Targeting Immune Cell Checkpoints during Sepsis


1
Department of Anesthesiology, Vanderbilt University Medical Center, Nashville, TN 37232, USA
2
Department of Pathology, Microbiology and Immunology, Vanderbilt University Medical Center, Nashville, TN 37232, USA
*
Author to whom correspondence should be addressed.
Int. J. Mol. Sci. 2017, 18(11), 2413; https://0-doi-org.brum.beds.ac.uk/10.3390/ijms18112413
Submission received: 18 October 2017
/
Revised: 10 November 2017
/
Accepted: 12 November 2017
/
Published: 14 November 2017
(This article belongs to the Special Issue Targeting Immune Checkpoints and Immunotherapy)
Abstract
:Immunosuppression is increasingly being recognized as one of the causes of increased morbidity and mortality during sepsis. Both innate and adaptive immune system dysfunction have been shown to cause an impaired ability to eradicate the primary infection and also lead to frequent occurrence of secondary opportunistic infections. Pre-clinical and clinical studies have shown that inhibitory immune checkpoint molecules, including programmed death-1 (PD-1), programmed death ligand-1 (PD-L1), cytotoxic T lymphocyte antigen-4 (CTLA-4), T cell membrane protein-3 (TIM-3), Lymphocyte activation-gene-3 (LAG-3) and 2B4, are upregulated during the course of sepsis. Engagement of these inhibitory molecules on various immune cells has been consistently shown to inhibit innate immune cell functions (e.g., phagocytosis, cytokine production and pathogen clearance) and also lead to impaired T cell competence. In numerous pre-clinical models of sepsis, therapeutic agents aimed at blocking engagement of inhibitory immune checkpoints on immune cells have been shown to improve innate and adaptive immune cell functions, increase host resistance to infection and significantly improve survival. Therefore, immunotherapy with immune cell checkpoint inhibitors holds significant potential for the future of sepsis therapy and merits further investigation.
Keywords:
sepsis; immunotherapy; T cell exhaustion; PD-1; PD-L1; CTLA-4; BTLA; TIM-3; LAG-3; 2B4; myeloid cells; immunosuppression1. Introduction
Sepsis is one of the most common causes of death among critically ill patients leading to estimated $14 billion in annual health care costs in the United States alone and much more worldwide [1]. Sepsis, defined as life-threatening organ dysfunction caused by dysregulated host responses to infection according to the third international consensus definition for sepsis and septic shock (Sepsis-3), accounts for more than 5.3 million deaths worldwide per annum [2,3]. No definitive therapy that targets the underlying pathobiology of sepsis exists. Thus, antibiotics, fluid resuscitation and organ support remain the mainstay of treatment. Recent clinical studies indicate that septic patients have a high one year mortality rate after hospital discharge, often due to the development of secondary infections [2,4]. Some investigators postulate that late infections in sepsis survivors stems from prolonged immunosuppression [5].
Classically, the time course of sepsis is characterized by pro-inflammatory and anti-inflammatory phases that occur during variable time points after sepsis. However, the typical biphasic initial pro-inflammatory phase followed by anti-inflammatory phase has been refuted by recent reports [6,7]. Some investigators propose that the pro-inflammatory and anti-inflammatory phases co-exist and that the prolonged presence of each phase defines a syndrome of chronic critical illness [8]. One of the hallmarks of sepsis is decreased ability to eradicate primary infection and increased susceptibility to secondary nosocomial infections often caused by opportunistic pathogens [9,10]. Sepsis often leads to significant multi-organ injury causing increased morbidity and mortality, as shown in numerous pre-clinical studies [11,12,13,14]. Immunosuppression is increasingly being recognized as a significant contributing factor for sepsis-induced morbidity and mortality, especially after hospital discharge [4,15,16]. This further highlights the pathological role of immunosuppression during sepsis, as a majority of deaths occur at delayed time points after the initial stabilization of septic patients with aggressive supportive therapy.
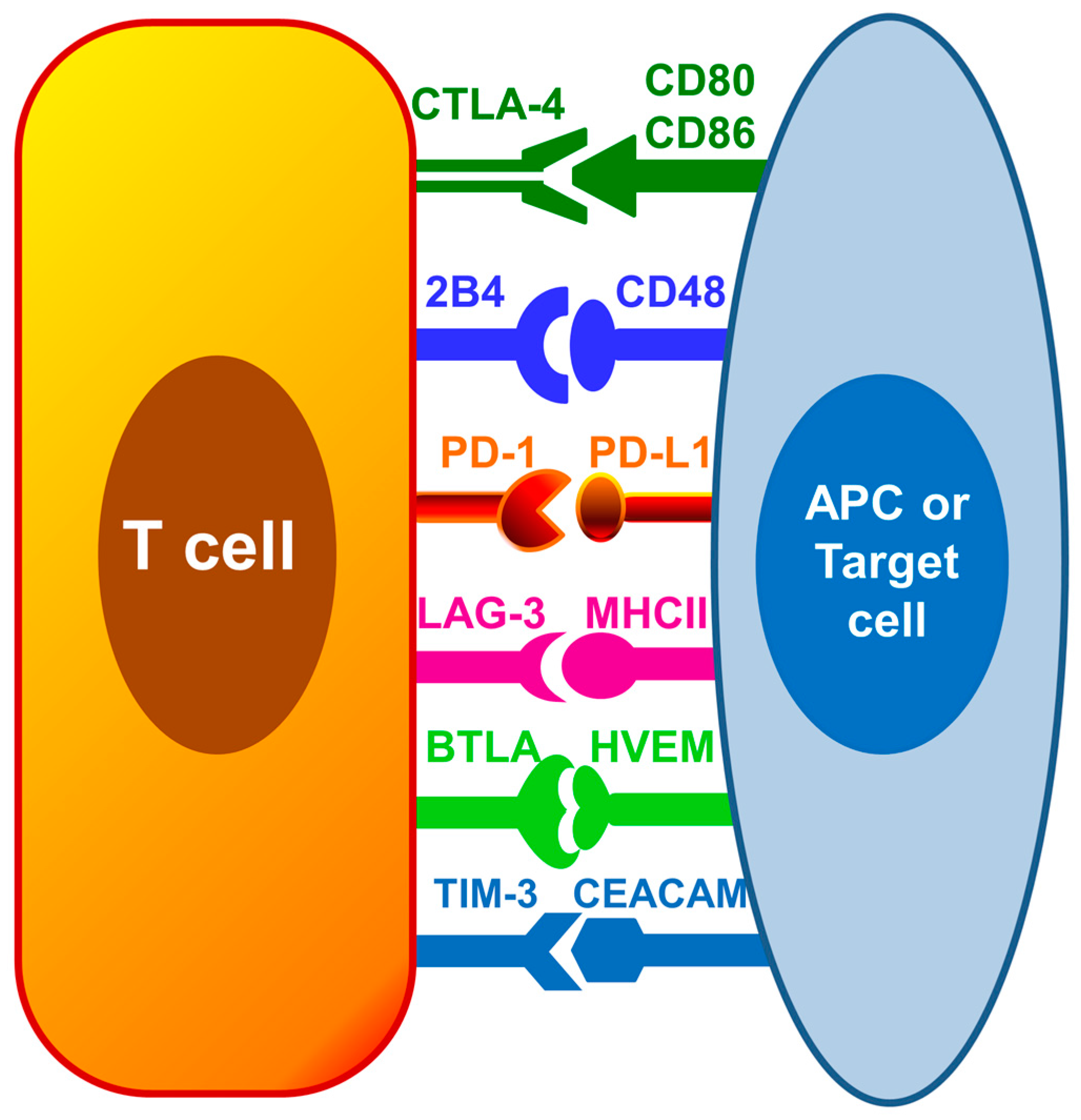
Therapies targeting immunosuppression are being intensively researched as a new approach for sepsis treatment. Immune checkpoint receptors including programmed death-1 (PD-1), programmed death ligand-1 (PD-L1), cytotoxic T lymphocyte antigen-4 (CTLA-4) and B and T lymphocyte attenuator (BTLA) have been shown to be increased on immune cells during sepsis and hypothesized to be one of the major contributors causing sepsis induced immune cell dysfunction [17]. These inhibitory immune regulators hinder the immune responses needed to clear off invading pathogens. The focus of this review is to discuss the current knowledge and recent advances regarding immune checkpoints during sepsis, and the future potential for novel immune checkpoints inhibitors, such as anti-PD1, anti-PD-L1 and anti-CTLA-4, as immunotherapeutic agents for restoring host immune response during sepsis. Figure 1 depicts the interaction among known inhibitor immune cell checkpoints on T cells and antigen presenting cells.
1.1. Sepsis-Induced Immunosuppression
New treatment protocols with aggressive supportive therapy rescue the majority of the septic patients during the early inflammatory phase, but survivors are then prone to develop an immunosuppressive phase [18]. Opportunistic pathogens such as Pseudomonas, Candida, Acinetobacter and Enterococcus are common culprits for secondary infections among septic patients [10]. Moreover, a high incidence of reactivation of latent viruses such as cytomegalovirus and herpes simplex virus has also been observed [19,20]. Immune cell dysfunction during sepsis not only causes a decreased capability to eradicate primary infection, but also increases the risk for such secondary infections. One of the major mechanisms for immunosuppression is hypothesized to be increased expression of immune regulatory checkpoints including PD-1, PD-L1, CTLA4 and BTLA, and targeting these negative regulators of immune responses has shown to improve host resistance to infections [15,17,21,22]. Although, immunosuppression during sepsis affects both innate and adaptive immune systems, T cell function is known to be the most significantly compromised during sepsis, as a result of the interaction between immune checkpoint receptors such as PD-1/PD-L1 [6,23,24]. The following sections will discuss the individual immune checkpoints and their known roles in sepsis immunopathology.
1.2. Programmed Death-1 (PD-1) and Its Ligands PD-L1/PD-L2
PD-1/PD-L1 axis is the most well characterized inhibitory immune checkpoint interaction that has been studied in sepsis immunopathology.
1.2.1. PD-1
Discovered in 1992, the programmed death-1 (PD-1) receptor (CD279) is a 228 amino acid, 50–55 kDa monomeric type I transmembrane glycoprotein belonging to the immunoglobulin superfamily, composed of an extracellular immunoglobulin Variable-type (V-type) extracellular domain, a transmembrane domain, and a cytoplasmic tail which executes the intracellular signaling [25,26]. The intracellular region of PD-1 receptor is composed of ITIM (immuno-receptor tyrosine-based inhibitory motif) and ITSM (immuno-receptor tyrosine-based switch motif) [27]. PD-1 protein is encoded by the Pdcd1 gene on chromosome 1 in mice and chromosome 2 in humans. Human and murine PD-1 proteins share approximately 60% amino acid identity [28]. T and B cells are the major leukocytes expressing PD-1 receptor, although it is also expressed on monocytes, natural killer cells, and dendritic cells [29]. Programmed death ligand, PD-L1 and PD-L2 are the known ligands for PD-1 receptor.
1.2.2. PD-L1 and PD-L2
PD-L1 (CD274) is also known as B7 homologue 1 or B7-H1, and PD-L2 is also known as B7-DC. PD-L1 is a 33 kDa transmembrane protein, first identified by Dong et al. in 1999 [30], and PD-L2 (CD273) is a 30 kDa transmembrane protein, first identified by Latchman et al. in 2001 [31]. Human and murine PD-L1 and PD-L2 share 69% and 70% amino acid identity, respectively [31]. PD-L1 is known to be expressed both on immune as well as non-immune cells. PD-L1 is not only constitutively expressed but also upregulated upon stimulation on dendritic cells, macrophages, T and B lymphocytes [29,32]. PD-L1 is also expressed in peripheral organs including heart, placenta, lung, liver, pancreas, kidney and tumor cells [29,32]. PD-L2 is more restricted in its distribution, found to be constitutively expressed in dendritic cells and monocytes, and transcripts have also been found in lung, placenta and liver [32].
1.3. Role of PD-1 and Its Ligands PD-L1 and PD-L2 in Immune Cell Dysfunction during Sepsis
It is well established that interaction of PD-1 with its ligands causes impaired T cell function. PD-1/PD-L1 induced T cell inhibition represents one of the major inhibitory receptor–ligand interactions studied during sepsis (Figure 2). PD-1 is known to be normally upregulated on the surface of activated CD4+ and CD8+ T cells to limit their hyper-activation and uncontrolled inflammation [33]. However, sustained up-regulation of PD-1 in the face of high antigen load as a result of severe infection, leads to impairment of both innate and adaptive immune responses [23,34]. The inhibitory immune checkpoint interaction often leads to a phenomenon known as T cell exhaustion. T cell exhaustion may lead to T cell dysfunction causing reduced effector T cell functions, decreased cytokine production, decreased proliferative capacity and apoptosis [34].
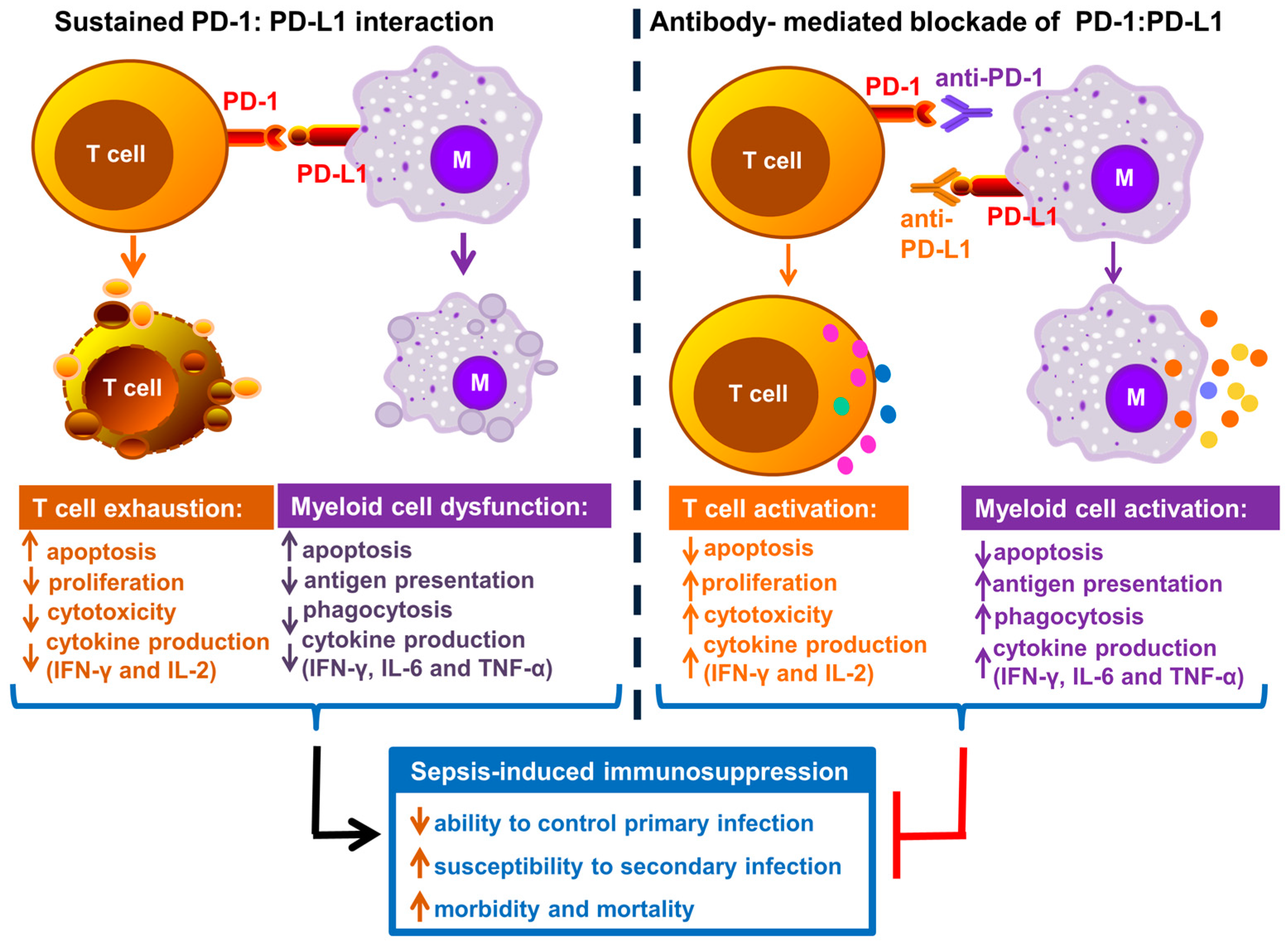
Over the past decade, numerous studies have shown a sustained increase in PD-1 and PD-L1 expression on various immune cells during sepsis. Figure 2 depicts the overview of immune cell dysfunction as a result of sustained PD-1–PD-L1 interaction during sepsis and inhibition of this interaction reverses sepsis induced immunosuppression and improves host resistance to infection.
In various pre-clinical studies employing different rodent models of sepsis such as cecal ligation and puncture (CLP) and burn wound infection with Pseudomonas aeruginosa, PD-1 expression has been shown to be upregulated on T cells and PD-L1 expression was increased on innate immune cells including monocytes, dendritic cells, Kuppfer cells and neutrophils [17,21,35,36,37,38,39,40,41]. The majority of these studies show that PD-1/PD-L1 axis stimulation during sepsis leads to T cell dysfunction and apoptosis, which is accompanied by increased pathogen burden, multi-organ injury, and mortality. PD-1 knockout has also been shown to improve survival in a neonatal model of cecal slurry-induced sepsis [42]. These findings are further strengthened by clinical studies which also reveal the roles of PD-1 and PD-L1 in immune cell dysfunction during sepsis. PD-1 expression on circulating T cells has been shown to significantly correlate with decreased T cell proliferation and increased secondary infections leading to higher mortality among septic shock patients [43]. Increased PD-L1 expression has also been correlated with increased T cell apoptosis, lymphopenia, and T cell dysfunction [44,45]. A recent notable study by Patera et al. showed that PD-L1 expression was significantly increased on suppressor phenotype subsets of neutrophils and monocytes, and increased PD-1 expression was observed on CD8+ T cells among septic patients [24]. Those alterations positively correlated with decreased phagocytic capacity of both neutrophils and monocytes, and decreased CD8+ T cell and natural killer (NK) cell function [24]. Wang et al. demonstrated that CLP-induced sepsis caused a significant increase in PD-1 expression on Kuppfer cells in the liver (a type of resident macrophages) and PD-1-deficient Kuppfer cells displayed increased phagocytic capacity and restoration of immune functions [40]. Huang et al. demonstrated a higher percentage of circulating neutrophils positive for PD-L1 expression, which correlated with lethal outcomes [38]. In a postmortem study involving sepsis patients, T cell function was shown to be evidently impaired in association with increased expression of PD-1 receptor and activation marker CD69, and a significant decrease in IL-7 receptor and CD28 expression (a co-stimulatory T cell receptor); and increased PD-L1 and PD-L2 expression on dendritic cells [23]. Taken together, these studies denote that PD-1/PD-L1 axis plays a critical role not only in T cell dysfunction, but also in innate immune cell impairment during sepsis.
Along with bacterial infections, bloodstream fungal infections with Candida has also been shown to cause an increased PD-1 expression on circulating CD4+ and CD8+ T cells and a decrease in co-stimulatory CD28 expression [46]. Other prospective clinical studies have shown that increased monocyte PD-L1 expression on days 3–4 after sepsis serves as an independent predictor of 28-day mortality in septic shock patients, and septic patients also demonstrated increased expression of PD-1 on CD4+ and CD8+ T lymphocytes [44,47]. As discussed earlier, PD-L2 expression is restricted to a handful of immune cells, including dendritic cells and monocytes, and there are limited studies addressing its direct role during sepsis-related immunopathology, as compared to PD-L1 which has been extensively studied during sepsis. Table 1 and Table 2 lists the summary of all the pre-clinical and clinical studies which implicate the inhibitory roles of PD-1, PD-L1/PD-L2 and other immune checkpoints during sepsis.
1.4. Does PD-L1 Play a Role in Organ Injury during Sepsis?
In addition to immunocytes, PD-L1 (but not PD-1) is also expressed in peripheral tissues including lung, liver, tissue endothelial cells and kidney [29,32]. Therefore, it is important to study the role of PD-L1, if any, with respect to organ injury during sepsis. There are limited studies which demonstrate this aspect of PD-L1. Wu et al. demonstrated that PD-L1 protein expression was significantly increased on intestinal colonic tissue among septic patients as well as in the intestinal tissue of mice subjected to CLP [53]. Furthermore, the preceding study also showed that increased PD-L1 expression on intestinal epithelial cells correlated with intestinal inflammation and increased permeability, signifying dysfunction of intestinal barrier function; and genetic deficiency of PD-L1 or blocking treatment with anti-PD-L1 antibody restored intestinal barrier function. Another study by the same group also showed that genetic deficiency of PD-L1 greatly reduces morphological intestinal injury and mortality induced by CLP in mice [38]. Therefore, it is reasonable to hypothesize that the inflammatory response in the local intestinal milieu is regulated, in part, by PD-1/PD-L1 axis, with PD-L1 on intestinal cells interacting with PD-1 expressed on leukocytes or non-immune cells that express PD-1, or a hitherto unknown binding partner for PD-L1 [38,53].
A study by Zhu et al. showed that CLP induced sepsis caused a significant increase in mRNA and protein levels of PD-L1 in the liver, which was associated with visible morphological damage; and treatment with anti-PD-L1 attenuated the sepsis induced liver injury [50]. From this study, it is not exactly clear if the increased PD-L1 expression was on hepatocytes or the liver resident Kupffer cells or infiltrating immune cells such as lymphocytes, monocytes and other cells. However, a study by Hutchins et al. utilizing a CLP model of sepsis, showed that sepsis caused an increase in expression of PD-L1 on liver sinusoidal endothelial cells, increased liver tissue vascular permeability and edema, and genetic deficiency of PD-L1 restored vascular barrier integrity and attenuated endothelial cell apoptosis [52]. Importantly, the preceding study also demonstrated an increased expression of PD-1 on F4/80+ Kupffer cells, which when depleted using clodronate liposomes, also significantly decreased the expression level of PD-L1 on liver sinusoidal endothelial cells. Therefore, it can be hypothesized that during sepsis, increased PD-1 on Kupffer cells might be interacting with increased PD-L1 on endothelial cells leading to detrimental effects on liver vascular permeability and potentially culminating in liver injury and failure. In a study by Zhang et al. evaluating the effectiveness of anti-PD-L1 antibody during CLP induced sepsis, it was noted that treatment with anti-PD-L1 decreased apoptosis of bronchial epithelial cells and alveolar epithelial cells in lungs, although the data was not shown [36]. Using postmortem lung tissue samples, Boomer et al. also showed that PD-L1 expression is detectable on lung parenchymal cells following sepsis [23].
Therefore, these studies demonstrate that PD-L1 plays a role in intestinal and liver injury during sepsis. Further studies need to be undertaken to uncover the exact role of PD-L1 in various organ injuries such as kidney, brain, lung, heart and others during sepsis. If PD-L1 indeed is detrimental for multi-organ function during sepsis, targeting PD-L1 would definitely open up a novel class of therapeutics for sepsis treatment.
1.5. Targeting PD-1 and PD-L1 during Sepsis
As discussed above, PD-1–PD-L1 interaction plays a significant role during sepsis-induced immunosuppression. Therefore, targeting either PD-1 or PD-L1 seems a logical approach to restore physiological immune responses and improve outcomes. Antibodies targeting PD-1 and PD-L1, which serve to block the PD-1–PD-L1 interaction, are being extensively studied in pre-clinical models to evaluate their therapeutic efficacy during sepsis. The advent of PD-1/PD-L1 pathway blocking antibodies has been a boon to the field of cancer therapy and numerous PD-1/PD-L1 blocking antibodies from various pharmaceutical companies have been approved by Food Drug Administration to treat human cancers [62]. These therapies have induced successful regression of advanced stage cancers and improved survival rate. Indeed, numerous pre-clinical studies have also shown that targeting PD-1 and PD-L1 during sepsis improves host resistance to infection, which merits further investigation. Table 3, Table 4 and Table 5 summarize the pre-clinical and clinical studies which have evaluated the therapeutic benefit of targeting numerous immune checkpoints.
1.5.1. Targeting PD-L1 during Sepsis
In a study by Zhang et al. treatment with anti-PD-L1 partially attenuated T cell apoptosis and depletion, decreased systemic inflammation, enhanced bacterial clearance, and improved survival [36]. A recent study by Shindo et al. demonstrated that a unique anti-PD-L1 peptide (termed as compound 8) doubled the survival rate in a two hit model of CLP-induced sepsis followed by Candida albicans-induced fungal infection [55]. Anti-PD-L1 has also been shown to attenuate liver injury in a murine model of CLP induced sepsis [50]. Anti-PD-L1 is effective not only in rodent models of bacterial sepsis but also in rodent models of fungal sepsis. In support of this, a study by Chang et al. showed that even a delayed treatment with anti-PD-L1 up to 24–48 h after the onset of fungal sepsis, reversed T cell dysfunction, increased MHC II on antigen presenting cells and significantly improved survival [51]. A more recent notable clinical study by Patera et al. demonstrated that ex vivo incubation of septic patient’s whole blood with anti-PD-L1 antibody significantly improved phagocytic function of neutrophils and monocytes, and restored CD8+ T cell and NK cell functions; with most beneficial effects seen among patient groups with lowest baseline function of these cells [24]. Another study by Chang et al. showed similar findings in that in ex vivo studies, anti-PD-L1 attenuated apoptosis and improved interferon-γ (IFN-γ) and intereukin-2 production by CD8+ T lymphocytes from septic patients [45]. A multicenter trial for evaluating the dose safety of anti-PD-L1 (BMS-936559 of Bristol–Myers Squibb) in patients with sepsis has recently been completed and results of the study are awaited (ClinicalTrial.gov# NCT02576457).
1.5.2. Targeting PD-1 during Sepsis
Along with anti-PD-L1, anti-PD1 has also been extensively tested for its therapeutic efficacy in preclinical models of sepsis. In a remarkable study by Brahmamdam et al. anti-PD1 antibody, even when administered 24 h after the onset of CLP induced sepsis, blocked apoptosis and depletion of T lymphocytes and dendritic cells; improved host immune responses; preserved delayed type hypersensitivity responses; and significantly improved survival [35]. Similar to anti-PD-L1, anti-PD1 is also protective during fungal sepsis. Studies by Shindo et al. in a two hit model of CLP followed by Candida albicans fungal sepsis showed that treatment with anti-PD1 improved MHC II expression on splenic macrophages and dendritic cells, and a combination therapy with interleukin-7 increased interferon-γ (IFN-γ) secretion by CD4+ and CD8+ T cells, and anti-PD-1 had no effect on either proliferation and CD28 expression on CD4+ and CD8+ T cells [64]. Furthermore, in another study, anti-PD1 was shown to be highly effective in restoring IFN-γ secretion by CD4+ and CD8+ T cells, MHC II expression on antigen presenting cells and significantly improving survival in a murine primary and two hit model of fungal sepsis [51]. It is noteworthy to mention that the preceding two studies by Shindo et al. and Chang et al. showed benefit with anti-PD1 even when it was administered late after the onset of sepsis, which strengthens its therapeutic implication [51,64]. In addition to animal studies, clinical studies have shown beneficial effects of anti-PD1. Patera et al. showed that anti-PD1 not only improves phagocytic function of innate immune cells like neutrophils and monocytes, but also restored T lymphocyte function, all isolated from the blood of septic patients [24]. Another clinical study by Chang et al. demonstrated that similar to anti-PD-L1, anti-PD1 attenuated apoptosis and improved IFN-γ and intereukin-2 production by CD8+ T lymphocytes from septic patients [45].
Therefore, overall the above studies show that blocking the PD-1/PD-L1 interaction with blocking antibodies against each restores immune function among immunosuppressed septic host and provides significant protection.
2. Cytotoxic T Lymphocyte Antigen-4 (CTLA-4)
CTLA-4 is also known as CD152, and it is a negative regulator of T cell function, which shares 30% homology to the T cell co-stimulatory molecule CD28 [34]. It is a dimeric cell surface glycoprotein expressed by both activated CD4+ and CD8+ T cells, and binds to its receptors CD80 (high affinity than CD86) and CD86 (relatively lower affinity than CD80) on antigen presenting cells [65]. CTLA-4 and CD28, both bind to CD80 and CD86, but CTLA-4 binding is of much higher affinity and thereby CTLA-4 counteracts CD28 induced co-stimulation of T cells. The mechanism of action of CTLA-4 to inhibit T lymphocyte proliferation and activation involves a reduction in IL-2 production and IL-2 receptor expression and arresting T cells at the G1 phase of the cell cycle [66,67]. Therefore, conditions which result in sustained upregulation of CTLA-4 compromise T cell immune response and render the host immunosuppressed. Therefore, inhibition of CTLA-4 with blocking antibodies against it might help restore T cell functions in such conditions. CTLA-4 is one of the first immune checkpoints to be clinically targeted in cancer therapy and Ipilimumab (Bristol–Myers Squibb, New York, NY, USA), a monoclonal CTLA-4 antibody, was approved by FDA in 2011 for the treatment of metastatic melanoma [68].
Using a murine model of CLP-induced sepsis, Inoue et al. demonstrated that CTLA-4 expression was progressively increased on both CD4+ and CD8+ T cells and regulatory T cells, starting at 24 h after induction of sepsis, along with T cell apoptosis and depletion [48]. In the same study, treatment with anti-CTLA-4 inhibited T cell apoptosis by more than 50% and significantly improved survival. Importantly, they also show that protective effect of anti-CTLA-4 is dose dependent and higher doses (200 µg per mouse) worsens the survival outcome as compared to significantly improved survival with lower doses (50 µg per mouse) in two different strain of mice (C57BL6 and CD-1 mice). Anti-CTLA-4 at a low dose (33 µg per mouse) also improves survival in a two hit model of sepsis, comprised of slowly progressive CLP-induced sepsis followed by infection with a fungus, Candida albicans [48]. Therefore, protective effects of anti-CTLA-4 are dose dependent. It is possible that at higher doses, anti-CTLA-4 drives T cell over activation leading to uncontrolled inflammation and deleterious effects of survival. Another study by Chang et al. using two model of sepsis including a primary Candida albicans fungal sepsis and a two hit model (CLP-induced sepsis followed by Candida albicans), demonstrated that anti-CTLA-4 increases T lymphocyte IFN-γ production, and significantly improves survival [51]. A prospective clinical study showed that as sepsis progressed, CTLA-4 expression was increased at day 7 on circulating T lymphocytes, as compared to the expression levels at the onset of sepsis [57]. As compared to PD-1–PD-L1 there are limited studies addressing the role of CTLA-4 during sepsis.
3. B and T Lymphocyte Attenuator (BTLA)
BTLA is another known inhibitory molecule, which is not only expressed on the surface of T cells but also on innate immune cells including monocytes, macrophages, and dendritic cells [34,69]. BTLA interacts with tumor necrosis factor superfamily molecule termed herpes virus entry mediator (HVEM) and known to cause inhibition of T cell exhaustion [34].
A study by Shubin et al. showed that BTLA expression on T cells correlated with increased mortality in a rodent model of sepsis [49]. In accordance with these findings, Shubin et al. further demonstrated that increased BTLA expression on peripheral CD4+ T cells among critically ill sepsis patients positively correlated with development of subsequent nosocomial infections [58]. Furthermore, the preceding study also showed that BTLA deficient mice (BTLA−/−) had increased numbers of CD4+ T cells in the spleen following sepsis and implicated a role for BTLA in apoptosis induced T cell loss. Another study by the same group, using a mouse model and CLP-induced sepsis, also showed that BTLA expression facilitates impairment of innate inflammatory cell activation and promotes MHC II reduction, increases bacterial burden following CLP, increases circulating interleukin-10 levels, and results in multi-organ injury and decreased survival; as compared to septic BTLA knockout mice [49]. Boomer et al. showed that HVEM (ligand for BTLA) is detectable in postmortem lungs of septic patients as compared to controls [23]. On the other hand, a prospective clinical study by Spec et al. showed that the levels of BTLA expression on immune cells was not statistically different among Candida infected septic patients as compared to control patients [46]. Interestingly, anti-BTLA monoclonal antibody is known to be having dual effects including blocking as well as potentiating effects on BTLA mediated effects [70,71]. Only one study has evaluated the effect of ant-BTLA antibody in a two hit model of hemorrhage followed by sepsis [54]. This study by Cheng et al. showed that, treatment with anti-BTLA (at a dose of 25 µg per gram body weight) caused excessive inflammatory immune responses, increased organ injury, leading to significantly increased morbidity and mortality [54]. These results demonstrate that anti-BTLA actually further potentiated BTLA actions in this model of sepsis. In an interesting study by Lange et al. soluble BTLA (sBTLA) levels in the plasma were founds to be significantly higher among sepsis patients as compared to controls and the levels also correlated with clinical severity of the disease [61]. Moreover, the relative risk of 28-day mortality among septic patients was five-fold higher among patients with baseline sBTLA levels of greater than 21 ng/mL, as compared to those with a level below this threshold, suggesting that sBTLA may be explored as a prognostic marker in sepsis [61]. This study has some major drawbacks as noted by the authors including limited plasma samples in certain later phases of the study period, due to missing samples or patients being transferred from the intensive care unit, leading to limitations on conclusions that could be drawn on dynamics of immune markers studied, and varied time points of sepsis onset among different subjects studied. Nonetheless, this is the first study implicating sBTLA as a prognostic marker during sepsis, and further in-depth studies are warranted to further decipher the role of sBTLA during sepsis.
4. T Cell Membrane Protein-3 (TIM-3), Lymphocyte Activation-Gene-3 (LAG-3) and 2B4
TIM-3, LAG3 and 2B4 are some of the other known T cell inhibitory molecules which can contribute to T cell exhaustion. TIM-3 interacts with CEACAM1 (carcinoembryonic antigen-related cell adhesion molecule 1) or Galectin 9, and LAG-3 interacts with antigen molecule presented in conjunction with major histocompatibility class II (MHC II) on antigen presenting cells, and 2B4 (also known as CD244) interacts with CD48 on antigen presenting cells [34].
As compared to other inhibitory molecules discussed above, TIM-3, LAG-3 and 2B4 have not been extensively investigated in sepsis yet. Cell surface expression of these inhibitory molecules is not as frequently altered as compared to other classical molecules including PD-1 and PD-L1 [34]. Interaction of TIM3 with its ligand galectin-9 has been shown to cause T cell death and tolerance in vivo [72]. A clinical study by Boomer et al. showed that expression of TIM-3 and LAG-3 was elevated on CD4+ T cells among septic patients and LAG-3 was more elevated on CD8+ T cells at the onset of acute sepsis phase as compared to TIM-3 [57]. Spec et al. demonstrated that there was no significant difference with respect to TIM-3 expression on CD4+ and CD8+ T cells during Candida sepsis among critically ill patients as compared to controls, although other inhibitory cell surface receptors including PD-1 and PD-L1 were upregulated and there was a trend towards increase in 2B4 expression on CD8+ T cells [46]. However, some recent pre-clinical studies have shown that blocking TIM-3 exacerbates sepsis. A study by Zhao et al. using CLP model of sepsis demonstrated that blocking TIM-3 signaling with soluble TIM-3-Immunoglubulin (sTIM-3-IgG) resulted in exacerbation of sepsis induced macrophage pro-inflammatory responses and lymphocyte apoptosis during acute phase of sepsis, and enhanced anti-inflammatory phenotype for macrophages and CD4+ T cells during late phase of sepsis [63]. Furthermore, the preceding study also showed that mice over-expressing TIM-3 attenuated sepsis induced immunosuppression and significantly improved survival, and similar results were obtained upon administering the TIM-3 ligand galectin-9. Previous studies from the same group had also shown that TIM-3 mRNA expression in human peripheral blood mononuclear cells is significantly lower in severe sepsis patients as compared to sepsis patients, and such downregulation of TIM-3 correlated with increased C-reactive protein levels, a clinical marker of patient’s inflammatory status [59]. The same study also showed that blocking TIM-3 signaling using anti-TIM-3 antibody or sTIM-3-IgG increased sepsis severity and significantly decreases survival in a CLP model of sepsis. This finding was correlated with the role of TIM-3 in negatively regulating toll like receptor-4 mediated responses of macrophages leading to inhibition of macrophage activation, and showed that TLR4 signaling pathway is an important mediator of TIM-3 related immune homeostatic mechanisms during sepsis [59]. Based on these studies, it seems that TIM-3 might play a permissive role for protection during sepsis induced immunosuppression, which merits further detailed investigation. However, in all these studies, TIM-3 antibody was administered starting at least one day before sepsis induction and it will be interesting to study the consequences of blocking TIM-3 signaling after sepsis induction. On the contrary, a clinical study by Ren et al. shows that TIM-3 expression on monocytes was significantly elevated among sepsis patients as compared to severe sepsis, septic shock and control patients; and soluble TIM-3 (sTIM-3) levels in the plasma of septic shock group was higher than just sepsis or severe sepsis groups, the levels of which correlated with eventual non-survivors [60]. Therefore, TIM-3 expression on monocytes and sTIM-3 exhibited opposite profiles among patients with varying sepsis severity and detailed mechanistic studies are therefore warranted to delineate the exact role of TIM-3 during sepsis.
In a recent study, Chen et al. demonstrated the inhibitory role of 2B4 during sepsis [56]. This study showed that 2B4 levels are significantly upregulated on T lymphocytes in both animal model of CLP induced sepsis and human sepsis patients as early as 24 h after sepsis induction, along with an increased expression of other T cell exhaustion markers including PD-1 and BTLA. Importantly, administration of anti-2B4 blocking antibody or genetic deficiency of 2B4 significantly improves survival after sepsis. Furthermore, immune cell specific conditional knockout of 2B4 also revealed that specific deletion of 2B4 expressed on CD4+ T cell was responsible for enhanced survival as compared to that expressed on CD8+ T cells and NK cells. Therefore, 2B4 could be a novel therapeutic target during sepsis and further studies are needed to discover the signaling mechanisms downstream of 2B4 in CD4+ T cell during sepsis.
5. Blockade of Immune Checkpoints during Sepsis: Is it always Appropriate?
Targeting immunosuppression has opened up a vast field of researching novel therapeutics for sepsis, an area of research which was marred by numerous failed clinical trials targeting pro-inflammatory mediators during sepsis [17,21,73]. However, taking lessons from previous clinical trials during sepsis, it is imperative to understand that no single targeted therapy will fit all the sepsis patients and individualized therapy is the extremely important. Stratifying septic patients based on immune cell phenotyping is one of the ways to determine specific patients who might benefit from immunotherapy. Majority of the clinical research up till now has relied on techniques such as flow cytometry to analyze cell surface expression of inhibitory molecules and ex vivo analysis of immune cells for production of cytokines are predominantly used to determine the state of immune system during sepsis. New technologies such as single-cell mass cytometry might well be the future for high content immune profiling techniques to characterize the phenotype and function of various immune cells during sepsis [74]. Lymphocyte expression of inhibitory receptors including PD-1, BTLA, CTLA-4, TIM-3, LAG-3 and 2B4; downregulation of HLA-DR on monocytes; and upregulation of PD-L1 on innate immune cells such as dendritic cells, monocytes, serve as markers for T cell exhaustion [23] and could very well guide the selection of patients for targeted therapies with individual or a combination of immune checkpoint inhibitors. Appropriate patient selection is the key to finding the right therapy. This was well demonstrated by a recent re-analysis of an original phase III clinical trial data which showed that infusion of recombinant human IL-1 receptor antagonist failed to reduce mortality among severe sepsis patients [75]. However, when the results of the same study were reanalyzed for subgroups of septic patients with characteristics of macrophage activation syndrome, there was a discernible significant positive impact of treatment on survival among this specific group of patients [76]. Another example is the use of anti-TIM-3 antibody during sepsis. Rodent models have shown that treatment with anti-TIM-3 potentiates inflammation and increases mortality during sepsis [63]. Additionally, combination therapies including immune checkpoint inhibitors and other therapies such as monophosphoryl lipid A, interleukin-7, interelukin-15, IFN-γ and FMS like tyrosine kinase-3 ligand might be more appropriate for specific patients and this will lead to better attenuation of sepsis induced immunosuppression [41,77,78,79,80].
Potential Side Effects of Blocking Immune Checkpoints
We should not forget that an unnecessary inhibition of immune checkpoints can lead to disruption of normal immune homeostasis and could cause inflammatory and auto-immune side effects. Under normal physiological circumstances, immune checkpoints help maintain self-tolerance and inhibit over activation of the T cell response against self-antigens. Clinical trials using immune checkpoint inhibitors have documented immune related adverse effects including fatigue, rash, nausea, pruritus, elevated liver enzymes, abnormal thyroid function, colitis and others [81,82]. Uncontrolled immune activation as a result of immune checkpoint blockade might affect multiple organ systems and result into irreversible lifelong disabilities such as vitiligo, gastro-intestinal disorders, endocrine disorders, polyarthritis, and other autoimmune disorders [82]. Pre-clinical studies in mice have shown that PD-1 deficiency causes an increased incidence of autoimmune pathologies such as lupus like syndromes, de novo type 1 diabetes, dilated cardiomyopathy and others [83,84]. Therefore, caution needs to be exercised when extending anti-PD-1/PD-L1 therapies for sepsis immunotherapy. Therapy that targets immune checkpoints has not been tested yet in septic patients and future research must carefully monitor and address the occurrence of any such adverse effects among septic patients. Once clinical trials are undertaken, subjects should be followed up after hospital discharge to understand not only the short term but also the long term adverse effects, as immune related adverse effects might not fully manifest for months or years. Furthermore, it is important that immune checkpoint blockade therapy should be targeted to patients who actually manifest an increased expression of these receptors. This will require careful monitoring of the phenotype of circulating immune cells among each of the septic patient before starting the therapy. The dose of individual immune checkpoint inhibitors also needs to be well titrated, as a higher dose might precipitate untoward effects and lead to a severe inflammatory response and increased mortality. For example, in murine model of CLP sepsis, anti-CTLA-4 antibody has been shown to be detrimental at higher doses [48]. Well monitored future clinical trials will be able to shed more light on the occurrence and prevalence of untoward effects in sepsis patients.
Hypothetically, targeting immune checkpoints could actually result in severe exaggeration of inflammatory responses if such a therapy is administered during an ongoing severe inflammatory phase of sepsis. Various pathological states including pro-inflammatory, anti-inflammatory and altered responses of immune cells are known to occur simultaneously in septic patients and non-specific boosting of the immune system functions could be detrimental during sepsis [85]. Moreover, murine models of sepsis might not correlate with the exact clinical scenario in septic patients and the majority of the information we have today is based on targeting immune checkpoints in rodent models. For example, the technique of most commonly used murine model of sepsis, CLP, is highly variable among different laboratories depending on the cecum ligation length, amount of fecal matter expressed, fluid/antibiotic resuscitation, and other variable factors. Nonetheless, rodent model do offer a great medium to undertake mechanistic studies during sepsis. A recent study by Patera et al. analyzed the responses to immune checkpoint inhibitors in circulating cells derived from sepsis patients [24], and further study of human patients need to be performed to grow our confidence in using immune checkpoint inhibitors for sepsis therapy before undertaking any larger clinical trials. Table 3, Table 4 and Table 5 summarize the pre-clinical and clinical studies which have evaluated the utility of blocking inhibitory immune cell checkpoints during sepsis.
6. Concluding Remarks
There is significant evidence to indicate that immunosuppression plays a detrimental role during sepsis based on numerous pre-clinical and clinical research studies. Targeting immune checkpoints which could potentially reverse innate and adaptive system hypo-responsiveness during sepsis could benefit sepsis patients. However, such a therapy needs to be individualized based on immune status of a particular patient; and cautious treatment with individual or a combination of immune checkpoint inhibitors could be the future of sepsis therapy.
Acknowledgments
This work was supported by the National Institute of Health grant NIH R01 GM66885 to Edward R. Sherwood and American Heart Association grant 16POST29920007 to Naeem K. Patil.
Author Contributions
Naeem K. Patil wrote the manuscript; Edward R. Sherwood, Yin Guo and Liming Luan critically revised the manuscript for important intellectual content; All authors have read and approved the manuscript.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
| PD-1 | Programmed death-1 |
| PD-L1 | Programmed death ligand-1 |
| PD-L2 | Programmed death ligand-2 |
| CTLA-4 | Cytotoxic T lymphocyte antign-4 |
| BTLA | B and T lymphocyte attenuator |
| HVEM | Herpes virus entry mediator |
| TIM-3 | T cell membrane protein-3 |
| CEACM | Carcinoembryonic antigen-related cell adhesion molecule |
| IFN-γ | Interferon-gamma |
| sTIM-3-Ig | Soluble TIM-3 Immunoglobulin |
| NK cell | Natural Killer Cell |
| CLP | Cecal ligation and puncture |
References
- Mayr, F.B.; Yende, S.; Angus, D.C. Epidemiology of severe sepsis. Virulence 2014, 5, 4–11. [Google Scholar] [CrossRef] [PubMed]
- Fleischmann, C.; Scherag, A.; Adhikari, N.K.; Hartog, C.S.; Tsaganos, T.; Schlattmann, P.; Angus, D.C.; Reinhart, K. International Forum of Acute Care Team. Assessment of Global incidence and mortality of hospital-treated sepsis. Current estimates and limitations. Am. J. Respir. Crit. Care Med. 2016, 193, 259–272. [Google Scholar] [CrossRef] [PubMed]
- Singer, M.; Deutschman, C.S.; Seymour, C.W.; Shankar-Hari, M.; Annane, D.; Bauer, M.; Bellomo, R.; Bernard, G.R.; Chiche, J.D.; Coopersmith, C.M.; et al. The third international consensus definitions for sepsis and septic shock (Sepsis-3). JAMA 2016, 315, 801–810. [Google Scholar] [CrossRef] [PubMed]
- Delano, M.J.; Ward, P.A. Sepsis-induced immune dysfunction: Can immune therapies reduce mortality? J. Clin. Investig. 2016, 126, 23–31. [Google Scholar] [CrossRef] [PubMed]
- Hotchkiss, R.S.; Sherwood, E.R. Immunology. Getting sepsis therapy right. Science 2015, 347, 1201–1202. [Google Scholar] [CrossRef] [PubMed]
- Boomer, J.S.; Green, J.M.; Hotchkiss, R.S. The changing immune system in sepsis: Is individualized immuno-modulatory therapy the answer? Virulence 2014, 5, 45–56. [Google Scholar] [CrossRef] [PubMed]
- Tang, B.M.; Huang, S.J.; McLean, A.S. Genome-wide transcription profiling of human sepsis: A systematic review. Crit. Care 2010, 14, R237. [Google Scholar] [CrossRef] [PubMed]
- Gentile, L.F.; Cuenca, A.G.; Efron, P.A.; Ang, D.; Bihorac, A.; McKinley, B.A.; Moldawer, L.L.; Moore, F.A. Persistent inflammation and immunosuppression: A common syndrome and new horizon for surgical intensive care. J. Trauma Acute Care Surg. 2012, 72, 1491–1501. [Google Scholar] [CrossRef] [PubMed]
- Monneret, G.; Venet, F.; Kullberg, B.J.; Netea, M.G. ICU-acquired immunosuppression and the risk for secondary fungal infections. Med. Mycol. 2011, 49 (Suppl. S1), S17–S23. [Google Scholar] [CrossRef] [PubMed]
- Otto, G.P.; Sossdorf, M.; Claus, R.A.; Rodel, J.; Menge, K.; Reinhart, K.; Bauer, M.; Riedemann, N.C. The late phase of sepsis is characterized by an increased microbiological burden and death rate. Crit. Care 2011, 15, R183. [Google Scholar] [CrossRef] [PubMed]
- Patil, N.K.; Parajuli, N.; MacMillan-Crow, L.A.; Mayeux, P.R. Inactivation of renal mitochondrial respiratory complexes and manganese superoxide dismutase during sepsis: Mitochondria-targeted antioxidant mitigates injury. Am. J. Physiol. Ren. Physiol. 2014, 306, F734–F743. [Google Scholar] [CrossRef] [PubMed]
- Abraham, E.; Singer, M. Mechanisms of sepsis-induced organ dysfunction. Crit. Care Med. 2007, 35, 2408–2416. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Luan, L.; Patil, N.K.; Wang, J.; Bohannon, J.K.; Rabacal, W.; Fensterheim, B.A.; Hernandez, A.; Sherwood, E.R. IL-15 Enables Septic Shock by Maintaining NK Cell Integrity and Function. J. Immunol. 2017, 198, 1320–1333. [Google Scholar] [CrossRef] [PubMed]
- Hotchkiss, R.S.; Karl, I.E. The pathophysiology and treatment of sepsis. N. Engl. J. Med. 2003, 348, 138–150. [Google Scholar] [CrossRef] [PubMed]
- Hotchkiss, R.S.; Coopersmith, C.M.; McDunn, J.E.; Ferguson, T.A. The sepsis seesaw: Tilting toward immunosuppression. Nat. Med. 2009, 15, 496–497. [Google Scholar] [CrossRef] [PubMed]
- Hotchkiss, R.S.; Opal, S. Immunotherapy for sepsis—A new approach against an ancient foe. N. Engl. J. Med. 2010, 363, 87–89. [Google Scholar] [CrossRef] [PubMed]
- Patil, N.K.; Bohannon, J.K.; Sherwood, E.R. Immunotherapy: A promising approach to reverse sepsis-induced immunosuppression. Pharmacol. Res. 2016, 111, 688–702. [Google Scholar] [CrossRef] [PubMed]
- Schorr, C.A.; Dellinger, R.P. The surviving sepsis campaign: Past, present and future. Trends Mol. Med. 2014, 20, 192–194. [Google Scholar] [CrossRef] [PubMed]
- Luyt, C.E.; Kaiser, L. Virus detection in patients with severe pneumonia: Still more questions than answers? Am. J. Respir. Crit. Care Med. 2012, 186, 301–302. [Google Scholar] [CrossRef] [PubMed]
- Walton, A.H.; Muenzer, J.T.; Rasche, D.; Boomer, J.S.; Sato, B.; Brownstein, B.H.; Pachot, A.; Brooks, T.L.; Deych, E.; Shannon, W.D.; et al. Reactivation of multiple viruses in patients with sepsis. PLoS ONE 2014, 9, e98819. [Google Scholar] [CrossRef] [PubMed]
- Hotchkiss, R.S.; Monneret, G.; Payen, D. Immunosuppression in sepsis: A novel understanding of the disorder and a new therapeutic approach. Lancet Infect. Dis. 2013, 13, 260–268. [Google Scholar] [CrossRef]
- Rudick, C.P.; Cornell, D.L.; Agrawal, D.K. Single versus combined immunoregulatory approach using PD-1 and CTLA-4 modulators in controlling sepsis. Expert Rev. Clin. Immunol. 2017, 13, 907–919. [Google Scholar] [CrossRef] [PubMed]
- Boomer, J.S.; To, K.; Chang, K.C.; Takasu, O.; Osborne, D.F.; Walton, A.H.; Bricker, T.L.; Jarman, S.D., 2nd; Kreisel, D.; Krupnick, A.S.; et al. Immunosuppression in patients who die of sepsis and multiple organ failure. JAMA 2011, 306, 2594–2605. [Google Scholar] [CrossRef] [PubMed]
- Patera, A.C.; Drewry, A.M.; Chang, K.; Beiter, E.R.; Osborne, D.; Hotchkiss, R.S. Frontline Science: Defects in immune function in patients with sepsis are associated with PD-1 or PD-L1 expression and can be restored by antibodies targeting PD-1 or PD-L1. J. Leukoc. Biol. 2016, 100, 1239–1254. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Schwartz, J.C.; Guo, X.; Bhatia, S.; Cao, E.; Lorenz, M.; Cammer, M.; Chen, L.; Zhang, Z.Y.; Edidin, M.A.; et al. Structural and functional analysis of the costimulatory receptor programmed death-1. Immunity 2004, 20, 337–347. [Google Scholar] [CrossRef]
- Ishida, Y.; Agata, Y.; Shibahara, K.; Honjo, T. Induced expression of PD-1, a novel member of the immunoglobulin gene superfamily, upon programmed cell death. EMBO J. 1992, 11, 3887–3895. [Google Scholar] [PubMed]
- Okazaki, T.; Maeda, A.; Nishimura, H.; Kurosaki, T.; Honjo, T. PD-1 immunoreceptor inhibits B cell receptor-mediated signaling by recruiting src homology 2-domain-containing tyrosine phosphatase 2 to phosphotyrosine. Proc. Natl. Acad. Sci. USA 2001, 98, 13866–13871. [Google Scholar] [CrossRef] [PubMed]
- Shinohara, T.; Taniwaki, M.; Ishida, Y.; Kawaichi, M.; Honjo, T. Structure and chromosomal localization of the human PD-1 gene (PDCD1). Genomics 1994, 23, 704–706. [Google Scholar] [CrossRef] [PubMed]
- Keir, M.E.; Butte, M.J.; Freeman, G.J.; Sharpe, A.H. PD-1 and its ligands in tolerance and immunity. Ann. Rev. Immunol. 2008, 26, 677–704. [Google Scholar] [CrossRef] [PubMed]
- Dong, H.; Zhu, G.; Tamada, K.; Chen, L. B7-H1, a third member of the B7 family, co-stimulates T-cell proliferation and interleukin-10 secretion. Nat. Med. 1999, 5, 1365–1369. [Google Scholar] [PubMed]
- Latchman, Y.; Wood, C.R.; Chernova, T.; Chaudhary, D.; Borde, M.; Chernova, I.; Iwai, Y.; Long, A.J.; Brown, J.A.; Nunes, R.; et al. PD-L2 is a second ligand for PD-1 and inhibits T cell activation. Nat. Immunol. 2001, 2, 261–268. [Google Scholar] [CrossRef] [PubMed]
- Collins, M.; Ling, V.; Carreno, B.M. The B7 family of immune-regulatory ligands. Genome Biol. 2005, 6, 223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crawford, A.; Wherry, E.J. The diversity of costimulatory and inhibitory receptor pathways and the regulation of antiviral T cell responses. Curr. Opin. Immunol. 2009, 21, 179–186. [Google Scholar] [CrossRef] [PubMed]
- Wherry, E.J.; Kurachi, M. Molecular and cellular insights into T cell exhaustion. Nat. Rev. Immunol. 2015, 15, 486–499. [Google Scholar] [CrossRef] [PubMed]
- Brahmamdam, P.; Inoue, S.; Unsinger, J.; Chang, K.C.; McDunn, J.E.; Hotchkiss, R.S. Delayed administration of anti-PD-1 antibody reverses immune dysfunction and improves survival during sepsis. J. Leukoc. Biol. 2010, 88, 233–240. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhou, Y.; Lou, J.; Li, J.; Bo, L.; Zhu, K.; Wan, X.; Deng, X.; Cai, Z. PD-L1 blockade improves survival in experimental sepsis by inhibiting lymphocyte apoptosis and reversing monocyte dysfunction. Crit. Care 2010, 14, R220. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Venet, F.; Wang, Y.L.; Lepape, A.; Yuan, Z.; Chen, Y.; Swan, R.; Kherouf, H.; Monneret, G.; Chung, C.S.; et al. PD-1 expression by macrophages plays a pathologic role in altering microbial clearance and the innate inflammatory response to sepsis. Proc. Natl. Acad. Sci. USA 2009, 106, 6303–6308. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Chen, Y.; Chung, C.S.; Yuan, Z.; Monaghan, S.F.; Wang, F.; Ayala, A. Identification of B7-H1 as a novel mediator of the innate immune/proinflammatory response as well as a possible myeloid cell prognostic biomarker in sepsis. J. Immunol. 2014, 192, 1091–1099. [Google Scholar] [CrossRef] [PubMed]
- McNab, F.W.; Berry, M.P.; Graham, C.M.; Bloch, S.A.; Oni, T.; Wilkinson, K.A.; Wilkinson, R.J.; Kon, O.M.; Banchereau, J.; Chaussabel, D.; et al. Programmed death ligand 1 is over-expressed by neutrophils in the blood of patients with active tuberculosis. Eur. J. Immunol. 2011, 41, 1941–1947. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Huang, X.; Chung, C.S.; Chen, Y.; Hutchins, N.A.; Ayala, A. Contribution of programmed cell death receptor (PD)-1 to Kupffer cell dysfunction in murine polymicrobial sepsis. Am. J. Physiol. Gastrointest. Liver Physiol. 2016, 311, G237–G245. [Google Scholar] [CrossRef] [PubMed]
- Patil, N.K.; Bohannon, J.K.; Luan, L.; Guo, Y.; Fensterheim, B.; Hernandez, A.; Wang, J.; Sherwood, E.R. Flt3 Ligand treatment attenuates T cell dysfunction and improves survival in a murine model of burn wound sepsis. Shock 2017, 47, 40–51. [Google Scholar] [CrossRef] [PubMed]
- Young, W.A.; Fallon, E.A.; Heffernan, D.S.; Efron, P.A.; Cioffi, W.G.; Ayala, A. Improved survival after induction of sepsis by cecal slurry in PD-1 knockout murine neonates. Surgery 2017, 161, 1387–1393. [Google Scholar] [CrossRef] [PubMed]
- Guignant, C.; Lepape, A.; Huang, X.; Kherouf, H.; Denis, L.; Poitevin, F.; Malcus, C.; Cheron, A.; Allaouchiche, B.; Gueyffier, F.; et al. Programmed death-1 levels correlate with increased mortality, nosocomial infection and immune dysfunctions in septic shock patients. Crit. Care 2011, 15, R99. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Li, J.; Lou, J.; Zhou, Y.; Bo, L.; Zhu, J.; Zhu, K.; Wan, X.; Cai, Z.; Deng, X. Upregulation of programmed death-1 on T cells and programmed death ligand-1 on monocytes in septic shock patients. Crit. Care 2011, 15, R70. [Google Scholar] [CrossRef] [PubMed]
- Chang, K.; Svabek, C.; Vazquez-Guillamet, C.; Sato, B.; Rasche, D.; Wilson, S.; Robbins, P.; Ulbrandt, N.; Suzich, J.; Green, J.; et al. Targeting the programmed cell death 1: Programmed cell death ligand 1 pathway reverses T cell exhaustion in patients with sepsis. Crit. Care 2014, 18, R3. [Google Scholar] [CrossRef] [PubMed]
- Spec, A.; Shindo, Y.; Burnham, C.A.; Wilson, S.; Ablordeppey, E.A.; Beiter, E.R.; Chang, K.; Drewry, A.M.; Hotchkiss, R.S. T cells from patients with Candida sepsis display a suppressive immunophenotype. Crit. Care 2016, 20, 15. [Google Scholar] [CrossRef] [PubMed]
- Shao, R.; Fang, Y.; Yu, H.; Zhao, L.; Jiang, Z.; Li, C.S. Monocyte programmed death ligand-1 expression after 3–4 days of sepsis is associated with risk stratification and mortality in septic patients: A prospective cohort study. Crit. Care 2016, 20, 124. [Google Scholar] [CrossRef] [PubMed]
- Inoue, S.; Bo, L.; Bian, J.; Unsinger, J.; Chang, K.; Hotchkiss, R.S. Dose-dependent effect of anti-CTLA-4 on survival in sepsis. Shock 2011, 36, 38–44. [Google Scholar] [CrossRef] [PubMed]
- Shubin, N.J.; Chung, C.S.; Heffernan, D.S.; Irwin, L.R.; Monaghan, S.F.; Ayala, A. BTLA expression contributes to septic morbidity and mortality by inducing innate inflammatory cell dysfunction. J. Leukoc. Biol. 2012, 92, 593–603. [Google Scholar] [CrossRef] [PubMed]
- Zhu, W.; Bao, R.; Fan, X.; Tao, T.; Zhu, J.; Wang, J.; Li, J.; Bo, L.; Deng, X. PD-L1 blockade attenuated sepsis-induced liver injury in a mouse cecal ligation and puncture model. Mediat. Inflamm. 2013, 2013, 361501. [Google Scholar] [CrossRef] [PubMed]
- Chang, K.C.; Burnham, C.A.; Compton, S.M.; Rasche, D.P.; Mazuski, R.J.; McDonough, J.S.; Unsinger, J.; Korman, A.J.; Green, J.M.; Hotchkiss, R.S. Blockade of the negative co-stimulatory molecules PD-1 and CTLA-4 improves survival in primary and secondary fungal sepsis. Crit. Care 2013, 17, R85. [Google Scholar] [CrossRef] [PubMed]
- Hutchins, N.A.; Wang, F.; Wang, Y.; Chung, C.S.; Ayala, A. Kupffer cells potentiate liver sinusoidal endothelial cell injury in sepsis by ligating programmed cell death ligand-1. J. Leukoc. Biol. 2013, 94, 963–970. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Chung, C.S.; Chen, Y.; Monaghan, S.F.; Patel, S.; Huang, X.; Heffernan, D.S.; Ayala, A. A novel role for programmed cell death receptor ligand-1 (PD-L1) in Sepsis-induced intestinal dysfunction. Mol. Med. 2016, 41, 21. [Google Scholar]
- Cheng, T.; Bai, J.; Chung, C.S.; Chen, Y.; Biron, B.M.; Ayala, A. Enhanced innate inflammation induced by anti-BTLA antibody in dual insult model of hemorrhagic shock/sepsis. Shock 2016, 45, 40–49. [Google Scholar] [CrossRef] [PubMed]
- Shindo, Y.; McDonough, J.S.; Chang, K.C.; Ramachandra, M.; Sasikumar, P.G.; Hotchkiss, R.S. Anti-PD-L1 peptide improves survival in sepsis. J. Surg. Res. 2017, 208, 33–39. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.W.; Mittal, R.; Klingensmith, N.J.; Burd, E.M.; Terhorst, C.; Martin, G.S.; Coopersmith, C.M.; Ford, M.L. Cutting Edge: 2B4-Mediated Coinhibition of CD4+ T Cells Underlies Mortality in Experimental Sepsis. J. Immunol. 2017, 199, 1961–1966. [Google Scholar] [CrossRef] [PubMed]
- Boomer, J.S.; Shuherk-Shaffer, J.; Hotchkiss, R.S.; Green, J.M. A prospective analysis of lymphocyte phenotype and function over the course of acute sepsis. Crit. Care 2012, 16, R112. [Google Scholar] [CrossRef] [PubMed]
- Shubin, N.J.; Monaghan, S.F.; Heffernan, D.S.; Chung, C.S.; Ayala, A. B and T lymphocyte attenuator expression on CD4+ T-cells associates with sepsis and subsequent infections in ICU patients. Crit. Care 2013, 17, R276. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Jiang, X.; Chen, G.; Xiao, Y.; Geng, S.; Kang, C.; Zhou, T.; Li, Y.; Guo, X.; Xiao, H.; et al. T cell Ig mucin-3 promotes homeostasis of sepsis by negatively regulating the TLR response. J. Immunol. 2013, 190, 2068–2079. [Google Scholar] [CrossRef] [PubMed]
- Ren, F.; Li, J.; Jiang, X.; Xiao, K.; Zhang, D.; Zhao, Z.; Ai, J.; Hou, C.; Jia, Y.; Han, G.; et al. Plasma soluble Tim-3 emerges as an inhibitor in sepsis: Sepsis contrary to membrane Tim-3 on monocytes. Tissue Antigens 2015, 86, 325–332. [Google Scholar] [CrossRef] [PubMed]
- Lange, A.; Sunden-Cullberg, J.; Magnuson, A.; Hultgren, O. Soluble B and T lymphocyte attenuator correlates to disease severity in sepsis and high levels are associated with an increased risk of mortality. PLoS ONE 2017, 12, e0169176. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Han, X. Anti-PD-1/PD-L1 therapy of human cancer: Past, present, and future. J. Clin. Investig. 2015, 125, 3384–3391. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Jiang, X.; Kang, C.; Xiao, Y.; Hou, C.; Yu, J.; Wang, R.; Xiao, H.; Zhou, T.; Wen, Z.; et al. Blockade of the T cell immunoglobulin and mucin domain protein 3 pathway exacerbates sepsis-induced immune deviation and immunosuppression. Clin. Exp. Immunol. 2014, 178, 279–291. [Google Scholar] [CrossRef] [PubMed]
- Shindo, Y.; Unsinger, J.; Burnham, C.A.; Green, J.M.; Hotchkiss, R.S. Interleukin-7 and anti-programmed cell death 1 antibody have differing effects to reverse sepsis-induced immunosuppression. Shock 2015, 43, 334–343. [Google Scholar] [CrossRef] [PubMed]
- Walker, L.S.; Sansom, D.M. The emerging role of CTLA4 as a cell-extrinsic regulator of T cell responses. Nat. Rev. Immunol. 2011, 11, 852–863. [Google Scholar] [CrossRef] [PubMed]
- Krummel, M.F.; Allison, J.P. CD28 and CTLA-4 have opposing effects on the response of T cells to stimulation. J. Exp. Med. 1995, 182, 459–465. [Google Scholar] [CrossRef] [PubMed]
- Walunas, T.L.; Lenschow, D.J.; Bakker, C.Y.; Linsley, P.S.; Freeman, G.J.; Green, J.M.; Thompson, C.B.; Bluestone, J.A. CTLA-4 can function as a negative regulator of T cell activation. Immunity 1994, 1, 405–413. [Google Scholar] [CrossRef]
- Postow, M.A.; Callahan, M.K.; Wolchok, J.D. Immune checkpoint blockade in cancer therapy. J. Clin. Oncol. 2015, 33, 1974–1982. [Google Scholar] [CrossRef] [PubMed]
- Murphy, T.L.; Murphy, K.M. Slow down and survive: Enigmatic immunoregulation by BTLA and HVEM. Ann. Rev. Immunol. 2010, 28, 389–411. [Google Scholar] [CrossRef] [PubMed]
- Lepenies, B.; Pfeffer, K.; Hurchla, M.A.; Murphy, T.L.; Murphy, K.M.; Oetzel, J.; Fleischer, B.; Jacobs, T. Ligation of B and T lymphocyte attenuator prevents the genesis of experimental cerebral malaria. J. Immunol. 2007, 179, 4093–4100. [Google Scholar] [CrossRef] [PubMed]
- Crawford, A.; Wherry, E.J. Editorial: Therapeutic potential of targeting BTLA. J. Leukoc. Biol. 2009, 86, 5–8. [Google Scholar] [CrossRef] [PubMed]
- Zhu, C.; Anderson, A.C.; Schubart, A.; Xiong, H.; Imitola, J.; Khoury, S.J.; Zheng, X.X.; Strom, T.B.; Kuchroo, V.K. The Tim-3 ligand galectin-9 negatively regulates T helper type 1 immunity. Nat. Immunol. 2005, 6, 1245–1252. [Google Scholar] [CrossRef] [PubMed]
- Leentjens, J.; Kox, M.; van der Hoeven, J.G.; Netea, M.G.; Pickkers, P. Immunotherapy for the adjunctive treatment of sepsis: From immunosuppression to immunostimulation. Time for a paradigm change? Am. J. Respir. Crit. Care Med. 2013, 187, 1287–1293. [Google Scholar] [CrossRef] [PubMed]
- Seshadri, A.; Brat, G.A.; Yorkgitis, B.K.; Keegan, J.; Dolan, J.; Salim, A.; Askari, R.; Lederer, J.A. Phenotyping the Immune Response to Trauma: A Multiparametric Systems Immunology Approach. Crit. Care Med. 2017, 45, 1523–1530. [Google Scholar] [CrossRef] [PubMed]
- Opal, S.M.; Fisher, C.J., Jr.; Dhainaut, J.F.; Vincent, J.L.; Brase, R.; Lowry, S.F.; Sadoff, J.C.; Slotman, G.J.; Levy, H.; Balk, R.A.; et al. Confirmatory interleukin-1 receptor antagonist trial in severe sepsis: A phase III, randomized, double-blind, placebo-controlled, multicenter trial. The Interleukin-1 Receptor Antagonist Sepsis Investigator Group. Crit. Care Med. 1997, 25, 1115–1124. [Google Scholar] [CrossRef] [PubMed]
- Shakoory, B.; Carcillo, J.A.; Chatham, W.W.; Amdur, R.L.; Zhao, H.; Dinarello, C.A.; Cron, R.Q.; Opal, S.M. Interleukin-1 Receptor Blockade Is Associated With Reduced Mortality in Sepsis Patients With Features of Macrophage Activation Syndrome: Reanalysis of a Prior Phase III Trial. Crit. Care Med. 2016, 44, 275–281. [Google Scholar] [CrossRef] [PubMed]
- Bohannon, J.K.; Luan, L.; Hernandez, A.; Afzal, A.; Guo, Y.; Patil, N.K.; Fensterheim, B.; Sherwood, E.R. Role of G-CSF in monophosphoryl lipid A-mediated augmentation of neutrophil functions after burn injury. J. Leukoc. Biol. 2016, 99, 629–640. [Google Scholar] [CrossRef] [PubMed]
- Inoue, S.; Unsinger, J.; Davis, C.G.; Muenzer, J.T.; Ferguson, T.A.; Chang, K.; Osborne, D.F.; Clark, A.T.; Coopersmith, C.M.; McDunn, J.E.; et al. IL-15 prevents apoptosis, reverses innate and adaptive immune dysfunction, and improves survival in sepsis. J. Immunol. 2010, 184, 1401–1409. [Google Scholar] [CrossRef] [PubMed]
- Unsinger, J.; McGlynn, M.; Kasten, K.R.; Hoekzema, A.S.; Watanabe, E.; Muenzer, J.T.; McDonough, J.S.; Tschoep, J.; Ferguson, T.A.; McDunn, J.E.; et al. IL-7 promotes T cell viability, trafficking, and functionality and improves survival in sepsis. J. Immunol. 2010, 184, 3768–3779. [Google Scholar] [CrossRef] [PubMed]
- Hernandez, A.; Bohannon, J.K.; Luan, L.; Fensterheim, B.A.; Guo, Y.; Patil, N.K.; McAdams, C.; Wang, J.; Sherwood, E.R. The role of MyD88- and TRIF-dependent signaling in monophosphoryl lipid A-induced expansion and recruitment of innate immunocytes. J. Leukoc. Biol. 2016, 100, 1311–1322. [Google Scholar] [CrossRef] [PubMed]
- Boutros, C.; Tarhini, A.; Routier, E.; Lambotte, O.; Ladurie, F.L.; Carbonnel, F.; Izzeddine, H.; Marabelle, A.; Champiat, S.; Berdelou, A.; et al. Safety profiles of anti-CTLA-4 and anti-PD-1 antibodies alone and in combination. Nat. Rev. Clin. Oncol. 2016, 13, 473–486. [Google Scholar] [CrossRef] [PubMed]
- Michot, J.M.; Bigenwald, C.; Champiat, S.; Collins, M.; Carbonnel, F.; Postel-Vinay, S.; Berdelou, A.; Varga, A.; Bahleda, R.; Hollebecque, A.; et al. Immune-related adverse events with immune checkpoint blockade: A comprehensive review. Eur. J. Cancer 2016, 54, 139–148. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, H.; Nose, M.; Hiai, H.; Minato, N.; Honjo, T. Development of lupus-like autoimmune diseases by disruption of the PD-1 gene encoding an ITIM motif-carrying immunoreceptor. Immunity 1999, 11, 141–151. [Google Scholar] [CrossRef]
- Okazaki, T.; Honjo, T. PD-1 and PD-1 ligands: From discovery to clinical application. Int. Immunol. 2007, 19, 813–824. [Google Scholar] [CrossRef] [PubMed]
- Cavaillon, J.M.; Eisen, D.; Annane, D. Is boosting the immune system in sepsis appropriate? Crit. Care 2014, 18, 216. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Inhibitory immune checkpoints on immune cells. Interaction among immune cell checkpoint receptors on T cells and antigen presenting cells (APCs) or target cells such as peripheral tissue epithelial cells inhibit leukocyte functions and may contribute to immune dysfunction. PD-1 = Programmed death-1; PD-L1 = Programmed death ligand-1; CTLA-4 = Cytotoxic T lymphocyte antigen-4; BTLA = B and T lymphocyte attenuator; HVEM = Herpes virus entry mediator; TIM-3 = T cell membrane protein-3; LAG-3 = Lymphocyte activation-gene-3; CEACAM = carcinoembryonic antigen-related cell adhesion molecule; MHC II = Major histocompatibility complex II.
Figure 1.
Inhibitory immune checkpoints on immune cells. Interaction among immune cell checkpoint receptors on T cells and antigen presenting cells (APCs) or target cells such as peripheral tissue epithelial cells inhibit leukocyte functions and may contribute to immune dysfunction. PD-1 = Programmed death-1; PD-L1 = Programmed death ligand-1; CTLA-4 = Cytotoxic T lymphocyte antigen-4; BTLA = B and T lymphocyte attenuator; HVEM = Herpes virus entry mediator; TIM-3 = T cell membrane protein-3; LAG-3 = Lymphocyte activation-gene-3; CEACAM = carcinoembryonic antigen-related cell adhesion molecule; MHC II = Major histocompatibility complex II.

Figure 2.
Graphical representation of PD-1–PD-L1 interaction leading to immune cell dysfunction and immunosuppression. PD-1–PD-L1 interaction leads to impaired T cell function (exhaustion) and antigen presenting cell (myeloid) dysfunction. Antibodies targeting each of these inhibitory molecules reverse sepsis induced immunosuppression and improve host resistance to infection. (M = antigen presenting or myeloid cell; PD-1 = Programmed cell death-1; PD-L1 = Programmed cell death ligand-1; IFN-γ = interferon-gamma; IL-2 = interleukin-2; IL-6 = inerleukin-6, upward arrows indicates an increase and downward arrows indicates a decrease).
Figure 2.
Graphical representation of PD-1–PD-L1 interaction leading to immune cell dysfunction and immunosuppression. PD-1–PD-L1 interaction leads to impaired T cell function (exhaustion) and antigen presenting cell (myeloid) dysfunction. Antibodies targeting each of these inhibitory molecules reverse sepsis induced immunosuppression and improve host resistance to infection. (M = antigen presenting or myeloid cell; PD-1 = Programmed cell death-1; PD-L1 = Programmed cell death ligand-1; IFN-γ = interferon-gamma; IL-2 = interleukin-2; IL-6 = inerleukin-6, upward arrows indicates an increase and downward arrows indicates a decrease).

{kind=link}
{kind=link}
Table 1.
Summary of pre-clinical studies showing alterations in expression of immune various checkpoints during sepsis.
Table 1.
Summary of pre-clinical studies showing alterations in expression of immune various checkpoints during sepsis.
| Reference | Sepsis Model | Alterations in Expression of Immune Checkpoints | Other Major Findings |
|---|---|---|---|
| Huang et al., 2009 [37] | Cecal Ligation and Puncture (CLP) | Increased PD-1 on peritoneal macrophages |
|
| Brahmamdam et al., 2010 [35] | CLP | Increased PD-1 on CD4+ and CD8+ splenic T cells |
|
| Zhang et al., 2010 [36] | CLP |
|
|
| Inoue et al., 2011 [48] | CLP | Increased CTLA-4 on splenic CD4+ and CD8+ T cells | Increased splenic T cell apoptosis and decreased survival rate |
| Shubin et al., 2012 [49] | CLP | Increased BTLA and HVEM on macrophages, monocytes, dendritic cells and neutrophils in peritoneum |
|
| Zhu et al., 2013 [50] | CLP | Increased PD-L1 in liver tissue |
|
| Chang et al., 2013 [51] |
| Increased PD-1 on splenic CD4+ and CD8+ T cells |
|
| Hutchins et al., 2013 [52] | CLP |
|
|
| Huang et al., 2014 [38] | CLP | Increased PD-L1 on macrophages, monocytes, T and Natural Killer T (NKT) cells and neutrophils |
|
| Wang et al., 2016 [40] | CLP | Increased PD-1 on liver Kupffer cells |
|
| Patil et al., 2016 [41] | Burn wound sepsis (Pseudomonas aeruginosa) |
|
|
| Wu et al., 2016 [53] | CLP | Increased PD-L1 intestinal epithelial cells |
|
| Cheng et al., 2016 [54] | Two hit model (hemorrhage + CLP) | Increased BTLA on peritoneal macrophages and dendritic cells; and in tissues—ileum, kidney, lung, liver and spleen |
|
| Shindo et al., 2017 [55] | Two hit model (CLP + fungal sepsis) | Increased PD-1 on splenic CD4+, NKT and NK cells Increased PD-L1 on CD4+, NKT and Natural Killer (NK) cells | Significantly decreased survival rate |
| Chen et al., 2017 [56] | CLP | Increased 2B4 on splenic CD4+ and CD8+ Increased PD-1 and BTLA on splenic CD4+ and CD8+ |
|
Table 2.
Summary of clinical studies showing alterations in expression of various immune checkpoints during sepsis.
Table 2.
Summary of clinical studies showing alterations in expression of various immune checkpoints during sepsis.
| Reference | Sample Size | Alterations in Expression of Immune Checkpoints | Any Other Major Clinical Findings |
|---|---|---|---|
| Guignant et al., 2011 [43] | 64 Patients, prospective study | Increased PD-1 and PD-L1 on CD4+ T cells, and higher PD-L1/PD-L2 on monocytes |
|
| Zhang et al., 2011 [44] | 19 Patients, prospective study | Increased PD-1 and CD4+ and CD8+ T cells, and higher PD-L1 on monocytes |
|
| Boomer et al., 2011 [23] | Postmortem study, 40 patients |
|
|
| Boomer et al., 2012 [57] | 24 Patients, prospective study |
| Impaired splenic T cell function (as measured by decreased IFN-γ production upon ex vivo stimulation of cells) |
| Shubin et al., 2013 [58] | 24 Patients, prospective study | Increased BTLA on circulating CD4+ T cells | Increased BTLA correlated with increased mortality |
| Yang et al., 2013 [59] | 26 Patients (12-sepsis,14-severe sepsis) | Increased TIM-3 mRNA in PBMC’s in sepsis patients as compared to severe sepsis patients | None |
| Chang et al., 2014 [45] | 43 Patients, Prospective study |
|
|
| Ren et al., 2015 [60] | Prospective study; 40-sepsis and42-severe sepsis patients18-septic shock pateints |
| Decreased soluble TIM-3 levels correlated with increased mortality |
| Patera et al., 2016 [24] | 17 Pateints, prospective study |
|
|
| Spec et al., 2016 [46] | 27 Candida fungal sepsis pateints, prospective study |
|
|
| Shao et al., 2016 [47] | 59 Patients, prospective study |
|
|
| Wu et al., 2016 [53] | Retrospective analysis | Increased PD-L1 on epithelial cells of colon | None |
| Lange et al., 2017 [61] | 101 Patients, prospective study | Increased plasma soluble BTLA levels (sBTLA) |
|
| Chen et al., 2017 [56] | 14 Patients, prospective study | Increased 2B4, PD-1 and CTLA-4 on CD4+ T cells | Decreased co-stimulatory ICOS and CD28 on CD4+ T cells |
Table 3.
Studies targeting immune checkpoints during sepsis with monoclonal antibodies (pre-clinical studies).
Table 3.
Studies targeting immune checkpoints during sepsis with monoclonal antibodies (pre-clinical studies).
| Reference | Sepsis Model | Antibody | Observed Therapeutic Effects |
|---|---|---|---|
| Zhang et al., 2010 [36] | CLP | anti-PD-L1, 200 µg, i.p. route, 24 h before and 2 h after CLP |
|
| Brahmamdam et al., 2010 [35] | CLP | anti-PD-1, 200 µg, i.v. route, 24 h after CLP |
|
| Inoue et al., 2011 [48] | CLP | anti-CTLA-4, 50 µg, i.p. route, 6 and 24 h after CLP; and 33 µg, i.p. after fungal sepsis |
|
| Zhu et al., 2013 [50] | CLP | anti-PD-L1, 50 µg, i.p. route, 1 h after CLP | Attenuation of liver injury (improved histology, and decreased ALT, AST) |
| Chang et al., 2013 [51] | Candida fungal sepsis, and two hit model (CLP + fungal sepsis) |
|
|
| Yang et al., 2013 [59] | CLP | anti-TIM-3, 200 µg, i.p. route, 1 day before and 1, 3, 5 and 7 days after CLP |
|
| Zhao et al., 2014 [63] | CLP | sTIM3-Ig to block TIM-3 signaling, 200 µg, i.p. route, 12 h before, and 48 and 96 h after CLP |
|
| Shindo et al., 2015 [64] | Two hit model (CLP + fungal sepsis) | anti-PD-1, 200 µg, i.p. route, on day 4 and 8 post CLP |
|
| Cheng et al., 2016 [54] | Two hit model (hemorrhage + CLP) | anti-BTLA-4, 25 µg/g administered just after CLP |
|
| Chen et al., 2017 [56] | CLP | anti-2B4, 250 µg, i.p. route, on days—0, 2, 4 and 6, after CLP |
|
| Shindo et al., 2017 [55] | Two hit model (CLP + fungal sepsis) | anti-PD-L1 peptide (compound 8), 3 mg/kg, s.c route, three times daily from days 5 to 13 after CLP | Significantly improved survival |
Table 4.
Pre-clinical studies employing immune checkpoint knockout/overexpression in mice.
| Reference | Sepsis Model | Animal Model | Observed Therapeutic Effects |
|---|---|---|---|
| Huang et al., 2009 [37] | CLP | PD-1 knockout |
|
| Shubin et al., 2012 [49] | CLP | BTLA knockout |
|
| Hutchins et al., 2013 [52] | CLP | PD-L1 knockout |
|
| Huang et al., 2014 [38] | CLP | PD-1 knockout |
|
| Zhao et al., 2014 [63] | CLP | TIM-3 overexpression |
|
| Wang et al., 2016 [40] | CLP | PD-1 knockout |
|
| Young et al., 2016 [42] | Neonatal sepsis model using cecal slurry | PD-1 knockout |
|
| Wu et al., 2016 [53] | CLP | PD-L1 knockout |
|
Table 5.
Evaluating the use of immune checkpoint inhibitors (Clinical studies—ex vivo treatment of isolated immune cells from septic patient’s blood with monoclonal antibodies).
Table 5.
Evaluating the use of immune checkpoint inhibitors (Clinical studies—ex vivo treatment of isolated immune cells from septic patient’s blood with monoclonal antibodies).
| Reference | Patient Population | Antibody Used | Observed Therapeutic Effects |
|---|---|---|---|
| Zhang et al., 2011 [44] | Prospective clinical study with 19 septic patients | anti-PD-L1 antibody |
|
| Chang et al., 2014 [45] | Prospective study with 43 septic patients | anti-PD-L1 antibody and anti-PD-1 antibody |
|
| Patera et al., 2013 [24] | Prospective study with 17 septic patients | anti-PD-L1 antibody and anti-PD-1 antibody |
|
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Patil, N.K.; Guo, Y.; Luan, L.; Sherwood, E.R. Targeting Immune Cell Checkpoints during Sepsis. Int. J. Mol. Sci. 2017, 18, 2413. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms18112413
AMA Style
Patil NK, Guo Y, Luan L, Sherwood ER. Targeting Immune Cell Checkpoints during Sepsis. International Journal of Molecular Sciences. 2017; 18(11):2413. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms18112413
Chicago/Turabian StylePatil, Naeem K., Yin Guo, Liming Luan, and Edward R. Sherwood. 2017. "Targeting Immune Cell Checkpoints during Sepsis" International Journal of Molecular Sciences 18, no. 11: 2413. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms18112413
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.