin, and Interleukin 33 in Respiratory Epithelial Cells Stimulated with PolyI:C and Human Parechovirus. Front. Immunol. 2016, 7, 440. [Google Scholar] [CrossRef] [PubMed]
- Schuijs, M.J.; Willart, M.A.; Vergote, K.; Gras, D.; Deswarte, K.; Ege, M.J.; Madeira, F.B.; Beyaert, R.; van Loo, G.; Bracher, F.; et al. Farm dust and endotoxin protect against allergy through A20 induction in lung epithelial cells. Science 2015, 349, 1106–1110. [Google Scholar] [CrossRef] [PubMed]
- Ganesan, S.; Comstock, A.T.; Sajjan, U.S. Barrier function of airway tract epithelium. Tissue Barriers 2013, 1, e24997. [Google Scholar] [CrossRef] [PubMed]
- Schleimer, R.P.; Kato, A.; Kern, R.; Kuperman, D.; Avila, P.C. Epithelium: At the interface of innate and adaptive immune responses. J. Allergy Clin. Immunol. 2007, 120, 1279–1284. [Google Scholar] [CrossRef] [PubMed]
- Whitsett, J.A.; Alenghat, T. Respiratory epithelial cells orchestrate pulmonary innate immunity. Nat. Immunol. 2015, 16, 27–35. [Google Scholar] [CrossRef] [PubMed]
- Lambrecht, B.N.; Hammad, H. The airway epithelium in asthma. Nat. Med. 2012, 18, 684–692. [Google Scholar] [CrossRef] [PubMed]
- Vroling, A.B.; Fokkens, W.J.; van Drunen, C.M. How epithelial cells detect danger: Aiding the immune response. Allergy 2008, 63, 1110–1123. [Google Scholar] [CrossRef] [PubMed]
- Deckers, J.; De Bosscher, K.; Lambrecht, B.N.; Hammad, H. Interplay between barrier epithelial cells and dendritic cells in allergic sensitization through the lung and the skin. Immunol. Rev. 2017, 278, 131–144. [Google Scholar] [CrossRef] [PubMed]
- Lambrecht, B.N.; Hammad, H. The role of dendritic and epithelial cells as master regulators of allergic airway inflammation. Lancet 2010, 376, 835–843. [Google Scholar] [CrossRef]
- Mellman, I. Dendritic cells: Master regulators of the immune response. Cancer Immunol. Res. 2013, 1, 145–149. [Google Scholar] [CrossRef] [PubMed]
- Mildner, A.; Jung, S. Development and Function of Dendritic Cell Subsets. Immunity 2014, 40, 642–656. [Google Scholar] [CrossRef] [PubMed]
- Swiecki, M.; Colonna, M. The multifaceted biology of plasmacytoid dendritic cells. Nat. Rev. Immunol. 2015, 15, 471–485. [Google Scholar] [CrossRef] [PubMed]
- Hammad, H.; Lambrecht, B.N. Dendritic cells and epithelial cells: Linking innate and adaptive immunity in asthma. Nat. Rev. Immunol. 2008, 8, 193–204. [Google Scholar] [CrossRef] [PubMed]
- Guilliams, M.; Ginhoux, F.; Jakubzick, C.; Naik, S.H.; Onai, N.; Schraml, B.U.; Segura, E.; Tussiwand, R.; Yona, S. Dendritic cells, monocytes and macrophages: A unified nomenclature based on ontogeny. Nat. Rev. Immunol. 2014, 14, 571–578. [Google Scholar] [CrossRef] [PubMed]
- Guilliams, M.; van de Laar, L. A Hitchhiker’s Guide to Myeloid Cell Subsets: Practical Implementation of a Novel Mononuclear Phagocyte Classification System. Front. Immunol. 2015, 6, 406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Upham, J.W.; Zhang, G.; Rate, A.; Yerkovich, S.T.; Kusel, M.; Sly, P.D.; Holt, P.G. Plasmacytoid dendritic cells during infancy are inversely associated with childhood respiratory tract infections and wheezing. J. Allergy Clin. Immunol. 2009, 124, 707–713. [Google Scholar] [CrossRef] [PubMed]
- Worbs, T.; Hammerschmidt, S.I.; Forster, R. Dendritic cell migration in health and disease. Nat. Rev. Immunol. 2017, 17, 30–48. [Google Scholar] [CrossRef] [PubMed]
- Kawai, T.; Akira, S. Toll-like receptors and their crosstalk with other innate receptors in infection and immunity. Immunity 2011, 34, 637–650. [Google Scholar] [CrossRef] [PubMed]
- Kawai, T.; Akira, S. The role of pattern-recognition receptors in innate immunity: Update on Toll-like receptors. Nat. Immunol. 2010, 11, 373–384. [Google Scholar] [CrossRef] [PubMed]
- Nhu, Q.M.; Shirey, K.; Teijaro, J.R.; Farber, D.L.; Netzel-Arnett, S.; Antalis, T.M.; Fasano, A.; Vogel, S.N. Novel signaling interactions between proteinase-activated receptor 2 and Toll-like receptors in vitro and in vivo. Mucosal Immunol. 2010, 3, 29–39. [Google Scholar] [CrossRef] [PubMed]
- Gieseler, F.; Ungefroren, H.; Settmacher, U.; Hollenberg, M.D.; Kaufmann, R. Proteinase-activated receptors (PARs)—Focus on receptor-receptor-interactions and their physiological and pathophysiological impact. Cell Commun. Signal. CCS 2013, 11, 86. [Google Scholar] [CrossRef] [PubMed]
- Bogiatzi, S.I.; Fernandez, I.; Bichet, J.C.; Marloie-Provost, M.A.; Volpe, E.; Sastre, X.; Soumelis, V. Cutting Edge: Proinflammatory and Th2 cytokines synergize to induce thymic stromal lymphopoietin production by human skin keratinocytes. J. Immunol. 2007, 178, 3373–3377. [Google Scholar] [CrossRef] [PubMed]
- Kato, A.; Favoreto, S., Jr.; Avila, P.C.; Schleimer, R.P. TLR3- and Th2 cytokine-dependent production of thymic stromal lymphopoietin in human airway epithelial cells. J. Immunol. 2007, 179, 1080–1087. [Google Scholar] [CrossRef] [PubMed]
- Kinoshita, H.; Takai, T.; Le, T.A.; Kamijo, S.; Wang, X.L.; Ushio, H.; Hara, M.; Kawasaki, J.; Vu, A.T.; Ogawa, T.; et al. Cytokine milieu modulates release of thymic stromal lymphopoietin from human keratinocytes stimulated with double-stranded RNA. J. Allergy Clin. Immunol. 2009, 123, 179–186. [Google Scholar] [CrossRef] [PubMed]
- Saenz, S.A.; Taylor, B.C.; Artis, D. Welcome to the neighborhood: Epithelial cell-derived cytokines license innate and adaptive immune responses at mucosal sites. Immunol. Rev. 2008, 226, 172–190. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Lu, R.; Zhao, G.; Pflugfelder, S.C.; Li, D.Q. TLR-mediated induction of pro-allergic cytokine IL-33 in ocular mucosal epithelium. Int. J. Biochem. Cell Biol. 2011, 43, 1383–1391. [Google Scholar] [CrossRef] [PubMed]
- Bulek, K.; Swaidani, S.; Aronica, M.; Li, X. Epithelium: The interplay between innate and Th2 immunity. Immunol. Cell Biol. 2010, 88, 257–268. [Google Scholar] [CrossRef] [PubMed]
- Sajjan, U.; Wang, Q.; Zhao, Y.; Gruenert, D.C.; Hershenson, M.B. Rhinovirus disrupts the barrier function of polarized airway epithelial cells. Am. J. Respir. Crit. Care Med. 2008, 178, 1271–1281. [Google Scholar] [CrossRef] [PubMed]
- Rezaee, F.; Meednu, N.; Emo, J.A.; Saatian, B.; Chapman, T.J.; Naydenov, N.G.; De Benedetto, A.; Beck, L.A.; Ivanov, A.I.; Georas, S.N. Polyinosinic:polycytidylic acid induces protein kinase D-dependent disassembly of apical junctions and barrier dysfunction in airway epithelial cells. J. Allergy Clin. Immunol. 2011, 128, 1216–1224. [Google Scholar] [CrossRef] [PubMed]
- Sun, R.; Zhang, Y.; Lv, Q.; Liu, B.; Jin, M.; Zhang, W.; He, Q.; Deng, M.; Liu, X.; Li, G.; et al. Toll-like receptor 3 (TLR3) induces apoptosis via death receptors and mitochondria by up-regulating the transactivating p63 isoform alpha (TAP63alpha). J. Biol. Chem. 2011, 286, 15918–15928. [Google Scholar] [CrossRef] [PubMed]
- Raetz, C.R.; Whitfield, C. Lipopolysaccharide endotoxins. Annu. Rev. Biochem. 2002, 71, 635–700. [Google Scholar] [CrossRef] [PubMed]
- Gereda, J.E.; Leung, D.Y.; Liu, A.H. Levels of environmental endotoxin and prevalence of atopic disease. JAMA 2000, 284, 1652–1653. [Google Scholar] [CrossRef] [PubMed]
- Tulic, M.K.; Wale, J.L.; Holt, P.G.; Sly, P.D. Modification of the inflammatory response to allergen challenge after exposure to bacterial lipopolysaccharide. Am. J. Respir. Cell Mol. Biol. 2000, 22, 604–612. [Google Scholar] [PubMed]
- Verhasselt, V.; Buelens, C.; Willems, F.; De Groote, D.; Haeffner-Cavaillon, N.; Goldman, M. Bacterial lipopolysaccharide stimulates the production of cytokines and the expression of costimulatory molecules by human peripheral blood dendritic cells: Evidence for a soluble CD14-dependent pathway. J. Immunol. 1997, 158, 2919–2925. [Google Scholar] [PubMed]
- Eisenbarth, S.C.; Piggott, D.A.; Huleatt, J.W.; Visintin, I.; Herrick, C.A.; Bottomly, K. Lipopolysaccharide-enhanced, toll-like receptor 4-dependent T helper cell type 2 responses to inhaled antigen. J. Exp. Med. 2002, 196, 1645–1651. [Google Scholar] [CrossRef] [PubMed]
- Lowe, A.P.; Thomas, R.S.; Nials, A.T.; Kidd, E.J.; Broadley, K.J.; Ford, W.R. LPS exacerbates functional and inflammatory responses to ovalbumin and decreases sensitivity to inhaled fluticasone propionate in a guinea pig model of asthma. Br. J. Pharmacol. 2015, 172, 2588–2603. [Google Scholar] [CrossRef] [PubMed]
- Langenkamp, A.; Messi, M.; Lanzavecchia, A.; Sallusto, F. Kinetics of dendritic cell activation: Impact on priming of TH1, TH2 and nonpolarized T cells. Nat. Immunol. 2000, 1, 311–316. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, H.; Yoshimura, K.; Jaffe, H.A.; Crystal, R.G. Interleukin-8 gene expression in human bronchial epithelial cells. J. Biol. Chem. 1991, 266, 19611–19617. [Google Scholar] [PubMed]
- Schulz, C.; Farkas, L.; Wolf, K.; Kratzel, K.; Eissner, G.; Pfeifer, M. Differences in LPS-induced activation of bronchial epithelial cells (BEAS-2B) and type II-like pneumocytes (A-549). Scand. J. Immunol. 2002, 56, 294–302. [Google Scholar] [CrossRef] [PubMed]
- Skerrett, S.J.; Liggitt, H.D.; Hajjar, A.M.; Ernst, R.K.; Miller, S.I.; Wilson, C.B. Respiratory epithelial cells regulate lung inflammation in response to inhaled endotoxin. Am. J. Physiol. Lung Cell. Mol. Physiol. 2004, 287, L143–L152. [Google Scholar] [CrossRef] [PubMed]
- Coornaert, B.; Carpentier, I.; Beyaert, R. A20: Central gatekeeper in inflammation and immunity. J. Biol. Chem. 2009, 284, 8217–8221. [Google Scholar] [CrossRef] [PubMed]
- Ha, T.; Hua, F.; Liu, X.; Ma, J.; McMullen, J.R.; Shioi, T.; Izumo, S.; Kelley, J.; Gao, X.; Browder, W.; et al. Lipopolysaccharide-induced myocardial protection against ischaemia/reperfusion injury is mediated through a PI3K/Akt-dependent mechanism. Cardiovasc. Res. 2008, 78, 546–553. [Google Scholar] [CrossRef] [PubMed]
- Ding, Y.; Li, L. Lipopolysaccharide preconditioning induces protection against lipopolysaccharide-induced neurotoxicity in organotypic midbrain slice culture. Neurosci. Bull. 2008, 24, 209–218. [Google Scholar] [CrossRef] [PubMed]
- Carlsten, C.; Ferguson, A.; Dimich-Ward, H.; Chan, H.; DyBuncio, A.; Rousseau, R.; Becker, A.; Chan-Yeung, M. Association between endotoxin and mite allergen exposure with asthma and specific sensitization at age 7 in high-risk children. Pediatr. Allergy Immunol. 2011, 22, 320–326. [Google Scholar] [CrossRef] [PubMed]
- Ganesh, V.; Baru, A.M.; Hesse, C.; Friedrich, C.; Glage, S.; Gohmert, M.; Janke, C.; Sparwasser, T. Salmonella enterica serovar Typhimurium infection-induced CD11b+ Gr1+ cells ameliorate allergic airway inflammation. Infect. Immun. 2014, 82, 1052–1063. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, D.; Keller, A.C.; Faquim-Mauro, E.L.; de Macedo, M.S.; Cunha, F.Q.; Lefort, J.; Vargaftig, B.B.; Russo, M. Bacterial lipopolysaccharide signaling through Toll-like receptor 4 suppresses asthma-like responses via nitric oxide synthase 2 activity. J. Immunol. 2003, 171, 1001–1008. [Google Scholar] [CrossRef] [PubMed]
- Von Mutius, E.; Vercelli, D. Farm living: Effects on childhood asthma and allergy. Nat. Rev. Immunol. 2010, 10, 861–868. [Google Scholar] [CrossRef] [PubMed]
- Ege, M.J.; Mayer, M.; Normand, A.C.; Genuneit, J.; Cookson, W.O.; Braun-Fahrlander, C.; Heederik, D.; Piarroux, R.; von Mutius, E. Exposure to environmental microorganisms and childhood asthma. N. Engl. J. Med. 2011, 364, 701–709. [Google Scholar] [CrossRef] [PubMed]
- Martikainen, M.V.; Kaario, H.; Karvonen, A.; Schroder, P.C.; Renz, H.; Kaulek, V.; Dalphin, J.C.; von Mutius, E.; Schaub, B.; Pekkanen, J.; et al. Farm exposures are associated with lower percentage of circulating myeloid dendritic cell subtype 2 at age 6. Allergy 2015, 70, 1278–1287. [Google Scholar] [CrossRef] [PubMed]
- Schroder, P.C.; Illi, S.; Casaca, V.I.; Lluis, A.; Bock, A.; Roduit, C.; Depner, M.; Frei, R.; Genuneit, J.; Pfefferle, P.I.; et al. A switch in regulatory T cells through farm exposure during immune maturation in childhood. Allergy 2017, 72, 604–615. [Google Scholar] [CrossRef] [PubMed]
- Lluis, A.; Depner, M.; Gaugler, B.; Saas, P.; Casaca, V.I.; Raedler, D.; Michel, S.; Tost, J.; Liu, J.; Genuneit, J.; et al. Increased regulatory T-cell numbers are associated with farm milk exposure and lower atopic sensitization and asthma in childhood. J. Allergy Clin. Immunol. 2014, 133, 551–559. [Google Scholar] [CrossRef] [PubMed]
- Wickens, K.; Lane, J.M.; Fitzharris, P.; Siebers, R.; Riley, G.; Douwes, J.; Smith, T.; Crane, J. Farm residence and exposures and the risk of allergic diseases in New Zealand children. Allergy 2002, 57, 1171–1179. [Google Scholar] [CrossRef] [PubMed]
- Bisgaard, H.; Hermansen, M.N.; Buchvald, F.; Loland, L.; Halkjaer, L.B.; Bonnelykke, K.; Brasholt, M.; Heltberg, A.; Vissing, N.H.; Thorsen, S.V.; et al. Childhood asthma after bacterial colonization of the airway in neonates. N. Engl. J. Med. 2007, 357, 1487–1495. [Google Scholar] [CrossRef] [PubMed]
- Hammad, H.; Chieppa, M.; Perros, F.; Willart, M.A.; Germain, R.N.; Lambrecht, B.N. House dust mite allergen induces asthma via Toll-like receptor 4 triggering of airway structural cells. Nat. Med. 2009, 15, 410–416. [Google Scholar] [CrossRef] [PubMed]
- Harada, A.; Sekido, N.; Akahoshi, T.; Wada, T.; Mukaida, N.; Matsushima, K. Essential involvement of interleukin-8 (IL-8) in acute inflammation. J. Leukoc. Biol. 1994, 56, 559–564. [Google Scholar] [PubMed]
- Fogli, L.K.; Sundrud, M.S.; Goel, S.; Bajwa, S.; Jensen, K.; Derudder, E.; Sun, A.; Coffre, M.; Uyttenhove, C.; Van Snick, J.; et al. T cell-derived IL-17 mediates epithelial changes in the airway and drives pulmonary neutrophilia. J. Immunol. 2013, 191, 3100–3111. [Google Scholar] [CrossRef] [PubMed]
- Pugin, J.; Schurer-Maly, C.C.; Leturcq, D.; Moriarty, A.; Ulevitch, R.J.; Tobias, P.S. Lipopolysaccharide activation of human endothelial and epithelial cells is mediated by lipopolysaccharide-binding protein and soluble CD14. Proc. Natl. Acad. Sci. USA 1993, 90, 2744–2748. [Google Scholar] [CrossRef] [PubMed]
- Pugin, J.; Ulevitch, R.J.; Tobias, P.S. A critical role for monocytes and CD14 in endotoxin-induced endothelial cell activation. J. Exp. Med. 1993, 178, 2193–2200. [Google Scholar] [CrossRef] [PubMed]
- Rittirsch, D.; Flierl, M.A.; Day, D.E.; Nadeau, B.A.; McGuire, S.R.; Hoesel, L.M.; Ipaktchi, K.; Zetoune, F.S.; Sarma, J.V.; Leng, L.; et al. Acute lung injury induced by lipopolysaccharide is independent of complement activation. J. Immunol. 2008, 180, 7664–7672. [Google Scholar] [CrossRef] [PubMed]
- Rojas, M.; Woods, C.R.; Mora, A.L.; Xu, J.; Brigham, K.L. Endotoxin-induced lung injury in mice: Structural, functional, and biochemical responses. Am. J. Physiol. Lung Cell. Mol. Physiol. 2005, 288, L333–L341. [Google Scholar] [CrossRef] [PubMed]
- Yao, H.; Sun, Y.; Song, S.; Qi, Y.; Tao, X.; Xu, L.; Yin, L.; Han, X.; Xu, Y.; Li, H.; et al. Protective Effects of Dioscin against Lipopolysaccharide-Induced Acute Lung Injury through Inhibition of Oxidative Stress and Inflammation. Front. Pharmacol. 2017, 8, 120. [Google Scholar] [CrossRef] [PubMed]
- Eutamene, H.; Theodorou, V.; Schmidlin, F.; Tondereau, V.; Garcia-Villar, R.; Salvador-Cartier, C.; Chovet, M.; Bertrand, C.; Bueno, L. LPS-induced lung inflammation is linked to increased epithelial permeability: Role of MLCK. Eur. Respir. J. 2005, 25, 789–796. [Google Scholar] [CrossRef] [PubMed]
- Bodey, G.P.; Bolivar, R.; Fainstein, V.; Jadeja, L. Infections Caused by Pseudomonas aeruginosa. Rev. Infect. Dis. 1983, 5, 279–313. [Google Scholar] [CrossRef] [PubMed]
- Da Silva, A.M.T.; Kaulbach, H.C.; Chuidian, F.S.; Lambert, D.R.; Suffredini, A.F.; Danner, R.L. Shock and Multiple-Organ Dysfunction after Self-Administration of Salmonella Endotoxin. N. Engl. J. Med. 1993, 328, 1457–1460. [Google Scholar] [CrossRef] [PubMed]
- De Jong, E.C.; Vieira, P.L.; Kalinski, P.; Schuitemaker, J.H.; Tanaka, Y.; Wierenga, E.A.; Yazdanbakhsh, M.; Kapsenberg, M.L. Microbial compounds selectively induce Th1 cell-promoting or Th2 cell-promoting dendritic cells in vitro with diverse th cell-polarizing signals. J. Immunol. 2002, 168, 1704–1709. [Google Scholar] [CrossRef] [PubMed]
- Schijns, V.E.; Haagmans, B.L.; Wierda, C.M.; Kruithof, B.; Heijnen, I.A.; Alber, G.; Horzinek, M.C. Mice lacking IL-12 develop polarized Th1 cells during viral infection. J. Immunol. 1998, 160, 3958–3964. [Google Scholar] [PubMed]
- Cella, M.; Salio, M.; Sakakibara, Y.; Langen, H.; Julkunen, I.; Lanzavecchia, A. Maturation, activation, and protection of dendritic cells induced by double-stranded RNA. J. Exp. Med. 1999, 189, 821–829. [Google Scholar] [CrossRef] [PubMed]
- Sigurs, N.; Gustafsson, P.M.; Bjarnason, R.; Lundberg, F.; Schmidt, S.; Sigurbergsson, F.; Kjellman, B. Severe respiratory syncytial virus bronchiolitis in infancy and asthma and allergy at age 13. Am. J. Respir. Crit. Care Med. 2005, 171, 137–141. [Google Scholar] [CrossRef] [PubMed]
- Sigurs, N.; Aljassim, F.; Kjellman, B.; Robinson, P.D.; Sigurbergsson, F.; Bjarnason, R.; Gustafsson, P.M. Asthma and allergy patterns over 18 years after severe RSV bronchiolitis in the first year of life. Thorax 2010, 65, 1045–1052. [Google Scholar] [CrossRef] [PubMed]
- Legg, J.P.; Hussain, I.R.; Warner, J.A.; Johnston, S.L.; Warner, J.O. Type 1 and type 2 cytokine imbalance in acute respiratory syncytial virus bronchiolitis. Am. J. Respir. Crit. Care Med. 2003, 168, 633–639. [Google Scholar] [CrossRef] [PubMed]
- Lynch, J.P.; Sikder, M.A.A.; Curren, B.F.; Werder, R.B.; Simpson, J.; Cuív, P.Ó.; Dennis, P.G.; Everard, M.L.; Phipps, S. The Influence of the Microbiome on Early-Life Severe Viral Lower Respiratory Infections and Asthma—Food for Thought? Front. Immunol. 2017, 8, 156. [Google Scholar] [CrossRef] [PubMed]
- Schwarze, J. Lung dendritic cells in respiratory syncytial virus bronchiolitis. Pediatr. Infect. Dis. J. 2008, 27, S89–S91. [Google Scholar] [CrossRef] [PubMed]
- Stein, R.T.; Sherrill, D.; Morgan, W.J.; Holberg, C.J.; Halonen, M.; Taussig, L.M.; Wright, A.L.; Martinez, F.D. Respiratory syncytial virus in early life and risk of wheeze and allergy by age 13 years. Lancet 1999, 354, 541–545. [Google Scholar] [CrossRef]
- Kotaniemi-Syrjänen, A.; Vainionpää, R.; Reijonen, T.M.; Waris, M.; Korhonen, K.; Korppi, M. Rhinovirus-induced wheezing in infancy-the first sign of childhood asthma? J. Allergy Clin. Immunol. 2003, 111, 66–71. [Google Scholar] [CrossRef] [PubMed]
- Kusel, M.M.; de Klerk, N.H.; Kebadze, T.; Vohma, V.; Holt, P.G.; Johnston, S.L.; Sly, P.D. Early-life respiratory viral infections, atopic sensitization, and risk of subsequent development of persistent asthma. J. Allergy Clin. Immunol. 2007, 119, 1105–1110. [Google Scholar] [CrossRef] [PubMed]
- Jackson, D.J.; Gangnon, R.E.; Evans, M.D.; Roberg, K.A.; Anderson, E.L.; Pappas, T.E.; Printz, M.C.; Lee, W.M.; Shult, P.A.; Reisdorf, E.; et al. Wheezing rhinovirus illnesses in early life predict asthma development in high-risk children. Am. J. Respir. Crit. Care Med. 2008, 178, 667–672. [Google Scholar] [CrossRef] [PubMed]
- Arruda, E.; Pitkäranta, A.; Witek, T.J.; Doyle, C.A.; Hayden, F.G. Frequency and natural history of rhinovirus infections in adults during autumn. J. Clin. Microbiol. 1997, 35, 2864–2868. [Google Scholar] [PubMed]
- Kennedy, J.L.; Turner, R.B.; Braciale, T.; Heymann, P.W.; Borish, L. Pathogenesis of Rhinovirus Infection. Curr. Opin. Virol. 2012, 2, 287–293. [Google Scholar] [CrossRef] [PubMed]
- Yoo, J.K.; Kim, T.S.; Hufford, M.M.; Braciale, T.J. Viral infection of the lung: Host response and sequelae. J. Allergy Clin. Immunol. 2013, 132, 1263–1276. [Google Scholar] [CrossRef] [PubMed]
- Kirchberger, S.; Majdic, O.; Steinberger, P.; Bluml, S.; Pfistershammer, K.; Zlabinger, G.; Deszcz, L.; Kuechler, E.; Knapp, W.; Stockl, J. Human rhinoviruses inhibit the accessory function of dendritic cells by inducing sialoadhesin and B7-H1 expression. J. Immunol. 2005, 175, 1145–1152. [Google Scholar] [CrossRef] [PubMed]
- Arruda, E.; Boyle, T.R.; Winther, B.; Pevear, D.C.; Gwaltney, J.M., Jr.; Hayden, F.G. Localization of human rhinovirus replication in the upper respiratory tract by in situ hybridization. J. Infect. Dis. 1995, 171, 1329–1333. [Google Scholar] [CrossRef] [PubMed]
- Mosser, A.G.; Brockman-Schneider, R.; Amineva, S.; Burchell, L.; Sedgwick, J.B.; Busse, W.W.; Gern, J.E. Similar frequency of rhinovirus-infectible cells in upper and lower airway epithelium. J. Infect. Dis. 2002, 185, 734–743. [Google Scholar] [CrossRef] [PubMed]
- Blaas, D.; Fuchs, R. Mechanism of human rhinovirus infections. Mol. Cell. Pediatr. 2016, 3, 21. [Google Scholar] [CrossRef] [PubMed]
- Xatzipsalti, M.; Kyrana, S.; Tsolia, M.; Psarras, S.; Bossios, A.; Laza-Stanca, V.; Johnston, S.L.; Papadopoulos, N.G. Rhinovirus viremia in children with respiratory infections. Am. J. Respir. Crit. Care Med. 2005, 172, 1037–1040. [Google Scholar] [CrossRef] [PubMed]
- Bartlett, N.W.; Walton, R.P.; Edwards, M.R.; Aniscenko, J.; Caramori, G.; Zhu, J.; Glanville, N.; Choy, K.J.; Jourdan, P.; Burnet, J.; et al. Mouse models of rhinovirus-induced disease and exacerbation of allergic airway inflammation. Nat. Med. 2008, 14, 199–204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jacobs, S.E.; Soave, R.; Shore, T.B.; Satlin, M.J.; Schuetz, A.N.; Magro, C.; Jenkins, S.G.; Walsh, T.J. Human rhinovirus infections of the lower respiratory tract in hematopoietic stem cell transplant recipients. Transpl. Infect. Dis. 2013, 15, 474–486. [Google Scholar] [CrossRef] [PubMed]
- De Boer, J.D.; Yang, J.; van den Boogaard, F.E.; Hoogendijk, A.J.; de Beer, R.; van der Zee, J.S.; Roelofs, J.J.; van’t Veer, C.; de Vos, A.F.; van der Poll, T. Mast cell-deficient kit mice develop house dust mite-induced lung inflammation despite impaired eosinophil recruitment. J. Innate Immun. 2014, 6, 219–226. [Google Scholar] [CrossRef] [PubMed]
- Boehlecke, B.; Hazucha, M.; Alexis, N.E.; Jacobs, R.; Reist, P.; Bromberg, P.A.; Peden, D.B. Low-dose airborne endotoxin exposure enhances bronchial responsiveness to inhaled allergen in atopic asthmatics. J. Allergy Clin. Immunol. 2003, 112, 1241–1243. [Google Scholar] [CrossRef] [PubMed]
- Catrysse, L.; Vereecke, L.; Beyaert, R.; van Loo, G. A20 in inflammation and autoimmunity. Trends Immunol. 2014, 35, 22–31. [Google Scholar] [CrossRef] [PubMed]
- Silva-Santos, B.; Serre, K.; Norell, H. γδ T cells in cancer. Nat. Rev. Immunol. 2015, 15, 683–691. [Google Scholar] [CrossRef] [PubMed]
- Nembrini, C.; Sichelstiel, A.; Kisielow, J.; Kurrer, M.; Kopf, M.; Marsland, B.J. Bacterial-induced protection against allergic inflammation through a multicomponent immunoregulatory mechanism. Thorax 2011, 66, 755–763. [Google Scholar] [CrossRef] [PubMed]
- Hauk, P.J.; Krawiec, M.; Murphy, J.; Boguniewicz, J.; Schiltz, A.; Goleva, E.; Liu, A.H.; Leung, D.Y. Neutrophilic airway inflammation and association with bacterial lipopolysaccharide in children with asthma and wheezing. Pediatr. Pulmonol. 2008, 43, 916–923. [Google Scholar] [CrossRef] [PubMed]
- Kaufmann, A.; Gemsa, D.; Sprenger, H. Differential desensitization of lipopolysaccharide-inducible chemokine gene expression in human monocytes and macrophages. Eur. J. Immunol. 2000, 30, 1562–1567. [Google Scholar] [CrossRef]
- Wagner, J.G.; Van Dyken, S.J.; Hotchkiss, J.A.; Harkema, J.R. Endotoxin enhancement of ozone-induced mucous cell metaplasia is neutrophil-dependent in rat nasal epithelium. Toxicol. Sci. 2001, 60, 338–347. [Google Scholar] [CrossRef] [PubMed]
- Kääriö, H.; Huttunen, K.; Karvonen, A.M.; Schaub, B.; von Mutius, E.; Pekkanen, J.; Hirvonen, M.R.; Roponen, M. Exposure to a farm environment is associated with T helper 1 and regulatory cytokines at age 4.5 years. Clin. Exp. Allergy 2016, 46, 71–77. [Google Scholar] [CrossRef] [PubMed]
- Giovannangelo, M.; Gehring, U.; Nordling, E.; Oldenwening, M.; Terpstra, G.; Bellander, T.; Hoek, G.; Heinrich, J.; Brunekreef, B. Determinants of house dust endotoxin in three European countries—The AIRALLERG study. Indoor Air 2007, 17, 70–79. [Google Scholar] [CrossRef] [PubMed]
- Rullo, V.E.; Rizzo, M.C.; Arruda, L.K.; Sole, D.; Naspitz, C.K. Daycare centers and schools as sources of exposure to mites, cockroach, and endotoxin in the city of Sao Paulo, Brazil. J. Allergy Clin. Immunol. 2002, 110, 582–588. [Google Scholar] [CrossRef] [PubMed]
- Ball, T.M.; Castro-Rodriguez, J.A.; Griffith, K.A.; Holberg, C.J.; Martinez, F.D.; Wright, A.L. Siblings, day-care attendance, and the risk of asthma and wheezing during childhood. N. Engl. J. Med. 2000, 343, 538–543. [Google Scholar] [CrossRef] [PubMed]
- Maier, R.M.; Palmer, M.W.; Andersen, G.L.; Halonen, M.J.; Josephson, K.C.; Maier, R.S.; Martinez, F.D.; Neilson, J.W.; Stern, D.A.; Vercelli, D.; et al. Environmental determinants of and impact on childhood asthma by the bacterial community in household dust. Appl. Environ. Microbiol. 2010, 76, 2663–2667. [Google Scholar] [CrossRef] [PubMed]
- Makela, M.J.; Puhakka, T.; Ruuskanen, O.; Leinonen, M.; Saikku, P.; Kimpimaki, M.; Blomqvist, S.; Hyypia, T.; Arstila, P. Viruses and bacteria in the etiology of the common cold. J. Clin. Microbiol. 1998, 36, 539–542. [Google Scholar] [PubMed]
- Fry, A.M.; Lu, X.; Olsen, S.J.; Chittaganpitch, M.; Sawatwong, P.; Chantra, S.; Baggett, H.C.; Erdman, D. Human rhinovirus infections in rural Thailand: Epidemiological evidence for rhinovirus as both pathogen and bystander. PLoS ONE 2011, 6, e17780. [Google Scholar] [CrossRef] [PubMed]
- Iwane, M.K.; Prill, M.M.; Lu, X.; Miller, E.K.; Edwards, K.M.; Hall, C.B.; Griffin, M.R.; Staat, M.A.; Anderson, L.J.; Williams, J.V.; et al. Human rhinovirus species associated with hospitalizations for acute respiratory illness in young US children. J. Infect. Dis. 2011, 204, 1702–1710. [Google Scholar] [CrossRef] [PubMed]
- Van Benten, I.; Koopman, L.; Niesters, B.; Hop, W.; van Middelkoop, B.; de Waal, L.; van Drunen, K.; Osterhaus, A.; Neijens, H.; Fokkens, W. Predominance of rhinovirus in the nose of symptomatic and asymptomatic infants. Pediatr. Allergy Immunol. 2003, 14, 363–370. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.J. Thymic stromal lymphopoietin: Master switch for allergic inflammation. J. Exp. Med. 2006, 203, 269–273. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.J.; Soumelis, V.; Watanabe, N.; Ito, T.; Wang, Y.H.; Malefyt Rde, W.; Omori, M.; Zhou, B.; Ziegler, S.F. TSLP: An epithelial cell cytokine that regulates T cell differentiation by conditioning dendritic cell maturation. Annu. Rev. Immunol. 2007, 25, 193–219. [Google Scholar] [CrossRef] [PubMed]
- Pritchard, A.L.; Carroll, M.L.; Burel, J.G.; White, O.J.; Phipps, S.; Upham, J.W. Innate IFNs and plasmacytoid dendritic cells constrain Th2 cytokine responses to rhinovirus: A regulatory mechanism with relevance to asthma. J. Immunol. 2012, 188, 5898–5905. [Google Scholar] [CrossRef] [PubMed]
- Wark, P.A.B.; Johnston, S.L.; Bucchieri, F.; Powell, R.; Puddicombe, S.; Laza-Stanca, V.; Holgate, S.T.; Davies, D.E. Asthmatic bronchial epithelial cells have a deficient innate immune response to infection with rhinovirus. J. Exp. Med. 2005, 201, 937–947. [Google Scholar] [CrossRef] [PubMed]
- Braun-Fahrlander, C. Does the ‘hygiene hypothesis’ provide an explanation for the relatively low prevalence of asthma in Bangladesh? Int. J. Epidemiol. 2002, 31, 488–489. [Google Scholar] [CrossRef] [PubMed]
- Kaiko, G.E.; Loh, Z.; Spann, K.; Lynch, J.P.; Lalwani, A.; Zheng, Z.; Davidson, S.; Uematsu, S.; Akira, S.; Hayball, J.; et al. Toll-like receptor 7 gene deficiency and early-life Pneumovirus infection interact to predispose toward the development of asthma-like pathology in mice. J. Allergy Clin. Immunol. 2013, 131, 1331–1339. [Google Scholar] [CrossRef] [PubMed]
- Davidson, S.; Kaiko, G.; Loh, Z.; Lalwani, A.; Zhang, V.; Spann, K.; Foo, S.Y.; Hansbro, N.; Uematsu, S.; Akira, S.; et al. Plasmacytoid Dendritic Cells Promote Host Defense against Acute Pneumovirus Infection via the TLR7–MyD88-Dependent Signaling Pathway. J. Immunol. 2011, 186, 5938–5948. [Google Scholar] [CrossRef] [PubMed]
- Arbour, N.C.; Lorenz, E.; Schutte, B.C.; Zabner, J.; Kline, J.N.; Jones, M.; Frees, K.; Watt, J.L.; Schwartz, D.A. TLR4 mutations are associated with endotoxin hyporesponsiveness in humans. Nat. Genet. 2000, 25, 187–191. [Google Scholar] [PubMed]
- Illi, S.; von Mutius, E.; Lau, S.; Nickel, R.; Niggemann, B.; Sommerfeld, C.; Wahn, U. The pattern of atopic sensitization is associated with the development of asthma in childhood. J. Allergy Clin. Immunol. 2001, 108, 709–714. [Google Scholar] [CrossRef] [PubMed]
- Kloepfer, K.M.; Lee, W.M.; Pappas, T.E.; Kang, T.J.; Vrtis, R.F.; Evans, M.D.; Gangnon, R.E.; Bochkov, Y.A.; Jackson, D.J.; Lemanske, R.F., Jr.; et al. Detection of pathogenic bacteria during rhinovirus infection is associated with increased respiratory symptoms and asthma exacerbations. J. Allergy Clin. Immunol. 2014, 133, 1301–1307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gereda, J.E.; Leung, D.Y.; Thatayatikom, A.; Streib, J.E.; Price, M.R.; Klinnert, M.D.; Liu, A.H. Relation between house-dust endotoxin exposure, type 1 T-cell development, and allergen sensitisation in infants at high risk of asthma. Lancet 2000, 355, 1680–1683. [Google Scholar] [CrossRef]
- Gensollen, T.; Iyer, S.S.; Kasper, D.L.; Blumberg, R.S. How colonization by microbiota in early life shapes the immune system. Science 2016, 352, 539–544. [Google Scholar] [CrossRef] [PubMed]
- Clemente, J.C.; Ursell, L.K.; Parfrey, L.W.; Knight, R. The impact of the gut microbiota on human health: An integrative view. Cell 2012, 148, 1258–1270. [Google Scholar] [CrossRef] [PubMed]
- Dominguez-Bello, M.G.; Costello, E.K.; Contreras, M.; Magris, M.; Hidalgo, G.; Fierer, N.; Knight, R. Delivery mode shapes the acquisition and structure of the initial microbiota across multiple body habitats in newborns. Proc. Natl. Acad. Sci. USA 2010, 107, 11971–11975. [Google Scholar] [CrossRef] [PubMed]
- Neu, J.; Rushing, J. Cesarean versus vaginal delivery: Long-term infant outcomes and the hygiene hypothesis. Clin. Perinatol. 2011, 38, 321–331. [Google Scholar] [CrossRef] [PubMed]
- Penders, J.; Gerhold, K.; Thijs, C.; Zimmermann, K.; Wahn, U.; Lau, S.; Hamelmann, E. New insights into the hygiene hypothesis in allergic diseases: Mediation of sibling and birth mode effects by the gut microbiota. Gut Microbes 2014, 5, 239–244. [Google Scholar] [CrossRef] [PubMed]
- Penders, J.; Gerhold, K.; Stobberingh, E.E.; Thijs, C.; Zimmermann, K.; Lau, S.; Hamelmann, E. Establishment of the intestinal microbiota and its role for atopic dermatitis in early childhood. J. Allergy Clin. Immunol. 2013, 132, 601–607. [Google Scholar] [CrossRef] [PubMed]
- Bisgaard, H.; Li, N.; Bonnelykke, K.; Chawes, B.L.; Skov, T.; Paludan-Muller, G.; Stokholm, J.; Smith, B.; Krogfelt, K.A. Reduced diversity of the intestinal microbiota during infancy is associated with increased risk of allergic disease at school age. J. Allergy Clin. Immunol. 2011, 128, 646–652. [Google Scholar] [CrossRef] [PubMed]
- Abrahamsson, T.R.; Jakobsson, H.E.; Andersson, A.F.; Bjorksten, B.; Engstrand, L.; Jenmalm, M.C. Low gut microbiota diversity in early infancy precedes asthma at school age. Clin. Exp. Allergy 2014, 44, 842–850. [Google Scholar] [CrossRef] [PubMed]
- Kukkonen, K.; Savilahti, E.; Haahtela, T.; Juntunen-Backman, K.; Korpela, R.; Poussa, T.; Tuure, T.; Kuitunen, M. Probiotics and prebiotic galacto-oligosaccharides in the prevention of allergic diseases: A randomized, double-blind, placebo-controlled trial. J. Allergy Clin. Immunol. 2007, 119, 192–198. [Google Scholar] [CrossRef] [PubMed]
- Kalliomäki, M.; Salminen, S.; Arvilommi, H.; Kero, P.; Koskinen, P.; Isolauri, E. Probiotics in primary prevention of atopic disease: A randomised placebo-controlled trial. Lancet 2001, 357, 1076–1079. [Google Scholar] [CrossRef]
- Elazab, N.; Mendy, A.; Gasana, J.; Vieira, E.R.; Quizon, A.; Forno, E. Probiotic Administration in Early Life, Atopy, and Asthma: A Meta-analysis of Clinical Trials. Pediatrics 2013, 132, e666–e676. [Google Scholar] [CrossRef] [PubMed]
- Kopp, M.V.; Hennemuth, I.; Heinzmann, A.; Urbanek, R. Randomized, Double-Blind, Placebo-Controlled Trial of Probiotics for Primary Prevention: No Clinical Effects of Lactobacillus GG Supplementation. Pediatrics 2008, 121, e850–e856. [Google Scholar] [CrossRef] [PubMed]
- Brouwer, M.L.; Wolt-Plompen, S.A.; Dubois, A.E.; van der Heide, S.; Jansen, D.F.; Hoijer, M.A.; Kauffman, H.F.; Duiverman, E.J. No effects of probiotics on atopic dermatitis in infancy: A randomized placebo-controlled trial. Clin. Exp. Allergy 2006, 36, 899–906. [Google Scholar] [CrossRef] [PubMed]
- Trompette, A.; Gollwitzer, E.S.; Yadava, K.; Sichelstiel, A.K.; Sprenger, N.; Ngom-Bru, C.; Blanchard, C.; Junt, T.; Nicod, L.P.; Harris, N.L.; et al. Gut microbiota metabolism of dietary fiber influences allergic airway disease and hematopoiesis. Nat. Med. 2014, 20, 159–166. [Google Scholar] [CrossRef] [PubMed]
- Arpaia, N.; Campbell, C.; Fan, X.; Dikiy, S.; van der Veeken, J.; deRoos, P.; Liu, H.; Cross, J.R.; Pfeffer, K.; Coffer, P.J.; et al. Metabolites produced by commensal bacteria promote peripheral regulatory T-cell generation. Nature 2013, 504, 451–455. [Google Scholar] [CrossRef] [PubMed]
- Marino, E.; Caruso, M.; Campagna, D.; Polosa, R. Impact of air quality on lung health: Myth or reality? Ther. Adv. Chronic Dis. 2015, 6, 286–298. [Google Scholar] [CrossRef] [PubMed]
- Gehring, U.; Wijga, A.H.; Brauer, M.; Fischer, P.; de Jongste, J.C.; Kerkhof, M.; Oldenwening, M.; Smit, H.A.; Brunekreef, B. Traffic-related air pollution and the development of asthma and allergies during the first 8 years of life. Am. J. Respir. Crit. Care Med. 2010, 181, 596–603. [Google Scholar] [CrossRef] [PubMed]
- Carlsten, C.; Dybuncio, A.; Becker, A.; Chan-Yeung, M.; Brauer, M. Traffic-related air pollution and incident asthma in a high-risk birth cohort. Occup. Environ. Med. 2011, 68, 291–295. [Google Scholar] [CrossRef] [PubMed]
- Quansah, R.; Jaakkola, M.S.; Hugg, T.T.; Heikkinen, S.A.; Jaakkola, J.J. Residential dampness and molds and the risk of developing asthma: A systematic review and meta-analysis. PLoS ONE 2012, 7, e47526. [Google Scholar] [CrossRef] [PubMed]
- Mendell, M.J.; Mirer, A.G.; Cheung, K.; Tong, M.; Douwes, J. Respiratory and allergic health effects of dampness, mold, and dampness-related agents: A review of the epidemiologic evidence. Environ. Health Perspect. 2011, 119, 748–756. [Google Scholar] [CrossRef] [PubMed]
- Liu, A.H.; Murphy, J.R. Hygiene hypothesis: Fact or fiction? J. Allergy Clin. Immunol. 2003, 111, 471–478. [Google Scholar] [CrossRef] [PubMed]
- Salvi, S.S.; Babu, K.S.; Holgate, S.T. Is asthma really due to a polarized T cell response toward a helper T cell type 2 phenotype? Am. J. Respir. Crit. Care Med. 2001, 164, 1343–1346. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.-M.; Kim, Y.-S.; Jeon, S.G.; Kim, Y.-K. Immunopathogenesis of Allergic Asthma: More than the Th2 Hypothesis. Allergy Asthma Immunol. Res. 2013, 5, 189–196. [Google Scholar] [CrossRef] [PubMed]
- Hellings, P.W.; Kasran, A.; Liu, Z.; Vandekerckhove, P.; Wuyts, A.; Overbergh, L.; Mathieu, C.; Ceuppens, J.L. Interleukin-17 orchestrates the granulocyte influx into airways after allergen inhalation in a mouse model of allergic asthma. Am. J. Respir. Cell Mol. Biol. 2003, 28, 42–50. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
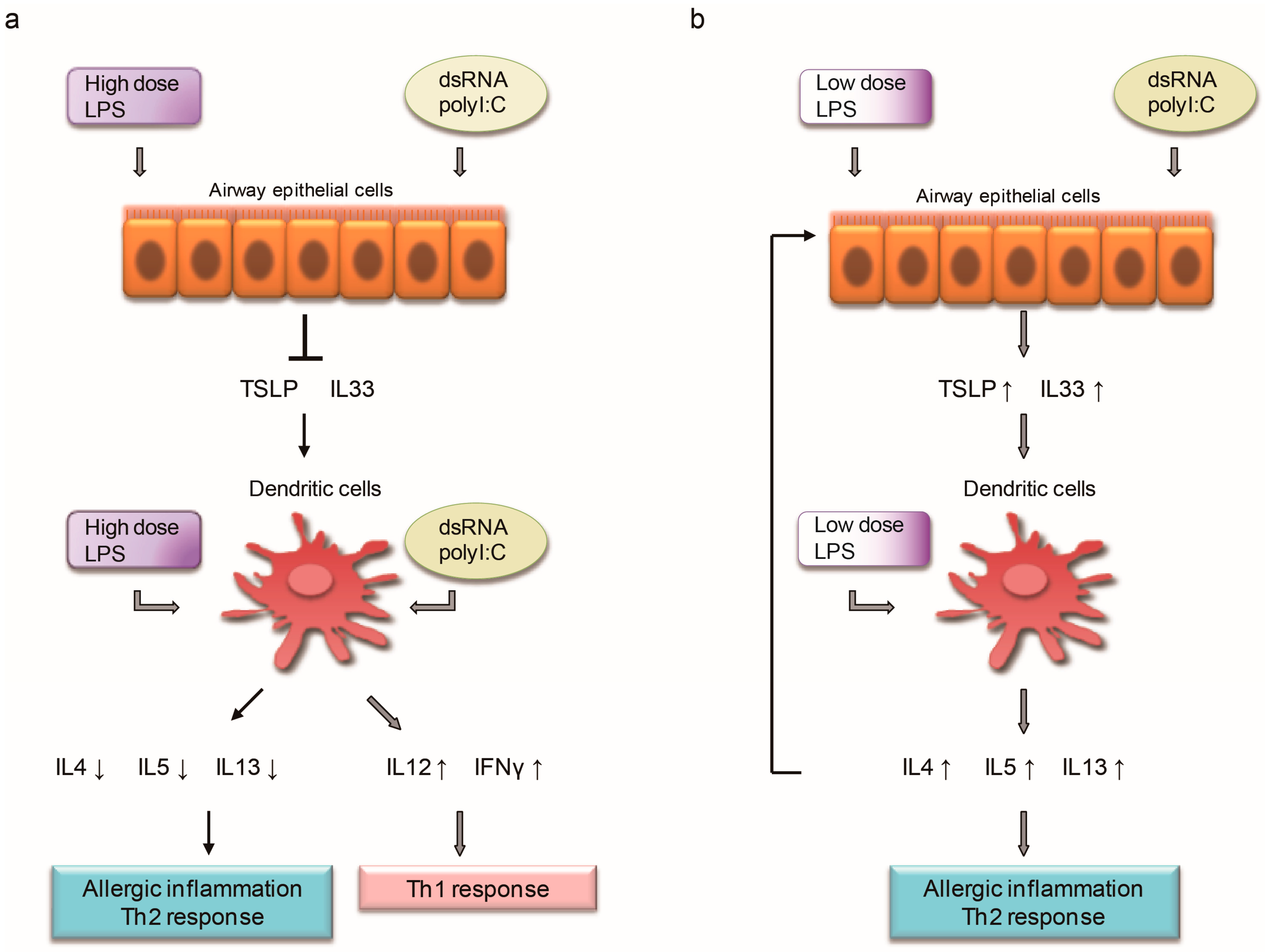{kind=link}
| Authors | Model | Origin of LPS | LPS Dose and Pathway Used | Allergen or Antigen | Allergen Dose and Pathway Used for Sensitization | Allergen Dose and Pathway Used for Challenging | Protocol | Result | Note |
|---|---|---|---|---|---|---|---|---|---|
| Tulic et al., 2000 [41] | male PVG rat | Salmonella typhimurium | 50 μg/mL inhaled | OVA | 100 μg/mL i.p. | Aerosolized 1% OVA at Day 11 after sensitization |
|
| Timing of LPS exposure determines protection or exacerbation of allergy. |
| Eisenbarth et al., 2002 [43] | BALB/cJ mice and BALB/cAnNCr | Escherichia coli | Concomitant use of 100 μg (high dose) or 0.1 μg LPS with OVA in sensitizing period | OVA | 100 μg OVA in 50 μL PBS intranasally, or 100 μg OVA in 2 mg Al(OH)3 intraperitoneally, with LPS depletion | 25 μg OVA intranasally | Sensitized on Days 0, 1 and 2, challenged on Days 14,15,18 and 19, killed on Day 21 | Mice exposed to LPS-depleted OVA showed no airway inflammatory responses after challenge; those sensitized with OVA containing low dose LPS demonstrated significant Th2 lung infiltrates; those exposed to PBS or low dose LPS alone did not generate pulmonary inflammation after challenge; those sensitized with OVA containing high dose LPS resulted in a Th1 associated response. | TLR4 signaling Is required for Th2 priming to inhaled antigens, and the dose of LPS during sensitizing period regulates the predominance of Th1 or Th2 response. |
| Lowe et al., 2015 [44] | Male Dunkin-Hartley guinea pigs (GPs) | LPS source not mentioned | Inhaled 30 μg/mL | OVA | Bil. i.p. injection of OVA,150 μg/mL and Al(OH)3 100 mg/mL normal saline | Sensitized GPs were exposed to inhaled OVA (300 μg/mL) on Day 21. | LPS (30 μg/mL) exposure was by two protocols: 72 and 24 h pre-OVA exposure, 48 h pre-OVA and co-administered with OVA by nebulizer, at rate of 0.3 mL/min for 1 h | LPS exposure 24 h before allergen challenge attenuates the early asthmatic response (EAR), whereas co-administered LPS does not influence the EAR. The addition of a second LPS exposure co-administered with OVA prolonged the EAR. Similarly, LPS exposure 24 h before allergen challenge diminished airway hyperreactivity (AHR) to histamine, whereas co-administered LPS prolonged the AHR | Emphasizing to the timing of LPS application |
| Langenkamp et al., 2000 [45] | Dendritic cells |
|
| TSST-1 as antigen | Pretreatment of dendritic cells with LPS, then after 8 or 48 h, 0.1 or 10 ng/mL TSST-1 was added to culture medium |
| This study explored DC’s priming tendency after LPS pre-stimulation, in response to superantigen TSST-1, which was an inflammatory response, but not allergic response. |
| Authors | Model | Origin of LPS | LPS Dose and Pathway Used | Allergen, Antigen or Stimulus | Allergen Dose and Pathway Used for Sensitization | Allergen Dose and Pathway Used for Challenging | Protocol | Result | Note |
|---|---|---|---|---|---|---|---|---|---|
| Carlsten et al., 2011 [52] | Human of age 7 | Home dust | Inhaled from environment | Dog allergen | Inhaled from environment | Inhaled from environment | Correlation study | Endotoxin was associated with decreased risk of sensitization to dog allergen. | HDM was also associated with decreased risk of sensitization to dog allergen, which needs further confirmatory studies. However, in Schuijs’ study below, HDM was noted to induce A20 also, though less pronounced than LPS. |
| Braun-Fahrlander et al., 2002 [3] | Human of age 6-13 | Home dust | Inhaled from environment | unspecified | Inhaled from environment | Inhaled from environment | Correlation study | Endotoxin levels in dust were inversely related to the incidence of hay fever, atopic asthma, and atopic sensitization. | |
| Schuijs et al., 2015 [10] |
| Ultrapure LPS purchased from Invivogen. Strain and species not specified. |
| HDM |
|
|
|
| TLR4 signaling in ECs induces attenuators of signaling such as A20. |
| Ganesh et al., 2014 [53] | BALB/c and DO11.10 mice | Salmonella enterica serovar typhimurium aroA strain SL 7207 | Intragastric inoculation with 0.5~1 × 109 CFU of whole S. typhimurium (SL 7207) | OVA | 10 μg OVA + adjuvant, i.p. | intranasal use of 30 μg of OVA | Mice were sensitized with OVA i.p. on Days 7, 8, 9, and 20, infected intragastrically with S. typhimurium on Days 0, 7, 20, and 27, challenged on Days 20, 24, 27, 30, and 34 by intranasal administration of of OVA | S. typhimurium infection in mice results in attenuation of cellular airway inflammation, reduced pathology and mucus production in the lungs, expansion of CD11b+Gr1+ myeloid cells, with no apparent diversion toward Th1. | This study used whole bacteria for experiment, instead of LPS only. |
| Rodriguez et al., 2003 [54] | C57BL/6J, BALB/c and C3H/HeJ mice | Salmonella abortus equi | LPS at a dose of 20 μg/animal was delivered intravenously concomitantly with a second OVA challenge | OVA | 4 μg OVA/1.6 mg aluminum hydroxide | 10 μg OVA/50 μL saline intranasally | Mice were immunized on Days 0 and 7, and challenged on Days 14 and 21 intranasally |
| Thus, systemic LPS displayed protective effect, while local LPS displayed pro-inflammatory effect with neutrophilia reaction. |
| Lin et al., 2016 [9] | H292 cell line | Escherichia coli | 0.3 to 30 μg/mL co-culture | polyI:C, HPeV1 | LPS pretreatment 2 h before polyI:C or HPeV1 co-culture with H292 cells. | The downstream production of TSLP and IL33 by stimulating H292 cells with polyI:C or HPeV1 was reduced with 30 μg/mL LPS pretreatment, but not 0.3 μg/mL LPS |
| Authors | Model | Origin of LPS | LPS Dose and Pathway Used | Allergen or Antigen | Allergen Dose and Pathway Used for Sensitization | Allergen Dose and Pathway Used for Challenging | Protocol | Result | Note |
|---|---|---|---|---|---|---|---|---|---|
| Rittirsch et al., 2008 [67] |
| Escherichia coli (serotype O111:B4) | 50 μg LPS in 40 μL PBS intratracheally, total 2,550,100 μg | nil | nil | nil | Permeability index checked from bronchoalveolar lavage at 0, 2, 4, 6, 8 h. |
| The LPS concentration used is 1250 μg/mL, as compared to the 0.3 and 30 μg/mL in cell line model [9], and the total LPS used is 50 μg, as compared with total 100 ng to 1 μg LPS in Schuijs’ study [10]. |
| Eutamene et al., 2005 [70] |
|
|
| nil | nil | nil |
|
| P. aeruginosa is a strong pathogen for airway [71], so total amount of LPS used is less, as compared with studies above. |
| Rojas et al., 2005 [68] | C57BL/6 male mice | Escherichia coli O111:B6 | Intraperitoneally with 1 mg/kg LPS | nil | nil | nil | Mice were inoculated intraperitoneally with 1 mg/kg of LPS from E. coli O111:B6. | Sublethal dose of i.p. LPS to mice caused rapid onset of interstitial pulmonary edema, inflammatory cell accumulation, and deposition of fibronectin and collagen in the lungs. | The scale of mg/kg is sublethal, compared to the protective dose scale of ng/mL to μg/mL. |
| Yao et al., 2017 [69] |
| LPS, source not specified |
| nil | nil | nil | Lung injury in mice and rats were induced by i.p. LPS. | Lung tissues revealed interstitial edema and hemorrhage, alveolar wall thickening, increased infiltration of neutrophils and macrophages in the lung parenchyma and alveolar spaces. | Again, the dose of causing acute lung injury is on the scale of mg/kg. |
| Taveira da Silva et al., 1993 [72] | Human | Salmonella minnesota | i.v. LPS | nil | nil | nil | The patient administered i.v. 1 mg of S. minnesota LPS, in sterile water, in an attempt to treat a tumor. | Septic shock syndrome induced, including a high-cardiac-output hypotension, disseminated intravascular coagulation, abnormalities of hepatic and renal function, and non-cardiogenic pulmonary edema. | 1 mg of purified LPS is equivalent to 15,000 ng/kg, thousands times higher than the usual dose of 4 ng/kg given to normal volunteers in experimental studies. Endothelial cells are much more sensitive to LPS than epithelial cells, with pg/mL level LPS activating endothelial cells in the presence of blood, compared to the relative resistance of respiratory epithelial cells to μg/mL level LPS [66]. |
| Pugin et al., 1993 [66] | Human umbilical vein endothelial cells (HUVEC) |
| Incubated with different dilutions of E. coli 0111:B4 or S. minnesota wild-type LPS, from 10−1 to 104 pg/mL | nil | nil | nil | HUVECs incubated with different dilutions of LPS for 6 h | In the presence of whole blood, 1000-fold less LPS was required to achieve the level of HUVEC activation (assessed by VCAM-1 upregulation) observed with plasma alone. | Endothelial cells are sensitive to ng/mL LPS in the absence of blood, but much more sensitive even to pg/mL LPS in the presence of blood. |
| Rodriguez et al., 2003 [54] | C57BL/6J, BALB/c and C3H/HeJ mice | Salmonella abortus equi | LPS at a dose of 20 μg/animal was delivered intranasally concomitantly with a second OVA challenge | OVA | 4 μg OVA/1.6 mg aluminum hydroxide | 10 μg OVA/50 μL saline intranasally | Mice were immunized on Days 0 and 7, and challenged on Days14 and 21 intranasally |
| Systemic LPS displayed protective effect, while local LPS displayed pro-inflammatory effect with neutrophilia reaction. |
| Hammad et al., 2009 [62] | Radiation-induced chimeric Tlr4-deficient mice with DCs deficient or ECs-like cells | Rhodobacter sphaeroides | 10 μg or 100 ng per mouse, in 80 μL PBS, intratracheal | HDM | nil | Intratracheal 100 μg HDM | 80 μL PBS intratracheal with HDM and LPS | TLR4 expression on lung structural cells, but not on DCs, is necessary and sufficient for lung DC activation and for priming of effector T helper responses to HDM. | TLR4 triggering on structural cells in the presence of HDM caused production of TSLP, GM-CSF, IL25 and IL33. The absence of TLR4 on structural cells, but not on hematopoietic cells, abolished HDM-driven allergic airway inflammation. |
| Cell Type | High Dose LPS | polyI:C or Virus |
|---|---|---|
| Epithelial cells | Minimal or no effect [31,46]. | Neutral because allergic inflammation due to TLRs pathway activation was blocked by pre-exposure to high dose LPS [9]. |
| Dendritic cells | Basically slight Th1 skewing due to high dose LPS with no IL12/IL4 skewing in the context [43,45] and less Th2-promoting mDC2s [57]. Unstimulated peripheral blood mononuclear cells produced more IL10, IL12 and IFNγ,indicating increased spontaneous production of Th1 and regulatory cytokines [103]. | Th1 predominant except RSV infection, which displays Th2 pattern [73,75]. |
| Net result: Th1 predominant # | ||
| Cell Type | Low Dose LPS | polyI:C or Virus |
|---|---|---|
| Epithelial cells | Minimal or no effect. | Th2 predominant due to TLR3 pathway activation with production of TSLP, IL33, IL25 etc. [31,32,33,34,35], even slightly enhanced by pre-exposure to low dose LPS [9]. |
| Dendritic cells | Basically slight Th2 skewing due to low dose LPS when no IL12/IL4 skewing in the context [43,45,57]. | Th1 or Th2 skewing, depending on the relative stimulatory force between Th2-prone allergic cytokines, such as TSLP and IL33 [112,113], and Th1-prone polyI:C or virus [73,75], except RSV, which displays Th2 pattern. |
| Net result:Th2 predominant # | ||
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lin, T.-H.; Su, H.-H.; Kang, H.-Y.; Chang, T.-H. The Interactive Roles of Lipopolysaccharides and dsRNA/Viruses on Respiratory Epithelial Cells and Dendritic Cells in Allergic Respiratory Disorders: The Hygiene Hypothesis. Int. J. Mol. Sci. 2017, 18, 2219. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms18102219
Lin T-H, Su H-H, Kang H-Y, Chang T-H. The Interactive Roles of Lipopolysaccharides and dsRNA/Viruses on Respiratory Epithelial Cells and Dendritic Cells in Allergic Respiratory Disorders: The Hygiene Hypothesis. International Journal of Molecular Sciences. 2017; 18(10):2219. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms18102219
Chicago/Turabian StyleLin, Tsang-Hsiung, Hsing-Hao Su, Hong-Yo Kang, and Tsung-Hsien Chang. 2017. "The Interactive Roles of Lipopolysaccharides and dsRNA/Viruses on Respiratory Epithelial Cells and Dendritic Cells in Allergic Respiratory Disorders: The Hygiene Hypothesis" International Journal of Molecular Sciences 18, no. 10: 2219. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms18102219