Collateral Damage Intended—Cancer-Associated Fibroblasts and Vasculature Are Potential Targets in Cancer Therapy


Abstract
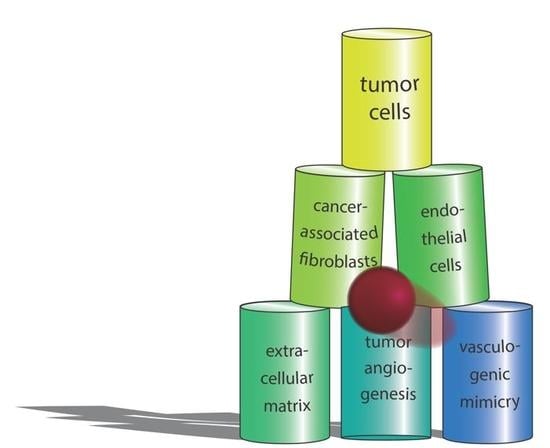
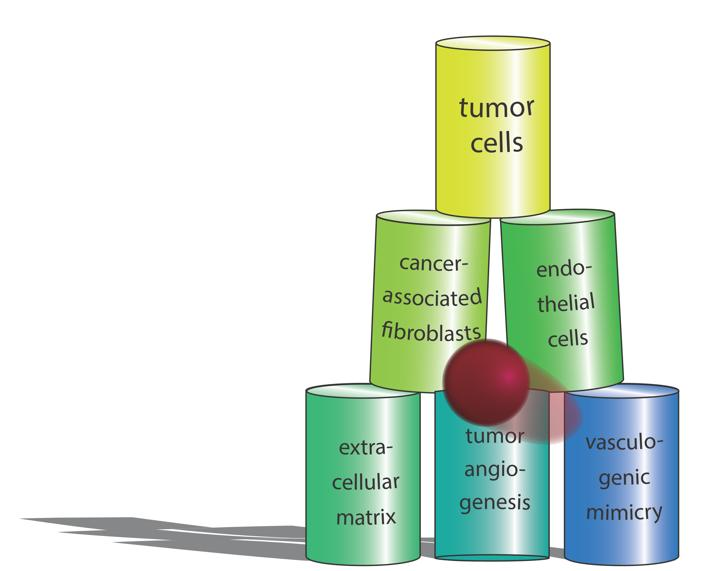
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
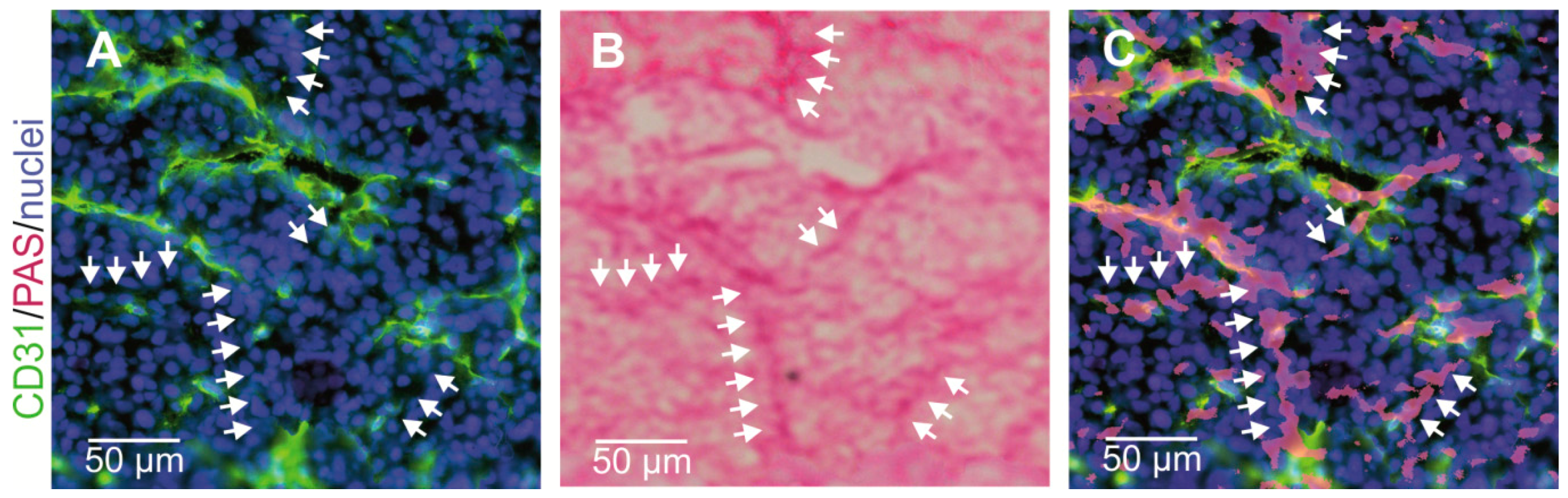{kind=link}
{kind=link}
1. Introduction
2. Setting the Stage: Cancer Cells Determine the Tumor Microenvironment via Metabolites and Cytokines, via Cell–Matrix and Cell–Cell Contacts
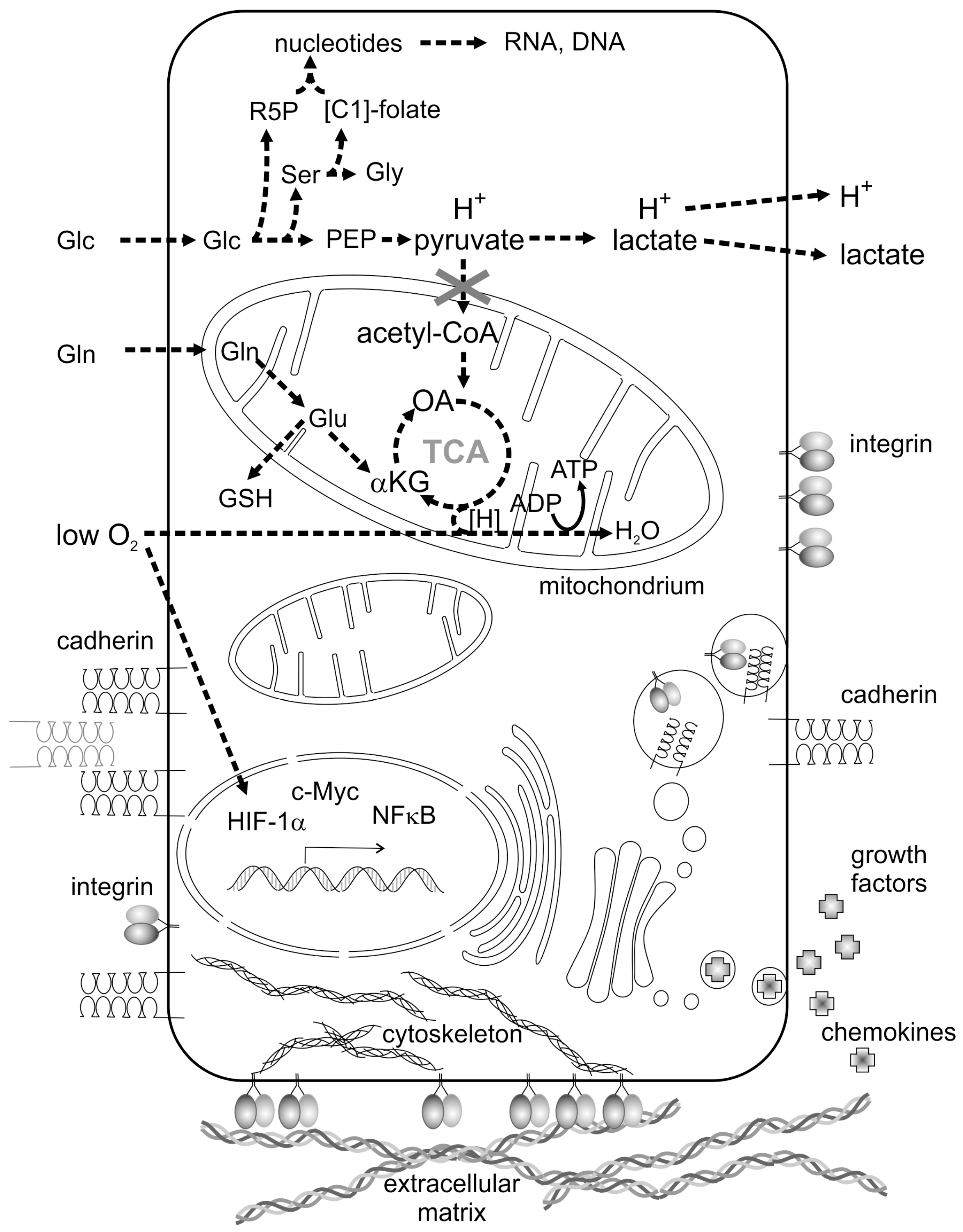
2.1. Metabolic Reprogramming of Cancer Cells
2.2. Cohesion, Adhesion and Soluble Mediators in the Communication between Tumor Cells
3. Stromal Fibroblasts, the Immediate Neighbors of Tumor Cells
3.1. CAFs Are Crucial for the Maintenance of a Pro-Tumorigenic TME
3.2. ECM Is a Means of Communication in the TME and Signals via Distinct Parameters: Qualitative and Quantitative Composition, Cross-Linkage of Supramolecular Structures, Tensional Status and Degradation
4. Interactions of Cancer Cells with Endothelial Cells
4.1. Tumor Vascularization
4.2. Soluble Factors Mediate CEC Interactions during Angiogenesis and Vasculogenesis
4.3. Direct Tumor Cell–Endothelial Cell Interaction and Integration of Tumor Cells in Mosaic Vessels
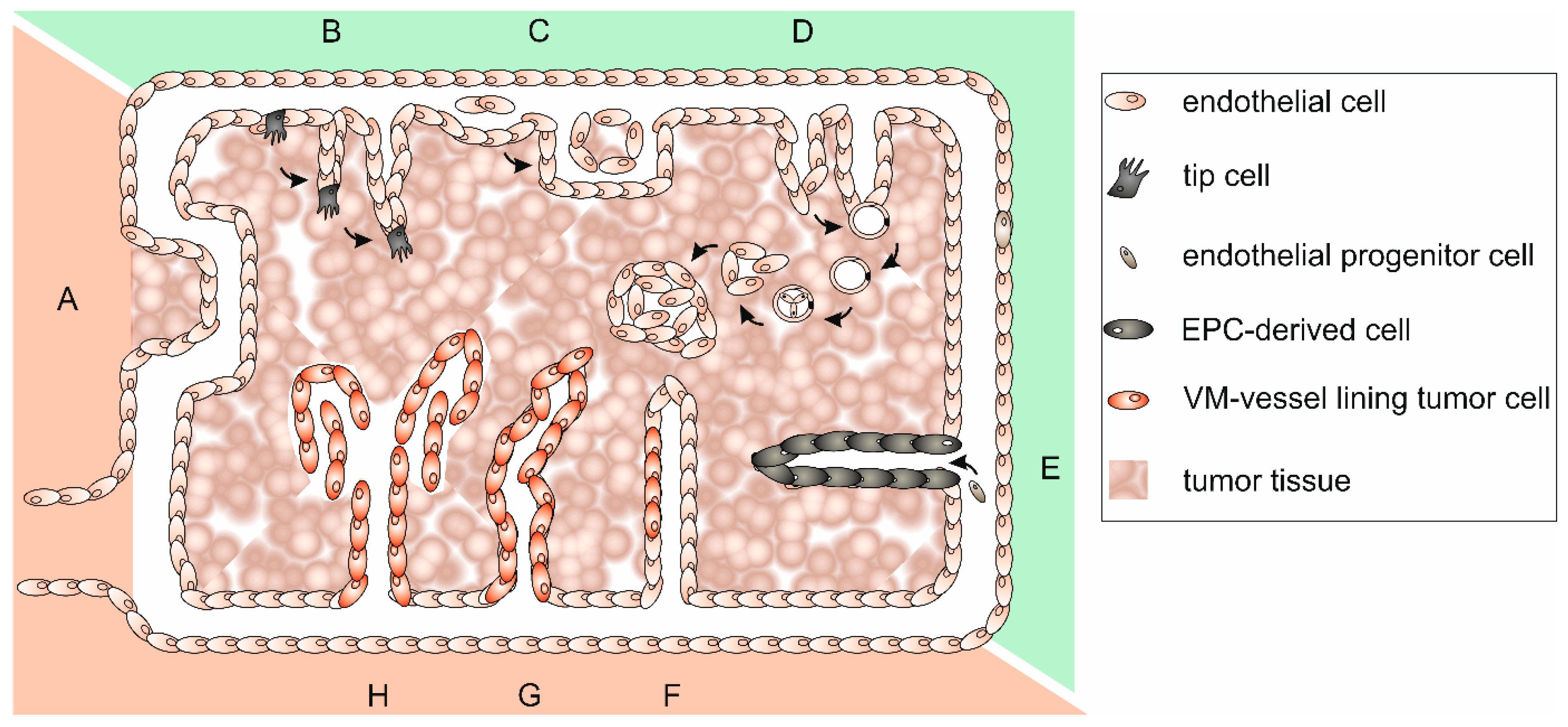
5. Tumor Cells Imitating Endothelial Cells in Vasculogenic Mimicry Vessels
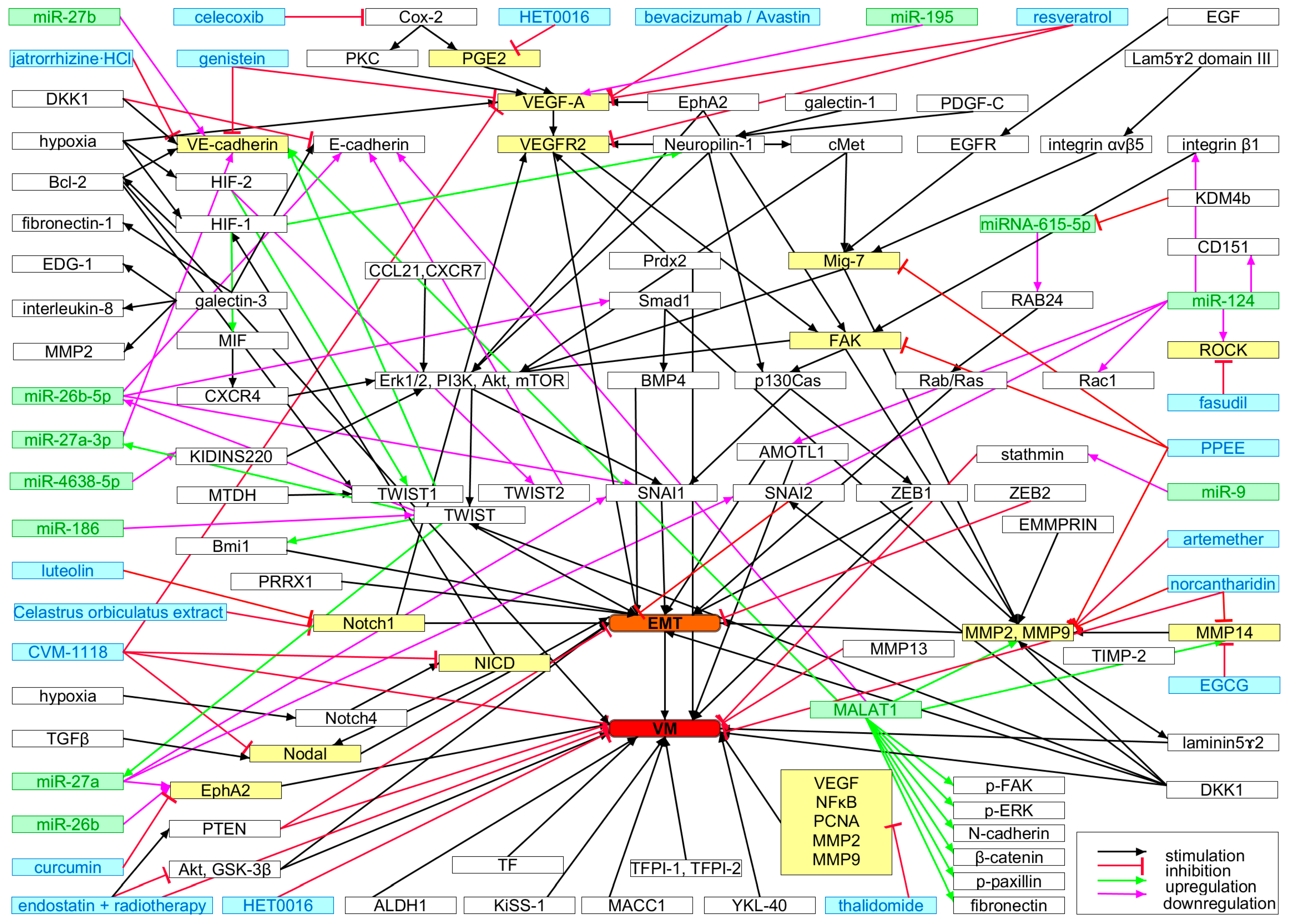
Vasculogenic Mimicry and Its Molecular Phenotypes
6. Perspective: New Cancer Therapies Targeting Tumor Vasculature and CAFs
6.1. Anti-Angiogenesis and Normalization of the Tumor Vasculature
6.2. VM Channels Are a Promising New Therapeutic Target
6.3. Therapeutic Potential of Targeting CAFs
7. Conclusions
Acknowledgments
Conflicts of Interest
Abbreviations
| Akt | Protein kinase B |
| AMOTL1 | Angiomotin-like protein 1 |
| ARF | ADP ribosylation factor |
| αSMA | α-Smooth muscle actin |
| Bcl-2 | B-cell lymphoma 2 |
| bFGF | Basic fibroblast growth factor |
| BM | Basement membrane |
| Bmi1 | B lymphoma Mo-MLV insertion region 1 homolog |
| BMP | Bone morphogenetic protein |
| CAF | Cancer-associated fibroblast |
| CD | Cluster of differentiation |
| CEC | Cancer-endothelial cell interaction |
| CHI3L1 | Chitinase-3-like protein 1 |
| CSC | Cancer stem-like cell |
| cMET | Hepatocyte growth factor receptor |
| c-Myc | Cellular Myelocytomatose (transcription factor) |
| COX-2 | Cyclooxygenase-2 |
| CXC | Cysteine-any amino acid-cyteine motif |
| CXCL12 | C-X-C motif chemokine 12 = stromal cell-derived factor 1 (SDF-1) |
| CXCR4 | C-X-C chemokine receptor type 4 |
| DKK1 | Dickkopf-related protein 1 |
| EC | Endothelial cell |
| ECM | Extracellular matrix |
| EDA | Extra-domain A fibronectin splice variant |
| EDB | Extra-domain B fibronectin splice variant |
| EDG-1 | Endothelial differentiation sphingolipid G-protein receptor-1 |
| EGCG | (−)-Epigallocatechin gallate |
| EGF(R) | Epidermal growth factor (receptor) |
| EMMPRIN | Extracellular matrix metalloproteinase inducer |
| EMT | Epithelial–mesenchymal transition |
| EndCC | Endothelial like cancer cell |
| EPC | Endothelial progenitor cell |
| EphA2 | Erythropoietin-producing human hepatocellular (EPH) receptor A2 |
| Erk | Extracellular signal–regulated kinase |
| FAK | Focal adhesion kinase |
| FGF(R) | Fibroblast growth factor (receptor) |
| GAPDH | Glyceraldehyde 3-phosphate dehydrogenase |
| Glc | Glucose |
| Gln | Glutamine |
| GLUT2 | Glucose transporter type 2 |
| GSH | Glutathione |
| HGF(R) | Hepatocyte growth factor (receptor), cMet |
| HIF | Hypoxia-inducible factor |
| HRE | Hypoxia-response element |
| IL | Interleukin |
| Jnk | c-Jun N-terminal kinase |
| KDM4b | Lysine-specific demethylase 4B |
| KIDINS220 | Kinase D-interacting substrate of 220 kDa |
| KiSS-1 | Kisspeptin |
| LAMC2 | Laminin subunit γ2 |
| Lam5g2 | Laminin-332 γ2chain |
| LOX | lysyl oxidase |
| MACC1 | Metastasis associated in colon cancer-1 |
| MALAT1 | Metastasis-associated lung adenocarcinoma transcript 1 |
| MCP1 | Monocyte chemotactic protein |
| Mig-7 | Migration-inducing gene 7 |
| miR | Micro RNA |
| MMP | Matrix metalloproteinase |
| MP | Microparticle |
| MRI | Magnetic resonance imaging |
| MTDH | Metadherin |
| NADPH + H+ | Nicotinamide adenine dinucleotide phosphate |
| NFκB | Nuclear factor κ-light-chain-enhancer of activated B cells |
| NICD | Notch intracellular domain |
| NRP1 | Neuropilin-1 |
| p130Cas | Cellular apoptosis susceptibility protein of 130 kDa |
| PAS | Periodic acid Schiff |
| PDGF | Platelet-derived growth factor |
| PEP | Phosphoenolpyruvate |
| PI3K | Phosphatidylinositol-4,5-bisphosphate 3-kinase |
| PK-M2 | pyruvate kinase isoform M2 |
| PPEE | Paris polyphylla ethanol extract |
| Prdx2 | Peroxiredoxin-2 |
| PRRX1 | Paired-related homeobox transcription factor 1 |
| ROCK | Rho-associated protein kinase |
| Rab | Ras superfamily of monomeric G protein |
| Rac1 | Ras-related C3 botulinum toxin substrate |
| RANKL | Receptor activator of nuclear factor κ-B ligand |
| Ras | Rat sarcoma protein |
| ROCK | Rho-associated protein kinase |
| ROS | Reactive oxygen species |
| Smad | Small body size/mothers against decapentaplegic protein |
| SNAI | snail family transcriptional repressor |
| TAM | Tumor-associated macrophage |
| TCA | Tricarboxylic acid |
| TF | Tissue factor |
| TFPI1 | Tissue factor pathway inhibitor |
| TGFβ1 | Transforming growth factor-β1 |
| TIE | Tyrosine kinase with immunoglobulin-like and EGF-like domains |
| TME | Tumor microenvironment |
| TNFα | Tumor necrosis factor α |
| VEGF(R) | Vascular endothelial growth factor (receptor) |
| VM | Vasculogenic mimicry |
| WHO | World Health Organization |
| Wnt | Wingless-related integration site |
| YAP | Yes-associated protein |
| YKL-40 | Human cartilage glycoprotein HC-gp39, Chitinase-3-like protein 1, CHI3L1 |
| ZEB | Zinc finger E-box-binding homeobox |
References
- Remon, J.; Pardo, N.; Martinez-Marti, A.; Cedres, S.; Navarro, A.; Martinez de Castro, A.M.; Felip, E. Immune-checkpoint inhibition in first-line treatment of advanced non-small cell lung cancer patients: Current status and future approaches. Lung Cancer 2017, 106, 70–75. [Google Scholar] [CrossRef] [PubMed]
- Force, J.; Salama, A.K. First-line treatment of metastatic melanoma: Role of nivolumab. Immunotargets Ther. 2017, 6, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Gajewski, T.F.; Schreiber, H.; Fu, Y.X. Innate and adaptive immune cells in the tumor microenvironment. Nat. Immunol. 2013, 14, 1014–1022. [Google Scholar] [CrossRef] [PubMed]
- Nakazawa, M.S.; Keith, B.; Simon, M.C. Oxygen availability and metabolic adaptations. Nat. Rev. Cancer 2016, 16, 663–673. [Google Scholar] [CrossRef] [PubMed]
- Yeung, S.J.; Pan, J.; Lee, M.H. Roles of p53, MYC and HIF-1 in regulating glycolysis—The seventh hallmark of cancer. Cell. Mol. Life Sci. 2008, 65, 3981–3999. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Zhang, H. Reprogramming of glucose, fatty acid and amino acid metabolism for cancer progression. Cell. Mol. Life Sci. 2016, 73, 377–392. [Google Scholar] [CrossRef] [PubMed]
- Lee, N.; Kim, D. Cancer metabolism: Fueling more than just growth. Mol. Cells 2016, 39, 847–854. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Outschoorn, U.E.; Peiris-Pages, M.; Pestell, R.G.; Sotgia, F.; Lisanti, M.P. Cancer metabolism: A therapeutic perspective. Nat. Rev. Clin. Oncol. 2017, 14, 11–31. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; Chen, X.; Wang, L.; Chen, S. The sweet trap in tumors: Aerobic glycolysis and potential targets for therapy. Oncotarget 2016, 7, 38908–38926. [Google Scholar] [CrossRef] [PubMed]
- Dayton, T.L.; Jacks, T.; Vander Heiden, M.G. PKM2, cancer metabolism, and the road ahead. EMBO Rep. 2016, 17, 1721–1730. [Google Scholar] [CrossRef] [PubMed]
- Dong, G.; Mao, Q.; Xia, W.; Xu, Y.; Wang, J.; Xu, L.; Jiang, F. PKM2 and cancer: The function of PKM2 beyond glycolysis (Review). Oncol. Lett. 2016, 11, 1980–1986. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Zhang, G.; Zhao, L.; Ma, Z.; Chen, H. Metabolic reprogramming in cancer cells: Glycolysis, glutaminolysis, and Bcl-2 proteins as novel therapeutic targets for cancer. World J. Surg. Oncol. 2016, 14, 15. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.; Yoon, J.H. Metabolic interplay between glycolysis and mitochondrial oxidation: The reverse Warburg effect and its therapeutic implication. World J. Biol. Chem. 2015, 6, 148–161. [Google Scholar] [CrossRef] [PubMed]
- Corbet, C.; Feron, O. Cancer cell metabolism and mitochondria: Nutrient plasticity for TCA cycle fueling. Biochim. Biophys. Acta 2017, 1868, 7–15. [Google Scholar] [CrossRef] [PubMed]
- De Vitto, H.; Perez-Valencia, J.; Radosevich, J.A. Glutamine at focus: Versatile roles in cancer. Tumor Biol. 2016, 37, 1541–1558. [Google Scholar] [CrossRef] [PubMed]
- Dang, L.; White, D.W.; Gross, S.; Bennett, B.D.; Bittinger, M.A.; Driggers, E.M.; Fantin, V.R.; Jang, H.G.; Jin, S.; Keenan, M.C.; et al. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature 2009, 462, 739–744. [Google Scholar] [CrossRef] [PubMed]
- Jin, L.; Alesi, G.N.; Kang, S. Glutaminolysis as a target for cancer therapy. Oncogene 2016, 35, 3619–3625. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Coussens, L.M. Accessories to the crime: Functions of cells recruited to the tumor microenvironment. Cancer Cell 2012, 21, 309–322. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Outschoorn, U.E.; Lisanti, M.P.; Sotgia, F. Catabolic cancer-associated fibroblasts transfer energy and biomass to anabolic cancer cells, fueling tumor growth. Semin. Cancer Biol. 2014, 25, 47–60. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Outschoorn, U.E.; Sotgia, F.; Lisanti, M.P. Metabolic asymmetry in cancer: A “balancing act” that promotes tumor growth. Cancer Cell 2014, 26, 5–7. [Google Scholar] [CrossRef] [PubMed]
- Rattigan, Y.I.; Patel, B.B.; Ackerstaff, E.; Sukenick, G.; Koutcher, J.A.; Glod, J.W.; Banerjee, D. Lactate is a mediator of metabolic cooperation between stromal carcinoma associated fibroblasts and glycolytic tumor cells in the tumor microenvironment. Exp. Cell Res. 2012, 318, 326–335. [Google Scholar] [CrossRef] [PubMed]
- Sotgia, F.; Martinez-Outschoorn, U.E.; Howell, A.; Pestell, R.G.; Pavlides, S.; Lisanti, M.P. Caveolin-1 and cancer metabolism in the tumor microenvironment: Markers, models, and mechanisms. Annu. Rev. Pathol. 2012, 7, 423–467. [Google Scholar] [CrossRef] [PubMed]
- Xing, Y.; Zhao, S.; Zhou, B.P.; Mi, J. Metabolic reprogramming of the tumour microenvironment. FEBS J. 2015, 282, 3892–3898. [Google Scholar] [CrossRef] [PubMed]
- Potente, M.; Carmeliet, P. The Link Between Angiogenesis and Endothelial Metabolism. Annu. Rev. Physiol. 2017, 79, 43–66. [Google Scholar] [CrossRef] [PubMed]
- Cantelmo, A.R.; Pircher, A.; Kalucka, J.; Carmeliet, P. Vessel pruning or healing: Endothelial metabolism as a novel target? Expert Opin. Ther. Targets 2017, 21, 239–247. [Google Scholar] [CrossRef] [PubMed]
- Behrens, J. Cadherins and catenins: Role in signal transduction and tumor progression. Cancer Metastasis Rev. 1999, 18, 15–30. [Google Scholar] [CrossRef]
- Le Bras, G.F.; Taubenslag, K.J.; Andl, C.D. The regulation of cell-cell adhesion during epithelial-mesenchymal transition, motility and tumor progression. Cell Adhes. Migr. 2012, 6, 365–373. [Google Scholar] [CrossRef] [PubMed]
- Gehler, S.; Ponik, S.M.; Riching, K.M.; Keely, P.J. Bi-directional signaling: Extracellular matrix and integrin regulation of breast tumor progression. Crit. Rev. Eukaryot. Gene Expr. 2013, 23, 139–157. [Google Scholar] [CrossRef] [PubMed]
- Xiong, J.; Balcioglu, H.E.; Danen, E.H. Integrin signaling in control of tumor growth and progression. Int. J. Biochem. Cell Biol. 2013, 45, 1012–1015. [Google Scholar] [CrossRef] [PubMed]
- Murphy, G.; Nagase, H. Localizing matrix metalloproteinase activities in the pericellular environment. FEBS J. 2011, 278, 2–15. [Google Scholar] [CrossRef] [PubMed]
- Papageorgis, P.; Stylianopoulos, T. Role of TGFβ in regulation of the tumor microenvironment and drug delivery (Review). Int. J. Oncol. 2015, 46, 933–943. [Google Scholar] [CrossRef] [PubMed]
- Renema, N.; Navet, B.; Heymann, M.F.; Lezot, F.; Heymann, D. RANK-RANKL signalling in cancer. Biosci. Rep. 2016, 36. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, K.; Umitsu, M.; De Silva, D.M.; Roy, A.; Bottaro, D.P. Hepatocyte growth factor/MET in cancer progression and biomarker discovery. Cancer Sci. 2017, 108, 296–307. [Google Scholar] [CrossRef] [PubMed]
- Katoh, M. FGFR inhibitors: Effects on cancer cells, tumor microenvironment and whole-body homeostasis (Review). Int. J. Mol. Med. 2016, 38, 3–15. [Google Scholar] [CrossRef] [PubMed]
- Khan, Z.; Marshall, J.F. The role of integrins in TGFβ activation in the tumour stroma. Cell Tissue Res. 2016, 365, 657–673. [Google Scholar] [CrossRef] [PubMed]
- Goel, H.L.; Mercurio, A.M. Enhancing integrin function by VEGF/neuropilin signaling: Implications for tumor biology. Cell Adhes. Migr. 2012, 6, 554–560. [Google Scholar] [CrossRef] [PubMed]
- Jeanes, A.; Gottardi, C.J.; Yap, A.S. Cadherins and cancer: How does cadherin dysfunction promote tumor progression? Oncogene 2008, 27, 6920–6929. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.G.; Grizzle, W.E. Exosomes and cancer: A newly described pathway of immune suppression. Clin. Cancer Res. 2011, 17, 959–964. [Google Scholar] [CrossRef] [PubMed]
- Henderson, M.C.; Azorsa, D.O. The genomic and proteomic content of cancer cell-derived exosomes. Front. Oncol. 2012, 2, 38. [Google Scholar] [CrossRef] [PubMed]
- Dinger, M.E.; Mercer, T.R.; Mattick, J.S. RNAs as extracellular signaling molecules. J. Mol. Endocrinol. 2008, 40, 151–159. [Google Scholar] [CrossRef] [PubMed]
- Rashed, M.H.; Bayraktar, E.; Helel, G.K.; Abd-Ellah, M.F.; Amero, P.; Chavez-Reyes, A.; Rodriguez-Aguayo, C. Exosomes: From garbage bins to promising therapeutic targets. Int. J. Mol. Sci. 2017, 18. [Google Scholar] [CrossRef] [PubMed]
- Thery, C.; Ostrowski, M.; Segura, E. Membrane vesicles as conveyors of immune responses. Nat. Rev. Immunol. 2009, 9, 581–593. [Google Scholar] [CrossRef] [PubMed]
- Paget, S. The distribution of secondary growths in cancer of the breast. Cancer Metastasis Rev. 1989, 8, 98–101. [Google Scholar] [CrossRef]
- Pietila, M.; Ivaska, J.; Mani, S.A. Whom to blame for metastasis, the epithelial-mesenchymal transition or the tumor microenvironment? Cancer Lett. 2016, 380, 359–368. [Google Scholar] [CrossRef] [PubMed]
- Heldin, C.H.; Rubin, K.; Pietras, K.; Ostman, A. High interstitial fluid pressure—An obstacle in cancer therapy. Nat. Rev. Cancer 2004, 4, 806–813. [Google Scholar] [CrossRef] [PubMed]
- Quail, D.F.; Joyce, J.A. Microenvironmental regulation of tumor progression and metastasis. Nat. Med. 2013, 19, 1423–1437. [Google Scholar] [CrossRef] [PubMed]
- Junttila, M.R.; de Sauvage, F.J. Influence of tumour micro-environment heterogeneity on therapeutic response. Nature 2013, 501, 346–354. [Google Scholar] [CrossRef] [PubMed]
- Butcher, D.T.; Alliston, T.; Weaver, V.M. A tense situation: Forcing tumour progression. Nat. Rev. Cancer 2009, 9, 108–122. [Google Scholar] [CrossRef] [PubMed]
- Fujii, S.; Fujihara, A.; Natori, K.; Abe, A.; Kuboki, Y.; Higuchi, Y.; Aizawa, M.; Kuwata, T.; Kinoshita, T.; Yasui, W.; et al. TEM1 expression in cancer-associated fibroblasts is correlated with a poor prognosis in patients with gastric cancer. Cancer Med. 2015, 4, 1667–1678. [Google Scholar] [CrossRef] [PubMed]
- Herrera, M.; Islam, A.B.; Herrera, A.; Martin, P.; Garcia, V.; Silva, J.; Garcia, J.M.; Salas, C.; Casal, I.; de Herreros, A.G.; et al. Functional heterogeneity of cancer-associated fibroblasts from human colon tumors shows specific prognostic gene expression signature. Clin. Cancer Res. 2013, 19, 5914–5926. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, M.; Ogawa, T.; Zhang, X.; Hanamura, N.; Kashikura, Y.; Takamura, M.; Yoneda, M.; Shiraishi, T. Role of stromal myofibroblasts in invasive breast cancer: Stromal expression of alpha-smooth muscle actin correlates with worse clinical outcome. Breast Cancer 2012, 19, 170–176. [Google Scholar] [CrossRef] [PubMed]
- Fujita, H.; Ohuchida, K.; Mizumoto, K.; Nakata, K.; Yu, J.; Kayashima, T.; Cui, L.; Manabe, T.; Ohtsuka, T.; Tanaka, M. alpha-Smooth muscle actin expressing stroma promotes an aggressive tumor biology in pancreatic ductal adenocarcinoma. Pancreas 2010. [Google Scholar] [CrossRef] [PubMed]
- Mueller, M.M.; Fusenig, N.E. Friends or foes—Bipolar effects of the tumour stroma in cancer. Nat. Rev. Cancer 2004, 4, 839–849. [Google Scholar] [CrossRef] [PubMed]
- Mattey, D.L.; Dawes, P.T.; Nixon, N.B.; Slater, H. Transforming growth factor b1 and interleukin 4 induced a smooth muscle actin expression and myofibroblast-like differentiation in human synovial fibroblasts in vitro: Modulation by basic fibroblast growth factor. Ann. Rheum. Dis. 1997, 56, 426–431. [Google Scholar] [CrossRef] [PubMed]
- Kalluri, R. The biology and function of fibroblasts in cancer. Nat. Rev. Cancer 2016, 16, 582–598. [Google Scholar] [CrossRef] [PubMed]
- Micallef, L.; Vedrenne, N.; Billet, F.; Coulomb, B.; Darby, I.A.; Desmouliere, A. The myofibroblast, multiple origins for major roles in normal and pathological tissue repair. Fibrogenes. Tissue Repair 2012, 5, S5. [Google Scholar] [CrossRef]
- Tomasek, J.J.; Gabbiani, G.; Hinz, B.; Chaponnier, C.; Brown, R.A. Myofibroblasts and mechano-regulation of connective tissue remodelling. Nat. Rev. Mol. Cell Biol. 2002, 3, 349–363. [Google Scholar] [CrossRef] [PubMed]
- Bhowmick, N.A.; Neilson, E.G.; Moses, H.L. Stromal fibroblasts in cancer initiation and progression. Nature 2004, 432, 332–337. [Google Scholar] [CrossRef] [PubMed]
- Polanska, U.M.; Orimo, A. Carcinoma-associated fibroblasts: Non-neoplastic tumour-promoting mesenchymal cells. J. Cell Physiol. 2013, 228, 1651–1657. [Google Scholar] [CrossRef] [PubMed]
- Marsh, T.; Pietras, K.; McAllister, S.S. Fibroblasts as architects of cancer pathogenesis. Biochim. Biophys. Acta 2013, 1832, 1070–1078. [Google Scholar] [CrossRef] [PubMed]
- Kalluri, R.; Zeisberg, M. Fibroblasts in cancer. Nat. Rev. Cancer 2006, 6, 392–401. [Google Scholar] [CrossRef] [PubMed]
- Webber, J.; Steadman, R.; Mason, M.D.; Tabi, Z.; Clayton, A. Cancer exosomes trigger fibroblast to myofibroblast differentiation. Cancer Res. 2010, 70, 9621–9630. [Google Scholar] [CrossRef] [PubMed]
- Dotto, G.P.; Weinberg, R.A.; Ariza, A. Malignant transformation of mouse primary keratinocytes by Harvey sarcoma virus and its modulation by surrounding normal cells. Proc. Natl. Acad. Sci. USA 1988, 85, 6389–6393. [Google Scholar] [CrossRef] [PubMed]
- Serini, G.; Gabbiani, G. Mechanisms of myofibroblast activity and phenotypic modulation. Exp. Cell Res. 1999, 250, 273–283. [Google Scholar] [CrossRef] [PubMed]
- Strutz, F.; Okada, H.; Lo, C.W.; Danoff, T.; Carone, R.L.; Tomaszewski, J.E.; Neilson, E.G. Identification and characterization of a fibroblast marker: FSP1. J. Cell Biol. 1995, 130, 393–405. [Google Scholar] [CrossRef] [PubMed]
- Sugimoto, H.; Mundel, T.M.; Kieran, M.W.; Kalluri, R. Identification of fibroblast heterogeneity in the tumor microenvironment. Cancer Biol. Ther. 2006, 5, 1640–1646. [Google Scholar] [CrossRef] [PubMed]
- Garin-Chesa, P.; Old, L.J.; Rettig, W.J. Cell surface glycoprotein of reactive stromal fibroblasts as a potential antibody target in human epithelial cancers. Proc. Natl. Acad. Sci. USA 1990, 87, 7235–7239. [Google Scholar] [CrossRef] [PubMed]
- Ni, W.D.; Yang, Z.T.; Cui, C.A.; Cui, Y.; Fang, L.Y.; Xuan, Y.H. Tenascin-C is a potential cancer-associated fibroblasts marker and predicts poor prognosis in prostate cancer. Biochem. Biophys. Res. Commun. 2017, 486, 607–612. [Google Scholar] [CrossRef] [PubMed]
- Orimo, A.; Weinberg, R.A. Stromal fibroblasts in cancer: A novel tumor-promoting cell type. Cell Cycle 2006, 5, 1597–1601. [Google Scholar] [CrossRef] [PubMed]
- Paunescu, V.; Bojin, F.M.; Tatu, C.A.; Gavriliuc, O.I.; Rosca, A.; Gruia, A.T.; Tanasie, G.; Bunu, C.; Crisnic, D.; Gherghiceanu, M.; et al. Tumour-associated fibroblasts and mesenchymal stem cells: More similarities than differences. J. Cell. Mol. Med. 2011, 15, 635–646. [Google Scholar] [CrossRef] [PubMed]
- Micke, P.; Ostman, A. Tumour-stroma interaction: Cancer-associated fibroblasts as novel targets in anti-cancer therapy? Lung Cancer 2004, 45 (Suppl. 2), S163–S175. [Google Scholar] [CrossRef] [PubMed]
- Mueller, L.; Goumas, F.A.; Affeldt, M.; Sandtner, S.; Gehling, U.M.; Brilloff, S.; Walter, J.; Karnatz, N.; Lamszus, K.; Rogiers, X.; et al. Stromal fibroblasts in colorectal liver metastases originate from resident fibroblasts and generate an inflammatory microenvironment. Am. J. Pathol. 2007, 171, 1608–1618. [Google Scholar] [CrossRef] [PubMed]
- Zeisberg, E.M.; Potenta, S.; Xie, L.; Zeisberg, M.; Kalluri, R. Discovery of endothelial to mesenchymal transition as a source for carcinoma-associated fibroblasts. Cancer Res. 2007, 67, 10123–10128. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Xiao, C.H.; Tan, L.D.; Wang, Q.S.; Li, X.Q.; Feng, Y.M. Cancer-associated fibroblasts induce epithelial-mesenchymal transition of breast cancer cells through paracrine TGF-β signalling. Br. J. Cancer 2014, 110, 724–732. [Google Scholar] [CrossRef] [PubMed]
- Fukumura, D.; Xavier, R.; Sugiura, T.; Chen, Y.; Park, E.C.; Lu, N.; Selig, M.; Nielsen, G.; Taksir, T.; Jain, R.K.; et al. Tumor induction of VEGF promoter activity in stromal cells. Cell 1998, 94, 715–725. [Google Scholar] [CrossRef]
- Pietras, K.; Pahler, J.; Bergers, G.; Hanahan, D. Functions of paracrine PDGF signaling in the proangiogenic tumor stroma revealed by pharmacological targeting. PLoS Med. 2008, 5, e19. [Google Scholar] [CrossRef] [PubMed]
- Orimo, A.; Gupta, P.B.; Sgroi, D.C.; Arenzana-Seisdedos, F.; Delaunay, T.; Naeem, R.; Carey, V.J.; Richardson, A.L.; Weinberg, R.A. Stromal fibroblasts present in invasive human breast carcinomas promote tumor growth and angiogenesis through elevated SDF-1/CXCL12 secretion. Cell 2005, 121, 335–348. [Google Scholar] [CrossRef] [PubMed]
- Suratt, B.T.; Petty, J.M.; Young, S.K.; Malcolm, K.C.; Lieber, J.G.; Nick, J.A.; Gonzalo, J.A.; Henson, P.M.; Worthen, G.S. Role of the CXCR4/SDF-1 chemokine axis in circulating neutrophil homeostasis. Blood 2004, 104, 565–571. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; DeBusk, L.M.; Fukuda, K.; Fingleton, B.; Green-Jarvis, B.; Shyr, Y.; Matrisian, L.M.; Carbone, D.P.; Lin, P.C. Expansion of myeloid immune suppressor Gr+CD11b+ cells in tumor-bearing host directly promotes tumor angiogenesis. Cancer Cell 2004, 6, 409–421. [Google Scholar] [CrossRef] [PubMed]
- Bergers, G.; Brekken, R.; McMahon, G.; Vu, T.H.; Itoh, T.; Tamaki, K.; Tanzawa, K.; Thorpe, P.; Itohara, S.; Werb, Z.; et al. Matrix metalloproteinase-9 triggers the angiogenic switch during carcinogenesis. Nat. Cell Biol. 2000, 2, 737–744. [Google Scholar] [CrossRef] [PubMed]
- Luga, V.; Zhang, L.; Viloria-Petit, A.M.; Ogunjimi, A.A.; Inanlou, M.R.; Chiu, E.; Buchanan, M.; Hosein, A.N.; Basik, M.; Wrana, J.L. Exosomes mediate stromal mobilization of autocrine Wnt-PCP signaling in breast cancer cell migration. Cell 2012, 151, 1542–1556. [Google Scholar] [CrossRef] [PubMed]
- Gkretsi, V.; Stylianou, A.; Papageorgis, P.; Polydorou, C.; Stylianopoulos, T. Remodeling components of the tumor microenvironment to enhance cancer therapy. Front. Oncol. 2015, 5, 214. [Google Scholar] [CrossRef] [PubMed]
- Torimura, T.; Ueno, T.; Inuzuka, S.; Kin, M.; Ohira, H.; Kimura, Y.; Majima, Y.; Sata, M.; Abe, H.; Tanikawa, K. The extracellular matrix in hepatocellular carcinoma shows different localization patterns depending on the differentiation and the histological pattern of tumors: Immunohistochemical analysis. J. Hepatol. 1994, 21, 37–46. [Google Scholar] [CrossRef]
- Miyazaki, K. Laminin-5 (laminin-332): Unique biological activity and role in tumor growth and invasion. Cancer Sci. 2006, 97, 91–98. [Google Scholar] [CrossRef] [PubMed]
- Giannelli, G.; Bergamini, C.; Fransvea, E.; Sgarra, C.; Antonaci, S. Laminin-5 with transforming growth factor-beta1 induces epithelial to mesenchymal transition in hepatocellular carcinoma. Gastroenterology 2005, 129, 1375–1383. [Google Scholar] [CrossRef] [PubMed]
- Ross, J.B.; Huh, D.; Noble, L.B.; Tavazoie, S.F. Identification of molecular determinants of primary and metastatic tumour re-initiation in breast cancer. Nat. Cell Biol. 2015, 17, 651–664. [Google Scholar] [CrossRef] [PubMed]
- Ghigna, C.; Valacca, C.; Biamonti, G. Alternative splicing and tumor progression. Curr. Genom. 2008, 9, 556–570. [Google Scholar] [CrossRef] [PubMed]
- Scarpino, S.; Stoppacciaro, A.; Pellegrini, C.; Marzullo, A.; Zardi, L.; Tartaglia, F.; Viale, G.; Ruco, L.P. Expression of EDA/EDB isoforms of fibronectin in papillary carcinoma of the thyroid. J. Pathol. 1999, 188, 163–167. [Google Scholar] [CrossRef]
- Hauptmann, S.; Zardi, L.; Siri, A.; Carnemolla, B.; Borsi, L.; Castellucci, M.; Klosterhalfen, B.; Hartung, P.; Weis, J.; Stocker, G.; et al. Extracellular matrix proteins in colorectal carcinomas. Expression of tenascin and fibronectin isoforms. Lab. Investig. 1995, 73, 172–182. [Google Scholar] [PubMed]
- Bordeleau, F.; Califano, J.P.; Negron Abril, Y.L.; Mason, B.N.; LaValley, D.J.; Shin, S.J.; Weiss, R.S.; Reinhart-King, C.A. Tissue stiffness regulates serine/arginine-rich protein-mediated splicing of the extra domain B-fibronectin isoform in tumors. Proc. Natl. Acad. Sci. USA 2015, 112, 8314–8319. [Google Scholar] [CrossRef] [PubMed]
- Serini, G.; Bochaton-Piallat, M.L.; Ropraz, P.; Geinoz, A.; Borsi, L.; Zardi, L.; Gabbiani, G. The fibronectin domain ED-A is crucial for myofibroblastic phenotype induction by transforming growth factor-beta1. J. Cell Biol. 1998, 142, 873–881. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, C.; McDonald, J.A. The roles of the myofibroblast in idiopathic pulmonary fibrosis. Ultrastructural and immunohistochemical features of sites of active extracellular matrix synthesis. Am. J. Pathol. 1991, 138, 1257–1265. [Google Scholar] [PubMed]
- Han, Z.; Zhou, Z.; Shi, X.; Wang, J.; Wu, X.; Sun, D.; Chen, Y.; Zhu, H.; Magi-Galluzzi, C.; Lu, Z.R. EDB Fibronectin specific peptide for prostate cancer targeting. Bioconjug. Chem. 2015, 26, 830–838. [Google Scholar] [CrossRef] [PubMed]
- Oskarsson, T.; Massague, J. Extracellular matrix players in metastatic niches. EMBO J. 2012, 31, 254–256. [Google Scholar] [CrossRef] [PubMed]
- Malanchi, I.; Santamaria-Martinez, A.; Susanto, E.; Peng, H.; Lehr, H.A.; Delaloye, J.F.; Huelsken, J. Interactions between cancer stem cells and their niche govern metastatic colonization. Nature 2011, 481, 85–89. [Google Scholar] [CrossRef] [PubMed]
- Oskarsson, T.; Acharyya, S.; Zhang, X.H.; Vanharanta, S.; Tavazoie, S.F.; Morris, P.G.; Downey, R.J.; Manova-Todorova, K.; Brogi, E.; Massague, J. Breast cancer cells produce tenascin C as a metastatic niche component to colonize the lungs. Nat. Med. 2011, 17, 867–874. [Google Scholar] [CrossRef] [PubMed]
- Egbert, M.; Ruetze, M.; Sattler, M.; Wenck, H.; Gallinat, S.; Lucius, R.; Weise, J.M. The matricellular protein periostin contributes to proper collagen function and is downregulated during skin aging. J. Dermatol. Sci. 2014, 73, 40–48. [Google Scholar] [CrossRef] [PubMed]
- Kii, I.; Nishiyama, T.; Li, M.; Matsumoto, K.; Saito, M.; Amizuka, N.; Kudo, A. Incorporation of tenascin-C into the extracellular matrix by periostin underlies an extracellular meshwork architecture. J. Biol. Chem. 2010, 285, 2028–2039. [Google Scholar] [CrossRef] [PubMed]
- Degen, M.; Brellier, F.; Kain, R.; Ruiz, C.; Terracciano, L.; Orend, G.; Chiquet-Ehrismann, R. Tenascin-W is a novel marker for activated tumor stroma in low-grade human breast cancer and influences cell behavior. Cancer Res. 2007, 67, 9169–9179. [Google Scholar] [CrossRef] [PubMed]
- Brellier, F.; Tucker, R.P.; Chiquet-Ehrismann, R. Tenascins and their implications in diseases and tissue mechanics. Scand. J. Med. Sci. Sports 2009, 19, 511–519. [Google Scholar] [CrossRef] [PubMed]
- Degen, M.; Brellier, F.; Schenk, S.; Driscoll, R.; Zaman, K.; Stupp, R.; Tornillo, L.; Terracciano, L.; Chiquet-Ehrismann, R.; Ruegg, C.; et al. Tenascin-W, a new marker of cancer stroma, is elevated in sera of colon and breast cancer patients. Int. J. Cancer 2008, 122, 2454–2461. [Google Scholar] [CrossRef] [PubMed]
- Scherberich, A.; Tucker, R.P.; Degen, M.; Brown-Luedi, M.; Andres, A.C.; Chiquet-Ehrismann, R. Tenascin-W is found in malignant mammary tumors, promotes alpha8 integrin-dependent motility and requires p38MAPK activity for BMP-2 and TNF-alpha induced expression in vitro. Oncogene 2005, 24, 1525–1532. [Google Scholar] [CrossRef] [PubMed]
- Akiri, G.; Sabo, E.; Dafni, H.; Vadasz, Z.; Kartvelishvily, Y.; Gan, N.; Kessler, O.; Cohen, T.; Resnick, M.; Neeman, M.; et al. Lysyl oxidase-related protein-1 promotes tumor fibrosis and tumor progression in vivo. Cancer Res. 2003, 63, 1657–1666. [Google Scholar] [PubMed]
- Smith-Mungo, L.I.; Kagan, H.M. Lysyl oxidase: Properties, regulation and multiple functions in biology. Matrix Biol. 1998, 16, 387–398. [Google Scholar] [CrossRef]
- Paszek, M.J.; Zahir, N.; Johnson, K.R.; Lakins, J.N.; Rozenberg, G.I.; Gefen, A.; Reinhart-King, C.A.; Margulies, S.S.; Dembo, M.; Boettiger, D.; et al. Tensional homeostasis and the malignant phenotype. Cancer Cell 2005, 8, 241–254. [Google Scholar] [CrossRef] [PubMed]
- Weaver, V.M.; Lelievre, S.; Lakins, J.N.; Chrenek, M.A.; Jones, J.C.; Giancotti, F.; Werb, Z.; Bissell, M.J. b4 integrin-dependent formation of polarized three-dimensional architecture confers resistance to apoptosis in normal and malignant mammary epithelium. Cancer Cell 2002, 2, 205–216. [Google Scholar] [CrossRef]
- White, D.E.; Kurpios, N.A.; Zuo, D.; Hassell, J.A.; Blaess, S.; Mueller, U.; Muller, W.J. Targeted disruption of b1-integrin in a transgenic mouse model of human breast cancer reveals an essential role in mammary tumor induction. Cancer Cell 2004, 6, 159–170. [Google Scholar] [CrossRef] [PubMed]
- Wipff, P.J.; Rifkin, D.B.; Meister, J.J.; Hinz, B. Myofibroblast contraction activates latent TGF-β1 from the extracellular matrix. J. Cell Biol. 2007, 179, 1311–1323. [Google Scholar] [CrossRef] [PubMed]
- Calvo, F.; Ege, N.; Grande-Garcia, A.; Hooper, S.; Jenkins, R.P.; Chaudhry, S.I.; Harrington, K.; Williamson, P.; Moeendarbary, E.; Charras, G.; et al. Mechanotransduction and YAP-dependent matrix remodelling is required for the generation and maintenance of cancer-associated fibroblasts. Nat. Cell Biol. 2013, 15, 637–646. [Google Scholar] [CrossRef] [PubMed]
- Sahai, E.; Marshall, C.J. ROCK and Dia have opposing effects on adherens junctions downstream of Rho. Nat. Cell Biol. 2002, 4, 408–415. [Google Scholar] [CrossRef] [PubMed]
- Burridge, K.; Wennerberg, K. Rho and Rac take center stage. Cell 2004, 116, 167–179. [Google Scholar] [CrossRef]
- Fan, Y.; Shen, B.; Tan, M.; Mu, X.; Qin, Y.; Zhang, F.; Liu, Y. TGF-β-induced upregulation of malat1 promotes bladder cancer metastasis by associating with suz12. Clin. Cancer Res. 2014, 20, 1531–1541. [Google Scholar] [CrossRef] [PubMed]
- Jodele, S.; Blavier, L.; Yoon, J.M.; DeClerck, Y.A. Modifying the soil to affect the seed: Role of stromal-derived matrix metalloproteinases in cancer progression. Cancer Metastasis Rev. 2006, 25, 35–43. [Google Scholar] [CrossRef] [PubMed]
- Fang, M.; Yuan, J.; Peng, C.; Li, Y. Collagen as a double-edged sword in tumor progression. Tumor Biol. 2014, 35, 2871–2882. [Google Scholar] [CrossRef] [PubMed]
- Bergamaschi, A.; Tagliabue, E.; Sorlie, T.; Naume, B.; Triulzi, T.; Orlandi, R.; Russnes, H.G.; Nesland, J.M.; Tammi, R.; Auvinen, P.; et al. Extracellular matrix signature identifies breast cancer subgroups with different clinical outcome. J. Pathol. 2008, 214, 357–367. [Google Scholar] [CrossRef] [PubMed]
- Monboisse, J.C.; Oudart, J.B.; Ramont, L.; Brassart-Pasco, S.; Maquart, F.X. Matrikines from basement membrane collagens: A new anti-cancer strategy. Biochim. Biophys. Acta 2014, 1840, 2589–2598. [Google Scholar] [CrossRef] [PubMed]
- Folkman, J. Antiangiogenesis in cancer therapy—Endostatin and its mechanisms of action. Exp. Cell Res. 2006, 312, 594–607. [Google Scholar] [CrossRef] [PubMed]
- Willis, C.D.; Poluzzi, C.; Mongiat, M.; Iozzo, R.V. Endorepellin laminin-like globular 1/2 domains bind Ig3–5 of vascular endothelial growth factor (VEGF) receptor 2 and block pro-angiogenic signaling by VEGFA in endothelial cells. FEBS J. 2013, 280, 2271–2284. [Google Scholar] [CrossRef] [PubMed]
- Billioux, A.; Modlich, U.; Bicknell, R. Angiogenesis. In The Cancer Handbook, 2nd ed.; Alison, M., Ed.; John Wiley & Sons: Hoboken, NJ, USA, 2007; Volume 1, pp. 144–154. [Google Scholar]
- Folkman, J. Looking for a good endothelial address. Cancer Cell 2002, 1, 113–115. [Google Scholar] [CrossRef]
- Hanahan, D.; Folkman, J. Patterns and emerging mechanisms of the angiogenic switch during tumorigenesis. Cell 1996, 86, 353–364. [Google Scholar] [CrossRef]
- Blouw, B.; Song, H.; Tihan, T.; Bosze, J.; Ferrara, N.; Gerber, H.-P.; Johnson, R.S.; Bergers, G. The hypoxic response of tumors is dependent on their microenvironment. Cancer Cell 2003, 4, 133–146. [Google Scholar] [CrossRef]
- Folkman, J.; Watson, K.; Ingber, D.; Hanahan, D. Induction of angiogenesis during the transition from hyperplasia to neoplasia. Nature 1989, 339, 58–61. [Google Scholar] [CrossRef] [PubMed]
- Weidner, N.; Semple, J.P.; Welch, W.R.; Folkman, J. Tumor angiogenesis and metastasis—Correlation in invasive breast carcinoma. N. Engl. J. Med. 1991, 324, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Kandel, J.; Bossy-Wetzel, E.; Radvanyi, F.; Klagsbrun, M.; Folkman, J.; Hanahan, D. Neovascularization is associated with a switch to the export of bFGF in the multistep development of fibrosarcoma. Cell 1991, 66, 1095–1104. [Google Scholar] [CrossRef]
- Niland, S.; Eble, J.A. Integrin-mediated cell-matrix interaction in physiological and pathological blood vessel formation. J. Oncol. 2012, 2012, 125278. [Google Scholar] [CrossRef] [PubMed]
- Sica, A.; Schioppa, T.; Mantovani, A.; Allavena, P. Tumour-associated macrophages are a distinct M2 polarised population promoting tumour progression: Potential targets of anti-cancer therapy. Eur. J. Cancer 2006, 42, 717–727. [Google Scholar] [CrossRef] [PubMed]
- Lin, E.Y.; Pollard, J.W. Tumor-associated macrophages press the angiogenic switch in breast cancer. Cancer Res. 2007, 67, 5064–5066. [Google Scholar] [CrossRef] [PubMed]
- Schmid, M.C.; Varner, J.A. Myeloid cell trafficking and tumor angiogenesis. Cancer Lett. 2007, 250, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Aplin, A.C.; Fogel, E.; Nicosia, R.F. MCP-1 promotes mural cell recruitment during angiogenesis in the aortic ring model. Angiogenesis 2010, 13, 219–226. [Google Scholar] [CrossRef] [PubMed]
- Hong, K.H.; Ryu, J.; Han, K.H. Monocyte chemoattractant protein-1-induced angiogenesis is mediated by vascular endothelial growth factor-A. Blood 2005, 105, 1405–1407. [Google Scholar] [CrossRef] [PubMed]
- Niu, J.; Azfer, A.; Zhelyabovska, O.; Fatma, S.; Kolattukudy, P.E. Monocyte chemotactic protein (MCP)-1 promotes angiogenesis via a novel transcription factor, MCP-1-induced protein (MCPIP). J. Biol. Chem. 2008, 283, 14542–14551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bergers, G.; Hanahan, D. Modes of resistance to anti-angiogenic therapy. Nat. Rev. Cancer 2008, 8, 592–603. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef]
- Nagy, J.A.; Chang, S.H.; Dvorak, A.M.; Dvorak, H.F. Why are tumour blood vessels abnormal and why is it important to know? Br. J. Cancer 2009, 100, 865–869. [Google Scholar] [CrossRef] [PubMed]
- Dome, B.; Hendrix, M.J.; Paku, S.; Tovari, J.; Timar, J. Alternative vascularization mechanisms in cancer: Pathology and therapeutic implications. Am. J. Pathol. 2007, 170, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Hillen, F.; Griffioen, A.W. Tumour vascularization: Sprouting angiogenesis and beyond. Cancer Metastasis Rev. 2007, 26, 489–502. [Google Scholar] [CrossRef] [PubMed]
- Nagy, J.A.; Dvorak, H.F. Heterogeneity of the tumor vasculature: The need for new tumor blood vessel type-specific targets. Clin. Exp. Metastasis 2012, 29, 657–662. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaspar, M.; Zardi, L.; Neri, D. Fibronectin as target for tumor therapy. Int. J. Cancer 2006, 118, 1331–1339. [Google Scholar] [CrossRef] [PubMed]
- Midulla, M.; Verma, R.; Pignatelli, M.; Ritter, M.A.; Courtenay-Luck, N.S.; George, A.J.T. Source of oncofetal ED-B-containing fibronectin: Implications of production by both tumor and endothelial cells. Cancer Res. 2000, 60, 164–169. [Google Scholar] [PubMed]
- Midwood, K.S.; Orend, G. The role of tenascin-C in tissue injury and tumorigenesis. J. Cell Commun. Signal. 2009, 3, 287–310. [Google Scholar] [CrossRef] [PubMed]
- Pezzolo, A.; Parodi, F.; Marimpietri, D.; Raffaghello, L.; Cocco, C.; Pistorio, A.; Mosconi, M.; Gambini, C.; Cilli, M.; Deaglio, S.; et al. Oct-4+/Tenascin C+ neuroblastoma cells serve as progenitors of tumor-derived endothelial cells. Cell Res. 2011, 21, 1470–1486. [Google Scholar] [CrossRef] [PubMed]
- Martina, E.; Chiquet-Ehrismann, R.; Brellier, F. Tenascin-W: An extracellular matrix protein associated with osteogenesis and cancer. Int. J. Biochem. Cell Biol. 2010, 42, 1412–1415. [Google Scholar] [CrossRef] [PubMed]
- Dudley, A.C. Tumor endothelial cells. Cold Spring Harb. Perspect. Med. 2012, 2, a006536. [Google Scholar] [CrossRef] [PubMed]
- Benjamin, L.E.; Hemo, I.; Keshet, E. A plasticity window for blood vessel remodelling is defined by pericyte coverage of the preformed endothelial network and is regulated by PDGF-B and VEGF. Development 1998, 125, 1591–1598. [Google Scholar] [PubMed]
- Burgers, A.C.; Lammert, E. Extraerythrocytic hemoglobin—A possible oxygen transporter in human malignant tumors. Med. Hypotheses 2011, 77, 580–583. [Google Scholar] [CrossRef] [PubMed]
- Maniotis, A.J.; Folberg, R.; Hess, A.; Seftor, E.A.; Gardner, L.M.; Pe’er, J.; Trent, J.M.; Meltzer, P.S.; Hendrix, M.J. Vascular channel formation by human melanoma cells in vivo and in vitro: Vasculogenic mimicry. Am. J. Pathol. 1999, 155, 739–752. [Google Scholar] [CrossRef]
- Sun, B.; Zhang, D.; Zhao, N.; Zhao, X. Epithelial-to-endothelial transition and cancer stem cells: Two cornerstones of vasculogenic mimicry in malignant tumors. Oncotarget 2017, 8, 30502–30510. [Google Scholar] [CrossRef] [PubMed]
- Dvorak, H.F.; Senger, D.R.; Dvorak, A.M. Fibrin as a component of the tumor stroma: Origins and biological significance. Cancer Metastasis Rev. 1983, 2, 41–73. [Google Scholar] [CrossRef]
- Dvorak, H.F. Tumors: Wounds that do not heal. Similarities between tumor stroma generation and wound healing. N. Engl. J. Med. 1986, 315, 1650–1659. [Google Scholar] [CrossRef] [PubMed]
- Dvorak, H.F.; Brown, L.F.; Detmar, M.; Dvorak, A.M. Vascular permeability factor/vascular endothelial growth factor, microvascular hyperpermeability, and angiogenesis. Am. J. Pathol. 1995, 146, 1029–1039. [Google Scholar] [PubMed]
- Jain, R.K. Normalizing tumor vasculature with anti-angiogenic therapy: A new paradigm for combination therapy. Nat. Med. 2001, 7, 987–989. [Google Scholar] [CrossRef] [PubMed]
- Holash, J.; Maisonpierre, P.C.; Compton, D.; Boland, P.; Alexander, C.R.; Zagzag, D.; Yancopoulos, G.D.; Wiegand, S.J. Vessel cooption, regression, and growth in tumors mediated by angiopoietins and VEGF. Science 1999, 284, 1994–1998. [Google Scholar] [CrossRef] [PubMed]
- Donnem, T.; Hu, J.; Ferguson, M.; Adighibe, O.; Snell, C.; Harris, A.L.; Gatter, K.C.; Pezzella, F. Vessel co-option in primary human tumors and metastases: An obstacle to effective anti-angiogenic treatment? Cancer Med. 2013, 2, 427–436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Asahara, T.; Murohara, T.; Sullivan, A.; Silver, M.; van der Zee, R.; Li, T.; Witzenbichler, B.; Schatteman, G.; Isner, J.M. Isolation of putative progenitor endothelial cells for angiogenesis. Science 1997, 275, 964–967. [Google Scholar] [CrossRef] [PubMed]
- Ruoslahti, E. Specialization of tumour vasculature. Nat. Rev. Cancer 2002, 2, 83–90. [Google Scholar] [CrossRef] [PubMed]
- Potente, M.; Gerhardt, H.; Carmeliet, P. Basic and therapeutic aspects of angiogenesis. Cell 2011, 146, 873–887. [Google Scholar] [CrossRef] [PubMed]
- Carmeliet, P. Angiogenesis in health and disease. Nat. Med. 2003, 9, 653–660. [Google Scholar] [CrossRef] [PubMed]
- Iruela-Arispe, M.L.; Davis, G.E. Cellular and molecular mechanisms of vascular lumen formation. Dev. Cell 2009, 16, 222–231. [Google Scholar] [CrossRef] [PubMed]
- Strilic, B.; Kucera, T.; Eglinger, J.; Hughes, M.R.; McNagny, K.M.; Tsukita, S.; Dejana, E.; Ferrara, N.; Lammert, E. The molecular basis of vascular lumen formation in the developing mouse aorta. Dev. Cell 2009, 17, 505–515. [Google Scholar] [CrossRef] [PubMed]
- Zovein, A.C.; Luque, A.; Turlo, K.A.; Hofmann, J.J.; Yee, K.M.; Becker, M.S.; Fassler, R.; Mellman, I.; Lane, T.F.; Iruela-Arispe, M.L. Beta1 integrin establishes endothelial cell polarity and arteriolar lumen formation via a Par3-dependent mechanism. Dev. Cell 2010, 18, 39–51. [Google Scholar] [CrossRef] [PubMed]
- Djonov, V.; Schmid, M.; Tschanz, S.A.; Burri, P.H. Intussusceptive angiogenesis: Its role in embryonic vascular network formation. Circ. Res. 2000, 86, 286–292. [Google Scholar] [CrossRef] [PubMed]
- Kurz, H.; Burri, P.H.; Djonov, V.G. Angiogenesis and vascular remodeling by intussusception: From form to function. News Physiol. Sci. 2003, 18, 65–70. [Google Scholar] [CrossRef] [PubMed]
- Gianni-Barrera, R.; Trani, M.; Reginato, S.; Banfi, A. To sprout or to split? VEGF, Notch and vascular morphogenesis. Biochem. Soc. Trans. 2011, 39, 1644–1648. [Google Scholar] [CrossRef] [PubMed]
- Nico, B.; Crivellato, E.; Guidolin, D.; Annese, T.; Longo, V.; Finato, N.; Vacca, A.; Ribatti, D. Intussusceptive microvascular growth in human glioma. Clin. Exp. Med. 2010, 10, 93–98. [Google Scholar] [CrossRef] [PubMed]
- Brat, D.J.; Van Meir, E.G. Glomeruloid microvascular proliferation orchestrated by VPF/VEGF: A new world of angiogenesis research. Am. J. Pathol. 2001, 158, 789–796. [Google Scholar] [CrossRef]
- Straume, O.; Chappuis, P.O.; Salvesen, H.B.; Halvorsen, O.J.; Haukaas, S.A.; Goffin, J.R.; Begin, L.R.; Foulkes, W.D.; Akslen, L.A. Prognostic importance of glomeruloid microvascular proliferation indicates an aggressive angiogenic phenotype in human cancers. Cancer Res. 2002, 62, 6808–6811. [Google Scholar] [PubMed]
- Lyden, D.; Hattori, K.; Dias, S.; Costa, C.; Blaikie, P.; Butros, L.; Chadburn, A.; Heissig, B.; Marks, W.; Witte, L.; et al. Impaired recruitment of bone-marrow-derived endothelial and hematopoietic precursor cells blocks tumor angiogenesis and growth. Nat. Med. 2001, 7, 1194–1201. [Google Scholar] [CrossRef] [PubMed]
- Reyes, M.; Dudek, A.; Jahagirdar, B.; Koodie, L.; Marker, P.H.; Verfaillie, C.M. Origin of endothelial progenitors in human postnatal bone marrow. J. Clin. Investig. 2002, 109, 337–346. [Google Scholar] [CrossRef] [PubMed]
- Ribatti, D. The involvement of endothelial progenitor cells in tumor angiogenesis. J. Cell. Mol. Med. 2004, 8, 294–300. [Google Scholar] [CrossRef] [PubMed]
- Moschetta, M.; Mishima, Y.; Sahin, I.; Manier, S.; Glavey, S.; Vacca, A.; Roccaro, A.M.; Ghobrial, I.M. Role of endothelial progenitor cells in cancer progression. Biochim. Biophys. Acta 2014, 1846, 26–39. [Google Scholar] [CrossRef] [PubMed]
- Ricci-Vitiani, L.; Pallini, R.; Biffoni, M.; Todaro, M.; Invernici, G.; Cenci, T.; Maira, G.; Parati, E.A.; Stassi, G.; Larocca, L.M.; et al. Tumour vascularization via endothelial differentiation of glioblastoma stem-like cells. Nature 2010, 468, 824–828. [Google Scholar] [CrossRef] [PubMed]
- Kirschmann, D.A.; Seftor, E.A.; Hardy, K.M.; Seftor, R.E.; Hendrix, M.J. Molecular pathways: Vasculogenic mimicry in tumor cells: Diagnostic and therapeutic implications. Clin. Cancer Res. 2012, 18, 2726–2732. [Google Scholar] [CrossRef] [PubMed]
- Folberg, R.; Maniotis, A.J. Vasculogenic mimicry. APMIS 2004, 112, 508–525. [Google Scholar] [CrossRef] [PubMed]
- Lin, A.Y.; Maniotis, A.J.; Valyi-Nagy, K.; Majumdar, D.; Setty, S.; Kadkol, S.; Leach, L.; Pe’er, J.; Folberg, R. Distinguishing Fibrovascular Septa From Vasculogenic Mimicry Patterns. Arch. Pathol. Lab. Med. 2005, 129, 884–892. [Google Scholar] [PubMed]
- El Hallani, S.; Boisselier, B.; Peglion, F.; Rousseau, A.; Colin, C.; Idbaih, A.; Marie, Y.; Mokhtari, K.; Thomas, J.L.; Eichmann, A.; et al. A new alternative mechanism in glioblastoma vascularization: Tubular vasculogenic mimicry. Brain 2010, 133, 973–982. [Google Scholar] [CrossRef] [PubMed]
- Bajcsy, P.; Lee, S.C.; Lin, A.; Folberg, R. Three-dimensional volume reconstruction of extracellular matrix proteins in uveal melanoma from fluorescent confocal laser scanning microscope images. J. Microsc. 2006, 221, 30–45. [Google Scholar] [CrossRef] [PubMed]
- Clarijs, R.; Otte-Holler, I.; Ruiter, D.J.; de Waal, R.M. Presence of a fluid-conducting meshwork in xenografted cutaneous and primary human uveal melanoma. Investig. Ophthalmol. Vis. Sci. 2002, 43, 912–918. [Google Scholar]
- Ahn, G.O.; Brown, J.M. Matrix metalloproteinase-9 is required for tumor vasculogenesis but not for angiogenesis: Role of bone marrow-derived myelomonocytic cells. Cancer Cell 2008, 13, 193–205. [Google Scholar] [CrossRef] [PubMed]
- Iivanainen, E.; Kahari, V.M.; Heino, J.; Elenius, K. Endothelial cell-matrix interactions. Microsc. Res. Tech. 2003, 60, 13–22. [Google Scholar] [CrossRef] [PubMed]
- Rundhaug, J.E. Matrix metalloproteinases and angiogenesis. J. Cell. Mol. Med. 2005, 9, 267–285. [Google Scholar] [CrossRef] [PubMed]
- Eble, J.A.; Niland, S. The extracellular matrix of blood vessels. Curr. Pharm. Des. 2009, 15, 1385–1400. [Google Scholar] [CrossRef] [PubMed]
- Andaloussi, S.E.L.; Mager, I.; Breakefield, X.O.; Wood, M.J. Extracellular vesicles: Biology and emerging therapeutic opportunities. Nat. Rev. Drug Discov. 2013, 12, 347–357. [Google Scholar] [CrossRef] [PubMed]
- Tirziu, D.; Giordano, F.J.; Simons, M. Cell communications in the heart. Circulation 2010, 122, 928–937. [Google Scholar] [CrossRef] [PubMed]
- Kedrin, D.; Gligorijevic, B.; Wyckoff, J.; Verkhusha, V.V.; Condeelis, J.; Segall, J.E.; van Rheenen, J. Intravital imaging of metastatic behavior through a mammary imaging window. Nat. Methods 2008, 5, 1019–1021. [Google Scholar] [CrossRef] [PubMed]
- Mierke, C.T.; Zitterbart, D.P.; Kollmannsberger, P.; Raupach, C.; Schlotzer-Schrehardt, U.; Goecke, T.W.; Behrens, J.; Fabry, B. Breakdown of the endothelial barrier function in tumor cell transmigration. Biophys. J. 2008, 94, 2832–2846. [Google Scholar] [CrossRef] [PubMed]
- Olsson, A.K.; Dimberg, A.; Kreuger, J.; Claesson-Welsh, L. VEGF receptor signalling—In control of vascular function. Nat. Rev. Mol. Cell Biol. 2006, 7, 359–371. [Google Scholar] [CrossRef] [PubMed]
- Weis, S.M.; Cheresh, D.A. alphaV integrins in angiogenesis and cancer. Cold Spring Harb. Perspect. Med. 2011, 1, a006478. [Google Scholar] [CrossRef] [PubMed]
- Dvorak, H.F. Vascular permeability factor/vascular endothelial growth factor: A critical cytokine in tumor angiogenesis and a potential target for diagnosis and therapy. J. Clin. Oncol. 2002, 20, 4368–4380. [Google Scholar] [CrossRef] [PubMed]
- Guo, F.; Wang, Y.; Liu, J.; Mok, S.C.; Xue, F.; Zhang, W. CXCL12/CXCR4: A symbiotic bridge linking cancer cells and their stromal neighbors in oncogenic communication networks. Oncogene 2016, 35, 816–826. [Google Scholar] [CrossRef] [PubMed]
- Teicher, B.A.; Fricker, S.P. CXCL12 (SDF-1)/CXCR4 pathway in cancer. Clin. Cancer Res. 2010, 16, 2927–2931. [Google Scholar] [CrossRef] [PubMed]
- Reymond, N.; d’Agua, B.B.; Ridley, A.J. Crossing the endothelial barrier during metastasis. Nat. Rev. Cancer 2013, 13, 858–870. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Lang, R.; Wei, J.; Fan, Y.; Cui, L.; Gu, F.; Guo, X.; Pringle, G.A.; Zhang, X.; Fu, L. Increased expression of SDF-1/CXCR4 is associated with lymph node metastasis of invasive micropapillary carcinoma of the breast. Histopathology 2009, 54, 741–750. [Google Scholar] [CrossRef] [PubMed]
- Iwasa, S.; Yanagawa, T.; Fan, J.; Katoh, R. Expression of CXCR4 and its ligand SDF-1 in intestinal-type gastric cancer is associated with lymph node and liver metastasis. Anticancer Res. 2009, 29, 4751–4758. [Google Scholar] [PubMed]
- Liang, J.J.; Zhu, S.; Bruggeman, R.; Zaino, R.J.; Evans, D.B.; Fleming, J.B.; Gomez, H.F.; Zander, D.S.; Wang, H. High levels of expression of human stromal cell-derived factor-1 are associated with worse prognosis in patients with stage II pancreatic ductal adenocarcinoma. Cancer Epidemiol. Biomark. Prev. 2010, 19, 2598–2604. [Google Scholar] [CrossRef] [PubMed]
- Thomas, R.M.; Kim, J.; Revelo-Penafiel, M.P.; Angel, R.; Dawson, D.W.; Lowy, A.M. The chemokine receptor CXCR4 is expressed in pancreatic intraepithelial neoplasia. Gut 2008, 57, 1555–1560. [Google Scholar] [CrossRef] [PubMed]
- Guo, L.; Cui, Z.M.; Zhang, J.; Huang, Y. Chemokine axes CXCL12/CXCR4 and CXCL16/CXCR6 correlate with lymph node metastasis in epithelial ovarian carcinoma. Chin. J. Cancer 2011, 30, 336–343. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Shi, X.; Shu, Z.; Xie, T.; Huang, K.; Wei, L.; Song, H.; Zhang, W.; Xue, X. Stromal cell-derived factor-1 (SDF-1)/CXCR4 axis enhances cellular invasion in ovarian carcinoma cells via integrin beta1 and beta3 expressions. Oncol. Res. 2013, 21, 217–225. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Zhang, J.; Cui, Z.M.; Zhao, J.; Zheng, Y. Expression of the CXCL12/CXCR4 and CXCL16/CXCR6 axes in cervical intraepithelial neoplasia and cervical cancer. Chin. J. Cancer 2013, 32, 289–296. [Google Scholar] [CrossRef] [PubMed]
- Murakami, T.; Maki, W.; Cardones, A.R.; Fang, H.; Tun Kyi, A.; Nestle, F.O.; Hwang, S.T. Expression of CXC chemokine receptor-4 enhances the pulmonary metastatic potential of murine B16 melanoma cells. Cancer Res. 2002, 62, 7328–7334. [Google Scholar] [PubMed]
- Hayes, J.; Peruzzi, P.P.; Lawler, S. MicroRNAs in cancer: Biomarkers, functions and therapy. Trends Mol. Med. 2014, 20, 460–469. [Google Scholar] [CrossRef] [PubMed]
- Kohlhapp, F.J.; Mitra, A.K.; Lengyel, E.; Peter, M.E. MicroRNAs as mediators and communicators between cancer cells and the tumor microenvironment. Oncogene 2015, 34, 5857–5868. [Google Scholar] [CrossRef] [PubMed]
- Wurdinger, T.; Tannous, B.A.; Saydam, O.; Skog, J.; Grau, S.; Soutschek, J.; Weissleder, R.; Breakefield, X.O.; Krichevsky, A.M. miR-296 regulates growth factor receptor overexpression in angiogenic endothelial cells. Cancer Cell 2008, 14, 382–393. [Google Scholar] [CrossRef] [PubMed]
- Boon, R.A.; Vickers, K.C. Intercellular transport of microRNAs. Arterioscler Thromb Vasc. Biol. 2013, 33, 186–192. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, G.; Wu, X.; Jiang, Z.; Kasman, I.; Yao, J.; Guan, Y.; Oeh, J.; Modrusan, Z.; Bais, C.; Sampath, D.; et al. Tumour-secreted miR-9 promotes endothelial cell migration and angiogenesis by activating the JAK-STAT pathway. EMBO J. 2012, 31, 3513–3523. [Google Scholar] [CrossRef] [PubMed]
- Carmeliet, P.; Jain, R.K. Angiogenesis in cancer and other diseases. Nature 2000, 407, 249–257. [Google Scholar] [CrossRef] [PubMed]
- Selek, L.; Dhobb, M.; van der Sanden, B.; Berger, F.; Wion, D. Existence of tumor-derived endothelial cells suggests an additional role for endothelial-to-mesenchymal transition in tumor progression. Int. J. Cancer 2011, 128, 1502–1503. [Google Scholar] [CrossRef] [PubMed]
- Ghiabi, P.; Jiang, J.; Pasquier, J.; Maleki, M.; Abu-Kaoud, N.; Rafii, S.; Rafii, A. Endothelial cells provide a notch-dependent pro-tumoral niche for enhancing breast cancer survival, stemness and pro-metastatic properties. PLoS ONE 2014, 9, e112424. [Google Scholar] [CrossRef] [PubMed]
- Gregory, L.A.; Ricart, R.A.; Patel, S.A.; Lim, P.K.; Rameshwar, P. microRNAs, Gap Junctional Intercellular Communication and Mesenchymal Stem Cells in Breast Cancer Metastasis. Curr. Cancer Ther. Rev. 2011, 7, 176–183. [Google Scholar] [CrossRef] [PubMed]
- Valiunas, V.; Polosina, Y.Y.; Miller, H.; Potapova, I.A.; Valiuniene, L.; Doronin, S.; Mathias, R.T.; Robinson, R.B.; Rosen, M.R.; Cohen, I.S.; et al. Connexin-specific cell-to-cell transfer of short interfering RNA by gap junctions. J. Physiol. 2005, 568, 459–468. [Google Scholar] [CrossRef] [PubMed]
- Kizana, E.; Cingolani, E.; Marban, E. Non-cell-autonomous effects of vector-expressed regulatory RNAs in mammalian heart cells. Gene Ther. 2009, 16, 1163–1168. [Google Scholar] [CrossRef] [PubMed]
- Leithe, E.; Sirnes, S.; Omori, Y.; Rivedal, E. Downregulation of gap junctions in cancer cells. Crit. Rev. Oncog. 2006, 12, 225–256. [Google Scholar] [CrossRef] [PubMed]
- Lopes-Bastos, B.M.; Jiang, W.G.; Cai, J. Tumour-Endothelial Cell Communications: Important and Indispensable Mediators of Tumour Angiogenesis. Anticancer Res. 2016, 36, 1119–1126. [Google Scholar] [PubMed]
- Pollmann, M.A.; Shao, Q.; Laird, D.W.; Sandig, M. Connexin 43 mediated gap junctional communication enhances breast tumor cell diapedesis in culture. Breast Cancer Res. 2005, 7, R522–R534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elzarrad, M.K.; Haroon, A.; Willecke, K.; Dobrowolski, R.; Gillespie, M.N.; Al-Mehdi, A.B. Connexin-43 upregulation in micrometastases and tumor vasculature and its role in tumor cell attachment to pulmonary endothelium. BMC Med. 2008, 6, 20. [Google Scholar] [CrossRef] [PubMed]
- Ito, A.; Katoh, F.; Kataoka, T.R.; Okada, M.; Tsubota, N.; Asada, H.; Yoshikawa, K.; Maeda, S.; Kitamura, Y.; Yamasaki, H.; et al. A role for heterologous gap junctions between melanoma and endothelial cells in metastasis. J. Clin. Investig. 2000, 105, 1189–1197. [Google Scholar] [CrossRef] [PubMed]
- Cai, J.; Jiang, W.G.; Mansel, R.E. Gap junctional communication and the tyrosine phosphorylation of connexin 43 in interaction between breast cancer and endothelial cells. Int. J. Mol. Med. 1998, 1, 273–278. [Google Scholar] [CrossRef] [PubMed]
- Zibara, K.; Awada, Z.; Dib, L.; El-Saghir, J.; Al-Ghadban, S.; Ibrik, A.; El-Zein, N.; El-Sabban, M. Anti-angiogenesis therapy and gap junction inhibition reduce MDA-MB-231 breast cancer cell invasion and metastasis in vitro and in vivo. Sci. Rep. 2015, 5, 12598. [Google Scholar] [CrossRef] [PubMed]
- Esser, S.; Lampugnani, M.G.; Corada, M.; Dejana, E.; Risau, W. Vascular endothelial growth factor induces VE-cadherin tyrosine phosphorylation in endothelial cells. J. Cell Sci. 1998, 111 Pt 13, 1853–1865. [Google Scholar] [PubMed]
- Lampugnani, M.G.; Corada, M.; Caveda, L.; Breviario, F.; Ayalon, O.; Geiger, B.; Dejana, E. The molecular organization of endothelial cell to cell junctions: Differential association of plakoglobin, beta-catenin, and alpha-catenin with vascular endothelial cadherin (VE-cadherin). J. Cell Biol. 1995, 129, 203–217. [Google Scholar] [CrossRef] [PubMed]
- Lampugnani, M.G.; Resnati, M.; Raiteri, M.; Pigott, R.; Pisacane, A.; Houen, G.; Ruco, L.P.; Dejana, E. A novel endothelial-specific membrane protein is a marker of cell-cell contacts. J. Cell Biol. 1992, 118, 1511–1522. [Google Scholar] [CrossRef] [PubMed]
- Wallez, Y.; Vilgrain, I.; Huber, P. Angiogenesis: The VE-cadherin switch. Trends Cardiovasc. Med. 2006, 16, 55–59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liao, F.; Li, Y.; O’Connor, W.; Zanetta, L.; Bassi, R.; Santiago, A.; Overholser, J.; Hooper, A.; Mignatti, P.; Dejana, E.; et al. Monoclonal antibody to vascular endothelial-cadherin is a potent inhibitor of angiogenesis, tumor growth, and metastasis. Cancer Res. 2000, 60, 6805–6810. [Google Scholar] [PubMed]
- Wessel, F.; Winderlich, M.; Holm, M.; Frye, M.; Rivera-Galdos, R.; Vockel, M.; Linnepe, R.; Ipe, U.; Stadtmann, A.; Zarbock, A.; et al. Leukocyte extravasation and vascular permeability are each controlled in vivo by different tyrosine residues of VE-cadherin. Nat. Immunol. 2014, 15, 223–230. [Google Scholar] [CrossRef] [PubMed]
- Gavard, J. Endothelial permeability and VE-cadherin: A wacky comradeship. Cell Adhes. Migr. 2014, 8, 158–164. [Google Scholar] [CrossRef]
- Potter, M.D.; Barbero, S.; Cheresh, D.A. Tyrosine phosphorylation of VE-cadherin prevents binding of p120- and beta-catenin and maintains the cellular mesenchymal state. J. Biol. Chem. 2005, 280, 31906–31912. [Google Scholar] [CrossRef] [PubMed]
- Peng, H.H.; Hodgson, L.; Henderson, A.J.; Dong, C. Involvement of phospholipase C signaling in melanoma cell-induced endothelial junction disassembly. Front. Biosci. 2005, 10, 1597–1606. [Google Scholar] [CrossRef] [PubMed]
- Haidari, M.; Zhang, W.; Caivano, A.; Chen, Z.; Ganjehei, L.; Mortazavi, A.; Stroud, C.; Woodside, D.G.; Willerson, J.T.; Dixon, R.A. Integrin alpha2beta1 mediates tyrosine phosphorylation of vascular endothelial cadherin induced by invasive breast cancer cells. J. Biol. Chem. 2012, 287, 32981–32992. [Google Scholar] [CrossRef] [PubMed]
- Aragon-Sanabria, V.; Pohler, S.E.; Eswar, V.J.; Bierowski, M.; Gomez, E.W.; Dong, C. VE-Cadherin Disassembly and Cell Contractility in the Endothelium are Necessary for Barrier Disruption Induced by Tumor Cells. Sci. Rep. 2017, 7, 45835. [Google Scholar] [CrossRef] [PubMed]
- McDonald, D.M.; Munn, L.; Jain, R.K. Vasculogenic mimicry: How convincing, how novel, and how significant? Am. J. Pathol. 2000, 156, 383–388. [Google Scholar] [CrossRef]
- Cao, Z.; Shang, B.; Zhang, G.; Miele, L.; Sarkar, F.H.; Wang, Z.; Zhou, Q. Tumor cell-mediated neovascularization and lymphangiogenesis contrive tumor progression and cancer metastasis. Biochim. Biophys. Acta 2013, 1836, 273–286. [Google Scholar] [CrossRef] [PubMed]
- Murphy, G.F.; Wilson, B.J.; Girouard, S.D.; Frank, N.Y.; Frank, M.H. Stem cells and targeted approaches to melanoma cure. Mol. Asp. Med. 2014, 39, 33–49. [Google Scholar] [CrossRef] [PubMed]
- Potgens, A.J.; van Altena, M.C.; Lubsen, N.H.; Ruiter, D.J.; de Waal, R.M. Analysis of the tumor vasculature and metastatic behavior of xenografts of human melanoma cell lines transfected with vascular permeability factor. Am. J. Pathol. 1996, 148, 1203–1217. [Google Scholar] [PubMed]
- Lammert, E.; Axnick, J. Vascular lumen formation. Cold Spring Harb. Perspect. Med. 2012, 2, a006619. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, H.; Shirakawa, K.; Kawamoto, S.; Saga, T.; Sato, N.; Hiraga, A.; Watanabe, I.; Heike, Y.; Togashi, K.; Konishi, J.; et al. Rapid accumulation and internalization of radiolabeled herceptin in an inflammatory breast cancer xenograft with vasculogenic mimicry predicted by the contrast-enhanced dynamic MRI with the macromolecular contrast agent G6-(1B4M-Gd)(256). Cancer Res. 2002, 62, 860–866. [Google Scholar] [PubMed]
- Shirakawa, K.; Kobayashi, H.; Heike, Y.; Kawamoto, S.; Brechbiel, M.W.; Kasumi, F.; Iwanaga, T.; Konishi, F.; Terada, M.; Wakasugi, H. Hemodynamics in vasculogenic mimicry and angiogenesis of inflammatory breast cancer xenograft. Cancer Res. 2002, 62, 560–566. [Google Scholar] [PubMed]
- Liu, Z.; Li, Y.; Zhao, W.; Ma, Y.; Yang, X. Demonstration of vasculogenic mimicry in astrocytomas and effects of Endostar on U251 cells. Pathol. Res. Pract. 2011, 207, 645–651. [Google Scholar] [CrossRef] [PubMed]
- Thies, A.; Mangold, U.; Moll, I.; Schumacher, U. PAS-positive loops and networks as a prognostic indicator in cutaneous malignant melanoma. J. Pathol. 2001, 195, 537–542. [Google Scholar] [CrossRef] [PubMed]
- Mihic-Probst, D.; Ikenberg, K.; Tinguely, M.; Schraml, P.; Behnke, S.; Seifert, B.; Civenni, G.; Sommer, L.; Moch, H.; Dummer, R. Tumor cell plasticity and angiogenesis in human melanomas. PLoS ONE 2012, 7, e33571. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, W.; Fan, Y.Z.; Zhang, W.Z.; Ge, C.Y. A pilot histomorphology and hemodynamic of vasculogenic mimicry in gallbladder carcinomas in vivo and in vitro. J. Exp. Clin. Cancer Res. 2011, 30, 46. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.Q.; Zheng, Q.H.; Chen, H.; Chen, L.; Xu, J.B.; Chen, M.Y.; Lu, D.; Wang, Z.H.; Tong, H.F.; Lin, S. Ginsenoside Rg3 inhibition of vasculogenic mimicry in pancreatic cancer through downregulation of VEcadherin/EphA2/MMP9/MMP2 expression. Int. J. Oncol. 2014, 45, 1065–1072. [Google Scholar] [CrossRef] [PubMed]
- Sun, B.; Zhang, S.; Zhang, D.; Du, J.; Guo, H.; Zhao, X.; Zhang, W.; Hao, X. Vasculogenic mimicry is associated with high tumor grade, invasion and metastasis, and short survival in patients with hepatocellular carcinoma. Oncol. Rep. 2006, 16, 693–698. [Google Scholar] [CrossRef] [PubMed]
- Tang, N.N.; Zhu, H.; Zhang, H.J.; Zhang, W.F.; Jin, H.L.; Wang, L.; Wang, P.; He, G.J.; Hao, B.; Shi, R.H. HIF-1alpha induces VE-cadherin expression and modulates vasculogenic mimicry in esophageal carcinoma cells. World J. Gastroenterol. 2014, 20, 17894–17904. [Google Scholar] [CrossRef] [PubMed]
- Sun, B.; Qie, S.; Zhang, S.; Sun, T.; Zhao, X.; Gao, S.; Ni, C.; Wang, X.; Liu, Y.; Zhang, L. Role and mechanism of vasculogenic mimicry in gastrointestinal stromal tumors. Hum. Pathol. 2008, 39, 444–451. [Google Scholar] [CrossRef] [PubMed]
- Baeten, C.I.; Hillen, F.; Pauwels, P.; de Bruine, A.P.; Baeten, C.G. Prognostic role of vasculogenic mimicry in colorectal cancer. Dis. Colon Rectum 2009, 52, 2028–2035. [Google Scholar] [CrossRef] [PubMed]
- Williamson, S.C.; Metcalf, R.L.; Trapani, F.; Mohan, S.; Antonello, J.; Abbott, B.; Leong, H.S.; Chester, C.P.; Simms, N.; Polanski, R.; et al. Vasculogenic mimicry in small cell lung cancer. Nat. Commun. 2016, 7, 13322. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Yu, L.; Wang, D.; Zhou, L.; Cheng, Z.; Chai, D.; Ma, L.; Tao, Y. Aberrant expression of CD133 in non-small cell lung cancer and its relationship to vasculogenic mimicry. BMC Cancer 2012, 12, 535. [Google Scholar] [CrossRef] [PubMed]
- Sood, A.K.; Seftor, E.A.; Fletcher, M.S.; Gardner, L.M.; Heidger, P.M.; Buller, R.E.; Seftor, R.E.; Hendrix, M.J. Molecular determinants of ovarian cancer plasticity. Am. J. Pathol. 2001, 158, 1279–1288. [Google Scholar] [CrossRef]
- Tang, H.S.; Feng, Y.J.; Yao, L.Q. Angiogenesis, vasculogenesis, and vasculogenic mimicry in ovarian cancer. Int. J. Gynecol. Cancer 2009, 19, 605–610. [Google Scholar] [CrossRef] [PubMed]
- Sharma, N.; Seftor, R.E.; Seftor, E.A.; Gruman, L.M.; Heidger, P.M., Jr.; Cohen, M.B.; Lubaroff, D.M.; Hendrix, M.J. Prostatic tumor cell plasticity involves cooperative interactions of distinct phenotypic subpopulations: Role in vasculogenic mimicry. Prostate 2002, 50, 189–201. [Google Scholar] [CrossRef] [PubMed]
- Cai, X.S.; Jia, Y.W.; Mei, J.; Tang, R.Y. Tumor blood vessels formation in osteosarcoma: Vasculogenesis mimicry. Chin. Med. J. 2004, 117, 94–98. [Google Scholar] [PubMed]
- Sun, B.; Zhang, S.; Zhao, X.; Zhang, W.; Hao, X. Vasculogenic mimicry is associated with poor survival in patients with mesothelial sarcomas and alveolar rhabdomyosarcomas. Int. J. Oncol. 2004, 25, 1609–1614. [Google Scholar] [CrossRef] [PubMed]
- Ria, R.; Reale, A.; De Luisi, A.; Ferrucci, A.; Moschetta, M.; Vacca, A. Bone marrow angiogenesis and progression in multiple myeloma. Am. J. Blood Res. 2011, 1, 76–89. [Google Scholar] [PubMed]
- Vacca, A.; Ria, R.; Reale, A.; Ribatti, D. Angiogenesis in multiple myeloma. Chem. Immunol. Allergy 2014, 99, 180–196. [Google Scholar] [CrossRef] [PubMed]
- Ellis, L.M.; Fidler, I.J. Finding the tumor copycat. Therapy fails, patients don’t. Nat. Med. 2010, 16, 974–975. [Google Scholar] [CrossRef] [PubMed]
- Pinto, M.P.; Sotomayor, P.; Carrasco-Avino, G.; Corvalan, A.H.; Owen, G.I. Escaping Antiangiogenic Therapy: Strategies Employed by Cancer Cells. Int. J. Mol. Sci. 2016, 17. [Google Scholar] [CrossRef] [PubMed]
- Huijbers, E.J.; van Beijnum, J.R.; Thijssen, V.L.; Sabrkhany, S.; Nowak-Sliwinska, P.; Griffioen, A.W. Role of the tumor stroma in resistance to anti-angiogenic therapy. Drug Resist. Updat 2016, 25, 26–37. [Google Scholar] [CrossRef] [PubMed]
- Kotyza, J. Chemokines in tumor proximal fluids. Biomed. Pap. Med. Fac. Univ. Palacky Olomouc. Czech Repub. 2017, 161, 41–49. [Google Scholar] [CrossRef] [PubMed]
- Pries, R.; Wollenberg, B. Cytokines in head and neck cancer. Cytokine Growth Factor Rev. 2006, 17, 141–146. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y. Future options of anti-angiogenic cancer therapy. Chin. J. Cancer 2016, 35, 21. [Google Scholar] [CrossRef] [PubMed]
- Rytlewski, J.A.; Alejandra Aldon, M.; Lewis, E.W.; Suggs, L.J. Mechanisms of tubulogenesis and endothelial phenotype expression by MSCs. Microvasc. Res. 2015, 99, 26–35. [Google Scholar] [CrossRef] [PubMed]
- Cheng, L.; Huang, Z.; Zhou, W.; Wu, Q.; Donnola, S.; Liu, J.K.; Fang, X.; Sloan, A.E.; Mao, Y.; Lathia, J.D.; et al. Glioblastoma stem cells generate vascular pericytes to support vessel function and tumor growth. Cell 2013, 153, 139–152. [Google Scholar] [CrossRef] [PubMed]
- Shenoy, A.K.; Jin, Y.; Luo, H.C.; Tang, M.; Pampo, C.; Shao, R.; Siemann, D.W.; Wu, L.Z.; Heldermon, C.D.; Law, B.K.; et al. Epithelial-to-mesenchymal transition confers pericyte properties on cancer cells. J. Clin. Investig. 2016, 126, 4174–4186. [Google Scholar] [CrossRef] [PubMed]
- Braeuer, R.R.; Watson, I.R.; Wu, C.J.; Mobley, A.K.; Kamiya, T.; Shoshan, E.; Bar-Eli, M. Why is melanoma so metastatic? Pigm. Cell Melanoma Res. 2014, 27. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.A.; Shi, M.; Li, J.Q.; Qian, C.N. Angiogenesis: Multiple masks in hepatocellular carcinoma and liver regeneration. Hepatol. Int. 2010, 4, 537–547. [Google Scholar] [CrossRef] [PubMed]
- Timar, J.; Tovari, J.; Raso, E.; Meszaros, L.; Bereczky, B.; Lapis, K. Platelet-mimicry of cancer cells: Epiphenomenon with clinical significance. Oncology 2005, 69, 185–201. [Google Scholar] [CrossRef] [PubMed]
- Kotiyal, S.; Bhattacharya, S. Epithelial Mesenchymal Transition and Vascular Mimicry in Breast Cancer Stem Cells. Crit. Rev. Eukaryot. Gene Expr. 2015, 25, 269–280. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.; Sun, B.C.; Zhu, D.W.; Zhao, X.L.; Sun, R.; Zhang, Y.H.; Zhang, D.F.; Dong, X.Y.; Gu, Q.; Li, Y.L.; et al. Notch4+cancer stem-like cells promote the metastatic and invasive ability of melanoma. Cancer Sci. 2016, 107, 1079–1091. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; Xu, S.; Gao, X.; Wang, J.; Xue, H.; Chen, Z.; Zhang, J.; Guo, X.; Qian, M.; Qiu, W.; et al. Macrophage migration inhibitory factor promotes vasculogenic mimicry formation induced by hypoxia via CXCR4/AKT/EMT pathway in human glioblastoma cells. Oncotarget 2017. [Google Scholar] [CrossRef]
- Priya, S.K.; Nagare, R.P.; Sneha, V.S.; Sidhanth, C.; Bindhya, S.; Manasa, P.; Ganesan, T.S. Tumour angiogenesis-Origin of blood vessels. Int. J. Cancer 2016, 139, 729–735. [Google Scholar] [CrossRef] [PubMed]
- Burrell, K.; Zadeh, G. Molecular Mechanisms of Tumor Angiogenesis. In Tumor Angiogenesis; Ran, S., Ed.; Intech Open: Rijeka, Croatia, 2012; pp. 275–296. [Google Scholar]
- Shahneh, F.Z.; Baradaran, B.; Zamani, F.; Aghebati-Maleki, L. Tumor angiogenesis and anti-angiogenic therapies. Hum. Antibodies 2013, 22, 15–19. [Google Scholar] [CrossRef] [PubMed]
- Plate, K.H.; Scholz, A.; Dumont, D.J. Tumor angiogenesis and anti-angiogenic therapy in malignant gliomas revisited. Acta Neuropathol. 2012, 124, 763–775. [Google Scholar] [CrossRef] [PubMed]
- Paulis, Y.W.J.; Soetekouw, P.M.; Verheul, H.M.; Tjan-Heijnen, V.C.; Griffioen, A.W. Signalling pathways in vasculogenic mimicry. Biochim. Biophys. Acta 2010, 1806, 18–28. [Google Scholar] [CrossRef] [PubMed]
- Delgado-Bellido, D.; Serrano-Saenz, S.; Fernandez-Cortes, M.; Oliver, F.J. Vasculogenic mimicry signaling revisited: Focus on non-vascular VE-cadherin. Mol. Cancer 2017, 16, 65. [Google Scholar] [CrossRef] [PubMed]
- Seftor, E.A.; Meltzer, P.S.; Schatteman, G.C.; Gruman, L.M.; Hess, A.R.; Kirschmann, D.A.; Seftor, R.E.; Hendrix, M.J. Expression of multiple molecular phenotypes by aggressive melanoma tumor cells: Role in vasculogenic mimicry. Crit. Rev. Oncol. Hematol. 2002, 44, 17–27. [Google Scholar] [CrossRef]
- Li, S.; Meng, W.; Guan, Z.; Guo, Y.; Han, X. The hypoxia-related signaling pathways of vasculogenic mimicry in tumor treatment. Biomed. Pharmacother. 2016, 80, 127–135. [Google Scholar] [CrossRef] [PubMed]
- Macklin, P.S.; McAuliffe, J.; Pugh, C.W.; Yamamoto, A. Hypoxia and HIF pathway in cancer and the placenta. Placenta 2017. [Google Scholar] [CrossRef] [PubMed]
- Bordeleau, F.; Mason, B.N.; Lollis, E.M.; Mazzola, M.; Zanotelli, M.R.; Somasegar, S.; Califano, J.P.; Montague, C.; LaValley, D.J.; Huynh, J.; et al. Matrix stiffening promotes a tumor vasculature phenotype. Proc. Natl. Acad. Sci. USA 2017, 114, 492–497. [Google Scholar] [CrossRef] [PubMed]
- Krock, B.L.; Skuli, N.; Simon, M.C. Hypoxia-induced angiogenesis: Good and evil. Genes Cancer 2011, 2, 1117–1133. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Zhu, D.M.; Zhou, X.G.; Yin, N.; Zhang, Y.; Zhang, Z.X.; Li, D.C.; Zhou, J. HIF-2alpha promotes the formation of vasculogenic mimicry in pancreatic cancer by regulating the binding of Twist1 to the VE-cadherin promoter. Oncotarget 2017, 8, 47801–47815. [Google Scholar] [CrossRef] [PubMed]
- Angara, K.; Rashid, M.H.; Shankar, A.; Ara, R.; Iskander, A.; Borin, T.F.; Jain, M.; Achyut, B.R.; Arbab, A.S. Vascular mimicry in glioblastoma following anti-angiogenic and anti-20-HETE therapies. Histol. Histopathol. 2017, 32, 917–928. [Google Scholar] [CrossRef] [PubMed]
- Pezzolo, A.; Marimpietri, D.; Raffaghello, L.; Cocco, C.; Pistorio, A.; Gambini, C.; Cilli, M.; Horenstein, A.; Malavasi, F.; Pistoia, V. Failure of anti tumor-derived endothelial cell immunotherapy depends on augmentation of tumor hypoxia. Oncotarget 2014, 5, 10368–10381. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Barral, A.; Orgaz, J.L.; Gomez, V.; del Peso, L.; Calzada, M.J.; Jimenez, B. Hypoxia negatively regulates antimetastatic PEDF in melanoma cells by a hypoxia inducible factor-independent, autophagy dependent mechanism. PLoS ONE 2012, 7, e32989. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, J.; Zhang, X.; Zhang, Y.; Zhu, D.; Zhang, L.; Li, Y.; Zhu, Y.; Li, D.; Zhou, J. HIF-2alpha promotes epithelial-mesenchymal transition through regulating Twist2 binding to the promoter of E-cadherin in pancreatic cancer. J. Exp. Clin. Cancer Res. 2016, 35, 26. [Google Scholar] [CrossRef] [PubMed]
- Alameddine, R.S.; Hamieh, L.; Shamseddine, A. From sprouting angiogenesis to erythrocytes generation by cancer stem cells: Evolving concepts in tumor microcirculation. BioMed Res. Int. 2014, 2014, 986768. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Yang, X.; Yang, Z.; Fei, F.; Li, S.; Qu, J.; Zhang, M.; Li, Y.; Zhang, X.; Zhang, S. Daughter Cells and Erythroid Cells Budding from PGCCs and Their Clinicopathological Significances in Colorectal Cancer. J. Cancer 2017, 8, 469–478. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Fu, Z.; Wei, J.; Guo, J.; Liu, M.; Du, K. Peroxiredoxin 2 is involved in vasculogenic mimicry formation by targeting VEGFR2 activation in colorectal cancer. Med. Oncol. 2015, 32, 414. [Google Scholar] [CrossRef] [PubMed]
- Dong, J.; Zhao, Y.; Huang, Q.; Fei, X.; Diao, Y.; Shen, Y.; Xiao, H.; Zhang, T.; Lan, Q.; Gu, X. Glioma stem/progenitor cells contribute to neovascularization via transdifferentiation. Stem Cell Rev. 2011, 7, 141–152. [Google Scholar] [CrossRef] [PubMed]
- Ren, H.; Du, P.; Ge, Z.; Jin, Y.; Ding, D.; Liu, X.; Zou, Q. TWIST1 and BMI1 in Cancer Metastasis and Chemoresistance. J. Cancer 2016, 7, 1074–1080. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.; Sun, B.; Zhao, X.; Wang, X.; Li, Y.; Qiu, Z.; Liu, T.; Gu, Q.; Dong, X.; Zhang, Y.; et al. Hypoxia promotes vasculogenic mimicry formation by the Twist1-Bmi1 connection in hepatocellular carcinoma. Int. J. Mol. Med. 2015, 36, 783–791. [Google Scholar] [CrossRef] [PubMed]
- Sun, T.; Sun, B.C.; Zhao, X.L.; Zhao, N.; Dong, X.Y.; Che, N.; Yao, Z.; Ma, Y.M.; Gu, Q.; Zong, W.K.; et al. Promotion of tumor cell metastasis and vasculogenic mimicry by way of transcription coactivation by Bcl-2 and Twist1: A study of hepatocellular carcinoma. Hepatology 2011, 54, 1690–1706. [Google Scholar] [CrossRef] [PubMed]
- Liang, Y.; Hu, J.; Li, J.; Liu, Y.; Yu, J.; Zhuang, X.; Mu, L.; Kong, X.; Hong, D.; Yang, Q.; et al. Epigenetic Activation of TWIST1 by MTDH Promotes Cancer Stem-like Cell Traits in Breast Cancer. Cancer Res. 2015, 75, 3672–3680. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Yang, Y.; Xu, S.; Ma, L.; He, M.; Zhang, Z. Slug signaling is up-regulated by CCL21/CCR7 [corrected] to induce EMT in human chondrosarcoma. Med. Oncol. 2015, 32, 478. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Sun, B.; Li, Y.; Zhao, X.; Zhao, X.; Gu, Q.; An, J.; Dong, X.; Liu, F.; Wang, Y. ZEB2 promotes vasculogenic mimicry by TGF-β1 induced epithelial-to-mesenchymal transition in hepatocellular carcinoma. Exp. Mol. Pathol. 2015, 98, 352–359. [Google Scholar] [CrossRef] [PubMed]
- Puisieux, A.; Brabletz, T.; Caramel, J. Oncogenic roles of EMT-inducing transcription factors. Nat. Cell Biol. 2014, 16, 488–494. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Lin, H.; Pan, J.; Mo, C.; Zhang, F.; Huang, B.; Wang, Z.; Chen, X.; Zhuang, J.; Wang, D.; et al. Vasculogenic Mimicry in Prostate Cancer: The Roles of EphA2 and PI3K. J. Cancer 2016, 7, 1114–1124. [Google Scholar] [CrossRef] [PubMed]
- Hess, A.R.; Seftor, E.A.; Seftor, R.E.B.; Hendrix, M.J.C. Phosphoinositide 3-kinase regulates membrane type 1-matrix metalloproteinase (MMP) and MMP-2 activity during melanoma cell vasculogenic mimicry. Cancer Res. 2003, 63, 4757–4762. [Google Scholar] [PubMed]
- Zhang, J.; Qiao, L.; Liang, N.; Xie, J.; Luo, H.; Deng, G.; Zhang, J. Vasculogenic mimicry and tumor metastasis. J. BUON 2016, 21, 533–541. [Google Scholar] [PubMed]
- Robertson, G.P. Mig-7 linked to vasculogenic mimicry. Am. J. Pathol. 2007, 170, 1454–1456. [Google Scholar] [CrossRef] [PubMed]
- Petty, A.P.; Garman, K.L.; Winn, V.D.; Spidel, C.M.; Lindsey, J.S. Overexpression of carcinoma and embryonic cytotrophoblast cell-specific Mig-7 induces invasion and vessel-like structure formation. Am. J. Pathol. 2007, 170, 1763–1780. [Google Scholar] [CrossRef] [PubMed]
- Hendrix, M.J.C.; Seftor, E.A.; Hess, A.R.; Seftor, R.E.B. Vasculogenic mimicry and tumour-cell plasticity: Lessons from melanoma. Nat. Rev. Cancer 2003, 3, 411–421. [Google Scholar] [CrossRef] [PubMed]
- Petty, A.P.; Wright, S.E.; Rewers-Felkins, K.A.; Yenderrozos, M.A.; Vorderstrasse, B.A.; Lindsey, J.S. Targeting Migration inducting gene-7 inhibits carcinoma cell invasion, early primary tumor growth, and stimulates monocyte oncolytic activity. Mol. Cancer Ther. 2009, 8, 2412–2423. [Google Scholar] [CrossRef] [PubMed]
- Sulzmaier, F.J.; Jean, C.; Schlaepfer, D.D. FAK in cancer: Mechanistic findings and clinical applications. Nat. Rev. Cancer 2014, 14, 598–610. [Google Scholar] [CrossRef] [PubMed]
- Van den Brule, F.A.; Buicu, C.; Baldet, M.; Sobel, M.E.; Cooper, D.N.; Marschal, P.; Castronovo, V. Galectin-1 modulates human melanoma cell adhesion to laminin. Biochem. Biophys. Res. Commun. 1995, 209, 760–767. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, S.H.; Ying, N.W.; Wu, M.H.; Chiang, W.F.; Hsu, C.L.; Wong, T.Y.; Jin, Y.T.; Hong, T.M.; Chen, Y.L. Galectin-1, a novel ligand of neuropilin-1, activates VEGFR-2 signaling and modulates the migration of vascular endothelial cells. Oncogene 2008, 27, 3746–3753. [Google Scholar] [CrossRef] [PubMed]
- Garin, M.I.; Chu, C.C.; Golshayan, D.; Cernuda-Morollon, E.; Wait, R.; Lechler, R.I. Galectin-1: A key effector of regulation mediated by CD4+CD25+ T cells. Blood 2007, 109, 2058–2065. [Google Scholar] [CrossRef] [PubMed]
- Cooper, D.N.; Massa, S.M.; Barondes, S.H. Endogenous muscle lectin inhibits myoblast adhesion to laminin. J. Cell Biol. 1991, 115, 1437–1448. [Google Scholar] [CrossRef] [PubMed]
- Mourad-Zeidan, A.A.; Melnikova, V.O.; Wang, H.; Raz, A.; Bar-Eli, M. Expression profiling of Galectin-3-depleted melanoma cells reveals its major role in melanoma cell plasticity and vasculogenic mimicry. Am. J. Pathol. 2008, 173, 1839–1852. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Sun, B.; Zhao, X.; Wang, X.; Zhang, D.; Gu, Q.; Liu, T. MMP-2 and MMP-13 affect vasculogenic mimicry formation in large cell lung cancer. J. Cell. Mol. Med. 2017. [Google Scholar] [CrossRef] [PubMed]
- Ruffini, F.; D’Atri, S.; Lacal, P.M. Neuropilin-1 expression promotes invasiveness of melanoma cells through vascular endothelial growth factor receptor-2-dependent and -independent mechanisms. Int. J. Oncol. 2013, 43, 297–306. [Google Scholar] [CrossRef] [PubMed]
- Ruffini, F.; Levati, L.; Graziani, G.; Caporali, S.; Atzori, M.G.; D’Atri, S.; Lacal, P.M. Platelet-derived growth factor-C promotes human melanoma aggressiveness through activation of neuropilin-1. Oncotarget 2017. [Google Scholar] [CrossRef] [PubMed]
- Lambrechts, D.; Lenz, H.J.; de Haas, S.; Carmeliet, P.; Scherer, S.J. Markers of response for the antiangiogenic agent bevacizumab. J. Clin. Oncol. 2013, 31, 1219–1230. [Google Scholar] [CrossRef] [PubMed]
- Graziani, G.; Lacal, P.M. Neuropilin-1 as Therapeutic Target for Malignant Melanoma. Front. Oncol. 2015, 5, 125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hardy, K.M.; Kirschmann, D.A.; Seftor, E.A.; Margaryan, N.V.; Postovit, L.M.; Strizzi, L.; Hendrix, M.J. Regulation of the embryonic morphogen Nodal by Notch4 facilitates manifestation of the aggressive melanoma phenotype. Cancer Res. 2010, 70, 10340–10350. [Google Scholar] [CrossRef] [PubMed]
- Jue, C.; Lin, C.; Zhisheng, Z.; Yayun, Q.; Feng, J.; Min, Z.; Haibo, W.; Youyang, S.; Hisamitsu, T.; Shintaro, I.; et al. Notch1 promotes vasculogenic mimicry in hepatocellular carcinoma by inducing EMT signaling. Oncotarget 2017, 8, 2501–2513. [Google Scholar] [CrossRef] [PubMed]
- Zhao, N.; Sun, H.; Sun, B.; Zhu, D.; Zhao, X.; Wang, Y.; Gu, Q.; Dong, X.; Liu, F.; Zhang, Y.; et al. miR-27a-3p suppresses tumor metastasis and VM by down-regulating VE-cadherin expression and inhibiting EMT: An essential role for Twist-1 in HCC. Sci. Rep. 2016, 6, 23091. [Google Scholar] [CrossRef] [PubMed]
- Zhao, N.; Sun, B.C.; Zhao, X.L.; Wang, Y.; Sun, H.Z.; Dong, X.Y.; Meng, J.; Gu, Q. Changes in microRNAs associated with Twist-1 and Bcl-2 overexpression identify signaling pathways. Exp. Mol. Pathol. 2015, 99, 524–532. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Lv, C.; Zhang, B.; Zhou, Q.; Cao, Z. MicroRNA-27b functions as a new inhibitor of ovarian cancer-mediated vasculogenic mimicry through suppression of VE-cadherin expression. RNA 2017. [Google Scholar] [CrossRef] [PubMed]
- Wu, N.; Zhao, X.; Liu, M.; Liu, H.; Yao, W.; Zhang, Y.; Cao, S.; Lin, X. Role of microRNA-26b in glioma development and its mediated regulation on EphA2. PLoS ONE 2011, 6, e16264. [Google Scholar] [CrossRef] [PubMed]
- An, L.; Liu, Y.; Wu, A.; Guan, Y. microRNA-124 inhibits migration and invasion by down-regulating ROCK1 in glioma. PLoS ONE 2013, 8, e69478. [Google Scholar] [CrossRef] [PubMed]
- Hunt, S.; Jones, A.V.; Hinsley, E.E.; Whawell, S.A.; Lambert, D.W. MicroRNA-124 suppresses oral squamous cell carcinoma motility by targeting ITGB1. FEBS Lett. 2011, 585, 187–192. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Chen, L.; Zhang, J.; Chen, H.; Fan, J.; Wang, K.; Luo, J.; Chen, Z.; Meng, Z.; Liu, L. Methylation-mediated silencing of the miR-124 genes facilitates pancreatic cancer progression and metastasis by targeting Rac1. Oncogene 2014, 33, 514–524. [Google Scholar] [CrossRef] [PubMed]
- Liang, Y.J.; Wang, Q.Y.; Zhou, C.X.; Yin, Q.Q.; He, M.; Yu, X.T.; Cao, D.X.; Chen, G.Q.; He, J.R.; Zhao, Q. MiR-124 targets Slug to regulate epithelial-mesenchymal transition and metastasis of breast cancer. Carcinogenesis 2013, 34, 713–722. [Google Scholar] [CrossRef] [PubMed]
- Wan, H.Y.; Li, Q.Q.; Zhang, Y.; Tian, W.; Li, Y.N.; Liu, M.; Li, X.; Tang, H. MiR-124 represses vasculogenic mimicry and cell motility by targeting amotL1 in cervical cancer cells. Cancer Lett. 2014, 355, 148–158. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Sun, B.C.; Zhao, X.L.; Zhao, N.; Sun, R.; Zhu, D.W.; Zhang, Y.H.; Li, Y.L.; Gu, Q.; Dong, X.Y.; et al. Twist1-related miR-26b-5p suppresses epithelial-mesenchymal transition, migration and invasion by targeting SMAD1 in hepatocellular carcinoma. Oncotarget 2016, 7, 24383–24401. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Sun, B.; Sun, H.; Zhao, X.; Wang, X.; Zhao, N.; Zhang, Y.; Li, Y.; Gu, Q.; Liu, F.; et al. Regulation of proliferation, angiogenesis and apoptosis in hepatocellular carcinoma by miR-26b-5p. Tumor Biol. 2016, 37, 10965–10979. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Wang, Y.; Deng, R.; Zhang, H.; Dou, J.; Yuan, H.; Hou, G.; Du, Y.; Chen, Q.; Yu, J. miR186 suppresses prostate cancer progression by targeting Twist1. Oncotarget 2016, 7, 33136–33151. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Shao, N.; Mao, X.Y.; Zhu, M.M.; Fan, W.F.; Shen, Z.X.; Xiao, R.; Wang, C.C.; Bao, W.P.; Xu, X.Y.; et al. MiR-4638–5p inhibits castration resistance of prostate cancer through repressing kidins220 expression and PI3K/AKT pathway activity. Oncotarget 2016, 7, 47444–47464. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Wang, X.; Liu, R.; Chen, L.; Yi, J.; Qi, B.; Shuang, Z.; Liu, M.; Li, X.; Li, S.; et al. KDM4B-mediated epigenetic silencing of miRNA-615–5p augments RAB24 to facilitate malignancy of hepatoma cells. Oncotarget 2017, 8, 17712–17725. [Google Scholar] [CrossRef] [PubMed]
- Gutschner, T.; Diederichs, S. The hallmarks of cancer: A long non-coding RNA point of view. RNA Biol. 2012, 9, 703–719. [Google Scholar] [CrossRef] [PubMed]
- Cao, M.X.; Jiang, Y.P.; Tang, Y.L.; Liang, X.H. The crosstalk between lncRNA and microRNA in cancer metastasis: Orchestrating the epithelial-mesenchymal plasticity. Oncotarget 2017, 8, 12472–12483. [Google Scholar] [CrossRef] [PubMed]
- Yu, C.; Xue, J.; Zhu, W.; Jiao, Y.; Zhang, S.; Cao, J. Warburg meets non-coding RNAs: The emerging role of ncRNA in regulating the glucose metabolism of cancer cells. Tumor Biol. 2015, 36, 81–94. [Google Scholar] [CrossRef] [PubMed]
- Beltran-Anaya, F.O.; Cedro-Tanda, A.; Hidalgo-Miranda, A.; Romero-Cordoba, S.L. Insights into the Regulatory Role of Non-coding RNAs in Cancer Metabolism. Front. Physiol. 2016, 7, 342. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Wu, Z.; Yuan, J.; Sun, L.; Lin, L.; Huang, N.; Bin, J.; Liao, Y.; Liao, W. Long non-coding RNA MALAT1 promotes gastric cancer tumorigenicity and metastasis by regulating vasculogenic mimicry and angiogenesis. Cancer Lett. 2017, 395, 31–44. [Google Scholar] [CrossRef] [PubMed]
- Ruf, W.; Seftor, E.A.; Petrovan, R.J.; Weiss, R.M.; Gruman, L.M.; Margaryan, N.V.; Seftor, R.E.; Miyagi, Y.; Hendrix, M.J.C. Differential role of tissue factor pathway inhibitors 1 and 2 in melanoma vasculogenic mimicry. Cancer Res. 2003, 63, 5381–5389. [Google Scholar] [PubMed]
- Kucera, T.; Lammert, E. Ancestral vascular tube formation and its adoption by tumors. Biol. Chem. 2009, 390, 985–994. [Google Scholar] [CrossRef] [PubMed]
- Qiao, L.; Liang, N.; Zhang, J.; Xie, J.; Liu, F.; Xu, D.; Yu, X.; Tian, Y. Advanced research on vasculogenic mimicry in cancer. J. Cell. Mol. Med. 2015, 19, 315–326. [Google Scholar] [CrossRef] [PubMed]
- Cao, Z.; Bao, M.; Miele, L.; Sarkar, F.H.; Wang, Z.; Zhou, Q. Tumour vasculogenic mimicry is associated with poor prognosis of human cancer patients: A systemic review and meta-analysis. Eur. J. Cancer 2013, 49, 3914–3923. [Google Scholar] [CrossRef] [PubMed]
- Guo, Q.; Yuan, Y.; Jin, Z.; Xu, T.; Gao, Y.; Wei, H.; Li, C.; Hou, W.; Hua, B. Association between tumor vasculogenic mimicry and the poor prognosis of gastric cancer in China: An updated systematic review and meta-analysis. BioMed Res. Int. 2016, 2016, 2408645. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Huang, J.; Yao, W.Y.; Ben, Q.W.; Chen, D.F.; He, X.Y.; Li, L.; Yuan, Y.Z. The origins of vacularization in tumors. Front. Biosci. (Landmark Ed.) 2012, 17, 2559–2565. [Google Scholar] [CrossRef] [PubMed]
- Tan, L.Y.; Mintoff, C.; Johan, M.Z.; Ebert, B.W.; Fedele, C.; Zhang, Y.F.; Szeto, P.; Sheppard, K.E.; McArthur, G.A.; Foster-Smith, E.; et al. Desmoglein 2 promotes vasculogenic mimicry in melanoma and is associated with poor clinical outcome. Oncotarget 2016. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.P.; Liao, Y.D.; Mai, D.M.; Xie, P.; Qiang, Y.Y.; Zheng, L.S.; Wang, M.Y.; Mei, Y.; Meng, D.F.; Xu, L.; et al. Tumor vasculogenic mimicry predicts poor prognosis in cancer patients: A meta-analysis. Angiogenesis 2016, 19, 191–200. [Google Scholar] [CrossRef] [PubMed]
- Zhu, B.; Zhou, L.; Yu, L.; Wu, S.; Song, W.; Gong, X.; Wang, D. Evaluation of the correlation of vasculogenic mimicry, ALDH1, KAI1 and microvessel density in the prediction of metastasis and prognosis in colorectal carcinoma. BMC Surg. 2017, 17, 47. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; Zhu, B.; Wu, S.; Zhou, L.; Song, W.; Gong, X.; Wang, D. Evaluation of the correlation of vasculogenic mimicry, ALDH1, KiSS-1, and MACC1 in the prediction of metastasis and prognosis in ovarian carcinoma. Diagn. Pathol. 2017, 12, 23. [Google Scholar] [CrossRef] [PubMed]
- Yao, L.; Zhang, D.; Zhao, X.; Sun, B.; Liu, Y.; Gu, Q.; Zhang, Y.; Zhao, X.; Che, N.; Zheng, Y.; et al. Dickkopf-1-promoted vasculogenic mimicry in non-small cell lung cancer is associated with EMT and development of a cancer stem-like cell phenotype. J. Cell. Mol. Med. 2016, 20, 1673–1685. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.B.; Xu, G.L.; Jia, W.D.; Li, J.S.; Ma, J.L.; Chen, K.; Wang, Z.H.; Ge, Y.S.; Ren, W.H.; Yu, J.H.; et al. Prognostic significance and mechanisms of patterned matrix vasculogenic mimicry in hepatocellular carcinoma. Med. Oncol. 2011, 28 (Suppl. 1), S228–S238. [Google Scholar] [CrossRef] [PubMed]
- Zununi Vahed, S.; Salehi, R.; Davaran, S.; Sharifi, S. Liposome-based drug co-delivery systems in cancer cells. Mater. Sci. Eng. C Mater. Biol. Appl. 2017, 71, 1327–1341. [Google Scholar] [CrossRef] [PubMed]
- Ferrara, N.; Gerber, H.P.; LeCouter, J. The biology of VEGF and its receptors. Nat. Med. 2003, 9, 669–676. [Google Scholar] [CrossRef] [PubMed]
- Vredenburgh, J.J.; Desjardins, A.; Herndon, J.E., 2nd; Dowell, J.M.; Reardon, D.A.; Quinn, J.A.; Rich, J.N.; Sathornsumetee, S.; Gururangan, S.; Wagner, M.; et al. Phase II trial of bevacizumab and irinotecan in recurrent malignant glioma. Clin. Cancer Res. 2007, 13, 1253–1259. [Google Scholar] [CrossRef] [PubMed]
- Van der Veldt, A.A.; Lubberink, M.; Bahce, I.; Walraven, M.; de Boer, M.P.; Greuter, H.N.; Hendrikse, N.H.; Eriksson, J.; Windhorst, A.D.; Postmus, P.E.; et al. Rapid decrease in delivery of chemotherapy to tumors after anti-VEGF therapy: Implications for scheduling of anti-angiogenic drugs. Cancer Cell 2012, 21, 82–91. [Google Scholar] [CrossRef] [PubMed]
- Siemann, D.W. The unique characteristics of tumor vasculature and preclinical evidence for its selective disruption by tumor-vascular disrupting agents. Cancer Treat. Rev. 2011, 37, 63–74. [Google Scholar] [CrossRef] [PubMed]
- Lin, Z.; Zhang, Q.; Luo, W. Angiogenesis inhibitors as therapeutic agents in cancer: Challenges and future directions. Eur. J. Pharmacol. 2016, 793, 76–81. [Google Scholar] [CrossRef] [PubMed]
- Mahase, S.; Rattenni, R.N.; Wesseling, P.; Leenders, W.; Baldotto, C.; Jain, R.; Zagzag, D. Hypoxia-mediated mechanisms associated with antiangiogenic treatment resistance in glioblastomas. Am. J. Pathol. 2017, 187, 940–953. [Google Scholar] [CrossRef] [PubMed]
- Pezzella, F.; Gatter, K.; Qian, C.N. Twenty years after: The beautiful hypothesis and the ugly facts. Chin. J. Cancer 2016, 35, 22. [Google Scholar] [CrossRef] [PubMed]
- Jain, R.K. Antiangiogenesis strategies revisited: From starving tumors to alleviating hypoxia. Cancer Cell 2014, 26, 605–622. [Google Scholar] [CrossRef] [PubMed]
- Folkman, J. Angiogenesis in cancer, vascular, rheumatoid and other disease. Nat. Med. 1995, 1, 27–31. [Google Scholar] [CrossRef] [PubMed]
- Boehm, T.; Folkman, J.; Browder, T.; O’Reilly, M.S. Antiangiogenic therapy of experimental cancer does not induce acquired drug resistance. Nature 1997, 390, 404–407. [Google Scholar] [CrossRef] [PubMed]
- Ribatti, D. The inefficacy of antiangiogenic therapies. J. Angiogenes Res. 2010, 2, 27. [Google Scholar] [CrossRef] [PubMed]
- Sennino, B.; McDonald, D.M. Controlling escape from angiogenesis inhibitors. Nat. Rev. Cancer 2012, 12, 699–709. [Google Scholar] [CrossRef] [PubMed]
- De Falco, S. Antiangiogenesis therapy: An update after the first decade. Korean J. Intern. Med. 2014, 29, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Jayson, G.C.; Kerbel, R.; Ellis, L.M.; Harris, A.L. Antiangiogenic therapy in oncology: Current status and future directions. Lancet 2016, 388, 518–529. [Google Scholar] [CrossRef]
- Crawford, Y.; Ferrara, N. Tumor and stromal pathways mediating refractoriness/resistance to anti-angiogenic therapies. Trends Pharmacol. Sci. 2009. [Google Scholar] [CrossRef] [PubMed]
- Francia, G.; Emmenegger, U.; Kerbel, R.S. Tumor-associated fibroblasts as “Trojan Horse” mediators of resistance to anti-VEGF therapy. Cancer Cell 2009, 15, 3–5. [Google Scholar] [CrossRef] [PubMed]
- Hendrix, M.J.; Seftor, E.A.; Seftor, R.E.; Chao, J.T.; Chien, D.S.; Chu, Y.W. Tumor cell vascular mimicry: Novel targeting opportunity in melanoma. Pharmacol. Ther. 2016, 159, 83–92. [Google Scholar] [CrossRef] [PubMed]
- Seftor, R.E.; Hess, A.R.; Seftor, E.A.; Kirschmann, D.A.; Hardy, K.M.; Margaryan, N.V.; Hendrix, M.J. Tumor cell vasculogenic mimicry: From controversy to therapeutic promise. Am. J. Pathol. 2012, 181, 1115–1125. [Google Scholar] [CrossRef] [PubMed]
- Chung, H.J.; Mahalingam, M. Angiogenesis, vasculogenic mimicry and vascular invasion in cutaneous malignant melanoma—Implications for therapeutic strategies and targeted therapies. Expert Rev. Anticancer Ther. 2014, 14, 621–639. [Google Scholar] [CrossRef] [PubMed]
- Mao, J.M.; Liu, J.; Guo, G.; Mao, X.G.; Li, C.X. Glioblastoma vasculogenic mimicry: Signaling pathways progression and potential anti-angiogenesis targets. Biomark. Res. 2015, 3, 8. [Google Scholar] [CrossRef] [PubMed]
- Sasanelli, F.; Hocking, A.; Pulford, E.; Irani, Y.; Klebe, S. Vasculogenic mimicry in vitro in tumour cells derived from metastatic malignant pleural effusions. Pathology 2017. [Google Scholar] [CrossRef] [PubMed]
- Racordon, D.; Valdivia, A.; Mingo, G.; Erices, R.; Aravena, R.; Santoro, F.; Bravo, M.L.; Ramirez, C.; Gonzalez, P.; Sandoval, A.; et al. Structural and functional identification of vasculogenic mimicry in vitro. Sci. Rep. 2017, 7, 6985. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.; Zhu, C.H.; Zhao, X.; Chen, C.; Zhang, H.L.; Yuan, H.H.; Deng, R.; Dou, J.Z.; Wang, Y.L.; Huang, J.; et al. Atypical ubiquitin E3 ligase complex Skp1-Pam-Fbxo45 controls the core epithelial-to-mesenchymal transition-inducing transcription factors. Oncotarget 2015, 6, 979–994. [Google Scholar] [CrossRef] [PubMed]
- Bianchi, M.E.; Agresti, A. HMG proteins: Dynamic players in gene regulation and differentiation. Curr. Opin. Genet. Dev. 2005, 15, 496–506. [Google Scholar] [CrossRef] [PubMed]
- Lotze, M.T.; DeMarco, R.A. Dealing with death: HMGB1 as a novel target for cancer therapy. Curr. Opin. Investig. Drugs 2003, 4, 1405–1409. [Google Scholar] [PubMed]
- Yin, H.; Shao, Y.; Chen, X. The effects of CD147 on the cell proliferation, apoptosis, invasion, and angiogenesis in glioma. Neurol. Sci. 2017, 38, 129–136. [Google Scholar] [CrossRef] [PubMed]
- Schultz, N.A.; Johansen, J.S. YKL-40-A Protein in the Field of Translational Medicine: A Role as a Biomarker in Cancer Patients? Cancers (Basel) 2010, 2, 1453–1491. [Google Scholar] [CrossRef] [PubMed]
- Shao, R.; Taylor, S.L.; Oh, D.S.; Schwartz, L.M. Vascular heterogeneity and targeting: The role of YKL-40 in glioblastoma vascularization. Oncotarget 2015, 6, 40507–40518. [Google Scholar] [CrossRef] [PubMed]
- Shao, R.; Francescone, R.; Ngernyuang, N.; Bentley, B.; Taylor, S.L.; Moral, L.; Yan, W. Anti-YKL-40 antibody and ionizing irradiation synergistically inhibit tumor vascularization and malignancy in glioblastoma. Carcinogenesis 2014, 35, 373–382. [Google Scholar] [CrossRef] [PubMed]
- Ribatti, D. Tumor refractoriness to anti-VEGF therapy. Oncotarget 2016, 7, 46668–46677. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Zhang, H.; Zhu, H.; Yang, X.; Yang, Y.; Yang, Y.; Min, H.; Chen, G.; Liu, J.; Lu, J.; et al. Endostatin combined with radiotherapy suppresses vasculogenic mimicry formation through inhibition of epithelial-mesenchymal transition in esophageal cancer. Tumor Biol. 2016, 37, 4679–4688. [Google Scholar] [CrossRef] [PubMed]
- Miyata, N.; Taniguchi, K.; Seki, T.; Ishimoto, T.; Sato-Watanabe, M.; Yasuda, Y.; Doi, M.; Kametani, S.; Tomishima, Y.; Ueki, T.; et al. HET0016, a potent and selective inhibitor of 20-HETE synthesizing enzyme. Br. J. Pharmacol. 2001, 133, 325–329. [Google Scholar] [CrossRef] [PubMed]
- Lv, H.; Li, Y.; Du, H.; Fang, J.; Song, X.; Zhang, J. The synthetic compound norcantharidiniInduced apoptosis in mantle cell lymphoma in vivo and in vitro through the PI3K-Akt-NF- kappa B signaling pathway. Evid. Based Complement. Alternat. Med. 2013, 2013, 461487. [Google Scholar] [CrossRef] [PubMed]
- Yeh, C.B.; Hsieh, M.J.; Hsieh, Y.H.; Chien, M.H.; Chiou, H.L.; Yang, S.F. Antimetastatic effects of norcantharidin on hepatocellular carcinoma by transcriptional inhibition of MMP-9 through modulation of NF-kB activity. PLoS ONE 2012, 7, e31055. [Google Scholar] [CrossRef] [PubMed]
- Yeh, C.B.; Hsieh, M.J.; Hsieh, Y.H.; Chien, M.H.; Chiou, H.L.; Yang, S.F. Correction: Antimetastatic effects of norcantharidin on hepatocellular carcinoma by transcriptional inhibition of MMP-9 through modulation of NF-kB activity. PLoS ONE 2017, 12, e0171900. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; You, D.; Lu, M.; He, Y.; Yan, S. Inhibitory effect of norcantharidin on melanoma tumor growth and vasculogenic mimicry by suppressing MMP-2 expression. Oncol. Lett. 2017, 13, 1660–1664. [Google Scholar] [CrossRef] [PubMed]
- Zhu, W.; Sun, W.; Zhang, J.T.; Liu, Z.Y.; Li, X.P.; Fan, Y.Z. Norcantharidin enhances TIMP-2 anti-vasculogenic mimicry activity for human gallbladder cancers through downregulating MMP-2 and MT1-MMP. Int. J. Oncol. 2015, 46, 627–640. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Li, M.; Gu, Y.; Liu, Z.; Xu, S.; Cui, Y.; Sun, B. Thalidomide influences growth and vasculogenic mimicry channel formation in melanoma. J. Exp. Clin. Cancer Res. 2008, 27, 60. [Google Scholar] [CrossRef] [PubMed]
- Guan, Y.Y.; Luan, X.; Lu, Q.; Liu, Y.R.; Sun, P.; Zhao, M.; Chen, H.Z.; Fang, C. Natural products with antiangiogenic and antivasculogenic mimicry activity. Mini. Rev. Med. Chem. 2016, 16, 1290–1302. [Google Scholar] [CrossRef] [PubMed]
- Su, S.J.; Yeh, T.M.; Chuang, W.J.; Ho, C.L.; Chang, K.L.; Cheng, H.L.; Liu, H.S.; Cheng, H.L.; Hsu, P.Y.; Chow, N.H. The novel targets for anti-angiogenesis of genistein on human cancer cells. Biochem. Pharmacol. 2005, 69, 307–318. [Google Scholar] [CrossRef] [PubMed]
- Cong, R.; Sun, Q.; Yang, L.; Gu, H.; Zeng, Y.; Wang, B. Effect of Genistein on vasculogenic mimicry formation by human uveal melanoma cells. J. Exp. Clin. Cancer Res. 2009, 28, 124. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Cao, Z.; Pan, Y.; Zhang, G.; Yang, P.; Guo, P.; Zhou, Q. Jatrorrhizine hydrochloride inhibits the proliferation and neovascularization of C8161 metastatic melanoma cells. Anticancer Drugs 2013, 24, 667–676. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.X.; He, Y.J.; Zhao, S.Z.; Wu, J.G.; Wang, J.T.; Zhu, L.M.; Lin, T.T.; Sun, B.C.; Li, X.R. Inhibition of tumor growth and vasculogenic mimicry by curcumin through down-regulation of the EphA2/PI3K/MMP pathway in a murine choroidal melanoma model. Cancer Biol. Ther. 2011, 11, 229–235. [Google Scholar] [CrossRef] [PubMed]
- Vartanian, A.A.; Burova, O.S.; Stepanova, E.V.; Baryshnikov, A.Y.; Lichinitser, M.R. Melanoma vasculogenic mimicry is strongly related to reactive oxygen species level. Melanoma Res. 2007, 17, 370–379. [Google Scholar] [CrossRef] [PubMed]
- Zang, M.; Hu, L.; Zhang, B.; Zhu, Z.; Li, J.; Zhu, Z.; Yan, M.; Liu, B. Luteolin suppresses angiogenesis and vasculogenic mimicry formation through inhibiting Notch1-VEGF signaling in gastric cancer. Biochem. Biophys. Res. Commun. 2017. [Google Scholar] [CrossRef] [PubMed]
- Jue, C.; Min, Z.; Zhisheng, Z.; Lin, C.; Yayun, Q.; Xuanyi, W.; Feng, J.; Haibo, W.; Youyang, S.; Tadashi, H.; et al. COE inhibits vasculogenic mimicry in hepatocellular carcinoma via suppressing Notch1 signaling. J. Ethnopharmacol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Yao, N.; Ren, K.; Wang, Y.; Jin, Q.; Lu, X.; Lu, Y.; Jiang, C.; Zhang, D.; Lu, J.; Wang, C.; et al. Paris polyphylla suppresses proliferation and vsculogenic mimicry of human osteosarcoma cells and inhibits tumor growth in vivo. Am. J. Chin. Med. 2017, 1–24. [Google Scholar] [CrossRef]
- Yamakawa, S.; Asai, T.; Uchida, T.; Matsukawa, M.; Akizawa, T.; Oku, N. (−)-Epigallocatechin gallate inhibits membrane-type 1 matrix metalloproteinase, MT1-MMP, and tumor angiogenesis. Cancer Lett. 2004, 210, 47–55. [Google Scholar] [CrossRef] [PubMed]
- Ying, M.; Chen, G.; Lu, W. Recent advances and strategies in tumor vasculature targeted nano-drug delivery systems. Curr. Pharm. Des. 2015, 21, 3066–3075. [Google Scholar] [CrossRef] [PubMed]
- Li, X.Y.; Zhao, Y.; Sun, M.G.; Shi, J.F.; Ju, R.J.; Zhang, C.X.; Li, X.T.; Zhao, W.Y.; Mu, L.M.; Zeng, F.; et al. Multifunctional liposomes loaded with paclitaxel and artemether for treatment of invasive brain glioma. Biomaterials 2014, 35, 5591–5604. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Wu, X.; Gao, Y.; Zhang, J.; Zhang, D.; Gu, S.; Zhu, G.; Liu, G.; Li, X. Aptamer-functionalized peptide H3CR5C as a novel nanovehicle for codelivery of fasudil and miRNA-195 targeting hepatocellular carcinoma. Int. J. Nanomed. 2016, 11, 3891–3905. [Google Scholar] [CrossRef] [PubMed]
- Ohuchida, K.; Mizumoto, K.; Murakami, M.; Qian, L.W.; Sato, N.; Nagai, E.; Matsumoto, K.; Nakamura, T.; Tanaka, M. Radiation to stromal fibroblasts increases invasiveness of pancreatic cancer cells through tumor-stromal interactions. Cancer Res. 2004, 64, 3215–3222. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Satyamoorthy, K.; Herlyn, M. N-cadherin-mediated intercellular interactions promote survival and migration of melanoma cells. Cancer Res. 2001, 61, 3819–3825. [Google Scholar] [PubMed]





© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cavaco, A.; Rezaei, M.; Niland, S.; Eble, J.A. Collateral Damage Intended—Cancer-Associated Fibroblasts and Vasculature Are Potential Targets in Cancer Therapy. Int. J. Mol. Sci. 2017, 18, 2355. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms18112355
Cavaco A, Rezaei M, Niland S, Eble JA. Collateral Damage Intended—Cancer-Associated Fibroblasts and Vasculature Are Potential Targets in Cancer Therapy. International Journal of Molecular Sciences. 2017; 18(11):2355. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms18112355
Chicago/Turabian StyleCavaco, Ana, Maryam Rezaei, Stephan Niland, and Johannes A. Eble. 2017. "Collateral Damage Intended—Cancer-Associated Fibroblasts and Vasculature Are Potential Targets in Cancer Therapy" International Journal of Molecular Sciences 18, no. 11: 2355. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms18112355