Infectious Agents in Atherosclerotic Cardiovascular Diseases through Oxidative Stress

 ,
,
Abstract
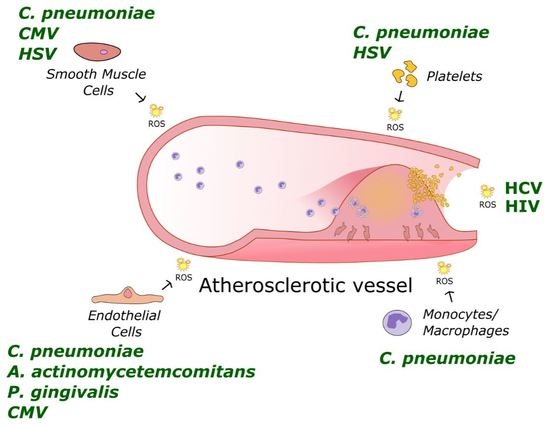
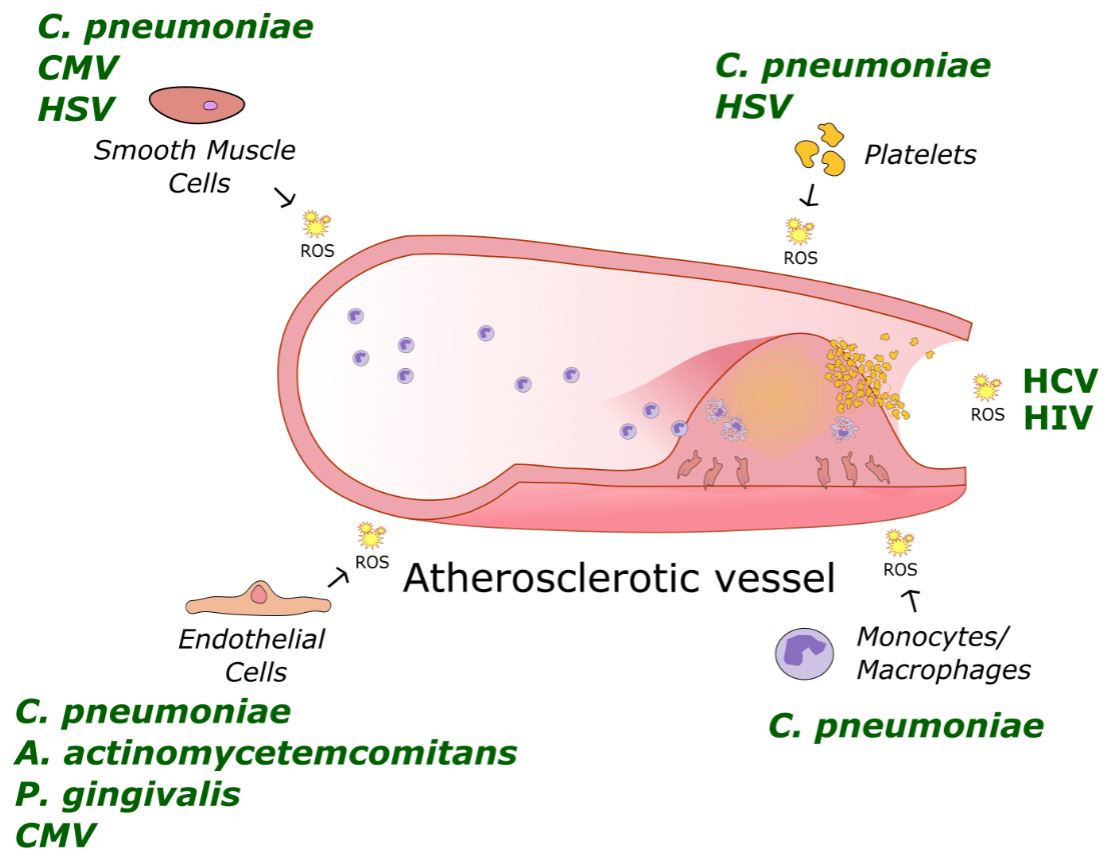
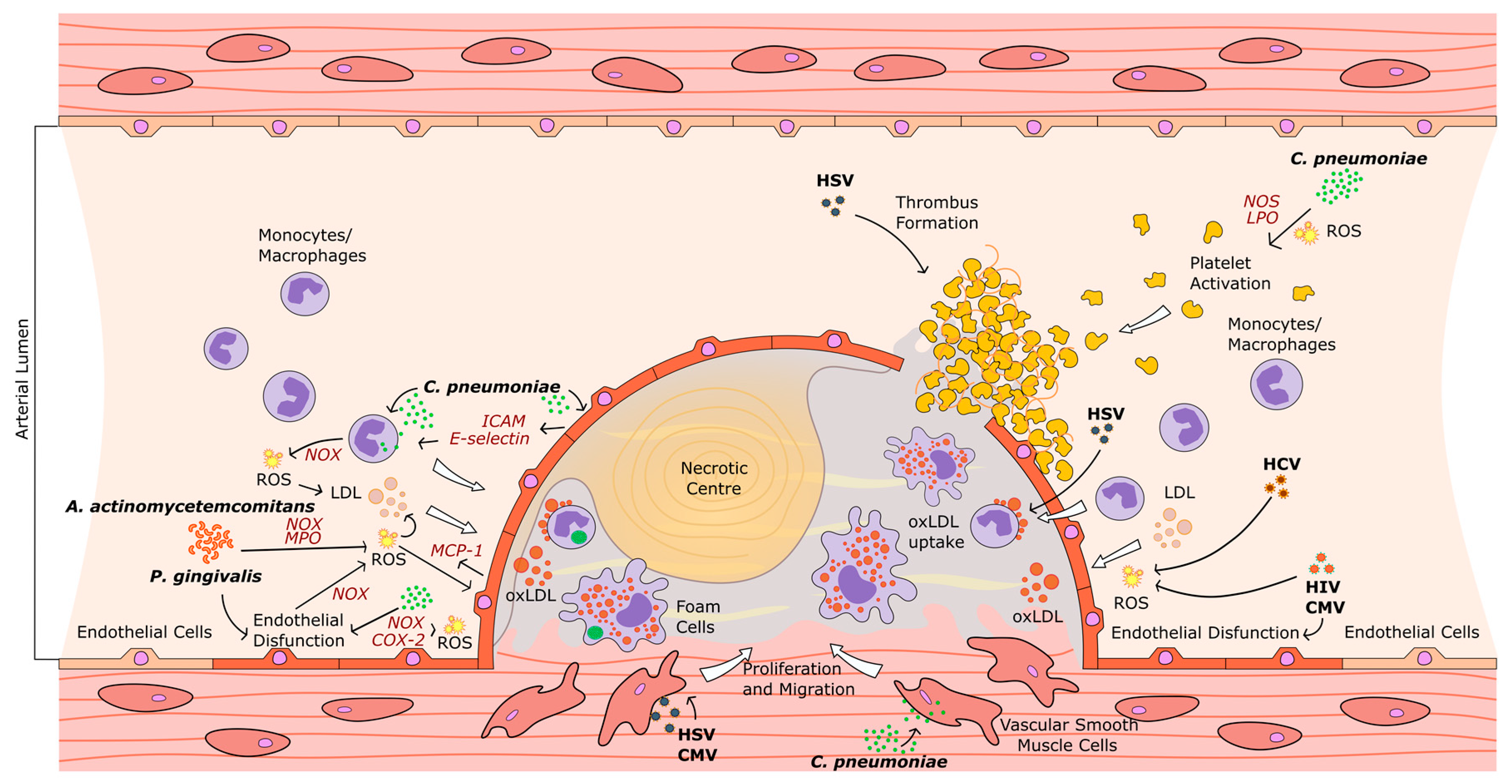
:
{kind=link}
{kind=link}
1. Introduction
2. Bacterial Infectious Agents
2.1. Periodontal Pathogens
2.2. Chlamydia pneumoniae
3. Viral Infectious Agents
3.1. HIV
3.2. HCV
3.3. Herpesviruses
3.3.1. Herpes Simplex Virus
3.3.2. Cytomegalovirus
4. Antioxidant Strategies in Infectious Agent-Mediated Atherosclerotic Cardiovascular Diseases
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- World Health Organization. Global Status Report on Noncommunicable Diseases 2010; World Health Organization: Geneva, Switzerland, 2011. [Google Scholar]
- Mangge, H.; Becker, K.; Fuchs, D.; Gostner, J.M. Antioxidants, inflammation and cardiovascular disease. World J. Cardiol. 2014, 6, 462–477. [Google Scholar] [CrossRef] [PubMed]
- He, F.; Zuo, L. Redox Roles of Reactive Oxygen Species in Cardiovascular Diseases. Int. J. Mol. Sci. 2015, 16, 27770–27780. [Google Scholar] [CrossRef] [PubMed]
- Kattoor, A.J.; Pothineni, N.V.K.; Palagiri, D.; Mehta, J.L. Oxidative Stress in Atherosclerosis. Curr. Atheroscler. Rep. 2017, 19, 42. [Google Scholar] [CrossRef] [PubMed]
- Förstermann, U.; Xia, N.; Li, H. Roles of Vascular Oxidative Stress and Nitric Oxide in the Pathogenesis of Atherosclerosis. Circ. Res. 2017, 120, 713–735. [Google Scholar] [CrossRef] [PubMed]
- Pant, S.; Deshmukh, A.; Gurumurthy, G.S.; Pothineni, N.V.; Watts, T.E.; Romeo, F.; Mehta, J.L. Inflammation and atherosclerosis—Revisited. J. Cardiovasc. Pharmacol. Ther. 2014, 19, 170–178. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.Y.; Li, C.J.; Hou, M.F.; Chu, P.Y. New insights into the role of inflammation in the pathogenesis of atherosclerosis. Int. J. Mol. Sci. 2017, 18, 2034. [Google Scholar] [CrossRef] [PubMed]
- Hulsmans, M.; Holvoet, P. The vicious circle between oxidative stress and inflammation in atherosclerosis. J. Cell. Mol. Med. 2010, 14, 70–78. [Google Scholar] [CrossRef] [PubMed]
- Lozhkin, A.; Vendrov, A.E.; Pan, H.; Wickline, S.A.; Madamanchi, N.R.; Runge, M.S. NADPH oxidase 4 regulates vascular inflammation in aging and atherosclerosis. J. Mol. Cell. Cardiol. 2017, 102, 10–21. [Google Scholar] [CrossRef] [PubMed]
- Korenaga, M.; Wang, T.; Li, Y.; Showalter, L.A.; Chan, T.; Sun, J.; Weinman, S.A. Hepatitis C virus core protein inhibits mitochondrial electron transport and increases reactive oxygen species (ROS) production. J. Biol. Chem. 2005, 280, 37481–37488. [Google Scholar] [CrossRef] [PubMed]
- Rosenfeld, M.E.; Campbell, L.A. Pathogens and atherosclerosis: Update on the potential contribution of multiple infectious organisms to the pathogenesis of atherosclerosis. Thromb. Haemost. 2011, 106, 858–867. [Google Scholar] [CrossRef] [PubMed]
- Porter, K.M.; Sutliff, R.L. HIV-1, reactive oxygen species, and vascular complications. Free Radic. Biol. Med. 2012, 53, 143–159. [Google Scholar] [CrossRef] [PubMed]
- Di Pietro, M.; Filardo, S.; De Santis, F.; Sessa, R. Chlamydia pneumoniae infection in atherosclerotic lesion development through oxidative stress: A brief overview. Int. J. Mol. Sci. 2013, 14, 15105–15120. [Google Scholar] [CrossRef] [PubMed]
- Kurita-Ochiai, T.; Yamamoto, M. Periodontal pathogens and atherosclerosis: Implications of inflammation and oxidative modification of LDL. Biomed Res. Int. 2014, 2014, 595981. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.L.; Liu, C.E.; Cho, W.L.; Kuo, C.L.; Cheng, W.L.; Huang, C.S.; Liu, C.S. Presence of cytomegalovirus DNA in leucocytes is associated with increased oxidative stress and subclinical atherosclerosis in healthy adults. Biomarkers 2014, 19, 109–113. [Google Scholar] [CrossRef] [PubMed]
- Sessa, R.; Di Pietro, M.; Filardo, S.; Turriziani, O. Infectious burden and atherosclerosis: A clinical issue. World J. Clin. Cases 2014, 16, 240–249. [Google Scholar] [CrossRef] [PubMed]
- Filardo, S.; Di Pietro, M.; Farcomeni, A.; Schiavoni, G.; Sessa, R. Chlamydia pneumoniae-mediated inflammation in atherosclerosis: A meta-analysis. Mediat. Inflamm. 2015, 2015, 378658. [Google Scholar] [CrossRef] [PubMed]
- Bengtsson, T.; Karlsson, H.; Gunnarsson, P.; Skoglund, C.; Elison, C.; Leanderson, P.; Lindahl, M. The periodontal pathogen Porphyromonas gingivalis cleaves apoB-100 and increases the expression of apoM in LDL in whole blood leading to cell proliferation. J. Intern. Med. 2008, 263, 558–571. [Google Scholar] [CrossRef] [PubMed]
- Börgeson, E.; Lönn, J.; Bergström, I.; Brodin, V.P.; Ramström, S.; Nayeri, F.; Särndahl, E.; Bengtsson, T. Lipoxin A4 inhibits Porphyromonas gingivalis-induced aggregation and reactive oxygen species production by modulating neutrophil-platelet interaction and CD11b expression. Infect. Immun. 2011, 79, 1489–1497. [Google Scholar] [CrossRef] [PubMed]
- Li, X.Y.; Wang, C.; Xiang, X.R.; Chen, F.C.; Yang, C.M.; Wu, J. Porphyromonas gingivalis lipopolysaccharide increases lipid accumulation by affecting CD36 and ATP-binding cassette transporter A1 in macrophages. Oncol. Rep. 2013, 30, 1329–1336. [Google Scholar] [CrossRef] [PubMed]
- Pollreisz, A.; Huang, Y.; Roth, G.A.; Cheng, B.; Kebschull, M.; Papapanou, P.N.; Schmidt, A.M.; Lalla, E. Enhanced monocyte migration and pro-inflammatory cytokine production by Porphyromonas gingivalis infection. J. Periodontal Res. 2010, 45, 239–245. [Google Scholar] [CrossRef] [PubMed]
- Bugueno, I.M.; Khelif, Y.; Seelam, N.; Morand, D.N.; Tenenbaum, H.; Davideau, J.L.; Huck, O. Porphyromonas gingivalis differentially modulates cell death profile in ox-LDL and TNF-α pre-treated endothelial cells. PLoS ONE 2016, 11, e0154590. [Google Scholar] [CrossRef] [PubMed]
- Shiheido, Y.; Maejima, Y.; Suzuki, J.I.; Aoyama, N.; Kaneko, M.; Watanabe, R.; Sakamaki, Y.; Wakayama, K.; Ikeda, Y.; Akazawa, H.; et al. Porphyromonas gingivalis, a periodontal pathogen, enhances myocardial vulnerability, thereby promoting post-infarct cardiac rupture. J. Mol. Cell. Cardiol. 2016, 99, 123–137. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, Y.; Kurita-Ochiai, T.; Kobayashi, R.; Suzuki, T.; Ando, T. Activation of the NLRP3 inflammasome in Porphyromonas gingivalis-accelerated atherosclerosis. Pathog. Dis. 2015, 73, ftv011. [Google Scholar] [CrossRef] [PubMed]
- Sessa, R.; Nicoletti, M.; Di Pietro, M.; Schiavoni, G.; Santino, I.; Zagaglia, C.; Del Piano, M.; Cipriani, P. Chlamydia pneumoniae and atherosclerosis: Current state and future prospectives. Int. J. Immunopathol. Pharmacol. 2009, 22, 9–14. [Google Scholar] [CrossRef] [PubMed]
- Schiavoni, G.; Di Pietro, M.; Ronco, C.; De Cal, M.; Cazzavillan, S.; Rassu, M.; Nicoletti, M.; Del Piano, M.; Sessa, R. Chlamydia pneumoniae infection as a risk factor for accelerated atherosclerosis in hemodialysis patients. J. Biol. Regul. Homeost. Agents 2010, 24, 367–375. [Google Scholar] [PubMed]
- Campbell, L.A.; Rosenfeld, M.E. Infection and Atherosclerosis Development. Arch. Med. Res. 2015, 46, 339–350. [Google Scholar] [CrossRef] [PubMed]
- Sessa, R.; Di Pietro, M.; Schiavoni, G.; Santino, I.; Benedetti-Valentini, F.; Perna, R.; Romano, S.; del Piano, M. Chlamydia pneumoniae DNA in patients with symptomatic carotid atherosclerotic disease. J. Vasc. Surg. 2003, 37, 1027–1031. [Google Scholar] [CrossRef] [PubMed]
- Sessa, R.; Di Pietro, M.; Schiavoni, G.; Petrucca, A.; Cipriani, P.; Zagaglia, C.; Nicoletti, M.; Santino, I.; del Piano, M. Measurement of Chlamydia pneumoniae bacterial load in peripheral blood mononuclear cells may be helpful to assess the state of chlamydial infection in patients with carotid atherosclerotic disease. Atherosclerosis 2007, 195, e224–e230. [Google Scholar] [CrossRef] [PubMed]
- Di Pietro, M.; Schiavoni, G.; Sessa, V.; Pallotta, F.; Costanzo, G.; Sessa, R. Chlamydia pneumoniae and osteoporosis-associated bone loss: A new risk factor? Osteoporos. Int. 2013, 24, 1677–1682. [Google Scholar] [CrossRef] [PubMed]
- Di Pietro, M.; Filardo, S.; De Santis, F.; Mastromarino, P.; Sessa, R. Chlamydia pneumoniae and oxidative stress in cardiovascular disease: State of the art and prevention strategies. Int. J. Mol. Sci. 2014, 16, 724–735. [Google Scholar] [CrossRef] [PubMed]
- Kreutmayer, S.; Csordas, A.; Kern, J.; Maass, V.; Almanzar, G.; Offterdinger, M.; Ollinger, R.; Maass, M.; Wick, G. Chlamydia pneumoniae infection acts as an endothelial stressor with the potential to initiate the earliest heat shock protein 60-dependent inflammatory stage of atherosclerosis. Cell Stress Chaperones 2013, 18, 259–268. [Google Scholar] [CrossRef] [PubMed]
- Rivera, J.; Walduck, A.K.; Strugnell, R.A.; Sobey, C.G.; Drummond, G.R. Chlamydia pneumoniae induces a pro-inflammatory phenotype in murine vascular smooth muscle cells independently of elevating reactive oxygen species. Clin. Exp. Pharmacol. Physiol. 2012, 39, 218–226. [Google Scholar] [CrossRef] [PubMed]
- Azenabor, A.A.; Yang, S.; Job, G.; Adedokun, O.O. Elicitation of reactive oxygen species in Chlamydia pneumoniae-stimulated macrophages: A Ca2+-dependent process involving simultaneous activation of NADPH oxidase and cytochrome oxidase genes. Med. Microbiol. Immunol. 2005, 194, 91–103. [Google Scholar] [CrossRef] [PubMed]
- Kälvegren, H.; Bylin, H.; Leanderson, P.; Richter, A.; Grenegård, M.; Bengtsson, T. Chlamydia pneumoniae induces nitric oxide synthase and lipoxygenase-dependent production of reactive oxygen species in platelets. Effects on oxidation of low density lipoproteins. Thromb. Haemost. 2005, 94, 327–335. [Google Scholar] [PubMed]
- Filardo, S.; Di Pietro, M.; Schiavoni, G.; Minniti, G.; Ortolani, E.; Romano, S.; Sessa, R. Chlamydia pneumoniae clinical isolate from gingival crevicular fluid: A potential atherogenic strain. Front. Cell. Infect. Microbiol. 2015, 5, 86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Evani, S.J.; Dallo, S.F.; Ramasubramanian, A.K. Biophysical and Biochemical Outcomes of Chlamydia pneumoniae Infection Promotes Pro-atherogenic Matrix Microenvironment. Front. Microbiol. 2016, 7, 1287. [Google Scholar] [CrossRef] [PubMed]
- Campbell, L.A.; Lee, A.W.; Rosenfeld, M.E.; Kuo, C.C. Chlamydia pneumoniae induces expression of pro-atherogenic factors through activation of the lectin-like oxidized LDL receptor-1. Pathog. Dis. 2013, 6, 1–6. [Google Scholar]
- Vielma, S.A.; Mironova, M.; Ku, J.R.; Lopes-Virella, M.F. Oxidized LDL further enhances expression of adhesion molecules in Chlamydophila pneumoniae-infected endothelial cells. J. Lipid Res. 2004, 5, 873–880. [Google Scholar] [CrossRef] [PubMed]
- Yaraei, K.; Campbell, L.A.; Zhu, X.; Liles, W.C.; Kuo, C.C.; Rosenfeld, M.E. Chlamydia pneumoniae augments the oxidized low-density lipoprotein-induced death of mouse macrophages by a caspase-independent pathway. Infect. Immun. 2005, 73, 4315–4322. [Google Scholar] [CrossRef] [PubMed]
- Nazzal, D.; Cantero, A.V.; Therville, N.; Segui, B.; Negre-Salvayre, A.; Thomsen, M.; Benoist, H. Chlamydia pneumoniae alters mildly oxidized low-density lipoprotein-induced cell death in human endothelial cells, leading to necrosis rather than apoptosis. J. Infect. Dis. 2006, 193, 136–145. [Google Scholar] [CrossRef] [PubMed]
- Kälvegren, H.; Andersson, J.; Grenegård, M.; Bengtsson, T. Platelet activation triggered by Chlamydia pneumoniae is antagonized by 12-lipoxygenase inhibitors but not cyclooxygenase inhibitors. Eur. J. Pharmacol. 2007, 566, 20–27. [Google Scholar] [CrossRef] [PubMed]
- Barbaro, G. Cardiovascular Manifestations of HIV Infection. Circulation 2002, 106, 1420–1425. [Google Scholar] [CrossRef] [PubMed]
- Lerman, A.; Burnett, J.C., Jr. Intact and altered endothelium in regulation of vasomotion. Circulation 1992, 86, 12–19. [Google Scholar]
- Lafon, M.E.; Gendrault, J.L.; Royer, C.; Jaeck, D.; Kirn, A.; Steffan, A.M. Human endothelial cells isolated from the hepatic sinusoids and the umbilical vein display a different permissiveness for HIV1. Res. Virol. 1993, 144, 99–104. [Google Scholar] [CrossRef]
- Funderburg, N.T.; Mayne, E.; Sieg, S.F.; Asaad, R.; Jiang, W.; Kalinowska, M.; Luciano, A.A.; Stevens, W.; Rodriguez, B.; Brenchley, J.M.; et al. Increased tissue factor expression on circulating monocytes in chronic HIV infection: Relationship to in vivo coagulation and immune activation. Blood 2010, 115, 161–167. [Google Scholar] [CrossRef] [PubMed]
- Mazzuca, P.; Caruso, A.; Caccuri, F. HIV-1 infection, microenvironment and endothelial cell dysfunction. New Microbiol. 2016, 39, 163–173. [Google Scholar] [PubMed]
- Gibellini, D.; Borderi, M.; Clò, A.; Morini, S.; Miserocchi, A.; Bon, I.; Ponti, C.; Re, M.C. HIV-related mechanisms in atherosclerosis and cardiovascular diseases. J. Cardiovasc. Med. 2013, 14, 780–790. [Google Scholar] [CrossRef] [PubMed]
- Toborek, M.L.Y.; Pu, H.; Malecki, A.; Flora, G.; Garrido, R.; Hennig, B.; Bauer, H.C.; Nath, A. HIV-Tat protein induced oxidative and inflammatory pathways in brain endothelium. J. Neurochem. 2003, 84, 169–179. [Google Scholar] [CrossRef] [PubMed]
- Gu, Y.; Wu, R.F.; Xu, Y.C.; Flores, S.C.; Terada, L.S. HIV Tat Activates c-Jun Amino-terminal Kinase through an Oxidant-Dependent Mechanism. Virology 2001, 286, 62–71. [Google Scholar] [CrossRef] [PubMed]
- Price, T.O.E.N.; Nakaoke, R.; Banks, W.A. HIV-1 viral proteins gp120 and Tat induce oxidative stress in brain endothelial cells. Brain Res. 2005, 1045, 57–63. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, A.; Zhang, X.; Manda, K.R.; Banks, W.A.; Ercal, N. HIV proteins (gp120 and Tat) and methamphetamine in oxidative stress-induced damage in the brain: Potential role of the thiol antioxidant N-acetylcysteine amide. Free Radic. Biol. Med. 2010, 8, 1388–1398. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.; Chi, D.S.; Li, C.; Hall, H.K.; Milhorn, D.M.; Krishnaswamy, G. HIV-1 Tat protein-induced VCAM-1 expression in human pulmonary artery endothelial cells and its signaling. Am. J. Physiol. Lung Cell. Mol. Physiol. 2000, 289, L252–L260. [Google Scholar] [CrossRef] [PubMed]
- Greenwood, A.J.; Hughes, J.; Wallace, G.; Seed, P.; Stanford, M.R.; Graham, E.M. Soluble intercellular adhesion molecule-1 (sICAM-1) and vascular cell adhesion molecule-1 (sVCAM-1) in patients with HIV/AIDS does not appear to correlate with cytomegalovirus retinitis. Int. J. STD AIDS 1998, 9, 713–714. [Google Scholar] [PubMed]
- Gibellini, D.; Borderi, M.; Clò, A.; Morini, S.; Miserocchi, A.; Bon, I.; Re, M.C. Antiretroviral molecules and cardiovascular diseases. New Microbiol. 2012, 35, 359–375. [Google Scholar] [PubMed]
- Adinolfi, L.E.; Restivo, L.; Zampino, R.; Guerrera, B.; Lonardo, A.; Ruggiero, L.; Riello, F.; Loria, P.; Florio, A. Chronic HCV infection is a risk of atherosclerosis. Role of HCV and HCV-related steatosis. Atherosclerosis 2012, 221, 496–502. [Google Scholar] [CrossRef] [PubMed]
- Petta, S.; Maida, M.; Macaluso, F.S.; Barbara, M.; Licata, A.; Craxì, A.; Cammà, C. Hepatitis C Virus Infection Is Associated with Increased Cardiovascular Mortality: A Meta-Analysis of Observational Studies. Gastroenterology 2016, 150, 145–155. [Google Scholar] [CrossRef] [PubMed]
- Boddi, M.; Abbate, R.; Chellini, B.; Giusti, B.; Giannini, C.; Pratesi, G.; Rossi, L.; Pratesi, C.; Gensini, G.F.; Paperetti, L.; et al. Hepatitis C virus RNA localization in human carotid plaques. J. Clin. Virol. 2010, 47, 72–75. [Google Scholar] [CrossRef] [PubMed]
- Petta, S.; Torres, D.; Fazio, G.; Cammà, C.; Cabibi, D.; Di Marco, V.; Licata, A.; Marchesini, G.; Mazzola, A.; Parrinello, G.; et al. Carotid atherosclerosis and chronic hepatitis C: A prospective study of risk associations. Hepatology 2012, 55, 1317–1323. [Google Scholar] [CrossRef] [PubMed]
- Branche, E.; Conzelmann, S.; Parisot, C.; Bedert, L.; Lévy, P.L.; Bartosch, B.; Clément, S.; Negro, F. Hepatitis C Virus Increases Occludin Expression via the Upregulation of Adipose Differentiation-Related Protein. PLoS ONE 2016, 11, e0146000. [Google Scholar] [CrossRef] [PubMed]
- Ivanov, A.V.; Bartosch, B.; Smirnova, O.A.; Isaguliants, M.G.; Kochetkov, S.N. HCV and oxidative stress in the liver. Viruses 2013, 5, 439–469. [Google Scholar] [CrossRef] [PubMed]
- Yazicioglu, G.; Isitan, F.; Altunbas, H.; Suleymanlar, I.; Ozdogan, M.; Balci, M.K.; Karayalcin, U. Insulin resistance in chronic hepatitis C. Int. J. Clin. Pract. 2004, 58, 1020–1022. [Google Scholar] [CrossRef] [PubMed]
- Koike, K.; Miyoshi, H. Oxidative stress and hepatitis C viral infection. Hepatol. Res. 2006, 34, 65–73. [Google Scholar] [CrossRef] [PubMed]
- Rutkowski, D.T.; Kaufman, R.J. A trip to the ER: Coping with stress. Trends Cell Biol. 2004, 14, 20–28. [Google Scholar] [CrossRef] [PubMed]
- Paracha, U.Z.; Fatima, K.; Alqahtani, M.; Chaudhary, A.; Abuzenadah, A.; Damanhouri, G.; Qadri, I. Oxidative stress and hepatitis C virus. Virol. J. 2013, 10, 251. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Monzon, C.; Majano, P.L.; Zubia, I.; Sanz, P.; Apolinario, A.; Moreno-Otero, R. Intrahepatic accumulation of nitrotyrosine in chronic viral hepatitis is associated with histological severity of liver disease. J. Hepatol. 2000, 32, 331–338. [Google Scholar] [CrossRef]
- Sheikh, M.Y.; Choi, J.; Qadri, I.; Friedman, J.E.; Sanyal, A.J. Hepatitis C virus infection: Molecular pathways to metabolic syndrome. Hepatology 2008, 47, 2127–2133. [Google Scholar] [CrossRef] [PubMed]
- Kohli, A.; Shaffer, A.; Sherman, A.; Kottilil, S. Treatment of hepatitis C: A systematic review. J. Am. Med. Assoc. 2014, 312, 631–640. [Google Scholar] [CrossRef] [PubMed]
- Pavone, P.; Tieghi, T.; d’Ettorre, G.; Lichtner, M.; Marocco, R.; Mezzaroma, I.; Passavanti, G.; Vittozzi, P.; Mastroianni, C.M.; Vullo, V. Rapid decline of fasting glucose in HCV diabetic patients treated with direct-acting antiviral agents. Clin. Microbiol. Infect. 2016, 22, 462.e1–462.e3. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Tang, Q.; Xu, L. Herpes simplex virus 2 infects human endothelial ECV304 cells and induces cell apoptosis synergistically with ox-LDL. J. Toxicol. Sci. 2014, 39, 909–917. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.P.; Sun, D.D.; Wang, Y.; Liu, W.; Yang, J. Herpes Simplex Virus Type 1 and Type 2 Infection Increases Atherosclerosis Risk: Evidence Based on a Meta-Analysis. Biomed Res. Int. 2016, 2016, 2630865. [Google Scholar] [CrossRef] [PubMed]
- Chirathaworn, C.; Pongpanich, A.; Poovorawan, Y. Herpes simplex virus 1 induced LOX-1 expression in an endothelial cell line, ECV 304. Viral Immunol. 2004, 17, 308–314. [Google Scholar] [CrossRef] [PubMed]
- Hajjar, D.P.; Pomerantz, K.B.; Falcone, D.J.; Weksler, B.B.; Grant, A.J. Herpes simplex virus infection in human arterial cells. Implications in arteriosclerosis. J. Clin. Investig. 1987, 80, 1317–1321. [Google Scholar] [CrossRef] [PubMed]
- Key, N.S.; Vercellotti, G.M.; Winkelmann, J.C.; Moldow, C.F.; Goodman, J.L.; Esmon, N.L.; Esmon, C.T.; Jacob, H.S. Infection of vascular endothelial cells with herpes simplex virus enhances tissue factor activity and reduces thrombomodulin expression. Proc. Natl. Acad. Sci. USA 1990, 87, 7095–7099. [Google Scholar] [CrossRef] [PubMed]
- Mendy, A.; Vieira, E.R.; Gasana, J. Seropositivity to herpes simplex virus type 2, but not type 1 is associated with premature cardiovascular diseases: A population-based cross-sectional study. Atherosclerosis 2013, 231, 18–21. [Google Scholar] [CrossRef] [PubMed]
- Van Dam-Mieras, M.C.; Bruggeman, C.A.; Muller, A.D.; Debie, W.H.; Zwaal, R.F. Induction of endothelial cell procoagulant activity by cytomegalovirus infection. Thromb. Res. 1987, 47, 69–75. [Google Scholar] [CrossRef]
- Speir, E.; Modali, R.; Huang, E.S.; Leon, M.B.; Shawl, F.; Finkel, T.; Epstein, S.E. Potential role of human cytomegalovirus and p53 interaction in coronary restenosis. Science 1994, 265, 391–394. [Google Scholar] [CrossRef] [PubMed]
- Swirski, F.K.; Nahrendorf, M. Leukocytes behavior in atherosclerosis, myocardial infarction, and heart failure. Science 2013, 339, 161–166. [Google Scholar] [CrossRef] [PubMed]
- Weis, M.; Kledal, T.N.; Lin, K.Y.; Panchal, S.N.; Gao, S.Z.; Valantine, H.A.; Mocarski, E.S.; Cooke, J.P. Cytomegalovirus infection impairs the nitric oxide synthase pathway: Role of asymmetric dimethylarginine in transplant arteriosclerosis. Circulation 2004, 109, 500–505. [Google Scholar] [CrossRef] [PubMed]
- Cai, Y.; Kurita-Ochiai, T.; Hashizume, T.; Yamamoto, M. Green tea epigallocatechin-3-gallate attenuates Porphyromonas gingivalis-induced atherosclerosis. Pathog. Dis. 2013, 67, 76–83. [Google Scholar] [CrossRef] [PubMed]
- Di Pietro, M.; de Santis, F.; Schiavoni, G.; Filardo, S.; Sessa, R. Resveratrol in Chlamydia pneumoniae induced foam cell formation and interleukin-17A sythesis. J. Biol. Regul. Homeost. Agents 2013, 27, 509–518. [Google Scholar] [PubMed]
- Mouithys-Mickalad, A.; Deby-Dupont, G.; Dogne, J.M.; de Leval, X.; Kohnen, S.; Navet, R.; Sluse, F.; Hoebeke, M.; Pirotte, B.; Lamy, M. Effects of COX-2 inhibitors on ROS produced by Chlamydia pneumoniae-primed human promonocytic cells (THP-1). Biochem. Biophys. Res. Commun. 2004, 325, 1122–1130. [Google Scholar] [CrossRef] [PubMed]
- Prochnau, D.; Rödel, J.; Prager, K.; Kuersten, D.; Heller, R.; Straube, E.; Figulla, H.R. Induced expression of lectin-like oxidized LDL receptor-1 in vascular smooth muscle cells following Chlamydia pneumoniae infection and its down-regulation by fluvastatin. Acta Microbiol. Immunol. Hung. 2010, 57, 147–155. [Google Scholar] [CrossRef] [PubMed]
- Choi, E.K.; Park, S.A.; Oh, W.M.; Kang, H.C.; Kuramitsu, H.K.; Kim, B.G.; Kang, I.C. Mechanisms of Porphyromonas gingivalis-induced monocyte chemoattractant protein-1 expression in endothelial cells. FEMS Immunol. Med. Microbiol. 2005, 44, 51–58. [Google Scholar] [CrossRef] [PubMed]
- Fukuoka, K.; Sawabe, A.; Sugimoto, T.; Koga, M.; Okuda, H.; Kitayama, T.; Shirai, M.; Komai, K.; Komemushi, S.; Matsuda, K. Inhibitory actions of several natural products on proliferation of rat vascular smooth muscle cells induced by Hsp60 from Chlamydia pneumoniae J138. J. Agric. Food Chem. 2004, 52, 6326–6329. [Google Scholar] [CrossRef] [PubMed]
- Stephensen, C.B.; Marquis, G.S.; Jacob, R.A.; Kruzich, L.A.; Douglas, S.D.; Wilson, C.M. Vitamins C and E in adolescents and young adults with HIV infection. Am. J. Clin. Nutr. 2006, 83, 870–879. [Google Scholar] [PubMed]
- Allard, J.P.; Aghdassi, E.; Chau, J.; Tam, C.; Kovacs, C.M.; Salit, I.E.; Walmsley, S.L. Effects of vitamin E and C supplementation on oxidative stress and viral load in HIV-infected subjects. Aids 1998, 12, 1653–1659. [Google Scholar] [CrossRef] [PubMed]
- Ungvari, Z.; Bagi, Z.; Feher, A.; Recchia, F.A.; Sonntag, W.E.; Pearson, K.; de Cabo, R.; Csiszar, A. Resveratrol confers endothelial protection via activation of the antioxidant transcription factor Nrf2. Am. J. Physiol. Heart Circ. Physiol. 2010, 299, H18–H24. [Google Scholar] [CrossRef] [PubMed]
- Zakkar, M.; Van der Heiden, K.; Luong le, A.; Chaudhury, H.; Cuhlmann, S.; Hamdulay, S.S.; Krams, R.; Edirisinghe, I.; Rahman, I.; Carlsen, H.; et al. Activation of Nrf2 in endothelial cells protects arteries from exhibiting a proinflammatory state. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 1851–1857. [Google Scholar] [CrossRef] [PubMed]
- Patrick, L. Hepatitis C: Epidemiology and review of complementary/alternative medicine treatments. Altern. Med. Rev. 1999, 4, 220–238. [Google Scholar] [PubMed]
- Melhem, A.; Stern, M.; Shibolet, O.; Israeli, E.; Ackerman, Z.; Pappo, O.; Hemed, N.; Rowe, M.; Ohana, H.; Zabrecky, G.; et al. Treatment of chronic hepatitis C virus infection via antioxidants: Results of a phase I clinical trial. J. Clin. Gastroenterol. 2005, 39, 737–742. [Google Scholar] [CrossRef] [PubMed]

© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Di Pietro, M.; Filardo, S.; Falasca, F.; Turriziani, O.; Sessa, R. Infectious Agents in Atherosclerotic Cardiovascular Diseases through Oxidative Stress. Int. J. Mol. Sci. 2017, 18, 2459. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms18112459
Di Pietro M, Filardo S, Falasca F, Turriziani O, Sessa R. Infectious Agents in Atherosclerotic Cardiovascular Diseases through Oxidative Stress. International Journal of Molecular Sciences. 2017; 18(11):2459. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms18112459
Chicago/Turabian StyleDi Pietro, Marisa, Simone Filardo, Francesca Falasca, Ombretta Turriziani, and Rosa Sessa. 2017. "Infectious Agents in Atherosclerotic Cardiovascular Diseases through Oxidative Stress" International Journal of Molecular Sciences 18, no. 11: 2459. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms18112459