Oxidative Stress in Human Atherothrombosis: Sources, Markers and Therapeutic Targets

 and
and 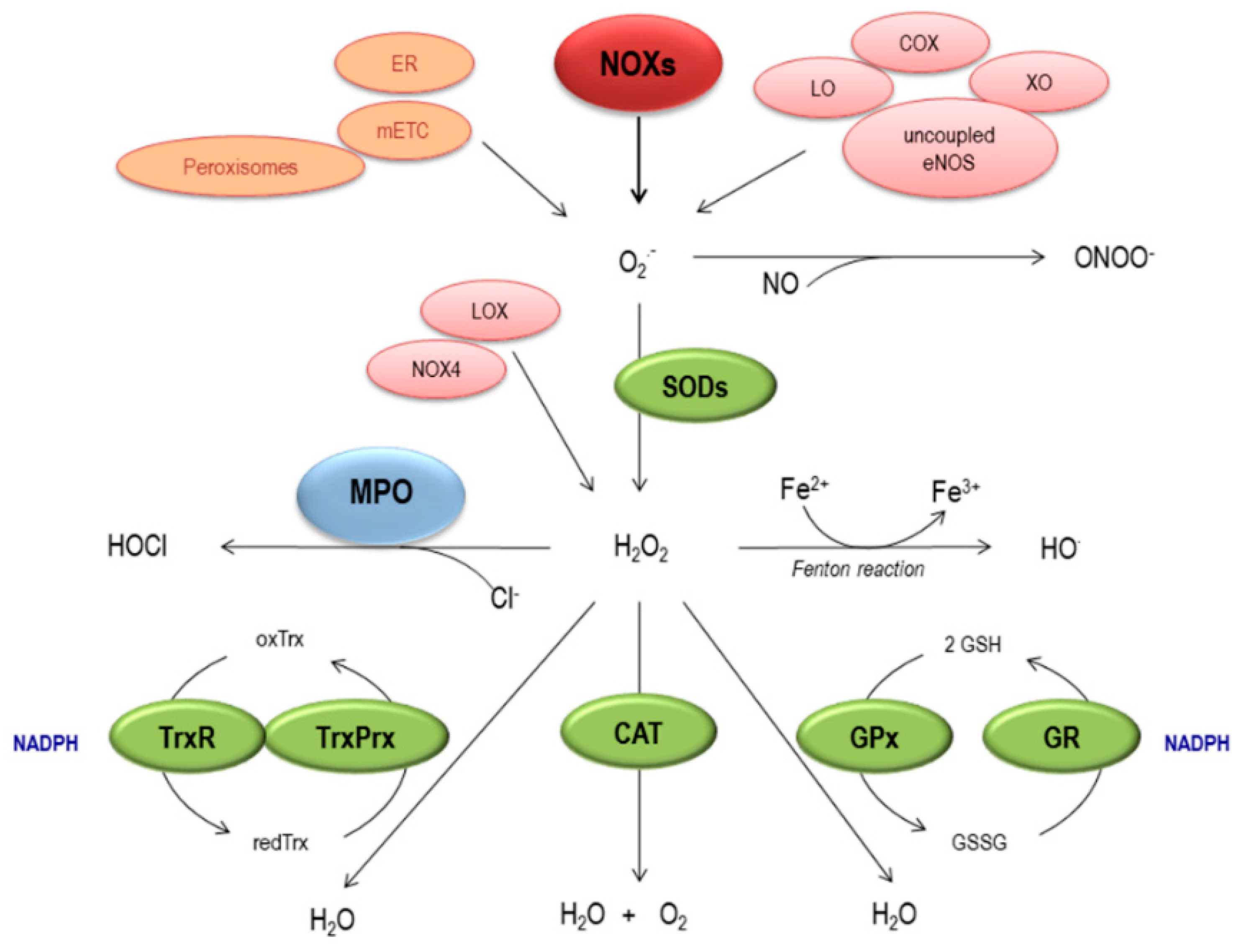{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Generation and Elimination of ROS
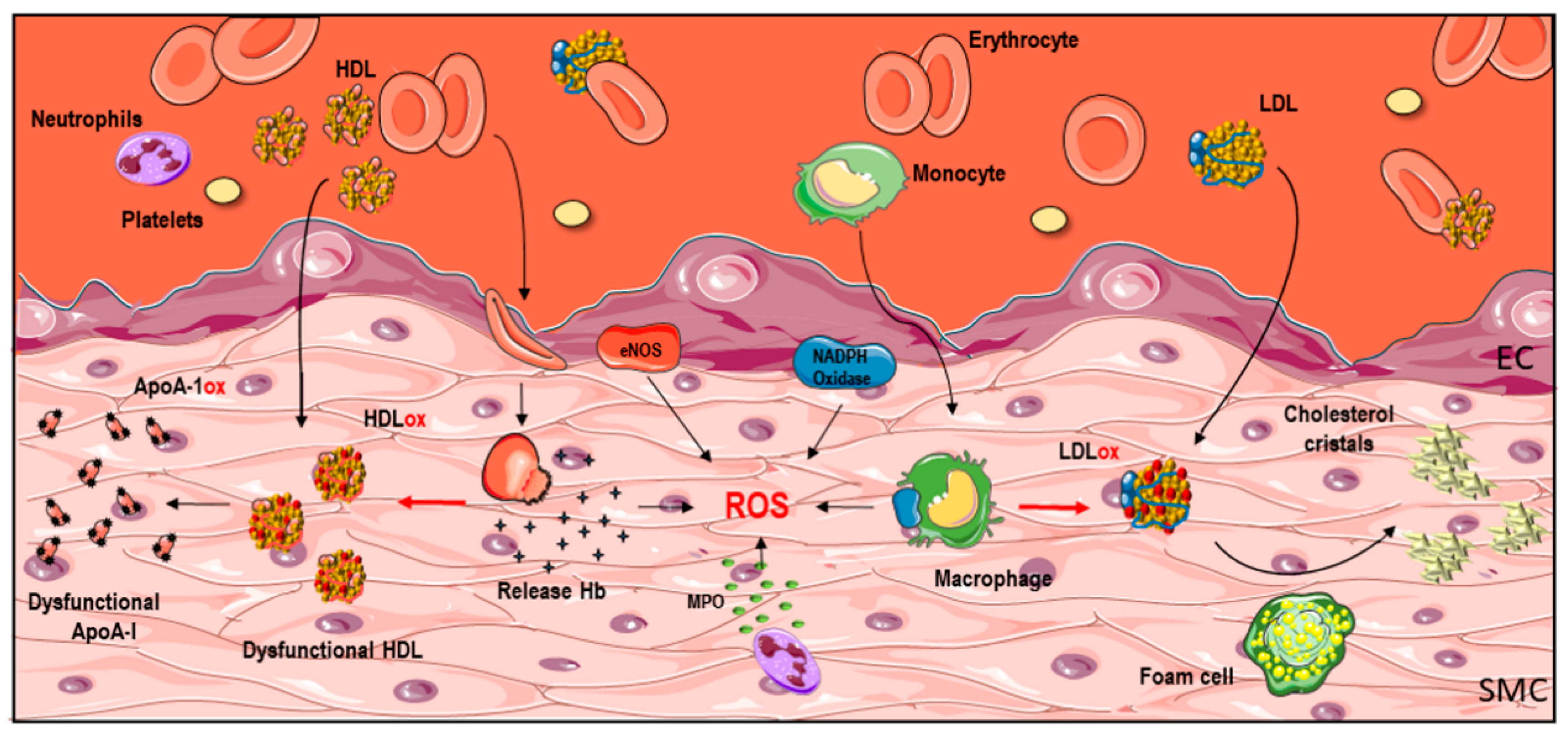
3. Sources of Oxidative Stress in Human Vascular Diseases
3.1. NADPH Oxidase
3.2. MPO
3.3. Iron
4. Markers of Oxidative Stress in Human Vascular Diseases
4.1. Oxidized LDL
4.2. Oxidized HDL
4.3. Antioxidants
5. Antioxidants as a Potential Therapeutic Strategy to Prevent Pathological Vascular Remodeling
6. Conclusions
Acknowledgments
Conflicts of Interest
References
- Bentzon, J.F.; Otsuka, F.; Virmani, R.; Falk, E. Mechanisms of plaque formation and rupture. Circ. Res. 2014, 114, 1852–1866. [Google Scholar] [CrossRef] [PubMed]
- Weintraub, N.L. Understanding abdominal aortic aneurysm. N. Engl. J. Med. 2009, 361, 1114–1116. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Wang, Q.; Zhu, J.; Xiao, Q.; Zhang, L. Reactive oxygen species: Key regulators in vascular health and diseases. Br. J. Pharmacol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Montezano, A.C.; Dulak-Lis, M.; Tsiropoulou, S.; Harvey, A.; Briones, A.M.; Touyz, R.M. Oxidative stress and human hypertension: Vascular mechanisms, biomarkers, and novel therapies. Can. J. Cardiol. 2015, 31, 631–641. [Google Scholar] [CrossRef] [PubMed]
- Konior, A.; Schramm, A.; Czesnikiewicz-Guzik, M.; Guzik, T.J. NADPH oxidases in vascular pathology. Antioxid. Redox Signal. 2014, 20, 2794–2814. [Google Scholar] [CrossRef] [PubMed]
- Violi, F.; Pignatelli, P. Clinical Application of NOX Activity and Other Oxidative Biomarkers in Cardiovascular Disease: A Critical Review. Antioxid. Redox Signal. 2015, 23, 514–532. [Google Scholar] [CrossRef] [PubMed]
- Violi, F.; Carnevale, R.; Loffredo, L.; Pignatelli, P.; Gallin, J.I. NADPH Oxidase-2 and Atherothrombosis: Insight from Chronic Granulomatous Disease. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 218–225. [Google Scholar] [CrossRef] [PubMed]
- Violi, F.; Loffredo, L.; Carnevale, R.; Pignatelli, P.; Pastori, D. Atherothrombosis and oxidative stress: Mechanisms and management in elderly. Antioxid. Redox Signal. 2017. [Google Scholar] [CrossRef] [PubMed]
- Steinberg, D. The LDL modification hypothesis of atherogenesis: An update. J. Lipid Res. 2009, 50, S376–S381. [Google Scholar] [CrossRef] [PubMed]
- Michel, J.B.; Martin-Ventura, J.L.; Nicoletti, A.; Ho-Tin-Noé, B. Pathology of human plaque vulnerability: Mechanisms and consequences of intraplaque haemorrhages. Atherosclerosis 2014, 234, 311–319. [Google Scholar] [CrossRef] [PubMed]
- Michel, J.B.; Martin-Ventura, J.L.; Egido, J.; Sakalihasan, N.; Treska, V.; Lindholt, J.; Allaire, E.; Thorsteinsdottir, U.; Cockerill, G.; Swedenborg, J. FAD EU consortium. Novel aspects of the pathogenesis of aneurysms of the abdominal aorta in humans. Cardiovasc. Res. 2011, 90, 18–27. [Google Scholar] [CrossRef] [PubMed]
- Farbstein, D.; Soloveichik, Y.Z.; Levy, N.S.; Levy, A.P. Genetics of redox systems and their relationship with cardiovascular disease. Curr. Atheroscler. Rep. 2011, 13, 215–224. [Google Scholar] [CrossRef] [PubMed]
- Dröge, W. Free radicals in the physiological control of cell function. Physiol. Rev. 2002, 82, 47–95. [Google Scholar] [CrossRef] [PubMed]
- García-Redondo, A.B.; Aguado, A.; Briones, A.M.; Salaices, M. NADPH oxidases and vascular remodeling in cardiovascular diseases. Pharmacol. Res. 2016, 114, 110–120. [Google Scholar] [CrossRef] [PubMed]
- Holmström, K.M.; Finkel, T. Cellular mechanisms and physiological consequences of redox-dependent signalling. Nat. Rev. Mol. Cell Biol. 2014, 15, 411–421. [Google Scholar] [CrossRef] [PubMed]
- Drummond, G.R.; Selemidis, S.; Griendling, K.K.; Sobey, C.G. Combating oxidative stress in vascular disease: NADPH oxidases as therapeutic targets. Nat. Rev. Drug Discov. 2011, 10, 453–471. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez, C.; Martínez-González, J.; Raposo, B.; Alcudia, J.F.; Guadall, A.; Badimon, L. Regulation of lysyl oxidase in vascular cells: Lysyl oxidase as a new player in cardiovascular diseases. Cardiovasc. Res. 2008, 79, 7–13. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Revelles, S.; García-Redondo, A.B.; Avendaño, M.S.; Varona, S.; Palao, T.; Orriols, M.; Roque, F.R.; Fortuño, A.; Touyz, R.M.; Martínez-González, J.; et al. Lysyl Oxidase Induces Vascular Oxidative Stress and Contributes to Arterial Stiffness and Abnormal Elastin Structure in Hypertension: Role of p38MAPK. Antioxid. Redox Signal. 2017, 27, 379–397. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Holmgren, A. The thioredoxin antioxidant system. Free Radic. Biol. Med. 2014, 66, 75–87. [Google Scholar] [CrossRef] [PubMed]
- Frijhoff, J.; Winyard, P.G.; Zarkovic, N.; Davies, S.S.; Stocker, R.; Cheng, D.; Knight, A.R.; Taylor, E.L.; Oettrich, J.; Ruskovska, T.; et al. Clinical Relevance of Biomarkers of Oxidative Stress. Antioxid. Redox Signal. 2015, 23, 1144–1170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, B.; Lu, Y.; Chen, Y.; Cheng, J. The role of Nrf2 in oxidative stress-induced endothelial injuries. J. Endocrinol. 2015, 225, R83–R99. [Google Scholar] [CrossRef] [PubMed]
- Egea, J.; Fabregat, I.; Frapart, Y.M.; Ghezzi, P.; Görlach, A.; Kietzmann, T.; Kubaichuk, K.; Knaus, U.G.; Lopez, M.G.; Olaso-Gonzalez, G.; et al. European contribution to the study of ROS: A summary of the findings and prospects for the future from the COST action BM1203 (EU-ROS). Redox Biol. 2017, 13, 94–162. [Google Scholar] [CrossRef] [PubMed]
- McCormick, M.L.; Gavrila, D.; Weintraub, N.L. Role of oxidative stress in the pathogenesis of abdominal aortic aneurysms. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 461–469. [Google Scholar] [CrossRef] [PubMed]
- Emeto, T.I.; Moxon, J.V.; Au, M.; Golledge, J. Oxidative stress and abdominal aortic aneurysm: Potential treatment targets. Clin. Sci. (Lond.) 2016, 130, 301–315. [Google Scholar] [CrossRef] [PubMed]
- Förstermann, U.; Xia, N.; Li, H. Roles of Vascular Oxidative Stress and Nitric Oxide in the Pathogenesis of Atherosclerosis. Circ. Res. 2017, 120, 713–735. [Google Scholar] [CrossRef] [PubMed]
- Kattoor, A.J.; Pothineni, N.V.K.; Palagiri, D.; Mehta, J.L. Oxidative Stress in Atherosclerosis. Curr. Atheroscler. Rep. 2017, 19, 42. [Google Scholar] [CrossRef] [PubMed]
- Nosalski, R.; Guzik, T.J. Perivascular adipose tissue inflammation in vascular disease. Br. J. Pharmacol. 2017, 174, 3496–3513. [Google Scholar] [CrossRef] [PubMed]
- Lassègue, B.; San Martín, A.; Griendling, K.K. Biochemistry, physiology, and pathophysiology of NADPH oxidases in the cardiovascular system. Circ. Res. 2012, 110, 1364–1390. [Google Scholar] [CrossRef] [PubMed]
- Schröder, K. NADPH oxidases in redox regulation of cell adhesion and migration. Antioxid. Redox Signal. 2014, 20, 2043–2058. [Google Scholar] [CrossRef] [PubMed]
- Montezano, A.C.; Touyz, R.M. Reactive oxygen species, vascular Noxs, and hypertension: Focus on translational and clinical research. Antioxid. Redox Signal. 2014, 20, 164–182. [Google Scholar] [CrossRef] [PubMed]
- Pignatelli, P.; Sanguigni, V.; Lenti, L.; Ferro, D.; Finocchi, A.; Rossi, P.; Violi, F. gp91phox-dependent expression of platelet CD40 ligand. Circulation 2004, 110, 1326–1329. [Google Scholar] [CrossRef] [PubMed]
- Guzik, T.J.; West, N.E.; Black, E.; McDonald, D.; Ratnatunga, C.; Pillai, R.; Channon, K.M. Vascular superoxide production by NAD(P)H oxidase: Association with endothelial dysfunction and clinical risk factors. Circ. Res. 2000, 86, E85–E90. [Google Scholar] [CrossRef] [PubMed]
- Kalinina, N.; Agrotis, A.; Tararak, E.; Antropova, Y.; Kanellakis, P.; Ilyinskaya, O.; Quinn, M.T.; Smirnov, V.; Bobik, A. Cytochrome b558-dependent NAD(P)H oxidase-phox units in smooth muscle and macrophages of atherosclerotic lesions. Arterioscler. Thromb. Vasc. Biol. 2002, 22, 2037–2043. [Google Scholar] [CrossRef] [PubMed]
- Sorescu, D.; Weiss, D.; Lassègue, B.; Clempus, R.E.; Szocs, K.; Sorescu, G.P.; Valppu, L.; Quinn, M.T.; Lambeth, J.D.; Vega, J.D.; et al. Superoxide production and expression of nox family proteins in human atherosclerosis. Circulation 2002, 105, 1429–1435. [Google Scholar] [CrossRef] [PubMed]
- Azumi, H.; Inoue, N.; Takeshita, S.; Rikitake, Y.; Kawashima, S.; Hayashi, Y.; Itoh, H.; Yokoyama, M. Expression of NADH/NADPH oxidase p22phox in human coronary arteries. Circulation 1999, 100, 1494–1498. [Google Scholar] [CrossRef] [PubMed]
- Gray, S.P.; Di Marco, E.; Kennedy, K.; Chew, P.; Okabe, J.; El-Osta, A.; Calkin, A.C.; Biessen, E.A.; Touyz, R.M.; Cooper, M.E.; et al. Reactive Oxygen Species Can Provide Atheroprotection via NOX4-Dependent Inhibition of Inflammation and Vascular Remodeling. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 295–307. [Google Scholar] [CrossRef] [PubMed]
- Guzik, T.J.; Chen, W.; Gongora, M.C.; Guzik, B.; Lob, H.E.; Mangalat, D.; Hoch, N.; Dikalov, S.; Rudzinski, P.; Kapelak, B.; et al. Calcium-dependent NOX5 nicotinamide adenine dinucleotide phosphate oxidase contributes to vascular oxidative stress in human coronary artery disease. J. Am. Coll. Cardiol. 2008, 52, 1803–1909. [Google Scholar] [CrossRef] [PubMed]
- Manea, A.; Manea, S.A.; Gan, A.M.; Constantin, A.; Fenyo, I.M.; Raicu, M.; Muresian, H.; Simionescu, M. Human monocytes and macrophages express NADPH oxidase 5; a potential source of reactive oxygen species in atherosclerosis. Biochem. Biophys. Res. Commun. 2015, 461, 172–179. [Google Scholar] [CrossRef] [PubMed]
- Guzik, T.J.; Mussa, S.; Gastaldi, D.; Sadowski, J.; Ratnatunga, C.; Pillai, R.; Channon, K.M. Mechanisms of increased vascular superoxide production in human diabetes mellitus: Role of NAD(P)H oxidase and endothelial nitric oxide synthase. Circulation 2002, 105, 1656–1662. [Google Scholar] [CrossRef] [PubMed]
- Zalba, G.; Beloqui, O.; San José, G.; Moreno, M.U.; Fortuño, A.; Díez, J. NADPH oxidase-dependent superoxide production is associated with carotid intima-media thickness in subjects free of clinical atherosclerotic disease. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 1452–1457. [Google Scholar] [CrossRef] [PubMed]
- Guzik, B.; Sagan, A.; Ludew, D.; Mrowiecki, W.; Chwała, M.; Bujak-Gizycka, B.; Filip, G.; Grudzien, G.; Kapelak, B.; Zmudka, K.; et al. Mechanisms of oxidative stress in human aortic aneurysms—Association with clinical risk factors for atherosclerosis and disease severity. Int. J. Cardiol. 2013, 168, 2389–2396. [Google Scholar] [CrossRef] [PubMed]
- Loffredo, L.; Carnevale, R.; Sanguigni, V.; Plebani, A.; Rossi, P.; Pignata, C.; De Mattia, D.; Finocchi, A.; Martire, B.; Pietrogrande, M.C.; et al. Does NADPH oxidase deficiency cause artery dilatation in humans? Antioxid. Redox Signal. 2013, 18, 1491–1496. [Google Scholar] [CrossRef] [PubMed]
- Violi, F.; Pignatelli, P.; Pignata, C.; Plebani, A.; Rossi, P.; Sanguigni, V.; Carnevale, R.; Soresina, A.; Finocchi, A.; Cirillo, E.; et al. Reduced atherosclerotic burden in subjects with genetically determined low oxidative stress. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 406–412. [Google Scholar] [CrossRef] [PubMed]
- Sibley, C.T.; Estwick, T.; Zavodni, A.; Huang, C.Y.; Kwan, A.C.; Soule, B.P.; Long Priel, D.A.; Remaley, A.T.; Rudman Spergel, A.K.; Turkbey, E.B.; et al. Assessment of atherosclerosis in chronic granulomatous disease. Circulation 2014, 130, 2031–2039. [Google Scholar] [CrossRef] [PubMed]
- Guzik, T.J.; West, N.E.; Black, E.; McDonald, D.; Ratnatunga, C.; Pillai, R.; Channon, K.M. Functional effect of the C242T polymorphism in the NAD(P)H oxidase p22phox gene on vascular superoxide production in atherosclerosis. Circulation 2000, 102, 1744–1747. [Google Scholar] [CrossRef] [PubMed]
- Arca, M.; Conti, B.; Montali, A.; Pignatelli, P.; Campagna, F.; Barillà, F.; Tanzilli, G.; Verna, R.; Vestri, A.; Gaudio, C.; Violi, F. C242T polymorphism of NADPH oxidase p22phox and recurrence of cardiovascular events in coronary artery disease. Arterioscler. Thromb. Vasc. Biol. 2008, 28, 752–757. [Google Scholar] [CrossRef] [PubMed]
- Galijasevic, S.; Maitra, D.; Lu, T.; Sliskovic, I.; Abdulhamid, I.; Abu-Soud, H.M. Myeloperoxidase interaction with peroxynitrite: Chloride deficiency and heme depletion. Free Radic. Biol. Med. 2009, 47, 431–439. [Google Scholar] [CrossRef] [PubMed]
- Shao, B.; Tang, C.; Heinecke, J.W.; Oram, J.F. Oxidation of apolipoprotein A-I by myeloperoxidase impairs the initial interactions with ABCA1 required for signaling and cholesterol export. J. Lipid Res. 2010, 51, 1849–1858. [Google Scholar] [CrossRef] [PubMed]
- Eiserich, J.P.; Baldus, S.; Brennan, M.L.; Ma, W.; Zhang, C.; Tousson, A.; Castro, L.; Lusis, A.J.; Nauseef, W.M.; White, C.R.; et al. Myeloperoxidase, a leukocyte-derived vascular NO oxidase. Science 2002, 296, 2391–2394. [Google Scholar] [CrossRef] [PubMed]
- Daugherty, A.; Dunn, J.L.; Rateri, D.L.; Heinecke, J.W. Myeloperoxidase, a catalyst for lipoprotein oxidation, is expressed in human atherosclerotic lesions. J. Clin. Investig. 1994, 94, 437–444. [Google Scholar] [CrossRef] [PubMed]
- Hazell, L.J.; Arnold, L.; Flowers, D.; Waeg, G.; Malle, E.; Stocker, R. Presence of hypochlorite-modified proteins in human atherosclerotic lesions. J. Clin. Investig. 1996, 97, 1535–1544. [Google Scholar] [CrossRef] [PubMed]
- Leclercq, A.; Houard, X.; Philippe, M.; Ollivier, V.; Sebbag, U.; Meilhac, O.; Michel, J.B. Involvement of intraplaque hemorrhage in atherothrombosis evolution via neutrophil protease enrichment. J. Leukoc. Biol. 2007, 82, 1420–1429. [Google Scholar] [CrossRef] [PubMed]
- Houard, X.; Touat, Z.; Ollivier, V.; Louedec, L.; Philippe, M.; Sebbag, U.; Meilhac, O.; Rossignol, P.; Michel, J.B. Mediators of neutrophil recruitment in human abdominal aortic aneurysms. Cardiovasc. Res. 2009, 82, 532–541. [Google Scholar] [CrossRef] [PubMed]
- Martin-Ventura, J.L.; Leclercq, A.; Blanco-Colio, L.M.; Egido, J.; Rossignol, P.; Meilhac, O.; Michel, J.B. Low plasma levels of HSP70 in patients with carotid atherosclerosis are associated with increased levels of proteolytic markers of neutrophil activation. Atherosclerosis 2007, 194, 334–341. [Google Scholar] [CrossRef] [PubMed]
- Brennan, M.L.; Penn, M.S.; Van Lente, F.; Nambi, V.; Shishehbor, M.H.; Aviles, R.J.; Goormastic, M.; Pepoy, M.L.; McErlean, E.S.; Topol, E.J.; et al. Prognostic value of myeloperoxidase in patients with chest pain. N. Engl. J. Med. 2003, 349, 1595–1604. [Google Scholar] [CrossRef] [PubMed]
- Kacprzak, M.; Zielinska, M. Prognostic value of myeloperoxidase concentration in patients with ST-segment elevation myocardial infarction treated with primary percutaneous coronary intervention. Int. J. Cardiol. 2016, 223, 452–457. [Google Scholar] [CrossRef] [PubMed]
- Khine, H.W.; Teiber, J.F.; Haley, R.W.; Khera, A.; Ayers, C.R.; Rohatgi, A. Association of the serum myeloperoxidase/high-density lipoprotein particle ratio and incident cardiovascular events in a multi-ethnic population: Observations from the Dallas Heart Study. Atherosclerosis 2017, 263, 156–162. [Google Scholar] [CrossRef] [PubMed]
- Stadler, N.; Lindner, R.A.; Davies, M.J. Direct detection and quantification of transition metal ions in human atherosclerotic plaques: Evidence for the presence of elevated levels of iron and copper. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 949–954. [Google Scholar] [CrossRef] [PubMed]
- Delbosc, S.; Bayles, R.G.; Laschet, J.; Ollivier, V.; Ho-Tin-Noé, B.; Touat, Z.; Deschildre, C.; Morvan, M.; Louedec, L.; Gouya, L.; et al. Erythrocyte Efferocytosis by the Arterial Wall Promotes Oxidation in Early-Stage Atheroma in Humans. Front. Cardiovasc. Med. 2017, 4, 43. [Google Scholar] [CrossRef] [PubMed]
- Nagy, E.; Eaton, J.W.; Jeney, V.; Soares, M.P.; Varga, Z.; Galajda, Z.; Szentmiklósi, J.; Méhes, G.; Csonka, T.; Smith, A.; et al. Red cells, hemoglobin, heme, iron, and atherogenesis. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 1347–1353. [Google Scholar] [CrossRef] [PubMed]
- Kraml, P. The role of iron in the pathogenesis of atherosclerosis. Physiol. Res. 2017, 66 (Suppl. 1), S55–S67. [Google Scholar] [PubMed]
- Sawada, H.; Hao, H.; Naito, Y.; Oboshi, M.; Hirotani, S.; Mitsuno, M.; Miyamoto, Y.; Hirota, S.; Masuyama, T. Aortic iron overload with oxidative stress and inflammation in human and murine abdominal aortic aneurysm. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 1507–1514. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, J.L. Iron and sex difference in heart disease risk. Lancet 1981, 1, 1293–1294. [Google Scholar] [CrossRef]
- Martinez-Pinna, R.; Lindholt, J.S.; Madrigal-Matute, J.; Blanco-Colio, L.M.; Esteban-Salan, M.; Torres-Fonseca, M.M.; Lefebvre, T.; Delbosc, S.; Laustsen, J.; Driss, F.; et al. From tissue iron retention to low systemic haemoglobin levels, new pathophysiological biomarkers of human abdominal aortic aneurysm. Thromb. Haemost. 2014, 112, 87–95. [Google Scholar] [CrossRef] [PubMed]
- Sarnak, M.J.; Tighiouart, H.; Manjunath, G.; MacLeod, B.; Griffith, J.; Salem, D.; Levey, A.S. Anemia as a risk factor for cardiovascular disease in The Atherosclerosis Risk in Communities (ARIC) study. J. Am. Coll. Cardiol. 2002, 40, 27–33. [Google Scholar] [CrossRef]
- Weiss, G. Iron metabolism in the anemia of chronic disease. Biochim. Biophys. Acta 2009, 1790, 682–693. [Google Scholar] [CrossRef] [PubMed]
- Griendling, K.K.; Touyz, R.M.; Zweier, J.L.; Dikalov, S.; Chilian, W.; Chen, Y.R.; Harrison, D.G.; Bhatnagar, A. American Heart Association Council on Basic Cardiovascular Sciences. Measurement of Reactive Oxygen Species, Reactive Nitrogen Species, and Redox-Dependent Signaling in the Cardiovascular System: A Scientific Statement from the American Heart Association. Circ. Res. 2016, 119, 39–75. [Google Scholar] [CrossRef] [PubMed]
- Downs, J.R.; Clearfield, M.; Weis, S.; Whitney, E.; Shapiro, D.R.; Beere, P.A.; Langendorfer, A.; Stein, E.A.; Kruyer, W.; Gotto, A.M., Jr. Primary prevention of acute coronary events with lovastatin in men and women with average cholesterol levels: Results of AFCAPS/TexCAPS. Air Force/Texas Coronary Atherosclerosis Prevention Study. JAMA 1998, 279, 1615–1622. [Google Scholar] [CrossRef] [PubMed]
- Parthasarathy, S.; Steinberg, D.; Witztum, J.L. The role of oxidized low-density lipoproteins in the pathogenesis of atherosclerosis. Annu. Rev. Med. 1992, 43, 219–225. [Google Scholar] [CrossRef] [PubMed]
- Ball, R.Y.; Bindman, J.P.; Carpenter, K.L.; Mitchinson, M.J. Oxidized low density lipoprotein induces ceroid accumulation by murine peritoneal macrophages in vitro. Atherosclerosis 1986, 60, 173–181. [Google Scholar] [CrossRef]
- Lee, F.Y.; Lee, T.S.; Pan, C.C.; Huang, A.L.; Chau, L.Y. Colocalization of iron and ceroid in human atherosclerotic lesions. Atherosclerosis 1998, 138, 281–288. [Google Scholar] [CrossRef]
- Haka, A.S.; Kramer, J.R.; Dasari, R.R.; Fitzmaurice, M. Mechanism of ceroid formation in atherosclerotic plaque: In situ studies using a combination of Raman and fluorescence spectroscopy. J. Biomed. Opt. 2011, 16, 011011. [Google Scholar] [CrossRef] [PubMed]
- Delbosc, S.; Diallo, D.; Dejouvencel, T.; Lamiral, Z.; Louedec, L.; Martin-Ventura, J.L.; Rossignol, P.; Leseche, G.; Michel, J.B.; Meilhac, O. Impaired high-density lipoprotein anti-oxidant capacity in human abdominal aortic aneurysm. Cardiovasc. Res. 2013, 100, 307–315. [Google Scholar] [CrossRef] [PubMed]
- Pappenheimer, A.M.; Victor, J. Ceroid Pigment in Human Tissues. Am. J. Pathol. 1946, 22, 395–413. [Google Scholar] [PubMed]
- Glavind, J.; Hartmann, S. The occurrence of peroxidized lipids in atheromatous human aortas. Experientia 1951, 7, 464. [Google Scholar] [CrossRef] [PubMed]
- Ylä-Herttuala, S.; Palinski, W.; Rosenfeld, M.E.; Parthasarathy, S.; Carew, T.E.; Butler, S.; Witztum, J.L.; Steinberg, D. Evidence for the presence of oxidatively modified low density lipoprotein in atherosclerotic lesions of rabbit and man. J. Clin. Investig. 1989, 84, 1086–1095. [Google Scholar] [CrossRef] [PubMed]
- Malle, E.; Marsche, G.; Arnhold, J.; Davies, M.J. Modification of low-density lipoprotein by myeloperoxidase-derived oxidants and reagent hypochlorous acid. Biochim. Biophys. Acta. 2006, 1761, 392–415. [Google Scholar] [CrossRef] [PubMed]
- Kotani, K.; Maekawa, M.; Kanno, T.; Kondo, A.; Toda, N.; Manabe, M. Distribution of immunoreactive malondialdehyde-modified low-density lipoprotein in human serum. Biochim. Biophys. Acta 1994, 1215, 121–125. [Google Scholar] [CrossRef]
- Fogelman, A.M.; Shechter, I.; Seager, J.; Hokom, M.; Child, J.S.; Edwards, P.A. Malondialdehyde alteration of low density lipoproteins leads to cholesteryl ester accumulation in human monocyte-macrophages. Proc. Natl. Acad. Sci. USA 1980, 77, 2214–2218. [Google Scholar] [CrossRef] [PubMed]
- Toshima, S.; Hasegawa, A.; Kurabayashi, M.; Itabe, H.; Takano, T.; Sugano, J.; Shimamura, K.; Kimura, J.; Michishita, I.; Suzuki, T.; et al. Circulating oxidized low density lipoprotein levels. A biochemical risk marker for coronary heart disease. Arterioscler. Thromb. Vasc. Biol. 2000, 20, 2243–2247. [Google Scholar] [CrossRef] [PubMed]
- Tanaga, K.; Bujo, H.; Inoue, M.; Mikami, K.; Kotani, K.; Takahashi, K.; Kanno, T.; Saito, Y. Increased circulating malondialdehyde-modified LDL levels in patients with coronary artery diseases and their association with peak sizes of LDL particles. Arterioscler. Thromb. Vasc. Biol. 2002, 22, 662–666. [Google Scholar] [CrossRef] [PubMed]
- Kotani, K.; Tashiro, J.; Yamazaki, K.; Nakamura, Y.; Miyazaki, A.; Bujo, H.; Saito, Y.; Kanno, T.; Maekawa, M. Investigation of MDA-LDL (malondialdehyde-modified low-density lipoprotein) as a prognostic marker for coronary artery disease in patients with type 2 diabetes mellitus. Clin. Chim. Acta 2015, 450, 145–150. [Google Scholar] [CrossRef] [PubMed]
- Amaki, T.; Suzuki, T.; Nakamura, F.; Hayashi, D.; Imai, Y.; Morita, H.; Fukino, K.; Nojiri, T.; Kitano, S.; Hibi, N.; et al. Circulating malondialdehyde modified LDL is a biochemical risk marker for coronary artery disease. Heart 2004, 90, 1211–1213. [Google Scholar] [CrossRef] [PubMed]
- Matsuo, Y.; Kubo, T.; Okumoto, Y.; Ishibashi, K.; Komukai, K.; Tanimoto, T.; Ino, Y.; Kitabata, H.; Hirata, K.; Imanishi, T.; et al. Circulating malondialdehyde-modified low-density lipoprotein levels are associated with the presence of thin-cap fibroatheromas determined by optical coherence tomography in coronary artery disease. Eur. Heart J. Cardiovasc. Imaging 2013, 14, 43–50. [Google Scholar] [CrossRef] [PubMed]
- Ito, T.; Fujita, H.; Tani, T.; Ohte, N. Malondialdehyde-modified low-density lipoprotein is a predictor of cardiac events in patients with stable angina on lipid-lowering therapy after percutaneous coronary intervention using drug-eluting stent. Atherosclerosis 2015, 239, 311–317. [Google Scholar] [CrossRef] [PubMed]
- Gounopoulos, P.; Merki, E.; Hansen, L.F.; Choi, S.H.; Tsimikas, S. Antibodies to oxidized low density lipoprotein: Epidemiological studies and potential clinical applications in cardiovascular disease. Minerva Cardioangiol. 2007, 55, 821–837. [Google Scholar] [PubMed]
- Ravandi, A.; Boekholdt, S.M.; Mallat, Z.; Talmud, P.J.; Kastelein, J.J.; Wareham, N.J.; Miller, E.R.; Benessiano, J.; Tedgui, A.; Witztum, J.L.; et al. Relationship of IgG and IgM autoantibodies and immune complexes to oxidized LDL with markers of oxidation and inflammation and cardiovascular events: Results from the EPIC-Norfolk Study. J. Lipid Res. 2011, 52, 1829–1836. [Google Scholar] [CrossRef] [PubMed]
- Tsimikas, S.; Willeit, P.; Willeit, J.; Santer, P.; Mayr, M.; Xu, Q.; Mayr, A.; Witztum, J.L.; Kiechl, S. Oxidation-specific biomarkers, prospective 15-year cardiovascular and stroke outcomes, and net reclassification of cardiovascular events. J. Am. Coll. Cardiol. 2012, 60, 2218–2229. [Google Scholar] [CrossRef] [PubMed]
- Bowry, V.W.; Stanley, K.K.; Stocker, R. High density lipoprotein is the major carrier of lipid hydroperoxides in human blood plasma from fasting donors. Proc. Natl. Acad. Sci. USA 1992, 89, 10316–10320. [Google Scholar] [CrossRef] [PubMed]
- Favari, E.; Chroni, A.; Tietge, U.J.; Zanotti, I.; Escolà-Gil, J.C.; Bernini, F. Cholesterol efflux and reverse cholesterol transport. Handb. Exp. Pharmacol. 2015, 224, 181–206. [Google Scholar] [PubMed]
- Murphy, A.J.; Woollard, K.J. High-density lipoprotein: A potent inhibitor of inflammation. Clin. Exp. Pharmacol. Physiol. 2010, 37, 710–718. [Google Scholar] [CrossRef] [PubMed]
- Mineo, C.; Deguchi, H.; Griffin, J.H.; Shaul, P.W. Endothelial and antithrombotic actions of HDL. Circ. Res. 2006, 98, 1352–1364. [Google Scholar] [CrossRef] [PubMed]
- Gordon, D.J.; Probstfield, J.L.; Garrison, R.J.; Neaton, J.D.; Castelli, W.P.; Knoke, J.D.; Jacobs, D.R., Jr.; Bangdiwala, S.; Tyroler, H.A. High-density lipoprotein cholesterol and cardiovascular disease. Four prospective American studies. Circulation 1989, 79, 8–15. [Google Scholar] [CrossRef] [PubMed]
- Golledge, J.; van Bockxmeer, F.; Jamrozik, K.; McCann, M.; Norman, P.E. Association between serum lipoproteins and abdominal aortic aneurysm. Am. J. Cardiol. 2010, 105, 1480–1484. [Google Scholar] [CrossRef] [PubMed]
- Stather, P.W.; Sidloff, D.A.; Dattani, N.; Gokani, V.J.; Choke, E.; Sayers, R.D.; Bown, M.J. Meta-analysis and meta-regression analysis of biomarkers for abdominal aortic aneurysm. Br. J. Surg. 2014, 101, 1358–1372. [Google Scholar] [CrossRef] [PubMed]
- Burillo, E.; Lindholt, J.S.; Molina-Sánchez, P.; Jorge, I.; Martinez-Pinna, R.; Blanco-Colio, L.M.; Tarin, C.; Torres-Fonseca, M.M.; Esteban, M.; Laustsen, J.; et al. ApoA-I/HDL-C levels are inversely associated with abdominal aortic aneurysm progression. Thromb. Haemost. 2015, 113, 1335–1346. [Google Scholar] [CrossRef] [PubMed]
- Burillo, E.; Andres, E.M.; Mateo-Gallego, R.; Fiddyment, S.; Jarauta, E.; Cenarro, A.; Civeira, F. High-density lipoprotein cholesterol increase and non-cardiovascular mortality: A meta-analysis. Heart 2010, 96, 1345–1351. [Google Scholar] [CrossRef] [PubMed]
- Karathanasis, S.K.; Freeman, L.A.; Gordon, S.M.; Remaley, A.T. The Changing Face of HDL and the Best Way to Measure It. Clin. Chem. 2017, 63, 196–210. [Google Scholar] [CrossRef] [PubMed]
- Khera, A.V.; Cuchel, M.; de la Llera-Moya, M.; Rodrigues, A.; Burke, M.F.; Jafri, K.; French, B.C.; Phillips, J.A.; Mucksavage, M.L.; Wilensky, R.L.; et al. Cholesterol efflux capacity, high-density lipoprotein function, and atherosclerosis. N. Engl. J. Med. 2011, 364, 127–135. [Google Scholar] [CrossRef] [PubMed]
- Rohatgi, A.; Khera, A.; Berry, J.D.; Givens, E.G.; Ayers, C.R.; Wedin, K.E.; Neeland, I.J.; Yuhanna, I.S.; Rader, D.R.; de Lemos, J.A.; et al. HDL cholesterol efflux capacity and incident cardiovascular events. N. Engl. J. Med. 2014, 371, 2383–2393. [Google Scholar] [CrossRef] [PubMed]
- O’Reilly, M.; Dillon, E.; Guo, W.; Finucane, O.; McMorrow, A.; Murphy, A.; Lyons, C.; Jones, D.; Ryan, M.; Gibney, M.; et al. High-Density Lipoprotein Proteomic Composition, and not Efflux Capacity, Reflects Differential Modulation of Reverse Cholesterol Transport by Saturated and Monounsaturated Fat Diets. Circulation 2016, 133, 1838–1850. [Google Scholar] [CrossRef] [PubMed]
- Ortiz-Muñoz, G.; Houard, X.; Martín-Ventura, J.L.; Ishida, B.Y.; Loyau, S.; Rossignol, P.; Moreno, J.A.; Kane, J.P.; Chalkley, R.J.; Burlingame, A.L.; et al. HDL antielastase activity prevents smooth muscle cell anoikis, a potential new antiatherogenic property. FASEB J. 2009, 23, 3129–3139. [Google Scholar] [CrossRef] [PubMed]
- Rosenson, R.S.; Brewer, H.B., Jr.; Ansell, B.J.; Barter, P.; Chapman, M.J.; Heinecke, J.W.; Kontush, A.; Tall, A.R.; Webb, N.R. Dysfunctional HDL and atherosclerotic cardiovascular disease. Nat. Rev. Cardiol. 2016, 13, 48–60. [Google Scholar] [CrossRef] [PubMed]
- Zheng, L.; Nukuna, B.; Brennan, M.L.; Sun, M.; Goormastic, M.; Settle, M.; Schmitt, D.; Fu, X.; Thomson, L.; Fox, P.L.; et al. Apolipoprotein A-I is a selective target for myeloperoxidase-catalyzed oxidation and functional impairment in subjects with cardiovascular disease. J. Clin. Investig. 2004, 114, 529–541. [Google Scholar] [CrossRef] [PubMed]
- Bergt, C.; Pennathur, S.; Fu, X.; Byun, J.; O’Brien, K.; McDonald, T.O.; Singh, P.; Anantharamaiah, G.M.; Chait, A.; Brunzell, J.; et al. The myeloperoxidase product hypochlorous acid oxidizes HDL in the human artery wall and impairs ABCA1-dependent cholesterol transport. Proc. Natl. Acad. Sci. USA 2004, 101, 13032–13037. [Google Scholar] [CrossRef] [PubMed]
- Shao, B.; Bergt, C.; Fu, X.; Green, P.; Voss, J.C.; Oda, M.N.; Oram, J.F.; Heinecke, J.W. Tyrosine 192 in apolipoprotein A-I is the major site of nitration and chlorination by myeloperoxidase, but only chlorination markedly impairs ABCA1-dependent cholesterol transport. J. Biol. Chem. 2005, 280, 5983–5993. [Google Scholar] [CrossRef] [PubMed]
- Chan, G.K.; Witkowski, A.; Gantz, D.L.; Zhang, T.O.; Zanni, M.T.; Jayaraman, S.; Cavigiolio, G. Myeloperoxidase-mediated Methionine Oxidation Promotes an Amyloidogenic Outcome for Apolipoprotein A-I. J. Biol. Chem. 2015, 290, 10958–10971. [Google Scholar] [CrossRef] [PubMed]
- DiDonato, J.A.; Huang, Y.; Aulak, K.S.; Even-Or, O.; Gerstenecker, G.; Gogonea, V.; Wu, Y.; Fox, P.L.; Tang, W.H.; Plow, E.F.; et al. Function and distribution of apolipoprotein A1 in the artery wall are markedly distinct from those in plasma. Circulation 2013, 128, 1644–1655. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; DiDonato, J.A.; Levison, B.S.; Schmitt, D.; Li, L.; Wu, Y.; Buffa, J.; Kim, T.; Gerstenecker, G.S.; Gu, X.; et al. An abundant dysfunctional apolipoprotein A1 in human atheroma. Nat. Med. 2014, 20, 193–203. [Google Scholar] [CrossRef] [PubMed]
- Yassine, H.N.; Jackson, A.M.; Reaven, P.D.; Nedelkov, D.; Nelson, R.W.; Lau, S.S.; Borchers, C.H. The Application of Multiple Reaction Monitoring to Assess Apo A-I Methionine Oxidations in Diabetes and Cardiovascular Disease. Transl. Proteom. 2014, 4–5, 18–24. [Google Scholar] [CrossRef] [PubMed]
- Harel, M.; Aharoni, A.; Gaidukov, L.; Brumshtein, B.; Khersonsky, O.; Meged, R.; Dvir, H.; Ravelli, R.B.; McCarthy, A.; Toker, L.; et al. Structure and evolution of the serum paraoxonase family of detoxifying and anti-atherosclerotic enzymes. Nat. Struct. Mol. Biol. 2004, 11, 412–419. [Google Scholar] [CrossRef] [PubMed]
- Deakin, S.P.; Bioletto, S.; Bochaton-Piallat, M.L.; James, R.W. HDL-associated paraoxonase-1 can redistribute to cell membranes and influence sensitivity to oxidative stress. Free Radic. Biol. Med. 2011, 50, 102–109. [Google Scholar] [CrossRef] [PubMed]
- Granér, M.; James, R.W.; Kahri, J.; Nieminen, M.S.; Syvänne, M.; Taskinen, M.R. Association of paraoxonase-1 activity and concentration with angiographic severity and extent of coronary artery disease. J. Am. Coll. Cardiol. 2006, 47, 2429–2435. [Google Scholar] [CrossRef] [PubMed]
- Mackness, B.; Durrington, P.; McElduff, P.; Yarnell, J.; Azam, N.; Watt, M.; Mackness, M. Low paraoxonase activity predicts coronary events in the Caerphilly Prospective Study. Circulation 2003, 107, 2775–2779. [Google Scholar] [CrossRef] [PubMed]
- Tang, W.H.; Hartiala, J.; Fan, Y.; Wu, Y.; Stewart, A.F.; Erdmann, J.; Kathiresan, S.; Roberts, R.; McPherson, R.; Allayee, H.; et al. Clinical and genetic association of serum paraoxonase and arylesterase activities with cardiovascular risk. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 2803–2812. [Google Scholar] [CrossRef] [PubMed]
- Kunutsor, S.K.; Bakker, S.J.; James, R.W.; Dullaart, R.P. Serum paraoxonase-1 activity and risk of incident cardiovascular disease: The PREVEND study and meta-analysis of prospective population studies. Atherosclerosis 2016, 245, 143–154. [Google Scholar] [CrossRef] [PubMed]
- Burillo, E.; Tarin, C.; Torres-Fonseca, M.M.; Fernandez-García, C.E.; Martinez-Pinna, R.; Martinez-Lopez, D.; Llamas-Granda, P.; Camafeita, E.; Lopez, J.A.; Vega de Ceniga, M.; et al. Paraoxonase-1 overexpression prevents experimental abdominal aortic aneurysm progression. Clin. Sci. (Lond.) 2016, 130, 1027–1038. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharyya, T.; Nicholls, S.J.; Topol, E.J.; Zhang, R.; Yang, X.; Schmitt, D.; Fu, X.; Shao, M.; Brennan, D.M.; Ellis, S.G.; et al. Relationship of paraoxonase 1 (PON1) gene polymorphisms and functional activity with systemic oxidative stress and cardiovascular risk. JAMA 2008, 299, 1265–1276. [Google Scholar] [CrossRef] [PubMed]
- Pietarinen-Runtti, P.; Lakari, E.; Raivio, K.O.; Kinnula, V.L. Expression of antioxidant enzymes in human inflammatory cells. Am. J. Physiol. Cell. Physiol. 2000, 278, 118–125. [Google Scholar]
- Ramos-Mozo, P.; Madrigal-Matute, J.; Martinez-Pinna, R.; Blanco-Colio, L.M.; Lopez, J.A.; Camafeita, E.; Meilhac, O.; Michel, J.B.; Aparicio, C.; Vega de Ceniga, M.; et al. Proteomic analysis of polymorphonuclear neutrophils identifies catalase as a novel biomarker of abdominal aortic aneurysm: Potential implication of oxidative stress in abdominal aortic aneurysm progression. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 3011–3019. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, S.; Inoue, N.; Azumi, H.; Seno, T.; Hirata, K.; Kawashima, S.; Hayashi, Y.; Itoh, H.; Yokozaki, H.; Yokoyama, M. Expressional changes of the vascular antioxidant system in atherosclerotic coronary arteries. J. Atheroscler. Thromb. 2002, 9, 184–190. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, H.; Nakamura, K.; Yodoi, J. Redox regulation of cellular activation. Annu. Rev. Immunol. 1997, 15, 351–369. [Google Scholar] [CrossRef] [PubMed]
- Couchie, D.; Vaisman, B.; Abderrazak, A.; Mahmood, D.F.D.; Hamza, M.M.; Canesi, F.; Diderot, V.; El Hadri, K.; Nègre-Salvayre, A.; Le Page, A.; et al. Human Plasma Thioredoxin-80 Increases With Age and in ApoE−/− Mice Induces Inflammation, Angiogenesis, and Atherosclerosis. Circulation 2017, 136, 464–475. [Google Scholar] [CrossRef] [PubMed]
- Okuda, M.; Inoue, N.; Azumi, H.; Seno, T.; Sumi, Y.; Hirata, K.; Kawashima, S.; Hayashi, Y.; Itoh, H.; Yodoi, J.; et al. Expression of glutaredoxin in human coronary arteries: Its potential role in antioxidant protection against atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2001, 21, 1483–1487. [Google Scholar] [CrossRef] [PubMed]
- Nishihira, K.; Yamashita, A.; Imamura, T.; Hatakeyama, K.; Sato, Y.; Nakamura, H.; Yodoi, J.; Ogawa, H.; Kitamura, K.; Asada, Y. Thioredoxin in coronary culprit lesions: Possible relationship to oxidative stress and intraplaque hemorrhage. Atherosclerosis 2008, 201, 360–367. [Google Scholar] [CrossRef] [PubMed]
- Furman, C.; Rundlöf, A.K.; Larigauderie, G.; Jaye, M.; Bricca, G.; Copin, C.; Kandoussi, A.M.; Fruchart, J.C.; Arnér, E.S.; Rouis, M. Thioredoxin reductase 1 is upregulated in atherosclerotic plaques: Specific induction of the promoter in human macrophages by oxidized low-density lipoproteins. Free Radic. Biol. Med. 2004, 37, 71–85. [Google Scholar] [CrossRef] [PubMed]
- Madrigal-Matute, J.; Fernandez-Garcia, C.E.; Blanco-Colio, L.M.; Burillo, E.; Fortuño, A.; Martinez-Pinna, R.; Llamas-Granda, P.; Beloqui, O.; Egido, J.; Zalba, G.; et al. Thioredoxin-1/peroxiredoxin-1 as sensors of oxidative stress mediated by NADPH oxidase activity in atherosclerosis. Free Radic. Biol. Med. 2015, 86, 352–361. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Pinna, R.; Ramos-Mozo, P.; Madrigal-Matute, J.; Blanco-Colio, L.M.; Lopez, J.A.; Calvo, E.; Camafeita, E.; Lindholt, J.S.; Meilhac, O.; Delbosc, S.; et al. Identification of peroxiredoxin-1 as a novel biomarker of abdominal aortic aneurysm. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 935–943. [Google Scholar] [CrossRef] [PubMed]
- Urbonavicius, S.; Lindholt, J.S.; Vorum, H.; Urbonaviciene, G.; Henneberg, E.W.; Honoré, B. Proteomic identification of differentially expressed proteins in aortic wall of patients with ruptured and nonruptured abdominal aortic aneurysms. J. Vasc. Surg. 2009, 49, 455–463. [Google Scholar] [CrossRef] [PubMed]
- Miller, F.J., Jr.; Sharp, W.J.; Fang, X.; Oberley, L.W.; Oberley, T.D.; Weintraub, N.L. Oxidative stress in human abdominal aortic aneurysms: A potential mediator of aneurysmal remodeling. Arterioscler. Thromb. Vasc. Biol. 2002, 22, 560–565. [Google Scholar] [CrossRef] [PubMed]
- Miyamoto, S.; Sakamoto, T.; Soejima, H.; Shimomura, H.; Kajiwara, I.; Kojima, S.; Hokamaki, J.; Sugiyama, S.; Yoshimura, M.; Ozaki, Y.; et al. Plasma thioredoxin levels and platelet aggregability in patients with acute myocardial infarction. Am. Heart J. 2003, 146, 465–471. [Google Scholar] [CrossRef]
- Hokamaki, J.; Kawano, H.; Soejima, H.; Miyamoto, S.; Kajiwara, I.; Kojima, S.; Sakamoto, T.; Sugiyama, S.; Yoshimura, M.; Nakamura, H.; et al. Plasma thioredoxin levels in patients with unstable angina. Int. J. Cardiol. 2005, 99, 225–231. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Pinna, R.; Lindholt, J.S.; Blanco-Colio, L.M.; Dejouvencel, T.; Madrigal-Matute, J.; Ramos-Mozo, P.; Vega de Ceniga, M.; Michel, J.B.; Egido, J.; Meilhac, O.; et al. Increased levels of thioredoxin in patients with abdominal aortic aneurysms (AAAs). A potential link of oxidative stress with AAA evolution. Atherosclerosis 2010, 212, 333–338. [Google Scholar] [CrossRef] [PubMed]
- Gori, T.; Münzel, T. Oxidative stress and endothelial dysfunction: Therapeutic implications. Ann. Med. 2011, 43, 259–272. [Google Scholar] [CrossRef] [PubMed]
- Stylos, E.; Chatziathanasiadou, M.V.; Tsiailanis, A.; Kellici, T.F.; Tsoumani, M.; Kostagianni, A.D.; Deligianni, M.; Tselepis, A.D.; Tzakos, A.G. Tailoring naringenin conjugates with amplified and triple antiplatelet activity profile: Rational design, synthesis, human plasma stability and in vitro evaluation. Biochim. Biophys. Acta 2017, 1861, 2609–2618. [Google Scholar] [CrossRef] [PubMed]
- Martinez de Lizarrondo, S.; Gakuba, C.; Herbig, B.A.; Repessé, Y.; Ali, C.; Denis, C.V.; Lenting, P.J.; Touzé, E.; Diamond, S.L.; Vivien, D.; et al. Potent Thrombolytic Effect of N-Acetylcysteine on Arterial Thrombi. Circulation 2017, 136, 646–660. [Google Scholar] [CrossRef] [PubMed]
- Furie, B.; Flaumenhaft, R. Thiol isomerases in thrombus formation. Circ. Res. 2014, 114, 1162–1173. [Google Scholar] [CrossRef] [PubMed]
- Pignatelli, P.; Carnevale, R.; Pastori, D.; Cangemi, R.; Napoleone, L.; Bartimoccia, S.; Nocella, C.; Basili, S.; Violi, F. Immediate antioxidant and antiplatelet effect of atorvastatin via inhibition of Nox2. Circulation 2012, 126, 92–103. [Google Scholar] [CrossRef] [PubMed]
- Briones, A.M.; Rodríguez-Criado, N.; Hernanz, R.; García-Redondo, A.B.; Rodrigues-Díez, R.R.; Alonso, M.J.; Egido, J.; Ruiz-Ortega, M.; Salaices, M. Atorvastatin prevents angiotensin II-induced vascular remodeling and oxidative stress. Hypertension 2009, 54, 142–149. [Google Scholar] [CrossRef] [PubMed]
- Wassmann, S.; Laufs, U.; Müller, K.; Konkol, C.; Ahlbory, K.; Bäumer, A.T.; Linz, W.; Böhm, M.; Nickenig, G. Cellular antioxidant effects of atorvastatin in vitro and in vivo. Arterioscler. Thromb. Vasc. Biol. 2002, 22, 300–305. [Google Scholar] [CrossRef] [PubMed]
- Nosalski, R.; McGinnigle, E.; Siedlinski, M.; Guzik, T.J. Novel Immune Mechanisms in Hypertension and Cardiovascular Risk. Curr. Cardiovasc. Risk Rep. 2017, 11, 12. [Google Scholar] [CrossRef] [PubMed]
- Schramm, A.; Matusik, P.; Osmenda, G.; Guzik, T.J. Targeting NADPH oxidases in vascular pharmacology. Vascul. Pharmacol. 2012, 56, 216–231. [Google Scholar] [CrossRef] [PubMed]
- Tousoulis, D.; Psaltopoulou, T.; Androulakis, E.; Papageorgiou, N.; Papaioannou, S.; Oikonomou, E.; Synetos, A.; Stefanadis, C. Oxidative stress and early atherosclerosis: Novel antioxidant treatment. Cardiovasc. Drugs Ther. 2015, 29, 75–88. [Google Scholar] [CrossRef] [PubMed]
- Chow, B.S.; Koulis, C.; Krishnaswamy, P.; Steckelings, U.M.; Unger, T.; Cooper, M.E.; Jandeleit-Dahm, K.A.; Allen, T.J. The angiotensin II type 2 receptor agonist Compound 21 is protective in experimental diabetes-associated atherosclerosis. Diabetologia 2016, 59, 1778–1790. [Google Scholar] [CrossRef] [PubMed]
- Sima, A.; Stancu, C.; Constantinescu, E.; Ologeanu, L.; Simionescu, M. The hyperlipemic hamster—A model for testing the anti-atherogenic effect of amlodipine. J. Cell. Mol. Med. 2001, 5, 153–162. [Google Scholar] [CrossRef] [PubMed]
- Tang, L.; Cong, Z.; Hao, S.; Li, P.; Huang, H.; Shen, Y.; Li, K.; Jing, H. Protective effect of melatonin on the development of abdominal aortic aneurysm in a rat model. J. Surg. Res. 2017, 209, 266–278.e1. [Google Scholar] [CrossRef] [PubMed]
- Xiong, W.; Mactaggart, J.; Knispel, R.; Worth, J.; Zhu, Z.; Li, Y.; Sun, Y.; Baxter, B.T.; Johanning, J. Inhibition of reactive oxygen species attenuates aneurysm formation in a murine model. Atherosclerosis 2009, 202, 128–134. [Google Scholar] [CrossRef] [PubMed]
- Kinoshita, H.; Matsumura, T.; Ishii, N.; Fukuda, K.; Senokuchi, T.; Motoshima, H.; Kondo, T.; Taketa, K.; Kawasaki, S.; Hanatani, S.; et al. Apocynin suppresses the progression of atherosclerosis in apoE-deficient mice by inactivation of macrophages. Biochem. Biophys. Res. Commun. 2013, 431, 124–130. [Google Scholar] [CrossRef] [PubMed]
- Tiyerili, V.; Camara, B.; Becher, M.U.; Schrickel, J.W.; Lütjohann, D.; Mollenhauer, M.; Baldus, S.; Nickenig, G.; Andrié, R.P. Neutrophil-derived myeloperoxidase promotes atherogenesis and neointima formation in mice. Int. J. Cardiol. 2016, 204, 29–36. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.W.; Blomkalns, A.L.; Ogbi, M.; Thomas, M.; Gavrila, D.; Neltner, B.S.; Cassis, L.A.; Thompson, R.W.; Weiss, R.M.; Lindower, P.D.; et al. Role of myeloperoxidase in abdominal aortic aneurysm formation: Mitigation by taurine. Am. J. Physiol. Heart Circ. Physiol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Minqin, R.; Rajendran, R.; Pan, N.; Tan, B.K.; Ong, W.Y.; Watt, F.; Halliwell, B. The iron chelator desferrioxamine inhibits atherosclerotic lesion development and decreases lesion iron concentrations in the cholesterol-fed rabbit. Free Radic. Biol. Med. 2005, 38, 1206–1211. [Google Scholar] [CrossRef] [PubMed]
- Chen, N.; Frishman, W.H. High-Density Lipoprotein Infusion Therapy and Atherosclerosis: Current Research and Future Directions. Cardiol. Rev. 2016, 24, 298–302. [Google Scholar] [CrossRef] [PubMed]
- Badimon, J.J.; Badimon, L.; Galvez, A.; Dische, R.; Fuster, V. High density lipoprotein plasma fractions inhibit aortic fatty streaks in cholesterol-fed rabbits. Lab. Investig. 1989, 60, 455–461. [Google Scholar] [PubMed]
- Badimon, J.J.; Badimon, L.; Fuster, V. Regression of atherosclerotic lesions by high density lipoprotein plasma fraction in the cholesterol-fed rabbit. J. Clin. Investig. 1990, 85, 1234–1241. [Google Scholar] [CrossRef] [PubMed]
- Rubin, E.M.; Krauss, R.M.; Spangler, E.A.; Verstuyft, J.G.; Clift, S.M. Inhibition of early atherogenesis in transgenic mice by human apolipoprotein AI. Nature 1991, 353, 265–267. [Google Scholar] [CrossRef] [PubMed]
- Krishna, S.M.; Seto, S.W.; Moxon, J.V.; Rush, C.; Walker, P.J.; Norman, P.E.; Golledge, J. Fenofibrate increases high-density lipoprotein and sphingosine 1 phosphate concentrations limiting abdominal aortic aneurysm progression in a mouse model. Am. J. Pathol. 2012, 181, 706–718. [Google Scholar] [CrossRef] [PubMed]
- Torsney, E.; Pirianov, G.; Charolidi, N.; Shoreim, A.; Gaze, D.; Petrova, S.; Laing, K.; Meisinger, T.; Xiong, W.; Baxter, B.T.; et al. Elevation of plasma high-density lipoproteins inhibits development of experimental abdominal aortic aneurysms. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 2678–2686. [Google Scholar] [CrossRef] [PubMed]
- Navab, M.; Anantharamaiah, G.M.; Hama, S.; Garber, D.W.; Chaddha, M.; Hough, G.; Lallone, R.; Fogelman, A.M. Oral administration of an Apo A-I mimetic Peptide synthesized from D-amino acids dramatically reduces atherosclerosis in mice independent of plasma cholesterol. Circulation 2002, 105, 290–292. [Google Scholar] [CrossRef] [PubMed]
- Navab, M.; Anantharamaiah, G.M.; Hama, S.; Hough, G.; Reddy, S.T.; Frank, J.S.; Garber, D.W.; Handattu, S.; Fogelman, A.M. D-4F and statins synergize to render HDL antiinflammatory in mice and monkeys and cause lesion regression in old apolipoprotein E-null mice. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 1426–1432. [Google Scholar] [CrossRef] [PubMed]
- Navab, M.; Anantharamaiah, G.M.; Reddy, S.T.; Hama, S.; Hough, G.; Grijalva, V.R.; Wagner, A.C.; Frank, J.S.; Datta, G.; Garber, D.; et al. Oral D-4F causes formation of pre-beta high-density lipoprotein and improves high-density lipoprotein-mediated cholesterol efflux and reverse cholesterol transport from macrophages in apolipoprotein E-null mice. Circulation 2004, 109, 3215–3220. [Google Scholar] [CrossRef] [PubMed]
- Hewing, B.; Parathath, S.; Barrett, T.; Chung, W.K.; Astudillo, Y.M.; Hamada, T.; Ramkhelawon, B.; Tallant, T.C.; Yusufishaq, M.S.; Didonato, J.A.; et al. Effects of native and myeloperoxidase-modified apolipoprotein a-I on reverse cholesterol transport and atherosclerosis in mice. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 779–789. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.F.; Pei, J.F.; Zhang, Y.; Zhang, R.; Wang, F.; Gao, P.; Zhang, Z.Q.; Wang, T.T.; She, Z.G.; Chen, H.Z.; et al. The Paraoxonase Gene Cluster Protects Against Abdominal Aortic Aneurysm Formation. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 291–300. [Google Scholar] [CrossRef] [PubMed]
- Tward, A.; Xia, Y.R.; Wang, X.P.; Shi, Y.S.; Park, C.; Castellani, L.W.; Lusis, A.J.; Shih, D.M. Decreased atherosclerotic lesion formation in human serum paraoxonase transgenic mice. Circulation 2002, 106, 484–490. [Google Scholar] [CrossRef] [PubMed]
- Guo, Z.; Van Remmen, H.; Yang, H.; Chen, X.; Mele, J.; Vijg, J.; Epstein, C.J.; Ho, Y.S.; Richardson, A. Changes in expression of antioxidant enzymes affect cell-mediated LDL oxidation and oxidized LDL-induced apoptosis in mouse aortic cells. Arterioscler. Thromb. Vasc. Biol. 2001, 21, 1131–1138. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Roberts, L.J.; Shi, M.J.; Zhou, L.C.; Ballard, B.R.; Richardson, A.; Guo, Z.M. Retardation of atherosclerosis by overexpression of catalase or both Cu/Zn-superoxide dismutase and catalase in mice lacking apolipoprotein E. Circ. Res. 2004, 95, 1075–1081. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wang, G.Z.; Rabinovitch, P.S.; Tabas, I. Macrophage mitochondrial oxidative stress promotes atherosclerosis and nuclear factor-κB-mediated inflammation in macrophages. Circ. Res. 2014, 114, 421–433. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wang, W.; Wang, N.; Tall, A.R.; Tabas, I. Mitochondrial Oxidative Stress Promotes Atherosclerosis and Neutrophil Extracellular Traps in Aged Mice. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 99–107. [Google Scholar] [CrossRef] [PubMed]
- Maiellaro-Rafferty, K.; Weiss, D.; Joseph, G.; Wan, W.; Gleason, R.L.; Taylor, W.R. Catalase overexpression in aortic smooth muscle prevents pathological mechanical changes underlying abdominal aortic aneurysm formation. Am. J. Physiol. Heart Circ. Physiol. 2011, 301, 355–362. [Google Scholar] [CrossRef] [PubMed]
- Parastatidis, I.; Weiss, D.; Joseph, G.; Taylor, W.R. Overexpression of catalase in vascular smooth muscle cells prevents the formation of abdominal aortic aneurysms. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 2389–2396. [Google Scholar] [CrossRef] [PubMed]
- Ho, Y.C.; Wu, M.L.; Gung, P.Y.; Chen, C.H.; Kuo, C.C.; Yet, S.F. Heme oxygenase-1 deficiency exacerbates angiotensin II-induced aorticaneurysm in mice. Oncotarget 2016, 7, 67760–67776. [Google Scholar] [CrossRef] [PubMed]



© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Martin-Ventura, J.L.; Rodrigues-Diez, R.; Martinez-Lopez, D.; Salaices, M.; Blanco-Colio, L.M.; Briones, A.M. Oxidative Stress in Human Atherothrombosis: Sources, Markers and Therapeutic Targets. Int. J. Mol. Sci. 2017, 18, 2315. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms18112315
Martin-Ventura JL, Rodrigues-Diez R, Martinez-Lopez D, Salaices M, Blanco-Colio LM, Briones AM. Oxidative Stress in Human Atherothrombosis: Sources, Markers and Therapeutic Targets. International Journal of Molecular Sciences. 2017; 18(11):2315. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms18112315
Chicago/Turabian StyleMartin-Ventura, Jose Luis, Raquel Rodrigues-Diez, Diego Martinez-Lopez, Mercedes Salaices, Luis Miguel Blanco-Colio, and Ana M. Briones. 2017. "Oxidative Stress in Human Atherothrombosis: Sources, Markers and Therapeutic Targets" International Journal of Molecular Sciences 18, no. 11: 2315. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms18112315