The Dark Side of the Epitranscriptome: Chemical Modifications in Long Non-Coding RNAs

Faculty of Medicine, Martin-Luther-University Halle-Wittenberg, 06120 Halle (Saale), Germany
*
Author to whom correspondence should be addressed.
†
These authors contributed equally to this work.
Int. J. Mol. Sci. 2017, 18(11), 2387; https://0-doi-org.brum.beds.ac.uk/10.3390/ijms18112387
Submission received: 1 October 2017
/
Revised: 5 November 2017
/
Accepted: 6 November 2017
/
Published: 10 November 2017
(This article belongs to the Collection Regulation by Non-coding RNAs)
Abstract
:The broad application of next-generation sequencing technologies in conjunction with improved bioinformatics has helped to illuminate the complexity of the transcriptome, both in terms of quantity and variety. In humans, 70–90% of the genome is transcribed, but only ~2% carries the blueprint for proteins. Hence, there is a huge class of non-translated transcripts, called long non-coding RNAs (lncRNAs), which have received much attention in the past decade. Several studies have shown that lncRNAs are involved in a plethora of cellular signaling pathways and actively regulate gene expression via a broad selection of molecular mechanisms. Only recently, sequencing-based, transcriptome-wide studies have characterized different types of post-transcriptional chemical modifications of RNAs. These modifications have been shown to affect the fate of RNA and further expand the variety of the transcriptome. However, our understanding of their biological function, especially in the context of lncRNAs, is still in its infancy. In this review, we will focus on three epitranscriptomic marks, namely pseudouridine (Ψ), N6-methyladenosine (m6A) and 5-methylcytosine (m5C). We will introduce writers, readers, and erasers of these modifications, and we will present methods for their detection. Finally, we will provide insights into the distribution and function of these chemical modifications in selected, cancer-related lncRNAs.
Keywords:
cancer; epitranscriptomics; lncRNA; noncoding RNA; 5-methylcytosine; m5C; N6-methyladenosine; m6A; pseudouridine1. Introduction
Non-genomically encoded modifications of macromolecules, ranging further than simple changes in the sequence of the single building blocks, play important roles in nearly all cellular processes. The need to regulate activities and abundances of working components led to mechanisms involving several layers of control. For example, post-translational modifications of proteins, like phosphorylation, acetylation, ubiquitination, glycosylation and methylation are well-known modifications that control the fate of proteins [1].
The first modified nucleotide in DNA was discovered in 1948 [2]. In the following decades, the research field of epigenetics evolved, before the term “epigenetics” was eventually coined in the 1990s. It has been redefined more than once since then [3]. Today we know plenty about the processes of imprinting, gene silencing, X-chromosome inactivation and the function of epigenetics in cancer development. Nevertheless, there is a plethora of information still to be unearthed.
In contrast to DNA and proteins, RNA was neglected for a long time and thought to be just an intermediary component on the way from the information stored inside the DNA double helix to the readily synthesized proteins that are to fulfill all important tasks inside the cell. This view changed in the 1980s, when catalytic functions of RNA molecules were brought to light [4]. Only then the field of non-coding RNA (ncRNA) came into being and slowly began to evolve. More and more classes of RNAs were described, possessing important functions while not coding for a peptide chain [5]. Surprisingly, it was revealed that a large fraction (70–90%) of the human genome is transcribed into RNA. If one takes into account that only 1–3% of the transcriptome carries the blueprint for the synthesis of proteins, it leaves us with the question whether or not the remaining non-coding transcripts are just “trash” [6,7].
NcRNAs are somewhat arbitrarily divided into two classes depending on their size: (1) small ncRNAs (<200 nucleotides (nt)); and (2) long ncRNAs (lncRNAs). Multiple types of small ncRNA (microRNAs (miRNAs), small interfering RNAs (siRNAs) and PIWI-interacting RNAs (piRNAs)) have been studied extensively, especially their role in development and carcinogenesis [8,9,10,11,12,13].
The group of long ncRNAs is highly heterogeneous and its members have an extensive variability in their cellular effects as well as their molecular influences. They are characterized by the lack of a functional open reading frame, meaning they encompass less than 100 amino acids [14,15,16,17]. It is their heterogeneity, which allows them to cover a broad spectrum of molecular and cellular functions by implementing different modes of action [18,19,20,21,22,23,24,25]. Of note, a recent analysis identified a consensus human transcriptome of 91,013 expressed, polyadenylated genes. Importantly, 58,648 genes (~68%) were classified as lncRNAs [26]. The lncRNAdb is a database comprising the growing number of functionally annotated lncRNAs [27].
In 1951, pseudouridine (Ψ), the first modification of a RNA base, was discovered [28], only shortly after the description of 5-methylcytosine (“epicytosine”) in DNA [2,29]. With time, more and more nucleotide modifications were described. Today, over 150 modifications are known and several online databases are keeping track of the progress on this front [30,31,32].
For years, research on RNA modifications focused mainly on transfer RNAs (tRNAs) as a result of their relative abundance and their small size, with ribosomal RNA (rRNA) following after technological advances in sequencing methodology were made [33,34,35,36]. Only after the emergence of next-generation sequencing (NGS) technology in the last couple of years, it was feasible to shift the scope of research towards “transcriptome-wide” modification studies. Nearly all experimental designs involve an enrichment step for polyadenylated (polyA) RNA or some other kind of selection step. Unsurprisingly, messenger RNAs (mRNAs) that make up only a small fraction of the transcribed RNA population are at the center of attention [37]. Today it is evident that RNA modifications are more prevalent and chemically diverse than their DNA counterparts [31]. They are highly dynamic and at least some are reversible, which makes them a critical component of the post-transcriptional gene regulatory landscape. It is becoming clear that RNA modifications and alterations of the RNA modification machinery can have detrimental effects in human disease [38].
This review will focus on the three most abundant RNA modifications, namely, pseudouridine (Ψ), N6-methyladenosine (m6A) and 5-methylcytosine (m5C) (Figure 1). After describing each modification, including the known interacting proteins in greater detail, and highlighting the detection methods for each, we will turn our focus on selected examples of cancer-related lncRNAs that have recently been shown to be part of the emerging epitranscriptome.
2. Discovery and Function of RNA Modifications
Post-transcriptional modifications of RNA molecules have been known for nearly 70 years and about 150 epitranscriptomic marks have been described in the last decades. Chromatographic methods were used in early studies and they are still a very valuable tool for detection today. They remain the gold standard, especially for quantification of RNA modifications. However, reliable transcriptome-wide mapping of the most prevalent alterations with the help of next-generation sequencing technology is the ultimate goal today.
In this paragraph, we will briefly summarize our current knowledge about the three most widespread RNA modifications, namely, pseudouridine (Ψ), N6-methyladenosine (m6A) and 5-methylcytosine (m5C), their interacting proteins and connections to disease states. Moreover, we will introduce current high-throughput detection methods of the aforementioned modifications and briefly discuss their benefits and limitations. In Table 1, we provide an overview of the currently known proteins involved in writing, reading and erasing the three epitranscriptomic marks.
2.1. Pseudouridine
Overall, 5-ribosyluracil (pseudouridine, Ψ) is the most abundant RNA modification, first described in 1951 and found in several classes of RNA, i.e., tRNA, rRNA, small nuclear RNA (snRNA), small nucleolar RNA (snoRNA), mRNA and ncRNA [28]. It is an isomer of the conventional RNA nucleoside uridine (see Figure 1). As a result of its high abundance, Ψ was even termed “the fifth nucleotide” [41,73]. Almost all tRNA molecules possess at least one Ψ residue and the TΨC loop is a characteristic feature of tRNAs.
Incorporated Ψ nucleosides enhance RNA’s ability for base stacking and make the sugar-phosphate backbone more rigid [74,75]. Ψ engages in classical Watson–Crick base pairing with adenosine like its non-modified isomer uridine, though its pairing with all other four bases is stronger than uridine’s. Interestingly, the conversion of uridine to Ψ in translation termination codons was able to suppress translation termination in yeast where pseudouridine-containing stop codons guided the incorporation of selected amino acids [76]. Importantly, altered Ψ distribution patterns in mRNAs and in ncRNAs could be observed in yeast and human cells after stress application [77]. This demonstrates how RNA modifications can expand the genetic code and permit more flexibility to adapt to environmental factors.
In humans, 13 proteins have been identified that contain a pseudouridine synthase domain. These so called pseudouridine synthases (PUS) fall into one of two categories: RNA-dependent or RNA-independent. PUS from the first category rely on other small RNAs that guide these enzymes to their respective target RNAs. In contrast, PUS from the latter category can fulfill their catalytic duty without these adaptor RNAs. Dyskerin, for example, associates with H/ACA snoRNAs, while PUS1 belongs to the snoRNA-independent group [73].
Very recently, it was shown that TruB pseudouridine synthase family member 1 (TRUB1), which is also known as PUS4, and PUS7 combine for about 60% of all reproducibly detected Ψ sites in mRNA [44]. Moreover, a consensus motif (GUUCNANNC) for pseudouridylation by TRUB1 could be identified. While the function of TRUB2-dependent pseudouridylation of mRNA remains an open question, it could be shown that TRUB1 can localize to the nuclear and cytoplasmic compartments. However, its catalytic activity was suggested to be restricted to the nucleus [44]. In contrast, several other pseudouridine synthases (PUS1, pseudouridylate synthase-like 1 (PUSL1), TRUB2, RNA pseudouridylate synthase domain containing 3 (RPUSD3) and RPUSD4) have been predicted or proven to be localized, at least partially, to mitochondria [46,78]. Consequently, multiple mitochondrial RNAs (mtRNAs) are modified by PUS enzymes, e.g., mt-tRNA (RPUSD4 and PUS1), mt-rRNA (RPUSD4) and mt-mRNA (TRUB2 and RPUSD3) [46,78,79]. Intriguingly, deregulation of snoRNAs and mutations in pseudouridine synthases are associated with different diseases like lung cancer, mitochondrial myopathy, sideroblastic anaemia, and dyskeratosis congenita [47,80,81].
Until today, specific reader or eraser proteins for Ψ have not been found. The reason for the absence of an eraser protein could be the fact that the formed C–C bond between the base and the sugar (Ψ) is significantly more inert than the C–N bond (uridine). The Ψ formation could, therefore, be irreversible [74]. Hence, pseudouridylation is likely “read” by structural changes of the RNA molecule itself, which originate from the different properties of Ψ compared to uridine. This could affect the stability of RNA molecules and their interactions with proteins without the need of mediating proteins that specifically read the Ψ residues. Structural functions within RNA molecules and altered base pairing properties of pseudouridine have been described [73,76,82].
Ψ Detection Methods
High-throughput and site-specific mapping of Ψ in RNA relies on the unique reaction of Ψ with N-cyclohexyl-N′-(2-morpholinoethyl)carbodiimide metho-p-toluenesulfonate (CMCT) and the downstream application of next-generation sequencing [83,84]. Pseudo-seq, Ψ-seq and PSI-seq are all based on this workflow [77,85,86]. Extracted RNA is fragmented and treated with CMCT, which forms a covalent bond with Ψ, U and G residues. However, only the Ψ-CMC product is stable under alkaline conditions whereas U- and G-reaction products are getting hydrolyzed. Subsequently, the CMC-modified RNA is reverse transcribed. Importantly, reverse transcription will terminate one nucleotide 3′ to pseudouridylated sites due to the bulky (CMC-) group attached to Ψ residues. Hence, next-generation sequencing of cDNA libraries constructed with or without CMCT treatment allows one to map Ψ positions in RNA transcripts by calculating stop rate differences between these two samples.
A related, yet more sensitive method, called CeU-Seq, was recently developed [87]. Here, a derivative of CMCT, to which biotin can be added via click chemistry, is used to modify Ψ residues within RNA molecules. Subsequent pull-down of biotin-labeled transcripts with streptavidin beads leads to an enrichment of modified RNA molecules over non-modified ones. This results in a better signal-to-noise ratio and improves detection of Ψ-modified RNA transcripts of low abundance.
Next to these sequencing-based methods, alternative strategies have been introduced or are currently under development to map pseudouridine in RNA. For example, site-specific cleavage and radioactive-labeling followed by ligation-assisted extraction and thin-layer chromatography (SCARLET) is a low-throughput method to validate different RNA base modifications, e.g., Ψ, and to determine the stoichiometry at individual nucleotide positions [87,88]. SCARLET is described in greater detail in the m6A detection section below.
Analyzing Ψ modifications in RNA by mass spectrometry is another, rather challenging approach, because there is no mass difference between uridine and Ψ [89]. Accordingly, there is a need for chemical labels that can be introduced, either directly into the cells by addition to the growth medium or through chemical reaction of the isolated RNA ex vivo. Hence, there are no high-throughput methods yet, but advances in the field (even towards label-free approaches) are continuously made [90].
2.2. N6-Methyladenosine
N6-methyladenosine (m6A) was first discovered in 1974 [91,92]. It is found in snoRNAs, tRNAs, rRNAs and other ncRNAs, and is the most widespread base modification of mRNA. It accounts for 0.2–0.6% of all adenosines in mammalian mRNA with about three sites per transcript [93]. There are two slightly differing consensus motifs proposed in which m6A occurs: RRACH [94,95] and DRACH [96] (with D = G, A, or U; R = G or A; and H = C, A, or U). However, the distribution of m6A in mRNA is not random, but follows a certain pattern; it is often located near stop codons and in the 3′-untranslated region (UTR) suggesting a regulatory role in cellular processes [57,97]. Indeed, m6A modifications have been shown to play important roles in RNA stability (mRNA and ncRNA) [56,98], mRNA translation [59,99,100], secondary structure formation (mRNA and lncRNA) [61,101,102], alternative splicing, and polyadenylation [93,103] as well as subcellular RNA location [62,104]. Very recently, a novel role for m6A in the UV-induced DNA damage response pathways was reported [105]. Importantly, the levels of m6A in mRNA are highly dynamic and the modification is reversible. In fact, several m6A writer, reader, and eraser proteins have been identified [57,97,106]. This reinforces the idea that m6A modifications serve important functions and might be involved in cell signaling networks.
2.2.1. m6A Writers
The m6A formation is catalyzed inside the nucleus by the m6A writer complex, which consists of the enzymatically active methyltransferase-like 3 (METTL3) protein and several interacting proteins [48]. Known interaction partners of METTL3 are: (a) methyltransferase-like 14 (METTL14); (b) Wilms’ tumor 1-associating protein (WTAP); (c) KIAA1429, also called vir like m6A methyltransferase associated (VIRMA); (d) RNA-binding motif protein 15 (RBM15), and; (e) RBM15B. METTL3 possesses a catalytically active methyltransferase domain and it is the principal m6A forming enzyme in polyadenylated mRNA, but it does not methylate rRNA [107]. METTL14, on the other hand, has a degenerated active site and it is not catalytically active in the heterodimer with METTL3 [108]. It binds to substrate RNA and forms extensive contacts with METTL3 whose enzymatic activity is enhanced by this molecular interaction [107,109]. Hence, METTL14 acts as a RNA adaptor protein, which greatly enhances the methyltransferase activity of the m6A writer complex. Knockdown of METTL3 or METTL4 in glioblastoma stem-like cells (GSCs) dramatically increased their growth and self-renewal. In addition, this depletion substantially increased GSC-initiated tumor progression [110].
WTAP is a crucial component of the writer complex [49,51]. One of its functions is to localize the METTL3-METTL14-complex to nuclear speckles [50].
KIAA1429 is associated with the writer complex and its depletion led to a decrease in m6A abundance in RNA [51]. However, its molecular function is still obscure.
RBM15 and its paralog RBM15B are components of the methyltransferase complex and they interact with METTL3 in a WTAP-dependent manner [53,54]. RBM15/15B use their RNA-binding domains to enable the binding of the writer complex to specific mRNAs and even specific sites within these. The lncRNA X-inactive specific transcript (XIST), for instance, is a target of RBM15/15B directed methylation [54].
Recently, METTL16 was described as a methyltransferase fulfilling its functions independently of the m6A writer complex surrounding METTL3 [55]. It is a conserved U6 snRNA methyltransferase, and it has evolved an additional function in vertebrates to control S-Adenosyl methionine (SAM) homeostasis by differentially methylating a hairpin structure inside the methionine adenosyltransferase 2A (MAT2A) mRNA thereby modulating alternative splicing [55].
2.2.2. m6A Readers
The m6A modifications in RNA transcripts are predominantly read by the eukaryotic initiation factor 3 (eIF3) or by proteins that contain a YTH (YT521-B homology) domain. There are additional RNA-binding proteins (RBPs) associating with m6A, which are not seen as classical m6A binders.
DF1, DF2 and DF3 belong to the YT521-B homology domain family (YTHDF) and represent one group of cytoplasmic m6A reader proteins [57]. DF1 is involved in modulating translation efficiency whereas DF2 is proposed to have a function in mRNA stability [56,99]. Additionally, it was reported that DF2 can localize to the nucleus after stress induction where it promotes cap-independent translation initiation [100].
A second group of m6A reader proteins are YTH domain-containing proteins (YTHDCs). YTHDC1 is a nuclear enriched protein that binds to protein-coding and non-coding transcripts. It is the major reader of nuclear m6A modifications [106]. YTHDC1 is a mediator of the X-chromosome silencing effect of XIST and was characterized as a regulator of mRNA splicing events [54,103]. YTHDC2’s functions are poorly defined. It is located inside the nucleus as well as in the cytoplasm and was shown to bind to select m6A sites in ncRNAs [54]. Tanabe et al. linked upregulated expression of YTHDC2 to metastasis in colon cancer [111].
Another important m6A binding protein is eIF3. In fact, adenosine methylation is a major mechanism by which eIF3 is recruited to mRNAs. After binding to m6A in the 5′-UTR, translation is initiated by eIF3 in a 5′-cap- and eukaryotic initiation factor 4E (eIF4E)-independent manner [59]. These findings suggest an alternative way of translation initiation mediated by m6A modifications in 5′-UTRs of mRNAs when eIF4-dependent initiation is hindered by specific cell states.
Finally, heterogeneous nuclear ribonucleoprotein C (hnRNP C) and hnRNP A2/B1 belong to the group of proteins with reported binding to m6A after changes in local and secondary structure of mRNA and lncRNA [61]. Binding of these proteins to m6A-containing transcripts has been shown to affect alternative splicing as well as miRNA biogenesis [60,61]. Interestingly, the well-characterized, AU-rich element (ARE) and poly(A)-binding protein human antigen R (HuR) preferentially binds to sequences that lack m6A modifications, and loss of m6A methylation enhances HuR binding, which increases target RNA stability [98]. Further research will be necessary to illuminate the connection between the binding of those proteins and cellular processes.
2.2.3. m6A Erasers
The nuclear α-ketoglutarate-dependent dioxygenase alkB homolog 5 (ALKBH5) protein was recently identified as a RNA demethylating enzyme [62]. Alkbh5-deficient mice show defects in spermatogenesis, but are otherwise viable indicating that Alkbh5 demethylase activity is not strictly required during development [62]. Redundant demethylation pathways might be in place as well. In contrast, ALKBH5-mediated demethylation of m6A transcripts seems to be crucial in certain cancers. For example, Zhang et al. could show that ALKBH5 protein levels are elevated in GSCs and its expression is a negative prognostic factor for glioblastoma (GBM) patients [112]. Furthermore, the authors reported that ALKBH5 demethylates nascent FOXM1 transcripts which results in enhanced FOXM1 expression. Interestingly, FOXM1-AS, a nuclear lncRNA, facilitates the interaction between ALKBH5 and nascent FOXM1 transcripts. Depletion of ALKBH5 and FOXM1-AS disrupted GSC tumorigenesis through the FOXM1 axis [112]. In addition to its role in GBM, ALKBH5 expression is reported to be induced by hypoxia in breast cancer cells. Knockdown of ALKBH5 expression in MDA-MB-231 human breast cancer cells significantly reduced their capacity for tumor initiation as a result of reduced numbers of breast cancer stem cells (BCSCs) [113].
Very recent publications spawned conflicting data concerning the fat mass- and obesity-associated protein (FTO), a member of the AlkB-related dioxygenase family, which was originally described as an eraser of m6A modifications in RNA [63]. Recently, Mauer et al. reported that FTO acts as an eraser for the closely related N6, 2′-O-dimethyladenosine (m6Am) modification, which was co-detected with m6A in previous studies [114]. Their refined detection technique made it possible to differentiate between these two modifications, allowing a more detailed examination of FTO’s substrate spectrum. As a result, the authors could show that m6A is not the preferred target for FTO in vivo and they concluded that FTO is the eraser protein for m6Am.
In contrast, other reports showed a substantial increase in mRNA m6A levels in GSCs treated with the FTO inhibitor MA2, which suppressed GSC-initiated tumorigenesis and prolonged the lifespan of GSC-engrafted mice [110]. Another recent publication shed light on the role of FTO in acute myeloid leukemia (AML). Li et al. indicated that FTO, as a m6A demethylase, plays a critical oncogenic role in AML [115]. FTO is highly expressed in certain AMLs and it enhances oncogene-mediated cell transformation and leukemogenesis. It does so by reducing m6A levels in specific mRNA transcripts [115].
2.2.4. m6A Detection
First high-throughput m6A mapping strategies were based on the immunoprecipitation of modified RNA molecules using m6A-specific antibodies coupled to the subsequent application of NGS technologies (m6A-seq [57], MeRIP-Seq [97]). Here, isolated and poly(A)-enriched RNA is fragmented to about 100 nt long fragments, which are immunoprecipitated with m6A-specific antibodies. Thereafter, cDNA libraries are constructed from m6A-containing samples as well as non-immunoprecipitated input control samples and subjected to sequencing. NGS reads are then mapped to the reference genome. Fragments containing m6A will be enriched and provide more reads. Through algorithm-based position calling, m6A positions can be determined. However, both methods provide a rather low resolution (100–200 nt), because peaks can be broad and identification of a single modified adenosine residue can be difficult. The same is true for m6A residues that lie in close proximity to each other. Therefore, true identification of specific m6A residues on a transcriptome-wide level is not possible with m6A-seq or MeRIP-Seq. Another drawback of these methods is the specificity of the available antibodies. These recognize m6A as well as N6,2′-O-dimethyladenosine (m6Am), which both contain the 6-methyladenine base. Therefore, it is not possible to distinguish between the prevalent m6A and m6Am, which is found close to the 5′-cap of mRNAs [114].
To circumvent some of the problems that arise with MeRIP-Seq and m6A-seq, m6A individual-nucleotide-resolution cross-linking and immunoprecipitation (miCLIP) was developed [96]. This method uses cross-linking–induced mutation site (CIMS) and cross-linking–induced truncation site (CITS) profiles generated during reverse transcription, due to the binding of specific antibodies at m6A residues and subsequent cross-linking by UV light, to identify precise positions of m6A-modified residues in RNA at single-nucleotide resolution. After next-generation sequencing and bioinformatical analysis of consensus motifs, identification of the modified residues is easier than in previously used methods.
Another very similar method, called photo-crosslinking-assisted m6A sequencing (PA-m6A-seq), was recently introduced [116]. PA-m6A-seq combines the incorporation of a photoactivatable ribonucleoside, 4-thiouridine (4-SU), into RNA and the immunoprecipitation with a m6A-specific antibody. By crosslinking the antibody to the introduced nucleotide and the subsequent transition of U/T to C during reverse transcription-polymerase chain reaction (RT-PCR), it is possible to narrow the resulting peaks after next generation sequencing to about 23 nt, which makes m6A position calling easier.
The m6A-level and isoform-characterization sequencing (m6A-LAIC-seq) uses a RNA immunoprecipitation protocol with m6A-specific antibodies and spike-in RNAs as internal standards coupled with whole-transcriptome sequencing to gain quantitative information about m6A modifications in poly(A)+ RNA fractions [93]. Using spike-in standards permits analysis of m6A levels per gene, but not the methylation stoichiometry of a single modified nucleotide.
One laborious, low-throughput method to confirm m6A sites at single-nucleotide resolution is site-specific cleavage and radioactive-labeling followed by ligation-assisted extraction and thin-layer chromatography (SCARLET) [88]. Moreover, it features the great advantage of quantifying the methylation status of a single modified nucleotide. The method relies on an induced, site-specific cut by RNase H followed by radioactive labeling of the resulting RNA fragments and a splint ligation to a single-stranded DNA oligo. After RNA digestion, gel purification, and Nuclease P1 treatment the radiolabeled mononucleotides are separated by thin layer chromatography and the methylation status can be analyzed quantitatively. Application of SCARLET is not limited to m6A, but can be applied to detect other RNA modifications as well, e.g., m5C and Ψ [87].
2.3. 5-Methylcytosine
5-methylcytosine (m5C) occurs in tRNA, rRNA, mRNA and lncRNA [72]. In mouse and human protein-coding transcripts, m5C sites are found about 100 nt downstream of the translation initiation site and in the UTRs [72,117,118]. Currently, two groups of m5C writers are known. The seven members of the NOP2/SUN RNA methyltransferase family member (NSUN) family, constituting the first group, methylate tRNA (NSUN2, NSUN6), rRNA (NSUN1, NSUN5), mRNA (NSUN2), ncRNA (NSUN2) as well as mt-rRNA (NSUN4) and mt-tRNA (NSUN3), respectively [64,66,67,68,69,119,120]. So far, NSUN7 substrates are obscure. However, mutations in the Nsun7 gene lead to sperm motility defects, and therefore subfertility or complete infertility in male mice [70]. Moreover, mutations inside the NSUN2 gene are linked with autosomal-recessive intellectual disability [121,122,123], and overexpression as well as increased copy numbers of NSUN2 have been detected in human cancers [65,124,125].
The second writer protein group for m5C has only one member so far, namely, DNA methyltransferase-2 (DNMT2), which was previously thought to methylate DNA [126]. However, DNMT2 was found to act on tRNA with three tRNA substrates currently known [71,127]. DNMT2 expression levels, similar to other tRNA methyltransferases, were found to be frequently altered in cancer cells [128]. Indeed, data from hundreds of tumor samples collected by the COSMIC database reveal an overexpression of DNMT2 in several human cancers [129]. Additionally, more than 60 somatic mutations have been detected. An in vitro follow-up study examined 13 mutations and found varying results concerning DNMT2’s methylating activity [130]. However, translation of these data to pathways inside the cell’s regulatory network is difficult and needs to be addressed in context of the respective cancer type.
Importantly, a m5C eraser is still to be identified. Also, the functions of m5C are not well understood yet, although a recent study suggests a role for m5C in RNA transport [72]. The Aly/REF export factor (ALYREF), a mRNA export adaptor protein, was identified as a m5C binding (reader) protein, which promotes selective mRNA export from the nucleus [72].
Taken together, these findings suggest that m5C modifications in transcripts and the proteins involved in this pathway are important to control the fate and function of RNAs. A dysregulation of this system might contribute to pathophysiological states. Hence, a more detailed mapping of m5C modifications, as well as the discovery and functional analysis of m5C interacting proteins could contribute to a better understanding of the underlying molecular disease mechanisms.
m5C Detection
m5C is the most extensively studied base modification in DNA. Owing to the different chemical characteristics of DNA and RNA, the standard method of bisulfite treatment followed by sequencing had to be adapted to be used for m5C detection in RNA [131]. Bisulfite treatment leads to a chemical conversion of unmodified cytosine to uracil, whereas the methylated base remains unaltered. This difference can be detected by Sanger sequencing or after library construction by next-generation sequencing. Widespread m5C modifications could be detected by this method, called Bisulfite-seq, in tRNA, mRNA and ncRNA at single nucleotide resolution [118]. However, Bisulfite-seq has some drawbacks: cytosines in double-stranded RNA regions can remain unmodified by bisulfite treatment and are later falsely called m5C residues. Aside from the structure-related issue, sites with alternative modifications of cytosine can be misidentified as m5C sites, because those modified bases are usually resistant to bisulfite treatment as well. This is especially true for the closely related hm5C modification, which cannot be distinguished from m5C through Bisulfite-seq [132]. Therefore, candidate sites should be validated with complementary methods. For example, an alternative, immunoprecipitation-based protocol was recently developed [133]. Fragmented RNA is immunoprecipitated with a m5C-specific antibody or a control antibody, followed by library preparation and NGS. This protocol was applied to the RNA of the archaeon Sulfolobus solfataricus, which verified the Bisulfite-seq results [133].
Another, indirect m5C mapping method, called 5-Azacytidine–mediated RNA immunoprecipitation, or Aza-IP, takes advantage of the random incorporation of 5-Azacytosine into RNA during RNA synthesis inside the cell [127,134]. Overexpression of (epitope-tagged) RNA methyltransferase enzymes (RMTs) allows the immunoprecipitation of those enzymes with a (tag-) specific antibody. Importantly, 5-Azacytosine is a suicide substrate for m5C-RMTs due to the covalent link formed between the examined methyltransferase and its substrate RNA, which allows stringent washing steps. The Aza-IP is concluded by RNA fragmentation, cDNA library construction, and NGS. Comparison of resulting reads between the samples with a control or a specific antibody allows mapping of m5C sites. Additionally, the modified cytosine residue is read as a guanosine instead of cytosine during sequencing. This facilitates a precise calling of the candidate modified nucleotide. Identification of direct targets of DNMT2 and NSUN2 could be achieved with this method.
Indeed, NSUN2-specific methylation sites have been previously identified using yet another m5C detection method called methylation iCLIP (miCLIP) [120]. This method is derived from individual-nucleotide-resolution cross-linking and immunoprecipitation (iCLIP) [135], and abstains from chemical modifications of the RNA. To achieve this, a C271A mutant of NSUN2 was used, which forms a stable bond with its target cytosine residue due to the lack of its second cysteine in the catalytic center. The stable protein-RNA-complex was immunoprecipitated and NGS-based m5C mapping followed. A high cytosine appearance at position +1 in the cDNA libraries corresponds to the first nucleotide of the sequence reads, which means that reverse transcription terminated at the cross-link site of the cytosine with its modifying protein. New mRNA and ncRNA transcripts (e.g., vault RNAs) were identified as methylation targets, aside from confirming already known tRNA targets of NSUN2. Thus, identification of direct targets of NSUN2 can be achieved with single-nucleotide resolution using this protocol.
In summary, we are just at the beginning of a long journey to fully comprehend the breadth, dynamics and molecular functions of RNA modifications in mammalian cells. Recently developed high-throughput mapping approaches will enable us to characterize the epitranscriptome in diverse cellular contexts. Nevertheless, further technological improvements are needed to enhance the resolution and sensitivity of these methods. The discovery of additional writers, readers and erasers of the epitranscriptome, as well as a detailed analysis of already known ones, will spawn new research directions and might open the door for novel therapeutic strategies.
3. RNA Modifications in Cancer-Related lncRNAs
Initial studies showed that RNA modifications have an impact on transcript localization, turnover and translation rates, thereby adding a new layer of gene expression control. However, most studies focused on mRNAs and much less is known about the functional relevance of RNA modifications in lncRNAs. Importantly, recent transcriptome-wide mapping studies revealed an overwhelming amount of RNA modifications in thousands of lncRNAs (Figure 2). Here, we will focus on a selection of lncRNAs with a well-established role in human cancers [20]. We will briefly introduce these lncRNAs and summarize our current knowledge about the RNA modifications previously identified in these non-coding transcripts.
3.1. MALAT1
Transcribed from chromosome 11 through RNA Pol II the metastasis associated lung adenocarcinoma transcript 1 (MALAT1), also known as NEAT2 (nuclear-enriched abundant transcript 2), is a highly conserved and extremely abundant long non-coding RNA of ~8 kb in size that localizes to nuclear speckles [136]. Despite its ubiquitous expression in healthy organs, its genomic inactivation in mice is compatible with life and development [137,138,139].
Originally, MALAT1 was identified in a subtractive hybridization screen for transcripts with an altered expression in stage I non-small cell lung cancers (NSCLCs) that either did or did not metastasize [140]. Follow-up studies on its cellular and molecular function in lung cancer established MALAT1 as a master regulator of metastasis and a potential therapeutic target [141,142]. Furthermore, MALAT1 has been found to control proliferation, migration and apoptosis in many different human cancers, e.g., pancreatic cancer, hepatoma and ovarian cancer [143,144,145,146]. Furthermore, its overexpression can increase drug resistance as shown for temozolomide in glioblastoma cells [147].
Mechanistically, MALAT1 is thought to fulfill its cellular functions by regulating gene expression levels as well as alternative splicing [142,143,148,149]. Interestingly, Wilusz et al. showed that MALAT1 undergoes a maturation process that yields a mature and stable transcript [150]. Here, the 3′-terminus of the MALAT1 is cleaved by RNase P at position A6690 that follows after an adenosine-rich tract. This produces two ncRNAs: a long, 5′-capped MALAT1 transcript with a short poly(A)-tail like moiety and a small, tRNA-like ncRNA, the so called MALAT1-associated small cytoplasmic RNA (mascRNA) [150]. While the MALAT1 transcript remains in the nucleus, the mature 61-nt mascRNA is exported to the cytoplasm where it might act as an immune regulator [151]. Importantly, processed MALAT1 transcripts contain a 3′-triple-helical RNA stability element consisting of a U-rich internal loop that associates with a downstream A-rich tract to protect the MALAT1 transcript from degradation. This triple helix is recognized and bound by the m6A writer METTL16 [152]. This raises the possibility of a m6A modification being present in this triple-helix. Alternatively, MALAT1 could serve a role as a regulator of RNA processing or modification events through guiding METTL16 onto its RNA targets.
It has recently been shown that MALAT1 can carry m6A modifications [88]. The authors used SCARLET to determine the m6A status of MALAT1 in different cell lines focusing on the largest m6A/MeRIP-Seq peak previously identified [57,97]. This peak contains seven predicted m6A-consensus motifs (RRACH), and four of these consistently carried methylated residues across the four different cell-types tested. However, the modification rate varied: two positions (A2515 and A2577) displayed the highest (41–67% and 51–88%, respectively) modification rate, followed by A2611 (13–49%) and A2720 (7–14%). Only a small fraction of MALAT1 molecules (2–3%) carried the m6A modification at the other predicted sites (A2674/2684/2698) in two out of four cell lines. Importantly, secondary structure prediction and mapping experiments demonstrated that the two residues with the highest m6A rate (i.e., A2515 and A2577) are located in hairpin stems. Subsequent structural mapping assays using methylated and unmethylated synthetic RNA oligonucleotides in conjunction with a set of structure-sensitive nucleases revealed that the presence of m6A in the hairpin stem increases the opening of the stem, i.e., reduces duplex stability [88]. In a later study, the authors could show that adenosine methylation at position A2577 destabilizes the hairpin stem, making the opposing U-tract more single-stranded and accessible for RNA-binding proteins, e.g., hnRNP C [61]. Furthermore, nuclear magnetic resonance and Förster resonance energy transfer studies demonstrated that the overall structure of the MALAT1 hairpin is maintained upon m6A modification, but the nucleobases of the hairpin stem are more flexible and solvent accessible [153]. These results support a model in which m6A regulates protein binding through its influence on RNA structure (“m6A switch”) [61]. While the MALAT1 hairpin stem is the first example of such an m6A-switch, changes induced by m6A modifications might apply to a much larger family of m6A-regulated RNA structures. Of note, modification of MALAT1 with m6A is highly dynamic and can be modulated by heat shock, UV and growth factor treatments in HepG2 cells [57]. Thus, it would be interesting to learn more about the functional significance of these conditional modifications.
Next to m6A, MALAT1 also contains several pseudouridine residues at positions U5160, U5590 and U3374 [77,87]. However, their impact on MALAT1 structure, protein interaction or molecular function are not known.
Additionally, Squires et al. identified several putative m5C sites within MALAT1 through RNA bisulfite conversion combined with NGS [118]. However, the enzymes responsible for the m5C modification of MALAT1 are unknown, but DNMT2 and NSUN2 could potentially be excluded, since MALAT1 was only slightly enriched after respective Aza-IPs in HeLa cells [127]. Hence, other m5C writers should be tested in the future.
3.2. HOTAIR
The Hox transcript antisense intergenic RNA (HOTAIR) is a long, intergenic ncRNA of ~2.2 kb that is transcribed from the antisense strand of the developmental HOXC gene cluster on chromosome 12 [154]. Dysregulated expression of HOTAIR, which promotes metastasis in several cancer types, is often found in human cancers, e.g., melanoma, breast, hepatocellular, gastric, colorectal or pancreatic carcinoma, and its expression is correlated with poor prognosis, e.g., in colorectal cancers [155,156,157,158,159,160]. Moreover, a recent study showed that HOTAIR can serve as a plasma-derived biomarker for the diagnosis and monitoring of NSCLC [161].
Mechanistically, HOTAIR is located in the nucleus and the characterization of the molecular interactions of this trans-acting ncRNA revealed two regions involved in direct interactions with chromatin-modifying complexes [162]. One interaction site is located in a ~300 nt region at the 5′-end, enabling the direct binding to the polycomb-repressive complex 2 (PRC2), a complex displaying histone methyltransferase activity. The interaction with HOTAIR is required for PRC2 occupancy and histone H3 lysine-27 trimethylation (H3K27me3) resulting in inhibition of gene expression across 40 kb of the HOXD gene locus [154,162]. The second, ~700 nt long interaction site, is located at the 3′-end of HOTAIR and is required for the interaction with the histone demethylase complex lysine specific demethylase 1 (LSD1)/co-repressor of RE1-silencing transcription factor (CoREST)/RE1 silencing transcription factor (REST) [162]. The ability of HOTAIR to tether these two distinct chromatin-modifying complexes enables coupled histone H3K27 methylation and lysine 4 demethylation (H3K4) to induce epigenetic gene silencing.
Interestingly, a previous study identified a specific cytosine methylation in HOTAIR at position C1683 occurring with complete penetrance (i.e., 100% modification rate) and present in all five cell lines tested [163]. However, Aza-IPs of DNMT2 and NSUN2 did not enrich HOTAIR suggesting that other methyltransferases might be responsible for this modification [127]. Importantly, since the methylated cytosine residue is located within the 700 nt LSD1 binding motif, it is tempting to speculate about a regulatory impact of the epitranscriptome on the epigenome. However, a methylation-dependent interaction between HOTAIR and LSD1 with downstream effects on histone H3 lysine 4 methylation changes has yet to be shown.
While additional chemical modification in HOTAIR have not been analyzed in more detail so far, Meyer et al. identified a single m6A peak region (126 nt) in the first half of HOTAIR, not overlapping with m5C, in HEK293T cells using MeRIP-Seq [97]. In contrast, Dominissini et al. did not find any m6A signal in HOTAIR using HepG2 cells or human brain tissue despite the presence of several DRACH consensus motifs [57].
Studies form Carlile et al. using Pseudo-Seq in HeLa cells, and Li et al. applying CeU-Seq in HEK293T cells, could not establish HOTAIR as a target for pseudouridylation [77,87]. However, additional cell systems and tissues should be analyzed to obtain a more comprehensive view about the chemical modifications and their putative functions in HOTAIR.
3.3. XIST
The process of X inactivation, i.e., the transcriptional silencing of one of the pair of X chromosomes, is initiated early in female mammalian development to provide dosage equivalence between males and females. XIST is a ~17 kb lncRNA that is expressed from a region called X inactivation center (XIC). XIST is essential for the initiation and spread of X-inactivation by coating the chromosome in cis [164,165,166,167]. Recently, three independent studies mapped the XIST RNA-protein interactome thereby providing further insights into the molecular mechanisms of XIST-mediated heterochromatinization [168,169,170,171]. Despite the use of distinct methodologies and different cellular systems, several overlapping proteins were identified in these studies including the previously described interactor hnRNP U as well as the newly identified binders SPEN and RBM15 [168,170,171,172]. However, the functional relevance of these interactions needs to be assessed in more detail.
In line with this, a recent study revealed a RBM15/METTL3/YTHDC1 pathway of m6A formation and recognition that is required for XIST-mediated transcriptional repression [54]. In detail, the authors could show that the high m6A modification rate (78 m6A residues) of XIST is dependent on RBM15 and its paralogue RBM15B, two RNA-binding proteins that link the m6A methylation complex to XIST through interaction with WTAP that in turn binds to the methyltransferase METTL3. Finally, m6A residues in XIST are recognized by YTHDC1 which leads to gene silencing. How exactly YTHDC1 binding to XIST leads to gene silencing remains unclear, but might involve additional molecular interactions between YTHDC1 and other proteins with well-established roles in the initiation of transcriptional silencing [54].
In addition to m6A, XIST was also shown to contain methylated cytosine residues [163]. A 5’-region of XIST, termed repeat A-region, consists of 8.5 repeats with 26 nt per full repeat and is required for the association with PRC2 [167]. Characterization of posttranscriptional chemical modifications in XIST revealed five methylated cytosines within repeat 8: C701, C702, C703, C711 and C712. The methylation rate of individual cytosine residues was between 19–24%, and a simultaneous modification of all five residues was detected in 19% of the sequences analyzed. Interestingly, non-methylated, but not methylated RNA oligonucleotides spanning the R8 tetra-loop and part of the inter-repeat helix of XIST were bound by PRC2 indicating that m5C, in contrast to m6A, can prevent XIST-protein interactions. However, no m5C modification was detected at the corresponding cytosines C668, 669, 670 and 678 in the A-region of mouse Xist, arguing against a conserved mechanism [163].
Finally, a third chemical modification in XIST, a pseudouridine residue at position U11249, was recently discovered [87]. However, the functional role of this modification is currently unknown.
Interestingly, X chromosome aneuploidies have long been associated with human cancers, but causality has not been established. A recent study in mice made a step forward. Here, deletion of Xist in the blood compartment of mice led to the development of a highly aggressive myeloproliferative neoplasm and myelodysplastic syndrome (mixed MPN/MDS) with 100% penetrance establishing a tumor-suppressive role of Xist [173]. Intriguingly, MDS is more common in women and XIST deletions and X chromosome duplications have been found in MPN, MDS, and myeloid cancers [174,175,176,177,178]. However, the association is not restricted to women, because extra X chromosomes are seen in acute lymphoblastic leukemias (ALL), AML, acute nonlymphoblastic leukemia (ANLL), adult T cell leukemia, chronic myeloid leukemia (CML), erythroleukemia and non-Hodgkin lymphoma of both sexes and ~60% of childhood ALL display extra X chromosomes and an extra X may be the only aneuploidy in some CML [173,179,180]. In contrast to these hematological cancers, XIST gene copy number amplifications and increased expression levels have been detected in other cancers, e.g., microsatellite-unstable colorectal carcinoma (CRC) [181]. Elevated expression of XIST was recently associated with poor survival in CRC patients, and knockdown of XIST inhibited proliferation, invasion, epithelial-mesenchymal transition (EMT) and CRC stem cell formation in vitro, as well as tumor growth and metastasis in vivo [182].
Hence, XIST might have context-dependent pro- or antitumor functions in human cancers and it would be interesting to know, if chemical modifications in XIST can shift the balance in one or the other direction.
3.4. SRA1
The steroid receptor RNA activator (SRA) is an example of a bifunctional gene that is active as a lncRNA (SRA1), yet also encoding a conserved protein (SRAP) [183]. SRA1 has a large number of isoforms, some of which display tissue-specific expression [184,185]. While most of the isoforms share a central core region that is necessary for its function as a coactivator, only some isoforms contain an open reading frame for SRAP production [183,186,187]. Both, the coding and the non-coding part of SRA have been described to be involved in the regulation of the transcriptional activity of different hormone receptors (androgen receptor, estrogen receptor, glucocorticoid receptor, thyroid hormone receptor, and retinoic acid receptor) in a cell-specific manner indicating potential anti-cancer targets [186,188]. However, the role of SRA1 in carcinogenesis is not fully understood yet. For example, transgenic overexpression of SRA1 in mice caused hyperplasia and morphological abnormalities in steroid hormone responsive tissues, but did not induce tumors and was accompanied by higher apoptosis rates. SRA1 also antagonized Ras-induced tumor formation [184].
Interestingly, the pseudouridine synthase Pus1 was previously identified as an interaction partner and coactivator of retinoic acid receptors (RARs), as well as other class I and II nuclear receptors in mouse cells [40]. Furthermore, Pus1 was shown to bind and modify SRA1, which is required for its role as a coactivator. In a subsequent study, the same authors identified a specific uridine residue in SRA1 (U206) whose modification by Pus1 (or Pus3) might induce a functional switch to allow SRA1 to act as coactivator or corepressor [189]. This could partially explain cell-type specific functions of SRA1.
Other chemical modifications of SRA1 have not been described or functionally analyzed so far. However, a close examination of transcriptome-wide m6A datasets warrants further investigations to clarify a putative link between the epitranscriptome and SRA1-dependent nuclear receptor signaling events [57,97].
3.5. Additional lncRNAs with Posttranscriptional Chemical Modifications
Mining published datasets for lncRNAs reveals a broad selection of chemically modified transcripts (Table 2). For example, Dominissini et al. mapped m6A to well-known lncRNAs, e.g., PVT1 and NEAT1 as well as uncharacterized lncRNA transcripts [57]. Having a closer look at m5C sites in lncRNAs, Squires et al. identified several putative target sites, e.g., in SNHG12, GAS5, TERC, RPPH1 and ANRIL [118]. However, only a few studies exist that have carefully mapped the position of modified residues in single transcripts, e.g., m6A in the lncRNA taurine up-regulated 1 (TUG1) (A1114) [88]. The same is true for pseudouridine residues in lncRNAs. Transcriptome-wide studies identified pseudouridine sites, e.g., in LRRC75A antisense RNA 1 (LRRC75C-AS1; U1537) and small nucleolar RNA Host Gene 1 (SNHG1; U1766) [77]. Individual studies focusing on specific lncRNAs mapped a pseudouridine at position U250 in RN7SK and Hussain et al. could identify RN7SK as a target for the m5C-introducing enzyme NSUN2 in HEK293T cells [77,120]. Kcnq1ot1, an imprinted lncRNA interacting with G9a and PcG proteins with elevated levels in patients with myocardial infarction and a function in transcriptional interference, contains a pseudouridine at position U64919 [87]. In addition, Li et al. also identified a heat shock-inducible pseudouridine (U19886) in Kcnq1ot1 as well as several additional pseudouridine sites within different lncRNAs, e.g., ST7-AS1 (U1138), ZFAS1 (U569), SNHG7 (U292), DICER1-AS1 (U463), including also many inducible sites, e.g., DLEU2L (U1379, H2O2-inducible), APTR (U1282, H2O2-inducible), or MAGI2-AS3 (U3659, heat shock-inducible) [87]. The transcriptome-wide mapping of pseudouridine in HEK293T cells by Schwartz et al. revealed a highly conserved position (U307) as well as a putative site at position U179 in the telomerase RNA component (TERC) [85].
Nevertheless, the relevance of these epitranscriptomic changes in lncRNAs are largely unknown and require additional validation, as well as functional follow-up studies.
4. Conclusions
The research field of epitranscriptomics has made huge strides in the last few years. High-throughput sequencing techniques enabled nearly transcriptome-wide modification detection and generated enormous amounts of data [191]. Validation of this data avalanche is tedious and more difficult than it seems on first sight, and the majority of modification sites should be treated as candidate sites [192]. New approaches and techniques are needed to validate modification data and to rush the field forward. Third-generation sequencing technology, improved chromatography methods and newly devised mass spectrometry protocols look promising to help gain new insights into the epitranscriptome landscape [193,194]. Information about the stoichiometry of each modified site will be needed to fully understand the importance of RNA modifications and their contribution to the highly dynamic cellular processes. Newly discovered binding proteins, be it writers, readers or erasers, will provide hints about which pathways are influenced or directed by RNA modifications, and will broaden our understanding of post-transcriptional regulatory mechanisms.
Furthermore, investigating the distribution and function of chemical modifications in lncRNAs, as well as their association with the relevant proteins in more detail, will contribute towards an integrative understanding of the multilayered gene expression control mechanisms active in mammalian cells (Figure 3). Intriguingly, some lncRNAs seem to have cell-type specific functions. For example, MALAT1 was shown to be important for cell proliferation, apoptosis or motility by regulating alternative splicing and gene expression in one cell system, but affecting the expression of different genes in other cell lines [143]. Hence, it would be interesting to study the impact of chemical modifications on the cell-type-specific functions of MALAT1 and other lncRNAs.
Moreover, recent large-scale ribosome footprinting studies have made the surprising and controversial observation that lncRNAs interact with ribosomes and suggest that lncRNAs are capable of translating short peptides [195,196,197,198,199,200]. However, others have reached different conclusions [201]. Interestingly, a recent study suggests that ribosomes are the default destination for the majority of cytoplasmic lncRNAs and they may also play a role in lncRNA turnover [202]. Given the previously described ability of pseudouridine to expand the genetic code [76], it is tempting to speculate about epitranscriptome-based mechanisms that regulate ribosome-bound transcript degradation or might convert non-coding RNAs into coding ones.
In summary, we are only starting to unravel the full breadth of the transcriptome, which comes in many (chemical) flavors. To date, more than 150 modifications have been identified in RNA, but only a handful can be currently mapped with high-throughput methods. This offers plenty of opportunities to discover novel regulatory principles. Moreover, proteins involved in the epitranscriptomic cascade might represent interesting therapeutic targets. However, our current knowledge about the epitranscriptomic changes that might occur during carcinogenesis, as well as their functional relevance on single molecule level, especially in lncRNAs, is still very limited. Hence, further molecular and mechanistic investigations are needed. These studies might pave the way for the development of novel therapeutics and might help to identify biomarkers for early cancer detection and therapy response.
Acknowledgments
We apologize to all scientists whose important work could not be cited in this review due to space constraints. The authors wish to thank Monika Hämmerle for critical reading of the manuscript. Research in the Gutschner lab is supported by funds from the intramural Wilhelm-Roux-Program of the Medical Faculty, Martin-Luther-University Halle-Wittenberg.
Author Contributions
Roland Jacob and Sindy Zander wrote the manuscript and prepared figures and tables. Tony Gutschner conceptualized the review and edited the manuscript.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
| 4-SU | 4-thiouridine |
| ALKBH5 | α-ketoglutarate-dependent dioxygenase alkB homolog 5 |
| ALL | Acute lymphoblastic leukemia |
| ALYREF | Aly/REF export factor |
| AML | Acute myeloid leukemia |
| ANLL | Acute nonlymphoblastic leukemia |
| ANRIL | Antisense Non-coding RNA in the INK4 Locus |
| APTR | Alu-mediated CDKN1A/p21 transcriptional regulator |
| ARE | AU-rich element |
| Aza-IP | 5-azacytidine-mediated RNA immunoprecipitation |
| CeU-Seq | N3-CMC-enriched pseudouridine sequencing |
| CIMS | Cross-linking–induced mutation site |
| CITS | Cross-linking–induced truncation site |
| CMCT | N-cyclohexyl-N′-(2-morpholinoethyl)carbodiimide metho-p-toluenesulfonate |
| CML | Chronic myeloid leukemia |
| CoREST | Co-repressor of RE1-silencing transcription factor |
| CRC | Colorectal cancer |
| DICER1-AS1 | DICER1 antisense RNA 1 |
| DKC1 | Dyskerin pseudouridine synthase 1 |
| DLEU2L | Deleted in lymphocytic leukemia 2-like |
| DNMT2 | DNA methyltransferase-2 |
| eIF3 | Eukaryotic initiation factor 3 |
| eIF4E | Eukaryotic initiation factor 4E |
| EMT | Epithelial-mesenchymal transition |
| FTO | Fat mass- and obesity-associated protein |
| GAS5 | Growth arrest specific 5 |
| GBM | Glioblastoma |
| GSC | Glioblastoma stem-like cells |
| H3K27 | Histone H3 lysine-27 |
| H3K4 | Histone H3 lysine-4 |
| hnRNP U | Heterogeneous nuclear ribonucleoprotein U |
| HNRNPA2B1 | Heterogeneous nuclear ribonucleoprotein A2/B1 |
| HNRNPC | Heterogeneous nuclear ribonucleoprotein C |
| HOTAIR | HOX antisense intergenic RNA |
| HOXC/D | Homeobox C/D |
| HuR | Human antigen R |
| iCLIP | Individual-nucleotide-resolution crosslinking and immunoprecipitation |
| lncRNA | Long non-coding RNA |
| LRRC75C-AS1 | LRRC75A antisense RNA1 |
| LSD1 | Lysine specific demethylase 1A |
| m5C | 5-methylcytosine |
| m6A | N6-methyladenosine |
| m6Am | N6,2′-O-dimethyladenosine |
| MAGI2-AS3 | MAGI2 antisense RNA 3 |
| MALAT1 | Metastasis Associated Lung Adenocarcinoma Transcript 1 |
| mascRNA | MALAT1-associated small cytoplasmic RNA |
| MAT2A | Methionine adenosyltransferase 2A |
| MDS | Myelodysplastic syndrome |
| MeRIP-Seq | M6A specific methylated RNA immunoprecipitation-sequencing |
| METTL | Methyltransferase-like protein |
| miRNA | MicroRNA |
| MPN | Myeloproliferative neoplasm |
| mRNA | Messenger RNA |
| MS | Mass spectrometry |
| mtRNA | Mitochondrial RNA |
| ncRNA | Non-coding RNA |
| NEAT1 | Nuclear paraspeckle assembly transcript 1 |
| NEAT2 | Nuclear-Enriched Abundant Transcript 2 |
| NGS | Next-generation sequencing |
| NSCLC | Non-small cell lung cancer |
| NSUN | NOP2/Sun RNA methyltransferase family member |
| nt | Nucleotide |
| PA-m6A-seq | Photo-crosslinking-assisted m6A sequencing |
| PcG | Polycomb group |
| piRNA | PIWI-interacting RNA |
| PRC2 | Polycomb-repressive complex 2 |
| PTM | Posttranslational modification |
| PUS | Pseudouridine synthase |
| PUS7L | Pseudouridylate synthase 7 like |
| PUSL1 | Pseudouridylate synthase-like 1 |
| PVT1 | Pvt1 oncogene |
| RAR | Retinoic acid receptor |
| RBP | RNA-binding protein |
| REST | RE1-silencing transcription factor |
| RBM15 | RNA-binding motif protein 15 |
| RMT | RNA methyltransferase |
| RN7SK | RNA 7SK small nuclear |
| RPPH1 | Ribonuclease P RNA component H1 |
| RPUSD | RNA pseudouridylate synthase domain containing |
| rRNA | Ribosomal RNA |
| RT-PCR | Reverse transcription-polymerase chain reaction |
| SAM | S-Adenosyl methionine |
| SCARLET | Site-specific cleavage and radioactive-labeling followed by ligation-assisted extraction and thin-layer chromatography |
| siRNA | Small interfering RNA |
| SNHG | Small nucleolar RNA host gene |
| snoRNA | Small nucleolar RNA |
| snRNA | Small nuclear RNA |
| SPEN | Spen family transcriptional repressor |
| SRA | Steroid receptor RNA activator |
| SRAP | Steroid receptor RNA activator protein |
| ST7-AS1 | ST7 antisense RNA 1 |
| TERC | Telomerase RNA component |
| tRNA | Transfer RNA |
| TRUB | TruB pseudouridine synthase family member |
| TUG1 | Taurine up-regulated 1 |
| UTR | Untranslated region |
| VIRMA | Vir like m6A methyltransferase associated |
| WTAP | Wilms’ tumor 1-associating protein |
| XIC | X inactivation center |
| XIST | X-inactive specific transcript |
| YTH | YT521-B homology |
| YTHDC | YTH domain-containing protein |
| YTHDF | YT521-B homology domain family |
| ZFAS1 | ZNFX1 antisense RNA 1 |
| Ψ | Pseudouridine |
References
- Prabakaran, S.; Lippens, G.; Steen, H.; Gunawardena, J. Post-translational modification: Nature’s escape from genetic imprisonment and the basis for dynamic information encoding. Wiley Interdiscip. Rev. Syst. Biol. Med. 2012, 4, 565–583. [Google Scholar] [CrossRef] [PubMed]
- Hotchkiss, R.D. The quantitative separation of purines, pyrimidines, and nucleosides by paper chromatography. J. Biol. Chem. 1948, 175, 315–332. [Google Scholar] [PubMed]
- Berger, S.L.; Kouzarides, T.; Shiekhattar, R.; Shilatifard, A. An operational definition of epigenetics. Genes Dev. 2009, 23, 781–783. [Google Scholar] [CrossRef] [PubMed]
- Kruger, K.; Grabowski, P.J.; Zaug, A.J.; Sands, J.; Gottschling, D.E.; Cech, T.R. Self-splicing RNA: Autoexcision and autocyclization of the ribosomal RNA intervening sequence of Tetrahymena. Cell 1982, 31, 147–157. [Google Scholar] [CrossRef]
- Cech, T.R.; Steitz, J.A. The noncoding RNA revolution-trashing old rules to forge new ones. Cell 2014, 157, 77–94. [Google Scholar] [CrossRef] [PubMed]
- ENCODE Project Consortium. An integrated encyclopedia of DNA elements in the human genome. Nature 2012, 489, 57–74. [Google Scholar]
- Knowling, S.; Morris, K.V. Non-coding RNA and antisense RNA. Nature’s trash or treasure? Biochimie 2011, 93, 1922–1927. [Google Scholar] [CrossRef] [PubMed]
- Heinemann, A.; Zhao, F.; Pechlivanis, S.; Eberle, J.; Steinle, A.; Diederichs, S.; Schadendorf, D.; Paschen, A. Tumor suppressive microRNAs miR-34a/c control cancer cell expression of ULBP2, a stress-induced ligand of the natural killer cell receptor NKG2D. Cancer Res. 2012, 72, 460–471. [Google Scholar] [CrossRef] [PubMed]
- Kahlert, C.; Klupp, F.; Brand, K.; Lasitschka, F.; Diederichs, S.; Kirchberg, J.; Rahbari, N.; Dutta, S.; Bork, U.; Fritzmann, J.; et al. Invasion front-specific expression and prognostic significance of microRNA in colorectal liver metastases. Cancer Sci 2011, 102, 1799–1807. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Roth, A.; Yu, M.; Morris, R.; Bersani, F.; Rivera, M.N.; Lu, J.; Shioda, T.; Vasudevan, S.; Ramaswamy, S.; et al. The IGF2 intronic miR-483 selectively enhances transcription from IGF2 fetal promoters and enhances tumorigenesis. Genes Dev. 2013, 27, 2543–2548. [Google Scholar] [CrossRef] [PubMed]
- Pichler, M.; Stiegelbauer, V.; Vychytilova-Faltejskova, P.; Ivan, C.; Ling, H.; Winter, E.; Zhang, X.; Goblirsch, M.; Wulf-Goldenberg, A.; Ohtsuka, M.; et al. Genome-Wide miRNA Analysis Identifies miR-188–3p as a Novel Prognostic Marker and Molecular Factor Involved in Colorectal Carcinogenesis. Clin. Cancer Res. 2017, 23, 1323–1333. [Google Scholar] [CrossRef] [PubMed]
- Winter, J.; Jung, S.; Keller, S.; Gregory, R.I.; Diederichs, S. Many roads to maturity: MicroRNA biogenesis pathways and their regulation. Nat. Cell Biol. 2009, 11, 228–234. [Google Scholar] [CrossRef] [PubMed]
- Kowalczyk, M.S.; Higgs, D.R.; Gingeras, T.R. Molecular biology: RNA discrimination. Nature 2012, 482, 310–311. [Google Scholar] [CrossRef] [PubMed]
- Dinger, M.E.; Pang, K.C.; Mercer, T.R.; Mattick, J.S. Differentiating protein-coding and noncoding RNA: Challenges and ambiguities. PLoS Comput. Biol. 2008, 4, e1000176. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.L.; Carmichael, G.G. Long noncoding RNAs in mammalian cells: What, where, and why? Wiley Interdiscip. Rev. RNA 2010, 1, 2–21. [Google Scholar] [CrossRef] [PubMed]
- Lipovich, L.; Johnson, R.; Lin, C.Y. MacroRNA underdogs in a microRNA world: Evolutionary, regulatory, and biomedical significance of mammalian long non-protein-coding RNA. Biochim. Biophys. Acta 2010, 1799, 597–615. [Google Scholar] [CrossRef] [PubMed]
- Ponting, C.P.; Oliver, P.L.; Reik, W. Evolution and functions of long noncoding RNAs. Cell 2009, 136, 629–641. [Google Scholar] [CrossRef] [PubMed]
- Dhamija, S.; Diederichs, S. From junk to master regulators of invasion: lncRNA functions in migration, EMT and metastasis. Int. J. Cancer 2016, 139, 269–280. [Google Scholar] [CrossRef] [PubMed]
- Diederichs, S.; Bartsch, L.; Berkmann, J.C.; Frose, K.; Heitmann, J.; Hoppe, C.; Iggena, D.; Jazmati, D.; Karschnia, P.; Linsenmeier, M.; et al. The dark matter of the cancer genome: Aberrations in regulatory elements, untranslated regions, splice sites, non-coding RNA and synonymous mutations. EMBO Mol. Med. 2016, 8, 442–457. [Google Scholar] [CrossRef] [PubMed]
- Gutschner, T.; Diederichs, S. The hallmarks of cancer: A long non-coding RNA point of view. RNA Biol. 2012, 9, 703–719. [Google Scholar] [CrossRef] [PubMed]
- Haemmerle, M.; Gutschner, T. Long non-coding RNAs in cancer and development: Where do we go from here? Int. J. Mol. Sci. 2015, 16, 1395–1405. [Google Scholar] [CrossRef] [PubMed]
- Shen, P.; Pichler, M.; Chen, M.; Calin, G.A.; Ling, H. To Wnt or Lose: The Missing Non-Coding Linc in Colorectal Cancer. Int. J. Mol. Sci. 2017, 18, 2003. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.C.; Chang, H.Y. Molecular mechanisms of long noncoding RNAs. Mol. Cell 2011, 43, 904–914. [Google Scholar] [CrossRef] [PubMed]
- Guttman, M.; Amit, I.; Garber, M.; French, C.; Lin, M.F.; Feldser, D.; Huarte, M.; Zuk, O.; Carey, B.W.; Cassady, J.P.; et al. Chromatin signature reveals over a thousand highly conserved large non-coding RNAs in mammals. Nature 2009, 458, 223–227. [Google Scholar] [CrossRef] [PubMed]
- Huarte, M.; Guttman, M.; Feldser, D.; Garber, M.; Koziol, M.J.; Kenzelmann-Broz, D.; Khalil, A.M.; Zuk, O.; Amit, I.; Rabani, M.; et al. A large intergenic noncoding RNA induced by p53 mediates global gene repression in the p53 response. Cell 2010, 142, 409–419. [Google Scholar] [CrossRef] [PubMed]
- Iyer, M.K.; Niknafs, Y.S.; Malik, R.; Singhal, U.; Sahu, A.; Hosono, Y.; Barrette, T.R.; Prensner, J.R.; Evans, J.R.; Zhao, S.; et al. The landscape of long noncoding RNAs in the human transcriptome. Nat. Genet. 2015, 47, 199–208. [Google Scholar] [CrossRef] [PubMed]
- Quek, X.C.; Thomson, D.W.; Maag, J.L.; Bartonicek, N.; Signal, B.; Clark, M.B.; Gloss, B.S.; Dinger, M.E. lncRNAdb v2.0: Expanding the reference database for functional long noncoding RNAs. Nucl. Acids Res. 2015, 43, D168–D173. [Google Scholar] [CrossRef] [PubMed]
- Cohn, W.E.; Volkin, E. Nucleoside-5’-Phosphates from Ribonucleic Acid. Nature 1951, 167, 483–484. [Google Scholar] [CrossRef]
- Wyatt, G.R. Occurrence of 5-methylcytosine in nucleic acids. Nature 1950, 166, 237–238. [Google Scholar] [CrossRef] [PubMed]
- Helm, M.; Alfonzo, J.D. Posttranscriptional RNA Modifications: Playing metabolic games in a cell’s chemical Legoland. Chem. Biol. 2014, 21, 174–185. [Google Scholar] [CrossRef] [PubMed]
- Machnicka, M.A.; Milanowska, K.; Osman Oglou, O.; Purta, E.; Kurkowska, M.; Olchowik, A.; Januszewski, W.; Kalinowski, S.; Dunin-Horkawicz, S.; Rother, K.M.; et al. MODOMICS: A database of RNA modification pathways--2013 update. Nucl. Acids Res. 2013, 41, D262–D267. [Google Scholar] [CrossRef] [PubMed]
- Cantara, W.A.; Crain, P.F.; Rozenski, J.; McCloskey, J.A.; Harris, K.A.; Zhang, X.; Vendeix, F.A.; Fabris, D.; Agris, P.F. The RNA Modification Database, RNAMDB: 2011 update. Nucl. Acids Res. 2011, 39, D195–D201. [Google Scholar] [CrossRef] [PubMed]
- Kirchner, S.; Ignatova, Z. Emerging roles of tRNA in adaptive translation, signalling dynamics and disease. Nat. Rev. Genet. 2015, 16, 98–112. [Google Scholar] [CrossRef] [PubMed]
- Torres, A.G.; Batlle, E.; Ribas de Pouplana, L. Role of tRNA modifications in human diseases. Trends Mol. Med. 2014, 20, 306–314. [Google Scholar] [CrossRef] [PubMed]
- Decatur, W.A.; Fournier, M.J. rRNA modifications and ribosome function. Trends Biochem. Sci. 2002, 27, 344–351. [Google Scholar] [CrossRef]
- Lafontaine, D.L.J. Noncoding RNAs in eukaryotic ribosome biogenesis and function. Nat. Struct. Mol. Biol. 2015, 22, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Frye, M.; Jaffrey, S.R.; Pan, T.; Rechavi, G.; Suzuki, T. RNA modifications: What have we learned and where are we headed? Nat. Rev. Genet. 2016, 17, 365–372. [Google Scholar] [CrossRef] [PubMed]
- Jonkhout, N.; Tran, J.; Smith, M.A.; Schonrock, N.; Mattick, J.S.; Novoa, E.M. The RNA modification landscape in human disease. RNA 2017. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Patton, J.R. Cloning and characterization of a mammalian pseudouridine synthase. RNA 1999, 5, 409–419. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Patton, J.R.; Davis, S.L.; Florence, B.; Ames, S.J.; Spanjaard, R.A. Regulation of nuclear receptor activity by a pseudouridine synthase through posttranscriptional modification of steroid receptor RNA activator. Mol. Cell 2004, 15, 549–558. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Ma, S.; Yi, C. Pseudouridine: The fifth RNA nucleotide with renewed interests. Curr. Opin. Chem. Biol. 2016, 33, 108–116. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Patton, J.R. Pseudouridine synthase 3 from mouse modifies the anticodon loop of tRNA. Biochemistry 2000, 39, 12723–12730. [Google Scholar] [CrossRef] [PubMed]
- Shaheen, R.; Han, L.; Faqeih, E.; Ewida, N.; Alobeid, E.; Phizicky, E.M.; Alkuraya, F.S. A homozygous truncating mutation in PUS3 expands the role of tRNA modification in normal cognition. Hum. Genet. 2016, 135, 707–713. [Google Scholar] [CrossRef] [PubMed]
- Safra, M.; Nir, R.; Farouq, D.; Vainberg Slutskin, I.; Schwartz, S. TRUB1 is the predominant pseudouridine synthase acting on mammalian mRNA via a predictable and conserved code. Genome Res. 2017, 27, 393–406. [Google Scholar] [CrossRef] [PubMed]
- McCleverty, C.J.; Hornsby, M.; Spraggon, G.; Kreusch, A. Crystal structure of human Pus10, a novel pseudouridine synthase. J. Mol. Biol. 2007, 373, 1243–1254. [Google Scholar] [CrossRef] [PubMed]
- Antonicka, H.; Choquet, K.; Lin, Z.Y.; Gingras, A.C.; Kleinman, C.L.; Shoubridge, E.A. A pseudouridine synthase module is essential for mitochondrial protein synthesis and cell viability. EMBO Rep. 2017, 18, 28–38. [Google Scholar] [CrossRef] [PubMed]
- Heiss, N.S.; Knight, S.W.; Vulliamy, T.J.; Klauck, S.M.; Wiemann, S.; Mason, P.J.; Poustka, A.; Dokal, I. X-linked dyskeratosis congenita is caused by mutations in a highly conserved gene with putative nucleolar functions. Nat. Genet. 1998, 19, 32–38. [Google Scholar] [CrossRef] [PubMed]
- Bokar, J.A.; Shambaugh, M.E.; Polayes, D.; Matera, A.G.; Rottman, F.M. Purification and cDNA cloning of the AdoMet-binding subunit of the human mRNA (N6-adenosine)-methyltransferase. RNA 1997, 3, 1233–1247. [Google Scholar] [PubMed]
- Liu, J.; Yue, Y.; Han, D.; Wang, X.; Fu, Y.; Zhang, L.; Jia, G.; Yu, M.; Lu, Z.; Deng, X.; et al. A METTL3-METTL14 complex mediates mammalian nuclear RNA N6-adenosine methylation. Nat. Chem. Biol. 2014, 10, 93–95. [Google Scholar] [CrossRef] [PubMed]
- Ping, X.L.; Sun, B.F.; Wang, L.; Xiao, W.; Yang, X.; Wang, W.J.; Adhikari, S.; Shi, Y.; Lv, Y.; Chen, Y.S.; et al. Mammalian WTAP is a regulatory subunit of the RNA N6-methyladenosine methyltransferase. Cell Res. 2014, 24, 177–189. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, S.; Mumbach, M.R.; Jovanovic, M.; Wang, T.; Maciag, K.; Bushkin, G.G.; Mertins, P.; Ter-Ovanesyan, D.; Habib, N.; Cacchiarelli, D.; et al. Perturbation of m6A writers reveals two distinct classes of mRNA methylation at internal and 5’ sites. Cell Rep. 2014, 8, 284–296. [Google Scholar] [CrossRef] [PubMed]
- Zhong, S.; Li, H.; Bodi, Z.; Button, J.; Vespa, L.; Herzog, M.; Fray, R.G. MTA is an Arabidopsis messenger RNA adenosine methylase and interacts with a homolog of a sex-specific splicing factor. Plant Cell 2008, 20, 1278–1288. [Google Scholar] [CrossRef] [PubMed]
- Horiuchi, K.; Kawamura, T.; Iwanari, H.; Ohashi, R.; Naito, M.; Kodama, T.; Hamakubo, T. Identification of Wilms’ tumor 1-associating protein complex and its role in alternative splicing and the cell cycle. J. Biol. Chem. 2013, 288, 33292–33302. [Google Scholar] [CrossRef] [PubMed]
- Patil, D.P.; Chen, C.K.; Pickering, B.F.; Chow, A.; Jackson, C.; Guttman, M.; Jaffrey, S.R. m6A RNA methylation promotes XIST-mediated transcriptional repression. Nature 2016, 537, 369–373. [Google Scholar] [CrossRef] [PubMed]
- Pendleton, K.E.; Chen, B.; Liu, K.; Hunter, O.V.; Xie, Y.; Tu, B.P.; Conrad, N.K. The U6 snRNA m6A Methyltransferase METTL16 Regulates SAM Synthetase Intron Retention. Cell 2017, 169, 824–835. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Lu, Z.; Gomez, A.; Hon, G.C.; Yue, Y.; Han, D.; Fu, Y.; Parisien, M.; Dai, Q.; Jia, G.; et al. N6-methyladenosine-dependent regulation of messenger RNA stability. Nature 2014, 505, 117–120. [Google Scholar] [CrossRef] [PubMed]
- Dominissini, D.; Moshitch-Moshkovitz, S.; Schwartz, S.; Salmon-Divon, M.; Ungar, L.; Osenberg, S.; Cesarkas, K.; Jacob-Hirsch, J.; Amariglio, N.; Kupiec, M.; et al. Topology of the human and mouse m6A RNA methylomes revealed by m6A-seq. Nature 2012, 485, 201–206. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.; Wang, X.; Liu, K.; Roundtree, I.A.; Tempel, W.; Li, Y.; Lu, Z.; He, C.; Min, J. Structural basis for selective binding of m6A RNA by the YTHDC1 YTH domain. Nat. Chem. Biol. 2014, 10, 927–929. [Google Scholar] [CrossRef] [PubMed]
- Meyer, K.D.; Patil, D.P.; Zhou, J.; Zinoviev, A.; Skabkin, M.A.; Elemento, O.; Pestova, T.V.; Qian, S.B.; Jaffrey, S.R. 5’ UTR m6A Promotes Cap-Independent Translation. Cell 2015, 163, 999–1010. [Google Scholar] [CrossRef] [PubMed]
- Alarcon, C.R.; Goodarzi, H.; Lee, H.; Liu, X.; Tavazoie, S.; Tavazoie, S.F. HNRNPA2B1 Is a Mediator of m(6)A-Dependent Nuclear RNA Processing Events. Cell 2015, 162, 1299–1308. [Google Scholar] [CrossRef] [PubMed]
- Liu, N.; Dai, Q.; Zheng, G.; He, C.; Parisien, M.; Pan, T. N6-methyladenosine-dependent RNA structural switches regulate RNA–protein interactions. Nature 2015, 518, 560–564. [Google Scholar] [CrossRef] [PubMed]
- Zheng, G.; Dahl, J.A.; Niu, Y.; Fedorcsak, P.; Huang, C.M.; Li, C.J.; Vagbo, C.B.; Shi, Y.; Wang, W.L.; Song, S.H.; et al. ALKBH5 is a mammalian RNA demethylase that impacts RNA metabolism and mouse fertility. Mol. Cell 2013, 49, 18–29. [Google Scholar] [CrossRef] [PubMed]
- Jia, G.; Fu, Y.; Zhao, X.; Dai, Q.; Zheng, G.; Yang, Y.; Yi, C.; Lindahl, T.; Pan, T.; Yang, Y.G.; et al. N6-methyladenosine in nuclear RNA is a major substrate of the obesity-associated FTO. Nat. Chem. Biol. 2011, 7, 885–887. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.; Yang, J.; Watzinger, P.; Kotter, P.; Entian, K.D. Yeast Nop2 and Rcm1 methylate C2870 and C2278 of the 25S rRNA, respectively. Nucl. Acids Res. 2013, 41, 9062–9076. [Google Scholar] [CrossRef] [PubMed]
- Frye, M.; Watt, F.M. The RNA methyltransferase Misu (NSun2) mediates Myc-induced proliferation and is upregulated in tumors. Curr. Biol. 2006, 16, 971–981. [Google Scholar] [CrossRef] [PubMed]
- Haag, S.; Sloan, K.E.; Ranjan, N.; Warda, A.S.; Kretschmer, J.; Blessing, C.; Hubner, B.; Seikowski, J.; Dennerlein, S.; Rehling, P.; et al. NSUN3 and ABH1 modify the wobble position of mt-tRNAMet to expand codon recognition in mitochondrial translation. EMBO J. 2016, 35, 2104–2119. [Google Scholar] [CrossRef] [PubMed]
- Metodiev, M.D.; Spahr, H.; Loguercio Polosa, P.; Meharg, C.; Becker, C.; Altmueller, J.; Habermann, B.; Larsson, N.G.; Ruzzenente, B. NSUN4 is a dual function mitochondrial protein required for both methylation of 12S rRNA and coordination of mitoribosomal assembly. PLoS Genet. 2014, 10, e1004110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schosserer, M.; Minois, N.; Angerer, T.B.; Amring, M.; Dellago, H.; Harreither, E.; Calle-Perez, A.; Pircher, A.; Gerstl, M.P.; Pfeifenberger, S.; et al. Methylation of ribosomal RNA by NSUN5 is a conserved mechanism modulating organismal lifespan. Nat. Commun. 2015, 6, 6158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haag, S.; Warda, A.S.; Kretschmer, J.; Gunnigmann, M.A.; Hobartner, C.; Bohnsack, M.T. NSUN6 is a human RNA methyltransferase that catalyzes formation of m5C72 in specific tRNAs. RNA 2015, 21, 1532–1543. [Google Scholar] [CrossRef] [PubMed]
- Harris, T.; Marquez, B.; Suarez, S.; Schimenti, J. Sperm motility defects and infertility in male mice with a mutation in Nsun7, a member of the Sun domain-containing family of putative RNA methyltransferases. Biol. Reprod. 2007, 77, 376–382. [Google Scholar] [CrossRef] [PubMed]
- Goll, M.G.; Kirpekar, F.; Maggert, K.A.; Yoder, J.A.; Hsieh, C.L.; Zhang, X.; Golic, K.G.; Jacobsen, S.E.; Bestor, T.H. Methylation of tRNAAsp by the DNA methyltransferase homolog Dnmt2. Science 2006, 311, 395–398. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Yang, Y.; Sun, B.F.; Chen, Y.S.; Xu, J.W.; Lai, W.Y.; Li, A.; Wang, X.; Bhattarai, D.P.; Xiao, W.; et al. 5-methylcytosine promotes mRNA export—NSUN2 as the methyltransferase and ALYREF as an m5C reader. Cell Res. 2017, 27, 606–625. [Google Scholar] [CrossRef] [PubMed]
- Spenkuch, F.; Motorin, Y.; Helm, M. Pseudouridine: Still mysterious, but never a fake (uridine)! RNA Biol. 2014, 11, 1540–1554. [Google Scholar] [CrossRef] [PubMed]
- Charette, M.; Gray, M.W. Pseudouridine in RNA: What, where, how, and why. IUBMB Life 2000, 49, 341–351. [Google Scholar] [PubMed]
- Davis, D.R. Stabilization of RNA stacking by pseudouridine. Nucl. Acids Res. 1995, 23, 5020–5026. [Google Scholar] [CrossRef] [PubMed]
- Karijolich, J.; Yu, Y.T. Converting nonsense codons into sense codons by targeted pseudouridylation. Nature 2011, 474, 395–398. [Google Scholar] [CrossRef] [PubMed]
- Carlile, T.M.; Rojas-Duran, M.F.; Zinshteyn, B.; Shin, H.; Bartoli, K.M.; Gilbert, W.V. Pseudouridine profiling reveals regulated mRNA pseudouridylation in yeast and human cells. Nature 2014, 515, 143–146. [Google Scholar] [CrossRef] [PubMed]
- Zaganelli, S.; Rebelo-Guiomar, P.; Maundrell, K.; Rozanska, A.; Pierredon, S.; Powell, C.A.; Jourdain, A.A.; Hulo, N.; Lightowlers, R.N.; Chrzanowska-Lightowlers, Z.M.; et al. The Pseudouridine Synthase RPUSD4 Is an Essential Component of Mitochondrial RNA Granules. J. Biol. Chem. 2017, 292, 4519–4532. [Google Scholar] [CrossRef] [PubMed]
- Patton, J.R.; Bykhovskaya, Y.; Mengesha, E.; Bertolotto, C.; Fischel-Ghodsian, N. Mitochondrial myopathy and sideroblastic anemia (MLASA): Missense mutation in the pseudouridine synthase 1 (PUS1) gene is associated with the loss of tRNA pseudouridylation. J. Biol. Chem. 2005, 280, 19823–19828. [Google Scholar] [CrossRef] [PubMed]
- Mei, Y.P.; Liao, J.P.; Shen, J.; Yu, L.; Liu, B.L.; Liu, L.; Li, R.Y.; Ji, L.; Dorsey, S.G.; Jiang, Z.R.; et al. Small nucleolar RNA 42 acts as an oncogene in lung tumorigenesis. Oncogene 2012, 31, 2794–2804. [Google Scholar] [CrossRef] [PubMed]
- Bykhovskaya, Y.; Casas, K.; Mengesha, E.; Inbal, A.; Fischel-Ghodsian, N. Missense mutation in pseudouridine synthase 1 (PUS1) causes mitochondrial myopathy and sideroblastic anemia (MLASA). Am. J. Hum. Genet. 2004, 74, 1303–1308. [Google Scholar] [CrossRef] [PubMed]
- Ge, J.; Yu, Y.T. RNA pseudouridylation: New insights into an old modification. Trends Biochem. Sci. 2013, 38, 210–218. [Google Scholar] [CrossRef] [PubMed]
- Bakin, A.; Ofengand, J. Four newly located pseudouridylate residues in Escherichia coli 23S ribosomal RNA are all at the peptidyltransferase center: Analysis by the application of a new sequencing technique. Biochemistry 1993, 32, 9754–9762. [Google Scholar] [CrossRef] [PubMed]
- Bakin, A.V.; Ofengand, J. Mapping of pseudouridine residues in RNA to nucleotide resolution. Methods Mol. Biol. 1998, 77, 297–309. [Google Scholar] [PubMed]
- Schwartz, S.; Bernstein, D.A.; Mumbach, M.R.; Jovanovic, M.; Herbst, R.H.; Leon-Ricardo, B.X.; Engreitz, J.M.; Guttman, M.; Satija, R.; Lander, E.S.; et al. Transcriptome-wide mapping reveals widespread dynamic-regulated pseudouridylation of ncRNA and mRNA. Cell 2014, 159, 148–162. [Google Scholar] [CrossRef] [PubMed]
- Lovejoy, A.F.; Riordan, D.P.; Brown, P.O. Transcriptome-wide mapping of pseudouridines: Pseudouridine synthases modify specific mRNAs in S. cerevisiae. PLoS ONE 2014, 9, e110799. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Zhu, P.; Ma, S.; Song, J.; Bai, J.; Sun, F.; Yi, C. Chemical pulldown reveals dynamic pseudouridylation of the mammalian transcriptome. Nat. Chem. Biol. 2015, 11, 592–597. [Google Scholar] [CrossRef] [PubMed]
- Liu, N.; Parisien, M.; Dai, Q.; Zheng, G.; He, C.; Pan, T. Probing N6-methyladenosine RNA modification status at single nucleotide resolution in mRNA and long noncoding RNA. RNA 2013, 19, 1848–1856. [Google Scholar] [CrossRef] [PubMed]
- Durairaj, A.; Limbach, P.A. Mass spectrometry of the fifth nucleoside: A review of the identification of pseudouridine in nucleic acids. Anal. Chim. Acta 2008, 623, 117–125. [Google Scholar] [CrossRef] [PubMed]
- Yamauchi, Y.; Nobe, Y.; Izumikawa, K.; Higo, D.; Yamagishi, Y.; Takahashi, N.; Nakayama, H.; Isobe, T.; Taoka, M. A mass spectrometry-based method for direct determination of pseudouridine in RNA. Nucl. Acids Res. 2016, 44, e59. [Google Scholar] [CrossRef] [PubMed]
- Desrosiers, R.; Friderici, K.; Rottman, F. Identification of methylated nucleosides in messenger RNA from Novikoff hepatoma cells. Proc. Natl. Acad. Sci. USA 1974, 71, 3971–3975. [Google Scholar] [CrossRef] [PubMed]
- Perry, R.P.; Kelley, D.E. Existence of methylated messenger RNA in mouse L cells. Cell 1974, 1, 37–42. [Google Scholar] [CrossRef]
- Molinie, B.; Wang, J.; Lim, K.S.; Hillebrand, R.; Lu, Z.X.; van Wittenberghe, N.; Howard, B.D.; Daneshvar, K.; Mullen, A.C.; Dedon, P.; et al. m6A-LAIC-seq reveals the census and complexity of the m6A epitranscriptome. Nat. Methods 2016, 13, 692–698. [Google Scholar] [CrossRef] [PubMed]
- Wei, C.M.; Moss, B. Nucleotide sequences at the N6-methyladenosine sites of HeLa cell messenger ribonucleic acid. Biochemistry 1977, 16, 1672–1676. [Google Scholar] [CrossRef] [PubMed]
- Csepany, T.; Lin, A.; Baldick, C.J., Jr.; Beemon, K. Sequence specificity of mRNA N6-adenosine methyltransferase. J. Biol. Chem. 1990, 265, 20117–20122. [Google Scholar] [PubMed]
- Linder, B.; Grozhik, A.V.; Olarerin-George, A.O.; Meydan, C.; Mason, C.E.; Jaffrey, S.R. Single-nucleotide-resolution mapping of m6A and m6Am throughout the transcriptome. Nat. Methods 2015, 12, 767–772. [Google Scholar] [CrossRef] [PubMed]
- Meyer, K.D.; Saletore, Y.; Zumbo, P.; Elemento, O.; Mason, C.E.; Jaffrey, S.R. Comprehensive analysis of mRNA methylation reveals enrichment in 3’ UTRs and near stop codons. Cell 2012, 149, 1635–1646. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Li, Y.; Toth, J.I.; Petroski, M.D.; Zhang, Z.; Zhao, J.C. N6-methyladenosine modification destabilizes developmental regulators in embryonic stem cells. Nat. Cell Biol. 2014, 16, 191–198. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Zhao, B.S.; Roundtree, I.A.; Lu, Z.; Han, D.; Ma, H.; Weng, X.; Chen, K.; Shi, H.; He, C. N6-methyladenosine Modulates Messenger RNA Translation Efficiency. Cell 2015, 161, 1388–1399. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Wan, J.; Gao, X.; Zhang, X.; Jaffrey, S.R.; Qian, S.B. Dynamic m6A mRNA methylation directs translational control of heat shock response. Nature 2015, 526, 591–594. [Google Scholar] [CrossRef] [PubMed]
- Kierzek, E.; Kierzek, R. The thermodynamic stability of RNA duplexes and hairpins containing N6-alkyladenosines and 2-methylthio-N6-alkyladenosines. Nucl. Acids Res. 2003, 31, 4472–4480. [Google Scholar] [CrossRef] [PubMed]
- Roost, C.; Lynch, S.R.; Batista, P.J.; Qu, K.; Chang, H.Y.; Kool, E.T. Structure and thermodynamics of N6-methyladenosine in RNA: A spring-loaded base modification. J. Am. Chem. Soc. 2015, 137, 2107–2115. [Google Scholar] [CrossRef] [PubMed]
- Xiao, W.; Adhikari, S.; Dahal, U.; Chen, Y.S.; Hao, Y.J.; Sun, B.F.; Sun, H.Y.; Li, A.; Ping, X.L.; Lai, W.Y.; et al. Nuclear m6A Reader YTHDC1 Regulates mRNA Splicing. Mol. Cell 2016, 61, 507–519. [Google Scholar] [CrossRef] [PubMed]
- Fustin, J.M.; Doi, M.; Yamaguchi, Y.; Hida, H.; Nishimura, S.; Yoshida, M.; Isagawa, T.; Morioka, M.S.; Kakeya, H.; Manabe, I.; et al. RNA-methylation-dependent RNA processing controls the speed of the circadian clock. Cell 2013, 155, 793–806. [Google Scholar] [CrossRef] [PubMed]
- Xiang, Y.; Laurent, B.; Hsu, C.H.; Nachtergaele, S.; Lu, Z.; Sheng, W.; Xu, C.; Chen, H.; Ouyang, J.; Wang, S.; et al. RNA m6A methylation regulates the ultraviolet-induced DNA damage response. Nature 2017, 543, 573–576. [Google Scholar] [CrossRef] [PubMed]
- Meyer, K.D.; Jaffrey, S.R. Rethinking m6A Readers, Writers, and Erasers. Annu. Rev. Cell Dev. Biol. 2017, 33, 319–342. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Feng, J.; Xue, Y.; Guan, Z.; Zhang, D.; Liu, Z.; Gong, Z.; Wang, Q.; Huang, J.; Tang, C.; et al. Structural basis of N6-adenosine methylation by the METTL3–METTL14 complex. Nature 2016, 534, 575–578. [Google Scholar] [CrossRef] [PubMed]
- Sledz, P.; Jinek, M. Structural insights into the molecular mechanism of the m6A writer complex. Elife 2016, 5, e18434. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Doxtader, K.A.; Nam, Y. Structural Basis for Cooperative Function of Mettl3 and Mettl14 Methyltransferases. Mol. Cell 2016, 63, 306–317. [Google Scholar] [CrossRef] [PubMed]
- Cui, Q.; Shi, H.; Ye, P.; Li, L.; Qu, Q.; Sun, G.; Sun, G.; Lu, Z.; Huang, Y.; Yang, C.G.; et al. m6A RNA Methylation Regulates the Self-Renewal and Tumorigenesis of Glioblastoma Stem Cells. Cell Rep. 2017, 18, 2622–2634. [Google Scholar] [CrossRef] [PubMed]
- Tanabe, A.; Tanikawa, K.; Tsunetomi, M.; Takai, K.; Ikeda, H.; Konno, J.; Torigoe, T.; Maeda, H.; Kutomi, G.; Okita, K.; et al. RNA helicase YTHDC2 promotes cancer metastasis via the enhancement of the efficiency by which HIF-1α mRNA is translated. Cancer Lett. 2016, 376, 34–42. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Zhao, B.S.; Zhou, A.; Lin, K.; Zheng, S.; Lu, Z.; Chen, Y.; Sulman, E.P.; Xie, K.; Bogler, O.; et al. m6A Demethylase ALKBH5 Maintains Tumorigenicity of Glioblastoma Stem-like Cells by Sustaining FOXM1 Expression and Cell Proliferation Program. Cancer Cell 2017, 31, 591–606. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Samanta, D.; Lu, H.; Bullen, J.W.; Zhang, H.; Chen, I.; He, X.; Semenza, G.L. Hypoxia induces the breast cancer stem cell phenotype by HIF-dependent and ALKBH5-mediated m6A-demethylation of NANOG mRNA. Proc. Natl. Acad. Sci. USA 2016, 113, E2047–E2056. [Google Scholar] [CrossRef] [PubMed]
- Mauer, J.; Luo, X.; Blanjoie, A.; Jiao, X.; Grozhik, A.V.; Patil, D.P.; Linder, B.; Pickering, B.F.; Vasseur, J.J.; Chen, Q.; et al. Reversible methylation of m6Am in the 5’ cap controls mRNA stability. Nature 2017, 541, 371–375. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Weng, H.; Su, R.; Weng, X.; Zuo, Z.; Li, C.; Huang, H.; Nachtergaele, S.; Dong, L.; Hu, C.; et al. FTO Plays an Oncogenic Role in Acute Myeloid Leukemia as a N6-Methyladenosine RNA Demethylase. Cancer Cell 2017, 31, 127–141. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.; Lu, Z.; Wang, X.; Fu, Y.; Luo, G.-Z.; Liu, N.; Han, D.; Dominissini, D.; Dai, Q.; Pan, T.; et al. High-Resolution N6-Methyladenosine (m6A) Map Using Photo-Crosslinking-Assisted m6A Sequencing. Angew. Chem. 2015, 54, 1587–1590. [Google Scholar] [CrossRef] [PubMed]
- Amort, T.; Rieder, D.; Wille, A.; Khokhlova-Cubberley, D.; Riml, C.; Trixl, L.; Jia, X.Y.; Micura, R.; Lusser, A. Distinct 5-methylcytosine profiles in poly(A) RNA from mouse embryonic stem cells and brain. Genome Biol. 2017, 18, 1. [Google Scholar] [CrossRef] [PubMed]
- Squires, J.E.; Patel, H.R.; Nousch, M.; Sibbritt, T.; Humphreys, D.T.; Parker, B.J.; Suter, C.M.; Preiss, T. Widespread occurrence of 5-methylcytosine in human coding and non-coding RNA. Nucl. Acids Res. 2012, 40, 5023–5033. [Google Scholar] [CrossRef] [PubMed]
- Brzezicha, B.; Schmidt, M.; Makalowska, I.; Jarmolowski, A.; Pienkowska, J.; Szweykowska-Kulinska, Z. Identification of human tRNA:m5C methyltransferase catalysing intron-dependent m5C formation in the first position of the anticodon of the pre-tRNA Leu (CAA). Nucl. Acids Res. 2006, 34, 6034–6043. [Google Scholar] [CrossRef] [PubMed]
- Hussain, S.; Sajini, A.A.; Blanco, S.; Dietmann, S.; Lombard, P.; Sugimoto, Y.; Paramor, M.; Gleeson, J.G.; Odom, D.T.; Ule, J.; et al. NSun2-mediated cytosine-5 methylation of vault noncoding RNA determines its processing into regulatory small RNAs. Cell Rep. 2013, 4, 255–261. [Google Scholar] [CrossRef] [PubMed]
- Abbasi-Moheb, L.; Mertel, S.; Gonsior, M.; Nouri-Vahid, L.; Kahrizi, K.; Cirak, S.; Wieczorek, D.; Motazacker, M.M.; Esmaeeli-Nieh, S.; Cremer, K.; et al. Mutations in NSUN2 cause autosomal-recessive intellectual disability. Am. J. Hum. Genet. 2012, 90, 847–855. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.A.; Rafiq, M.A.; Noor, A.; Hussain, S.; Flores, J.V.; Rupp, V.; Vincent, A.K.; Malli, R.; Ali, G.; Khan, F.S.; et al. Mutation in NSUN2, which encodes an RNA methyltransferase, causes autosomal-recessive intellectual disability. Am. J. Hum. Genet. 2012, 90, 856–863. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinez, F.J.; Lee, J.H.; Lee, J.E.; Blanco, S.; Nickerson, E.; Gabriel, S.; Frye, M.; Al-Gazali, L.; Gleeson, J.G. Whole exome sequencing identifies a splicing mutation in NSUN2 as a cause of a Dubowitz-like syndrome. J. Med. Genet. 2012, 49, 380–385. [Google Scholar] [CrossRef] [PubMed]
- Yi, J.; Gao, R.; Chen, Y.; Yang, Z.; Han, P.; Zhang, H.; Dou, Y.; Liu, W.; Wang, W.; Du, G.; et al. Overexpression of NSUN2 by DNA hypomethylation is associated with metastatic progression in human breast cancer. Oncotarget 2017, 8, 20751–20765. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, M.; Hirata, S.; Sato, S.; Koga, S.; Fujii, M.; Qi, G.; Ogawa, I.; Takata, T.; Shimamoto, F.; Tatsuka, M. Frequent increased gene copy number and high protein expression of tRNA (cytosine-5-)-methyltransferase (NSUN2) in human cancers. DNA Cell Biol. 2012, 31, 660–671. [Google Scholar] [CrossRef] [PubMed]
- Kaiser, S.; Jurkowski, T.P.; Kellner, S.; Schneider, D.; Jeltsch, A.; Helm, M. The RNA methyltransferase Dnmt2 methylates DNA in the structural context of a tRNA. RNA Biol. 2016, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Khoddami, V.; Cairns, B.R. Identification of direct targets and modified bases of RNA cytosine methyltransferases. Nat. Biotechnol. 2013, 31, 458–464. [Google Scholar] [CrossRef] [PubMed]
- Jeltsch, A.; Ehrenhofer-Murray, A.; Jurkowski, T.P.; Lyko, F.; Reuter, G.; Ankri, S.; Nellen, W.; Schaefer, M.; Helm, M. Mechanism and biological role of Dnmt2 in Nucleic Acid Methylation. RNA Biol. 2016, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Forbes, S.A.; Beare, D.; Boutselakis, H.; Bamford, S.; Bindal, N.; Tate, J.; Cole, C.G.; Ward, S.; Dawson, E.; Ponting, L.; et al. COSMIC: Somatic cancer genetics at high-resolution. Nucl. Acids Res. 2017, 45, D777–D783. [Google Scholar] [CrossRef] [PubMed]
- Elhardt, W.; Shanmugam, R.; Jurkowski, T.P.; Jeltsch, A. Somatic cancer mutations in the DNMT2 tRNA methyltransferase alter its catalytic properties. Biochimie 2015, 112, 66–72. [Google Scholar] [CrossRef] [PubMed]
- Schaefer, M.; Pollex, T.; Hanna, K.; Lyko, F. RNA cytosine methylation analysis by bisulfite sequencing. Nucl. Acids Res. 2009, 37, e12. [Google Scholar] [CrossRef] [PubMed]
- Shafik, A.; Schumann, U.; Evers, M.; Sibbritt, T.; Preiss, T. The emerging epitranscriptomics of long noncoding RNAs. Biochim. Biophys. Acta 2016, 1859, 59–70. [Google Scholar] [CrossRef] [PubMed]
- Edelheit, S.; Schwartz, S.; Mumbach, M.R.; Wurtzel, O.; Sorek, R. Transcriptome-wide mapping of 5-methylcytidine RNA modifications in bacteria, archaea, and yeast reveals m5C within archaeal mRNAs. PLoS Genet. 2013, 9, e1003602. [Google Scholar] [CrossRef] [PubMed]
- Khoddami, V.; Cairns, B.R. Transcriptome-wide target profiling of RNA cytosine methyltransferases using the mechanism-based enrichment procedure Aza-IP. Nat. Protoc. 2014, 9, 337–361. [Google Scholar] [CrossRef] [PubMed]
- Konig, J.; Zarnack, K.; Rot, G.; Curk, T.; Kayikci, M.; Zupan, B.; Turner, D.J.; Luscombe, N.M.; Ule, J. iCLIP reveals the function of hnRNP particles in splicing at individual nucleotide resolution. Nat. Struct. Mol. Biol. 2010, 17, 909–915. [Google Scholar] [CrossRef] [PubMed]
- Hutchinson, J.N.; Ensminger, A.W.; Clemson, C.M.; Lynch, C.R.; Lawrence, J.B.; Chess, A. A screen for nuclear transcripts identifies two linked noncoding RNAs associated with SC35 splicing domains. BMC Genom. 2007, 8, 39. [Google Scholar] [CrossRef] [PubMed]
- Eissmann, M.; Gutschner, T.; Hammerle, M.; Gunther, S.; Caudron-Herger, M.; Gross, M.; Schirmacher, P.; Rippe, K.; Braun, T.; Zornig, M.; et al. Loss of the abundant nuclear non-coding RNA MALAT1 is compatible with life and development. RNA Biol. 2012, 9, 1076–1087. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, S.; Ip, J.Y.; Shioi, G.; Tripathi, V.; Zong, X.; Hirose, T.; Prasanth, K.V. Malat1 is not an essential component of nuclear speckles in mice. RNA 2012, 18, 1487–1499. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Arun, G.; Mao, Y.S.; Lazar, Z.; Hung, G.; Bhattacharjee, G.; Xiao, X.; Booth, C.J.; Wu, J.; Zhang, C.; et al. The lncRNA Malat1 is dispensable for mouse development but its transcription plays a cis-regulatory role in the adult. Cell Rep. 2012, 2, 111–123. [Google Scholar] [CrossRef] [PubMed]
- Ji, P.; Diederichs, S.; Wang, W.; Boing, S.; Metzger, R.; Schneider, P.M.; Tidow, N.; Brandt, B.; Buerger, H.; Bulk, E.; et al. MALAT-1, a novel noncoding RNA, and thymosin beta4 predict metastasis and survival in early-stage non-small cell lung cancer. Oncogene 2003, 22, 8031–8041. [Google Scholar] [CrossRef] [PubMed]
- Gutschner, T.; Baas, M.; Diederichs, S. Noncoding RNA gene silencing through genomic integration of RNA destabilizing elements using zinc finger nucleases. Genome Res. 2011, 21, 1944–1954. [Google Scholar] [CrossRef] [PubMed]
- Gutschner, T.; Hammerle, M.; Eissmann, M.; Hsu, J.; Kim, Y.; Hung, G.; Revenko, A.; Arun, G.; Stentrup, M.; Gross, M.; et al. The noncoding RNA MALAT1 is a critical regulator of the metastasis phenotype of lung cancer cells. Cancer Res. 2013, 73, 1180–1189. [Google Scholar] [CrossRef] [PubMed]
- Gutschner, T.; Hammerle, M.; Diederichs, S. MALAT1—A paradigm for long noncoding RNA function in cancer. J. Mol. Med. 2013, 91, 791–801. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Zhu, Y.; Pang, J.; Weng, X.; Feng, X.; Guo, Y. Knockdown of long non-coding RNA MALAT1 inhibits growth and motility of human hepatoma cells via modulation of miR-195. J. Cell Biochem. 2017. [Google Scholar] [CrossRef] [PubMed]
- Pang, E.J.; Yang, R.; Fu, X.B.; Liu, Y.F. Overexpression of long non-coding RNA MALAT1 is correlated with clinical progression and unfavorable prognosis in pancreatic cancer. Tumour Biol. 2015, 36, 2403–2407. [Google Scholar] [CrossRef] [PubMed]
- Pa, M.; Naizaer, G.; Seyiti, A.; Kuerbang, G. Long Noncoding RNA MALAT1 Functions as a Sponge of MiR-200c in Ovarian Cancer. Oncol. Res. 2017. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Yuan, X.; Yan, D.; Li, D.; Guan, F.; Dong, Y.; Wang, H.; Liu, X.; Yang, B. Long Non-Coding RNA MALAT1 Decreases the Sensitivity of Resistant Glioblastoma Cell Lines to Temozolomide. Cell. Physiol. Biochem. 2017, 42, 1192–1201. [Google Scholar] [CrossRef] [PubMed]
- Bernard, D.; Prasanth, K.V.; Tripathi, V.; Colasse, S.; Nakamura, T.; Xuan, Z.; Zhang, M.Q.; Sedel, F.; Jourdren, L.; Coulpier, F.; et al. A long nuclear-retained non-coding RNA regulates synaptogenesis by modulating gene expression. EMBO J. 2010, 29, 3082–3093. [Google Scholar] [CrossRef] [PubMed]
- Tripathi, V.; Ellis, J.D.; Shen, Z.; Song, D.Y.; Pan, Q.; Watt, A.T.; Freier, S.M.; Bennett, C.F.; Sharma, A.; Bubulya, P.A.; et al. The nuclear-retained noncoding RNA MALAT1 regulates alternative splicing by modulating SR splicing factor phosphorylation. Mol. Cell 2010, 39, 925–938. [Google Scholar] [CrossRef] [PubMed]
- Wilusz, J.E.; Freier, S.M.; Spector, D.L. 3’ end processing of a long nuclear-retained noncoding RNA yields a tRNA-like cytoplasmic RNA. Cell 2008, 135, 919–932. [Google Scholar] [CrossRef] [PubMed]
- Gast, M.; Schroen, B.; Voigt, A.; Haas, J.; Kuehl, U.; Lassner, D.; Skurk, C.; Escher, F.; Wang, X.; Kratzer, A.; et al. Long noncoding RNA MALAT1-derived mascRNA is involved in cardiovascular innate immunity. J. Mol. Cell Biol. 2016, 8, 178–181. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.A.; Kinzig, C.G.; DeGregorio, S.J.; Steitz, J.A. Methyltransferase-like protein 16 binds the 3′-terminal triple helix of MALAT1 long noncoding RNA. Proc. Natl. Acad. Sci. USA 2016, 113, 14013–14018. [Google Scholar] [CrossRef] [PubMed]
- Zhou, K.I.; Parisien, M.; Dai, Q.; Liu, N.; Diatchenko, L.; Sachleben, J.R.; Pan, T. N6-Methyladenosine Modification in a Long Noncoding RNA Hairpin Predisposes Its Conformation to Protein Binding. J. Mol. Biol. 2016, 428, 822–833. [Google Scholar] [CrossRef] [PubMed]
- Rinn, J.L.; Kertesz, M.; Wang, J.K.; Squazzo, S.L.; Xu, X.; Brugmann, S.A.; Goodnough, L.H.; Helms, J.A.; Farnham, P.J.; Segal, E.; et al. Functional demarcation of active and silent chromatin domains in human HOX loci by noncoding RNAs. Cell 2007, 129, 1311–1323. [Google Scholar] [CrossRef] [PubMed]
- Cai, B.; Song, X.Q.; Cai, J.P.; Zhang, S. HOTAIR: A cancer-related long non-coding RNA. Neoplasma 2014, 61, 379–391. [Google Scholar] [CrossRef] [PubMed]
- Gupta, R.A.; Shah, N.; Wang, K.C.; Kim, J.; Horlings, H.M.; Wong, D.J.; Tsai, M.C.; Hung, T.; Argani, P.; Rinn, J.L.; et al. Long non-coding RNA HOTAIR reprograms chromatin state to promote cancer metastasis. Nature 2010, 464, 1071–1076. [Google Scholar] [CrossRef] [PubMed]
- Kogo, R.; Shimamura, T.; Mimori, K.; Kawahara, K.; Imoto, S.; Sudo, T.; Tanaka, F.; Shibata, K.; Suzuki, A.; Komune, S.; et al. Long noncoding RNA HOTAIR regulates polycomb-dependent chromatin modification and is associated with poor prognosis in colorectal cancers. Cancer Res. 2011, 71, 6320–6326. [Google Scholar] [CrossRef] [PubMed]
- Richtig, G.; Ehall, B.; Richtig, E.; Aigelsreiter, A.; Gutschner, T.; Pichler, M. Function and Clinical Implications of Long Non-Coding RNAs in Melanoma. Int. J. Mol. Sci. 2017, 18, 715. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Zhang, L.; Zheng, S. Role of the long non-coding RNA HOTAIR in hepatocellular carcinoma. Oncol. Lett. 2017, 14, 1233–1239. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Kong, D.; Chen, Q.; Ping, Y.; Pang, D. Oncogenic long noncoding RNA landscape in breast cancer. Mol. Cancer 2017, 16, 129. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Wang, Y.; Liu, X.; Luo, P.; Jing, W.; Zhu, M.; Tu, J. Identification of Circulating Long Noncoding RNA HOTAIR as a Novel Biomarker for Diagnosis and Monitoring of Non-Small Cell Lung Cancer. Technol. Cancer Res. Treat. 2017. [Google Scholar] [CrossRef] [PubMed]
- Tsai, M.C.; Manor, O.; Wan, Y.; Mosammaparast, N.; Wang, J.K.; Lan, F.; Shi, Y.; Segal, E.; Chang, H.Y. Long noncoding RNA as modular scaffold of histone modification complexes. Science 2010, 329, 689–693. [Google Scholar] [CrossRef] [PubMed]
- Amort, T.; Souliere, M.F.; Wille, A.; Jia, X.Y.; Fiegl, H.; Worle, H.; Micura, R.; Lusser, A. Long non-coding RNAs as targets for cytosine methylation. RNA Biol. 2013, 10, 1003–1008. [Google Scholar] [CrossRef] [PubMed]
- Brockdorff, N.; Ashworth, A.; Kay, G.F.; McCabe, V.M.; Norris, D.P.; Cooper, P.J.; Swift, S.; Rastan, S. The product of the mouse Xist gene is a 15 kb inactive X-specific transcript containing no conserved ORF and located in the nucleus. Cell 1992, 71, 515–526. [Google Scholar] [CrossRef]
- Brown, C.J.; Ballabio, A.; Rupert, J.L.; Lafreniere, R.G.; Grompe, M.; Tonlorenzi, R.; Willard, H.F. A gene from the region of the human X inactivation centre is expressed exclusively from the inactive X chromosome. Nature 1991, 349, 38–44. [Google Scholar] [CrossRef] [PubMed]
- Penny, G.D.; Kay, G.F.; Sheardown, S.A.; Rastan, S.; Brockdorff, N. Requirement for Xist in X chromosome inactivation. Nature 1996, 379, 131–137. [Google Scholar] [CrossRef] [PubMed]
- Wutz, A. Gene silencing in X-chromosome inactivation: Advances in understanding facultative heterochromatin formation. Nat. Rev. Genet. 2011, 12, 542–553. [Google Scholar] [CrossRef] [PubMed]
- Chu, C.; Zhang, Q.C.; da Rocha, S.T.; Flynn, R.A.; Bharadwaj, M.; Calabrese, J.M.; Magnuson, T.; Heard, E.; Chang, H.Y. Systematic discovery of Xist RNA binding proteins. Cell 2015, 161, 404–416. [Google Scholar] [CrossRef] [PubMed]
- Da Rocha, S.T.; Heard, E. Novel players in X inactivation: Insights into Xist-mediated gene silencing and chromosome conformation. Nat. Struct. Mol. Biol. 2017, 24, 197–204. [Google Scholar] [CrossRef] [PubMed]
- McHugh, C.A.; Chen, C.K.; Chow, A.; Surka, C.F.; Tran, C.; McDonel, P.; Pandya-Jones, A.; Blanco, M.; Burghard, C.; Moradian, A.; et al. The Xist lncRNA interacts directly with SHARP to silence transcription through HDAC3. Nature 2015, 521, 232–236. [Google Scholar] [CrossRef] [PubMed]
- Minajigi, A.; Froberg, J.; Wei, C.; Sunwoo, H.; Kesner, B.; Colognori, D.; Lessing, D.; Payer, B.; Boukhali, M.; Haas, W.; et al. Chromosomes. A comprehensive Xist interactome reveals cohesin repulsion and an RNA-directed chromosome conformation. Science 2015, 349. [Google Scholar] [CrossRef] [PubMed]
- Hasegawa, Y.; Brockdorff, N.; Kawano, S.; Tsutui, K.; Tsutui, K.; Nakagawa, S. The matrix protein hnRNP U is required for chromosomal localization of Xist RNA. Dev. Cell 2010, 19, 469–476. [Google Scholar] [CrossRef] [PubMed]
- Yildirim, E.; Kirby, J.E.; Brown, D.E.; Mercier, F.E.; Sadreyev, R.I.; Scadden, D.T.; Lee, J.T. Xist RNA is a potent suppressor of hematologic cancer in mice. Cell 2013, 152, 727–742. [Google Scholar] [CrossRef] [PubMed]
- Bennett, J.M.; Orazi, A. Diagnostic criteria to distinguish hypocellular acute myeloid leukemia from hypocellular myelodysplastic syndromes and aplastic anemia: Recommendations for a standardized approach. Haematologica 2009, 94, 264–268. [Google Scholar] [CrossRef] [PubMed]
- Dewald, G.W.; Brecher, M.; Travis, L.B.; Stupca, P.J. Twenty-six patients with hematologic disorders and X chromosome abnormalities. Frequent idic(X)(q13) chromosomes and Xq13 anomalies associated with pathologic ringed sideroblasts. Cancer Genet. Cytogenet. 1989, 42, 173–185. [Google Scholar] [CrossRef]
- Dierlamm, J.; Michaux, L.; Criel, A.; Wlodarska, I.; Zeller, W.; Louwagie, A.; Michaux, J.L.; Mecucci, C.; van den Berghe, H. Isodicentric (X)(q13) in haematological malignancies: Presentation of five new cases, application of fluorescence in situ hybridization (FISH) and review of the literature. Br. J. Haematol. 1995, 91, 885–891. [Google Scholar] [CrossRef] [PubMed]
- Paulsson, K.; Haferlach, C.; Fonatsch, C.; Hagemeijer, A.; Andersen, M.K.; Slovak, M.L.; Johansson, B.; Foundation, M.D.S. The idic(X)(q13) in myeloid malignancies: Breakpoint clustering in segmental duplications and association with TET2 mutations. Hum. Mol. Genet. 2010, 19, 1507–1514. [Google Scholar] [CrossRef] [PubMed]
- Rack, K.A.; Chelly, J.; Gibbons, R.J.; Rider, S.; Benjamin, D.; Lafreniere, R.G.; Oscier, D.; Hendriks, R.W.; Craig, I.W.; Willard, H.F.; et al. Absence of the XIST gene from late-replicating isodicentric X chromosomes in leukaemia. Hum. Mol. Genet. 1994, 3, 1053–1059. [Google Scholar] [CrossRef] [PubMed]
- Heinonen, K.; Mahlamaki, E.; Riikonen, P.; Meltoranta, R.L.; Rahiala, J.; Perkkio, M. Acquired X-chromosome aneuploidy in children with acute lymphoblastic leukemia. Med. Pediatr. Oncol. 1999, 32, 360–365. [Google Scholar] [CrossRef]
- Yamamoto, K.; Nagata, K.; Kida, A.; Hamaguchi, H. Acquired gain of an X chromosome as the sole abnormality in the blast crisis of chronic neutrophilic leukemia. Cancer Genet. Cytogenet. 2002, 134, 84–87. [Google Scholar] [CrossRef]
- Lassmann, S.; Weis, R.; Makowiec, F.; Roth, J.; Danciu, M.; Hopt, U.; Werner, M. Array CGH identifies distinct DNA copy number profiles of oncogenes and tumor suppressor genes in chromosomal- and microsatellite-unstable sporadic colorectal carcinomas. J. Mol. Med. 2007, 85, 293–304. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.L.; Chen, L.Z.; Lu, Y.X.; Zhang, D.S.; Zeng, Z.L.; Pan, Z.Z.; Huang, P.; Wang, F.H.; Li, Y.H.; Ju, H.Q.; et al. Long noncoding RNA XIST expedites metastasis and modulates epithelial-mesenchymal transition in colorectal cancer. Cell Death Dis. 2017, 8, e3011. [Google Scholar] [CrossRef] [PubMed]
- Leygue, E. Steroid receptor RNA activator (SRA1): Unusual bifaceted gene products with suspected relevance to breast cancer. Nucl. Recept. Signal. 2007, 5, e006. [Google Scholar] [CrossRef] [PubMed]
- Lanz, R.B.; Chua, S.S.; Barron, N.; Soder, B.M.; DeMayo, F.; O Malley, B.W. Steroid receptor RNA activator stimulates proliferation as well as apoptosis in vivo. Mol. Cell. Biol. 2003, 23, 7163–7176. [Google Scholar] [CrossRef] [PubMed]
- Lanz, R.B.; McKenna, N.J.; Onate, S.A.; Albrecht, U.; Wong, J.; Tsai, S.Y.; Tsai, M.J.; O’Malley, B.W. A steroid receptor coactivator, SRA, functions as an RNA and is present in an SRC-1 complex. Cell 1999, 97, 17–27. [Google Scholar] [CrossRef]
- Hube, F.; Guo, J.; Chooniedass-Kothari, S.; Cooper, C.; Hamedani, M.K.; Dibrov, A.A.; Blanchard, A.A.; Wang, X.; Deng, G.; Myal, Y.; et al. Alternative splicing of the first intron of the steroid receptor RNA activator (SRA) participates in the generation of coding and noncoding RNA isoforms in breast cancer cell lines. DNA Cell Biol. 2006, 25, 418–428. [Google Scholar] [CrossRef] [PubMed]
- Lanz, R.B.; Razani, B.; Goldberg, A.D.; O’Malley, B.W. Distinct RNA motifs are important for coactivation of steroid hormone receptors by steroid receptor RNA activator (SRA). Proc. Natl. Acade. Sci. USA 2002, 99, 16081–16086. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Wu, H.T.; Zhu, N.; Shi, Y.N.; Liu, Z.; Ao, B.X.; Liao, D.F.; Zheng, X.L.; Qin, L. Steroid receptor RNA activator: Biologic function and role in disease. Clin. Chim. Acta 2016, 459, 137–146. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Patton, J.R.; Ghosh, S.K.; Fischel-Ghodsian, N.; Shen, L.; Spanjaard, R.A. Pus3p- and Pus1p-dependent pseudouridylation of steroid receptor RNA activator controls a functional switch that regulates nuclear receptor signaling. Mol. Endocrinol. 2007, 21, 686–699. [Google Scholar] [CrossRef] [PubMed]
- Kim, N.K.; Theimer, C.A.; Mitchell, J.R.; Collins, K.; Feigon, J. Effect of pseudouridylation on the structure and activity of the catalytically essential P6.1 hairpin in human telomerase RNA. Nucl. Acids Res. 2010, 38, 6746–6756. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Xiong, X.; Yi, C. Epitranscriptome sequencing technologies: Decoding RNA modifications. Nat. Methods 2016, 14, 23–31. [Google Scholar] [CrossRef] [PubMed]
- Helm, M.; Motorin, Y. Detecting RNA modifications in the epitranscriptome: Predict and validate. Nat. Rev. Genet. 2017, 18, 275–291. [Google Scholar] [CrossRef] [PubMed]
- Glasner, H.; Riml, C.; Micura, R.; Breuker, K. Label-free, direct localization and relative quantitation of the RNA nucleobase methylations m6A, m5C, m3U, and m5U by top-down mass spectrometry. Nucl. Acids Res. 2017, 45, 8014–8025. [Google Scholar] [CrossRef] [PubMed]
- Rose, R.E.; Quinn, R.; Sayre, J.L.; Fabris, D. Profiling ribonucleotide modifications at full-transcriptome level: A step toward MS-based epitranscriptomics. RNA 2015, 21, 1361–1374. [Google Scholar] [CrossRef] [PubMed]
- Anderson, D.M.; Anderson, K.M.; Chang, C.L.; Makarewich, C.A.; Nelson, B.R.; McAnally, J.R.; Kasaragod, P.; Shelton, J.M.; Liou, J.; Bassel-Duby, R.; et al. A micropeptide encoded by a putative long noncoding RNA regulates muscle performance. Cell 2015, 160, 595–606. [Google Scholar] [CrossRef] [PubMed]
- Bazzini, A.A.; Johnstone, T.G.; Christiano, R.; Mackowiak, S.D.; Obermayer, B.; Fleming, E.S.; Vejnar, C.E.; Lee, M.T.; Rajewsky, N.; Walther, T.C.; et al. Identification of small ORFs in vertebrates using ribosome footprinting and evolutionary conservation. EMBO J. 2014, 33, 981–993. [Google Scholar] [CrossRef] [PubMed]
- Calviello, L.; Mukherjee, N.; Wyler, E.; Zauber, H.; Hirsekorn, A.; Selbach, M.; Landthaler, M.; Obermayer, B.; Ohler, U. Detecting actively translated open reading frames in ribosome profiling data. Nat. Methods 2016, 13, 165–170. [Google Scholar] [CrossRef] [PubMed]
- Ingolia, N.T.; Brar, G.A.; Stern-Ginossar, N.; Harris, M.S.; Talhouarne, G.J.; Jackson, S.E.; Wills, M.R.; Weissman, J.S. Ribosome profiling reveals pervasive translation outside of annotated protein-coding genes. Cell Rep. 2014, 8, 1365–1379. [Google Scholar] [CrossRef] [PubMed]
- Ingolia, N.T.; Lareau, L.F.; Weissman, J.S. Ribosome profiling of mouse embryonic stem cells reveals the complexity and dynamics of mammalian proteomes. Cell 2011, 147, 789–802. [Google Scholar] [CrossRef] [PubMed]
- Juntawong, P.; Girke, T.; Bazin, J.; Bailey-Serres, J. Translational dynamics revealed by genome-wide profiling of ribosome footprints in Arabidopsis. Proc. Natl. Acad. Sci. USA 2014, 111, E203–E212. [Google Scholar] [CrossRef] [PubMed]
- Guttman, M.; Russell, P.; Ingolia, N.T.; Weissman, J.S.; Lander, E.S. Ribosome profiling provides evidence that large noncoding RNAs do not encode proteins. Cell 2013, 154, 240–251. [Google Scholar] [CrossRef] [PubMed]
- Carlevaro-Fita, J.; Rahim, A.; Guigo, R.; Vardy, L.A.; Johnson, R. Cytoplasmic long noncoding RNAs are frequently bound to and degraded at ribosomes in human cells. RNA 2016, 22, 867–882. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Selected chemical modifications present in RNA.

Figure 2.
Integrated data analysis of three m5C, two m6A and three Ψ sequencing studies highlighting the total amount of modifications in lncRNAs (a) as well as the number of individual lncRNAs that contain the respective chemically modified nucleotide (b) (adapted from [132]).
Figure 2.
Integrated data analysis of three m5C, two m6A and three Ψ sequencing studies highlighting the total amount of modifications in lncRNAs (a) as well as the number of individual lncRNAs that contain the respective chemically modified nucleotide (b) (adapted from [132]).

Figure 3.
Putative information flow impacting chemical RNA modifications. Internal and external signals lead to epitranscriptomic changes, which are applied by writer and eraser proteins, and subsequently conveyed by reader proteins. Some functions of these epitranscriptomic marks have already been shown while additional mechanisms can be envisioned.
Figure 3.
Putative information flow impacting chemical RNA modifications. Internal and external signals lead to epitranscriptomic changes, which are applied by writer and eraser proteins, and subsequently conveyed by reader proteins. Some functions of these epitranscriptomic marks have already been shown while additional mechanisms can be envisioned.


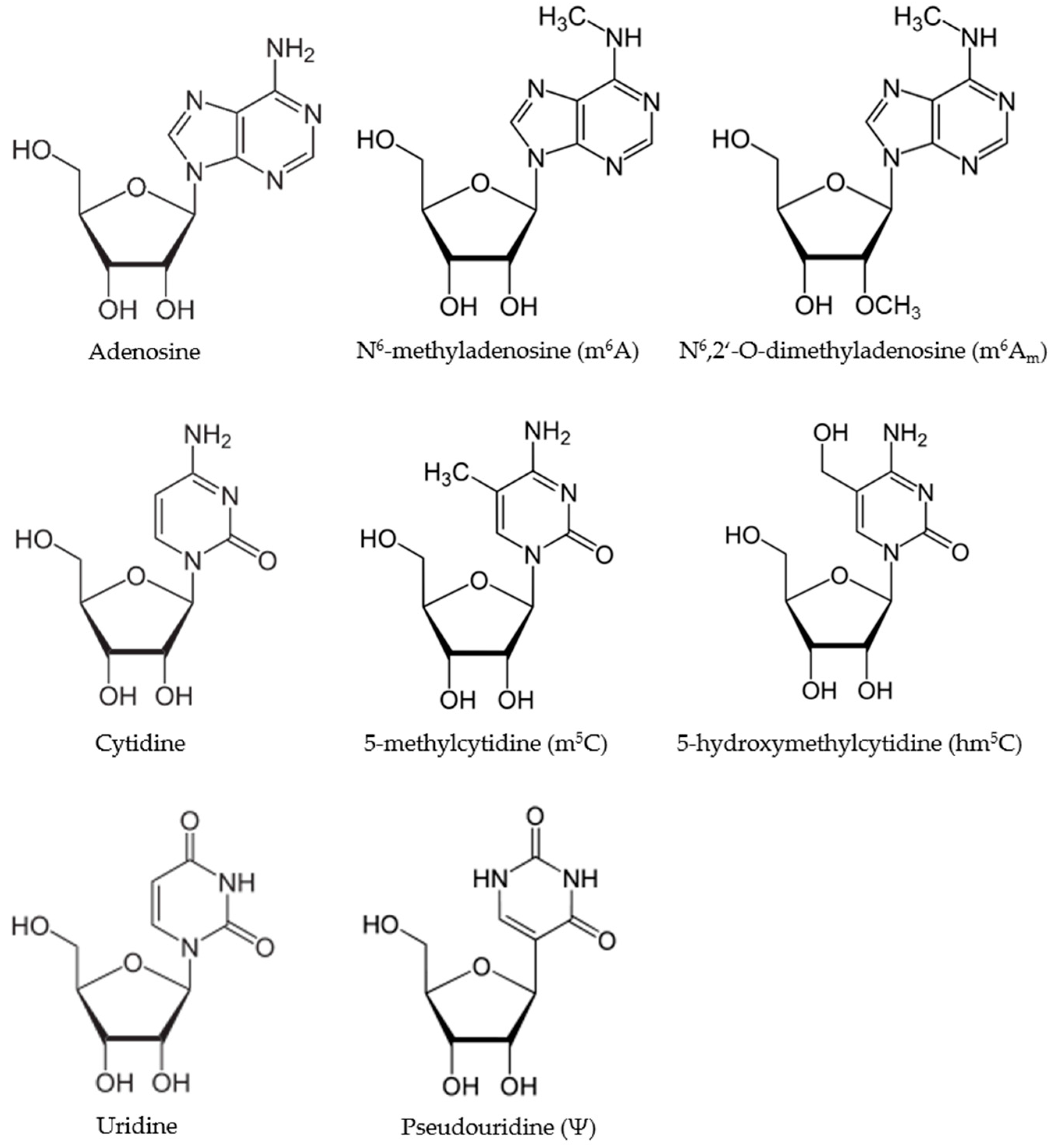{kind=link}
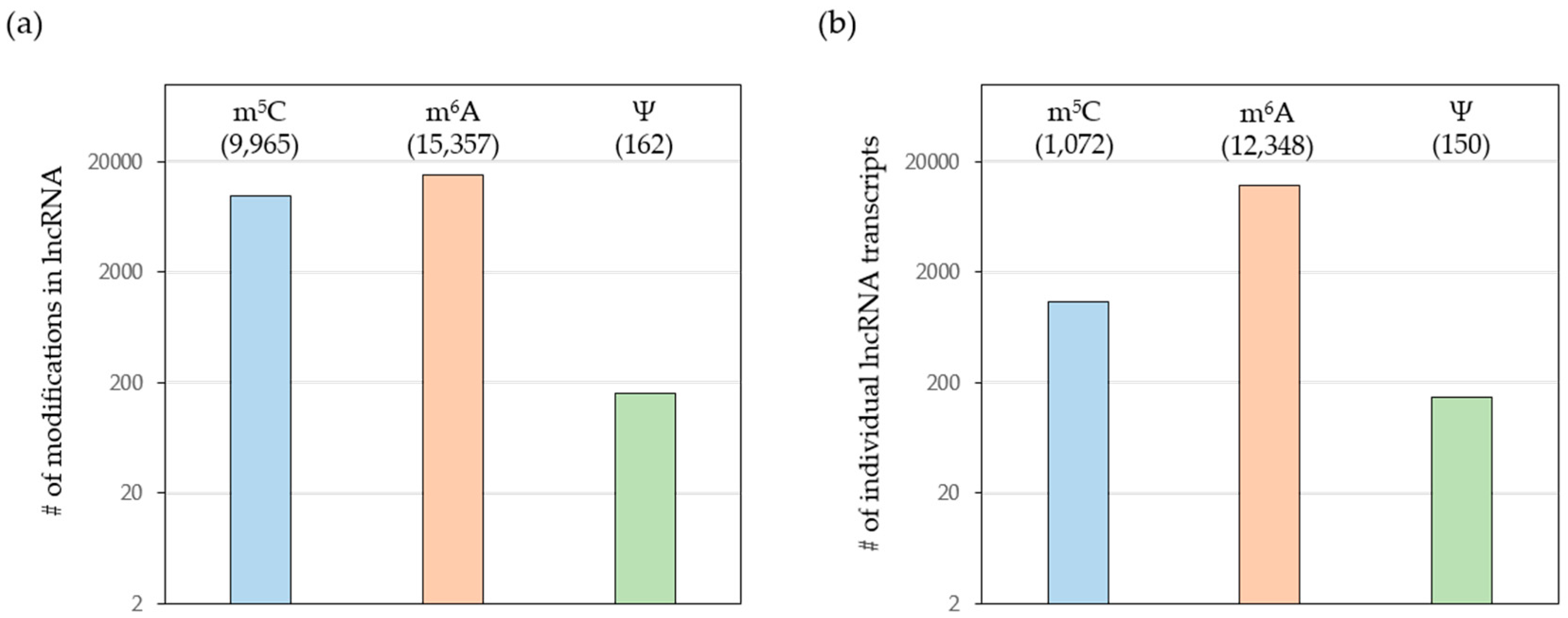{kind=link}
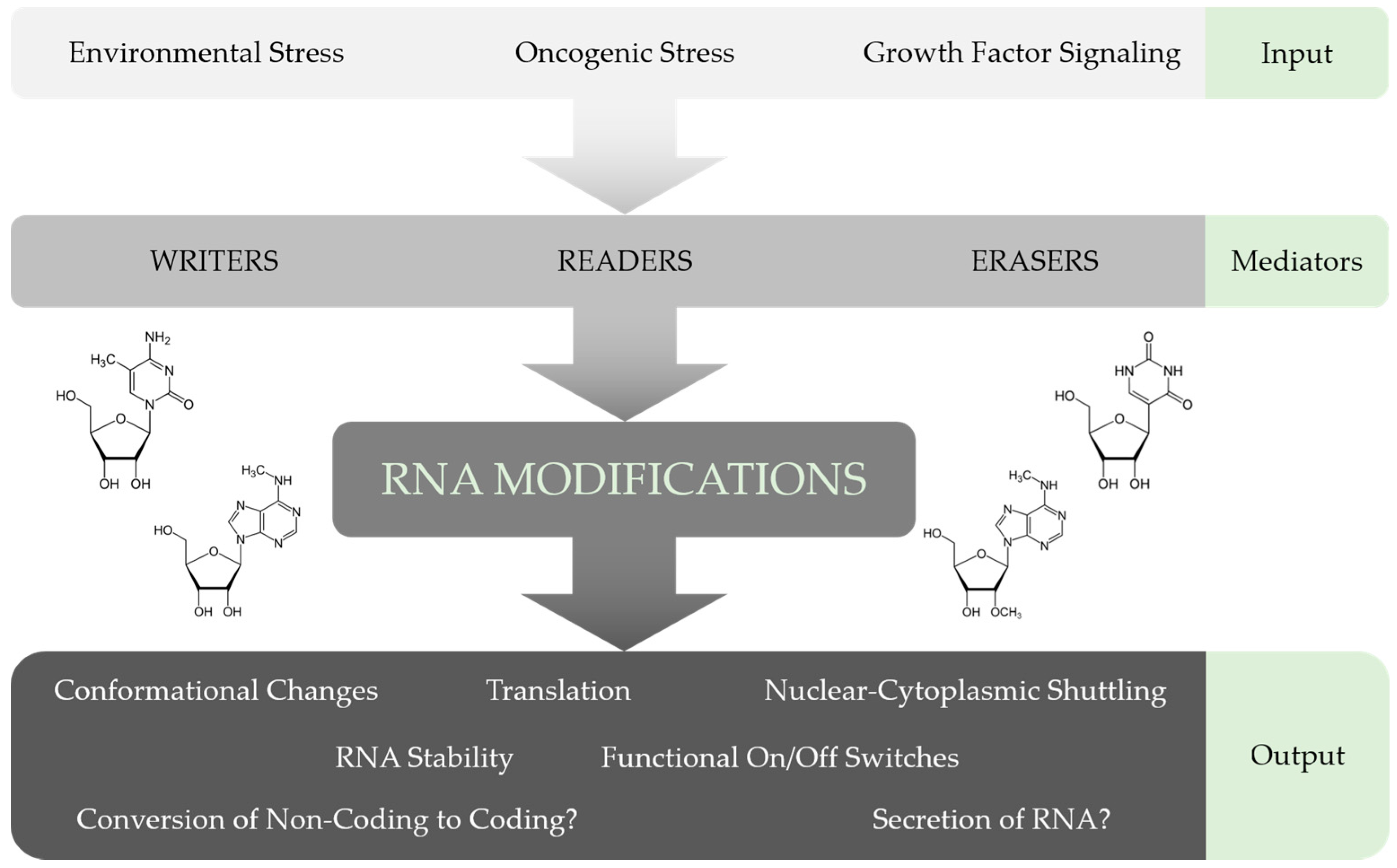{kind=link}
Table 1.
Known writer, reader, and eraser proteins for chemical RNA modifications.
| RNA Modification | Writer | Reader | Eraser |
|---|---|---|---|
| Ψ | PUS1 [39,40] PUSL1 [41] PUS3 [42,43] PUS7 [44] PUS7L [41] PUS10 [45] RPUSD1 [41] RPUSD2 [41] RPUSD3 [46] RPUSD4 [46] TRUB1 [44] TRUB2 [46] DKC1 [47] | ||
| m6A | METTL3 [48] METTL14 [49,50,51] WTAP [49,50,51,52] KIAA1429 [51,53] RBM15 [53,54] RBM15B [54] METTL16 [55] | YTHDF1 [56] YTHDF2 [57] YTHDF3 [57] YTHDC1 [58] YTHDC2 [54] eIF3 [59] HNRNPA2B1 [60] HNRNPC [61] | ALKBH5 [62] FTO [63] |
| m5C | NSUN1 [64] NSUN2 [65] NSUN3 [66] NSUN4 [67] NSUN5 [68] NSUN6 [69] NSUN7 [70] DNMT2 [71] | ALYREF [72] |
Table 2.
Selected lncRNAs and their recently identified chemical modifications.
| lncRNA | Modification | No. of Modified Residues | Reference |
|---|---|---|---|
| ANRIL | m6A | 1 | [97] |
| m5C | 2 | [118] | |
| DICER1-AS1 | m6A | 2 | [97] |
| Ψ | 1 | [87] | |
| GAS5 | m5C | 2 | [118] |
| HOTAIR | m6A | 1 | [97] |
| m5C | 1 | [163] | |
| Kcnq1ot1 | Ψ | 1 | [87] |
| LRRC75A-AS1 | Ψ | 1 | [77] |
| MALAT1 | m6A | 3 | [57] |
| 3 | [97] | ||
| m5C | 7 | [118] | |
| Ψ | 3 | [77] | |
| 3 | [87] | ||
| NEAT1 | m6A | 1 | [57] |
| m5C | 7 | [118] | |
| PVT1 | m6A | 2 | [57] |
| 1 | [97] | ||
| m5C | 1 | [118] | |
| 1 | [120] | ||
| Ψ | 1 | [77] | |
| RPPH1 | m5C | 4 | [118] |
| 1 | [127] | ||
| 1 | [120] | ||
| SNHG1 | Ψ | 1 | [77] |
| SNHG7 | Ψ | 1 | [87] |
| SNHG12 | m5C | 2 | [118] |
| SRA1 | m6A | 1 | [57] |
| 4 | [97] | ||
| Ψ | 1 | [40,189] | |
| ST7-AS1 | Ψ | 1 | [87] |
| TERC | m5C | 3 | [118] |
| Ψ | 2 | [85] | |
| 6 | [190] | ||
| TUG1 | m6A | 1 | [88] |
| 11 | [97] | ||
| XIST | m6A | 1 | [57] |
| 14 | [97] | ||
| m5C | 5 | [163] | |
| Ψ | 1 | [87] | |
| ZFAS1 | Ψ | 1 | [85] |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Jacob, R.; Zander, S.; Gutschner, T. The Dark Side of the Epitranscriptome: Chemical Modifications in Long Non-Coding RNAs. Int. J. Mol. Sci. 2017, 18, 2387. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms18112387
AMA Style
Jacob R, Zander S, Gutschner T. The Dark Side of the Epitranscriptome: Chemical Modifications in Long Non-Coding RNAs. International Journal of Molecular Sciences. 2017; 18(11):2387. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms18112387
Chicago/Turabian StyleJacob, Roland, Sindy Zander, and Tony Gutschner. 2017. "The Dark Side of the Epitranscriptome: Chemical Modifications in Long Non-Coding RNAs" International Journal of Molecular Sciences 18, no. 11: 2387. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms18112387
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.