Altered Aconitase 2 Activity in Huntington’s Disease Peripheral Blood Cells and Mouse Model Striatum


Abstract
:1. Introduction
2. Results
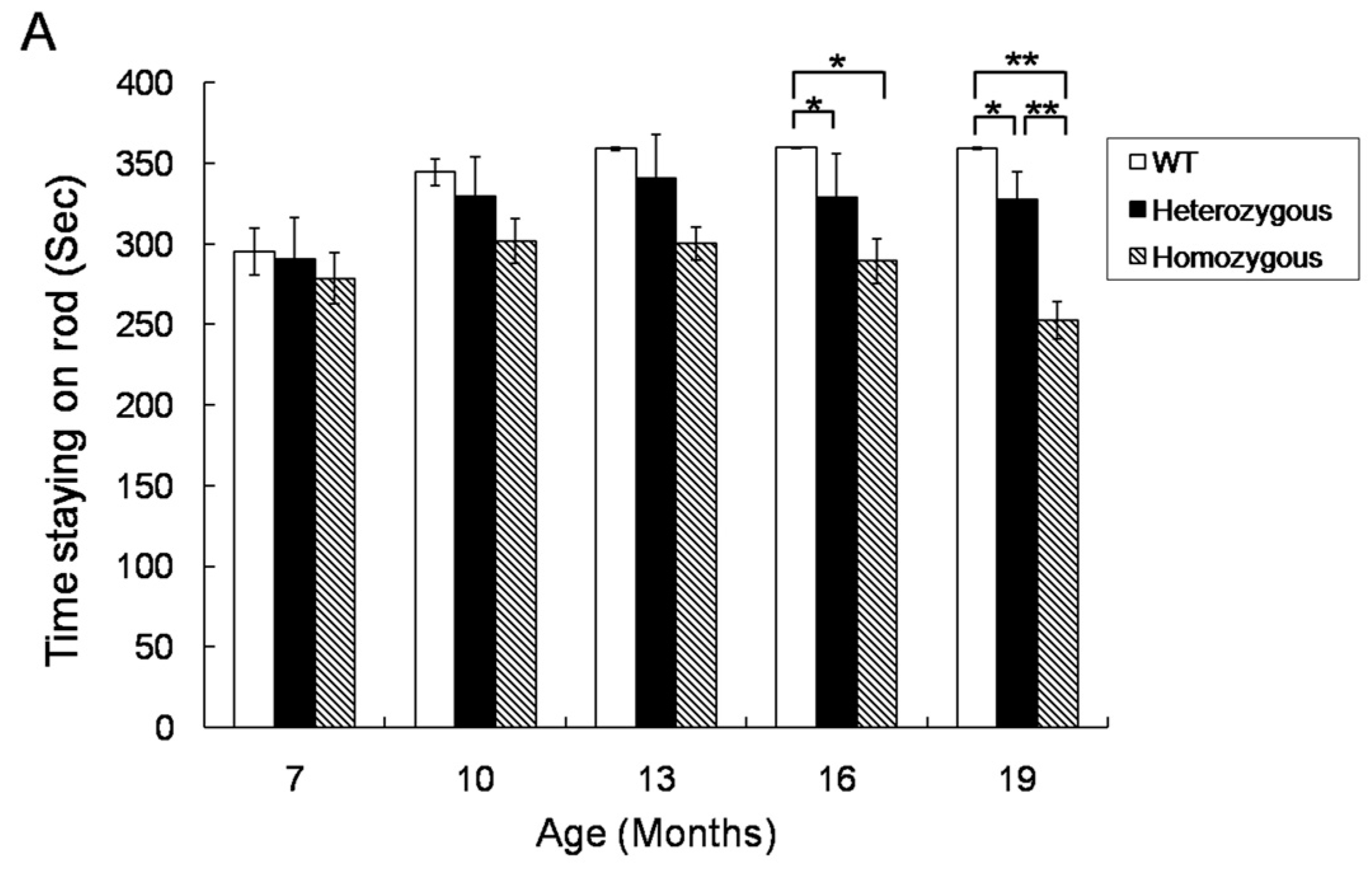
2.1. Rotarod Performance of Hdh(CAG)150 and R6/2 Mice
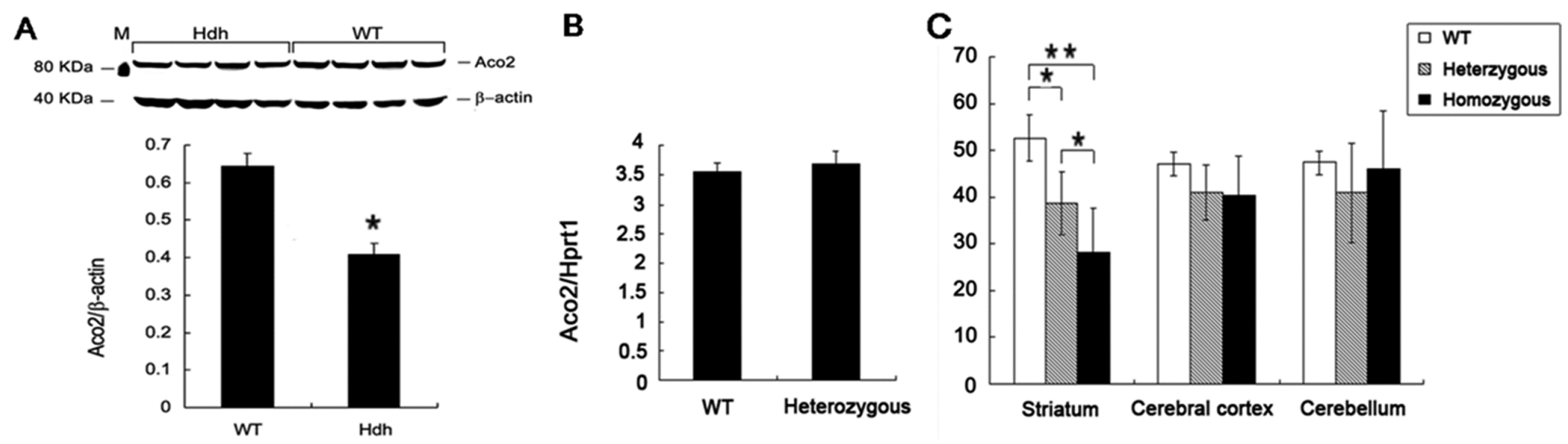
2.2. Decreased Aco2 in the Striatum of Hdh(CAG)150 Mice at 16 Months of Age
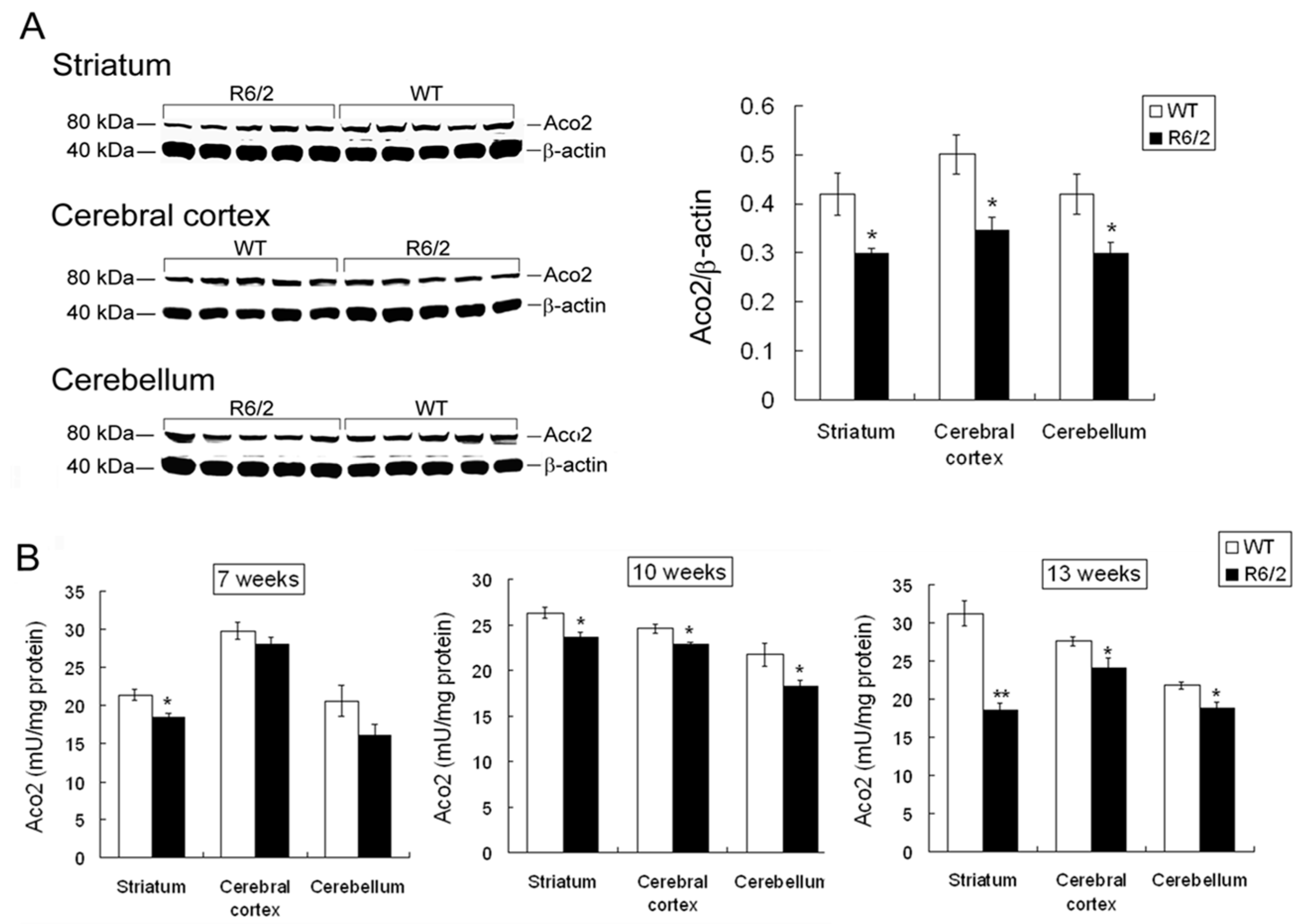
2.3. Decreased Aco2 in Brain Regions of R6/2 Mice from 7 to 13 Weeks of Age
2.4. Aco2 Activity in the Striatum of R6/2 Mice Was Restored by Treatment with NAC
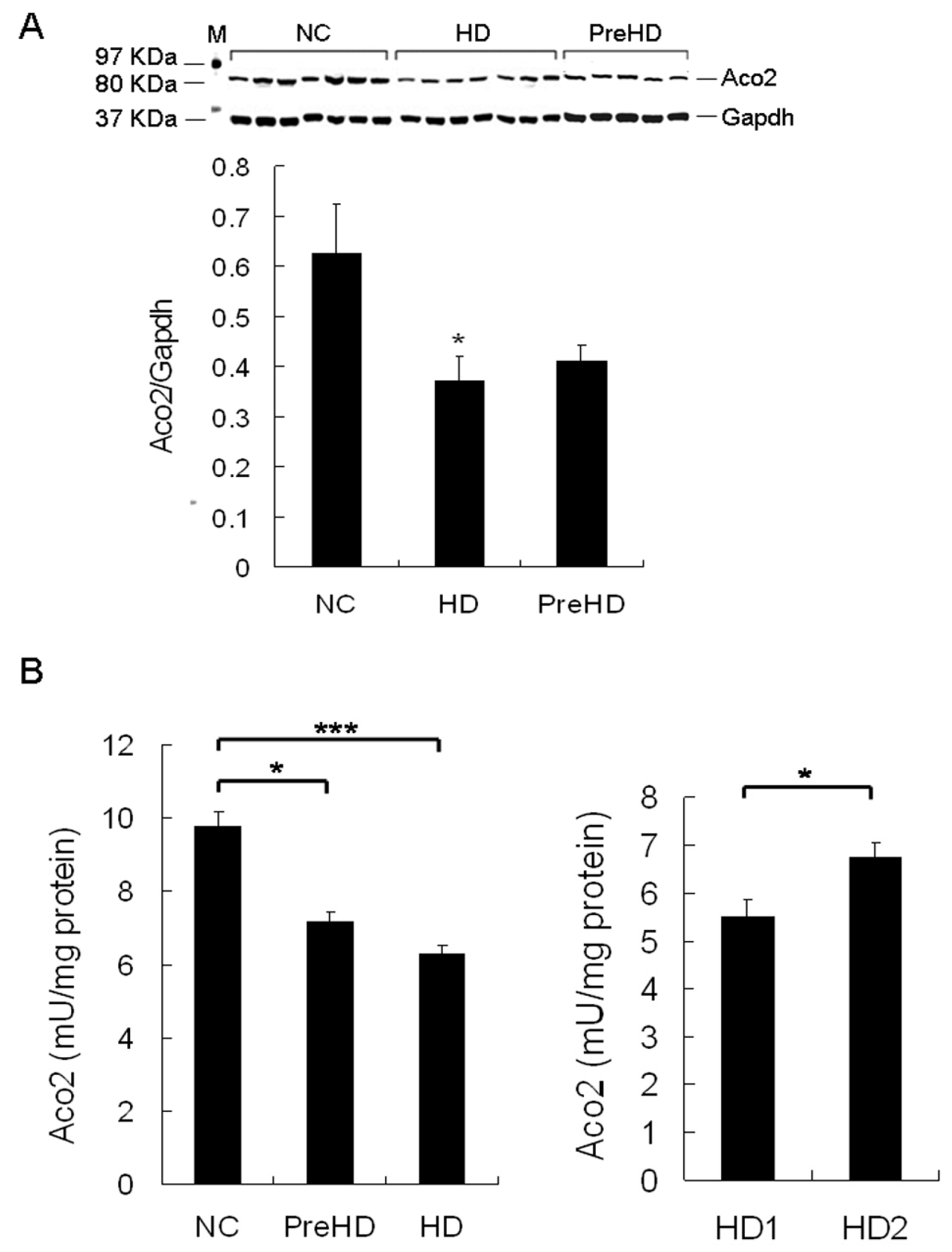
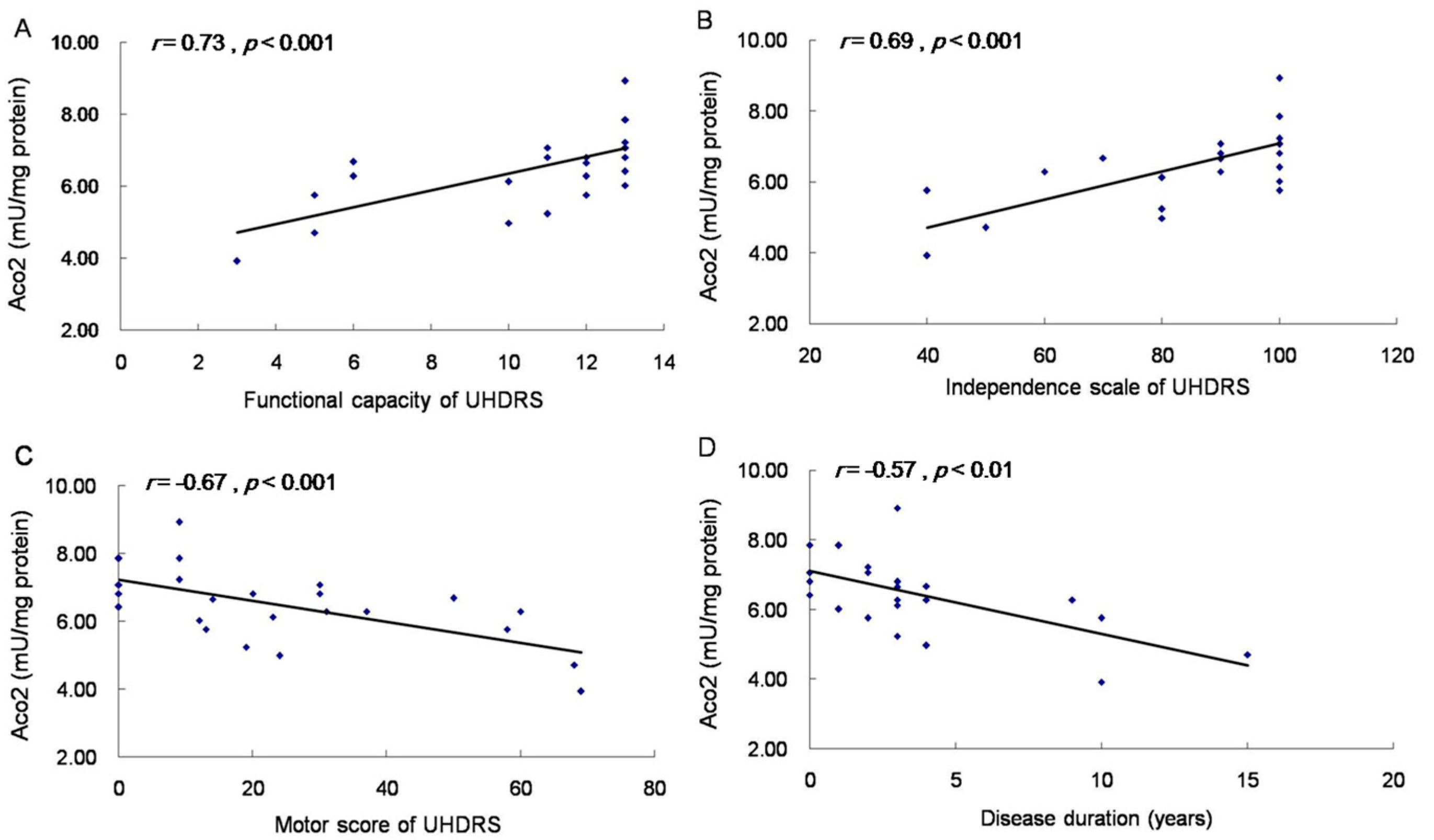
2.5. Aco2 Activity of PBMC Was Decreased in HD Patients and PreHD Carriers
3. Discussion
4. Materials and Methods
4.1. Animals and NAC Treatment
4.2. Rotarod Performance
4.3. HD Patients and PBMC Preparation
4.4. Western Blot Analysis
4.5. RNA Isolation and Quantitative Real-Time PCR (QRT-PCR)
4.6. Mitochondria Isolation from PBMC of HD Patients and Brain Tissues of Mice for Aco2 Activities
4.7. Assay for Aco2 Activity
4.8. Statistical Analysis
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- HDCRG (The Huntington’s Disease Collaborative Research Group). A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. Cell 1993, 72, 971–983. [Google Scholar]
- DiFiglia, M.; Sapp, E.; Chase, K.O.; Davies, S.W.; Bates, G.P.; Vonsattel, J.P.; Aronin, N. Aggregation of huntingtin in neuronal intranuclear inclusions and dystrophic neurites in brain. Science 1997, 277, 1990–1993. [Google Scholar] [CrossRef] [PubMed]
- Di Prospero, N.A.; Fischbeck, K.H. Therapeutics development for triplet repeat expansion diseases. Nat. Rev. Genet. 2005, 6, 756–765. [Google Scholar] [CrossRef] [PubMed]
- Valera, A.G.; Diaz-Hernandez, M.; Hernandez, F.; Ortega, Z.; Lucas, J.J. The ubiquitin-proteasome system in Huntington’s disease. Neuroscientist 2005, 11, 583–594. [Google Scholar] [CrossRef] [PubMed]
- Cha, J.H. Transcriptional signatures in Huntington’s disease. Prog. Neurobiol. 2007, 83, 228–248. [Google Scholar] [CrossRef] [PubMed]
- Stack, E.C.; Matson, W.R.; Ferrante, R.J. Evidence of oxidant damage in Huntington’s disease: Translational strategies using antioxidants. Ann. N. Y. Acad. Sci. 2008, 1147, 79–92. [Google Scholar] [CrossRef] [PubMed]
- Browne, S.E. Mitochondria and Huntington’s disease pathogenesis: Insight from genetic and chemical models. Ann. N. Y. Acad. Sci. 2008, 1147, 358–382. [Google Scholar] [CrossRef] [PubMed]
- Giorgini, F.; Muchowski, P.J. Connecting the dots in Huntington’s disease with protein interaction networks. Genome Biol. 2005, 6, 210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dalrymple, A.; Wild, E.J.; Joubert, R.; Sathasivam, K.; Bjorkqvist, M.; Petersen, A.; Jackson, G.S.; Isaacs, J.D.; Kristiansen, M.; Bates, G.P.; et al. Proteomic profiling of plasma in Huntington’s disease reveals neuroinflammatory activation and biomarker candidates. J. Proteome Res. 2007, 6, 2833–2840. [Google Scholar] [CrossRef] [PubMed]
- Björkqvist, M.; Wild, E.J.; Thiele, J.; Silvestroni, A.; Andre, R.; Lahiri, N.; Raibon, E.; Lee, R.V.; Benn, C.L.; Soulet, D.; et al. A novel pathogenic pathway of immune activation detectable before clinical onset in Huntington’s disease. J. Exp. Med. 2008, 205, 1869–1877. [Google Scholar] [CrossRef] [PubMed]
- Sapp, E.; Kegel, K.B.; Aronin, N.; Hashikawa, T.; Uchiyama, Y.; Tohyama, K.; Bhide, P.G.; Vonsattel, J.P.; DiFiglia, M. Early and progressive accumulation of reactive microglia in the Huntington disease brain. J. Neuropathol. Exp. Neurol. 2001, 60, 161–172. [Google Scholar] [CrossRef] [PubMed]
- Pavese, N.; Gerhard, A.; Tai, Y.F.; Ho, A.K.; Turkheimer, F.; Barker, R.A.; Brooks, D.J.; Piccini, P. Microglial activation correlates with severity in Huntington disease: A clinical and PET study. Neurology 2006, 66, 1638–1643. [Google Scholar] [CrossRef] [PubMed]
- Tai, Y.F.; Pavese, N.; Gerhard, A.; Tabrizi, S.J.; Barker, R.A.; Brooks, D.J.; Piccini, P. Microglial activation in presymptomatic Huntington’s disease gene carriers. Brain 2007, 130, 1759–1766. [Google Scholar] [CrossRef] [PubMed]
- Hsiao, H.Y.; Chern, Y. Targeting Glial Cells to Elucidate the Pathogenesis of Huntington’s Disease. Mol. Neurobiol. 2010, 14, 248–255. [Google Scholar] [CrossRef] [PubMed]
- Mangiarini, L.; Sathasivam, K.; Seller, M.; Cozens, B.; Harper, A.; Hetherington, C.; Lawton, M.; Trottier, Y.; Lehrach, H.; Davies, S.W.; et al. Exon 1 of the HD gene with an expanded CAG repeat is sufficient to cause a progressive neurological phenotype in transgenic mice. Cell 1996, 87, 493–506. [Google Scholar] [CrossRef]
- Lin, C.H.; Tallaksen-Greene, S.; Chien, W.M.; Cearley, J.A.; Jackson, W.S.; Crouse, A.B.; Ren, S.; Li, X.J.; Albin, R.L.; Detloff, P.J. Neurological abnormalities in a knock-in mouse model of Huntington’s disease. Hum. Mol. Genet. 2001, 10, 137–144. [Google Scholar] [CrossRef] [PubMed]
- Menalled, L.B.; Chesselet, M.F. Mouse models of Huntington’s disease. Trends Pharmacol. Sci. 2002, 23, 32–39. [Google Scholar] [CrossRef]
- Heng, M.Y.; Tallaksen-Greene, S.J.; Detloff, P.J.; Albin, R.L. Longitudinal evaluation of the Hdh (CAG)150 knock-in murine model of Huntington’s disease. J. Neurosci. 2007, 27, 8989–8998. [Google Scholar] [CrossRef] [PubMed]
- Tabrizi, S.J.; Workman, J.; Hart, P.E.; Mangiarini, L.; Mahal, A.; Bates, G.; Cooper, J.M.; Schapira, A.H. Mitochondrial dysfunction and free radical damage in the Huntington R6/2 transgenic mouse. Ann. Neurol. 2000, 47, 80–86. [Google Scholar] [CrossRef]
- Tabrizi, S.J.; Cleeter, M.W.; Xuereb, J.; Taanman, J.W.; Cooper, J.M.; Schapira, A.H. Biochemical abnormalities and excitotoxicity in Huntington’s disease brain. Ann. Neurol. 1999, 45, 25–32. [Google Scholar] [CrossRef]
- Sorolla, M.A.; Reverter-Branchat, G.; Tamarit, J.; Ferrer, I.; Ros, J.; Cabiscol, E. Proteomic and oxidative stress analysis in human brain samples of Huntington disease. Free Radic. Biol. Med. 2008, 45, 667–678. [Google Scholar] [CrossRef] [PubMed]
- Chiang, M.C.; Juo, C.G.; Chang, H.H.; Chen, H.M.; Yi, E.C.; Chern, Y. Systematic uncovering of multiple pathways underlying the pathology of Huntington disease by an acid-cleavable isotope-coded affinity tag approach. Mol. Cell. Proteom. 2007, 6, 781–797. [Google Scholar] [CrossRef] [PubMed]
- Huntington Study Group. Unified Huntington’s Disease Rating Scale: Reliability and Consistency. Mov. Disord. 1996, 11, 136–142. [Google Scholar]
- Li, J.Y.; Popovic, N.; Brundin, P. The use of the R6 transgenic mouse models of Huntington’s disease in attempts to develop novel therapeutic strategies. NeuroRx 2005, 2, 447–464. [Google Scholar] [CrossRef] [PubMed]
- Bogdanov, M.B.; Andreassen, O.A.; Dedeoglu, A.; Ferrante, R.J.; Beal, M.F. Increased oxidative damage to DNA in a transgenic mouse model of Huntington’s disease. J. Neurochem. 2001, 79, 1246–1249. [Google Scholar] [CrossRef] [PubMed]
- Colin, E.; Regulier, E.; Perrin, V.; Durr, A.; Brice, A.; Aebischer, P.; Deglon, N.; Humbert, S.; Saudou, F. Akt is altered in an animal model of Huntington’s disease and in patients. Eur. J. Neurosci. 2005, 21, 1478–1488. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Amante, D.J.; Moody, J.P.; Edgerly, C.K.; Bordiuk, O.L.; Smith, K.; Matson, S.A.; Matson, W.R.; Scherzer, C.R.; Rosas, H.D.; et al. Reduced creatine kinase as a central and peripheral biomarker in Huntington’s disease. Biochim. Biophys. Acta 2010, 1802, 673–681. [Google Scholar] [CrossRef] [PubMed]
- Zabel, C.; Mao, L.; Woodman, B.; Rohe, M.; Wacker, M.A.; Klaere, Y.; Koppelstaetter, A.; Nebrich, G.; Klein, O.; Grams, S.; et al. A large number of protein expression changes occur early in life and precede phenotype onset in a mouse model for Huntington’s disease. Mol. Cell. Proteom. 2009, 8, 720–734. [Google Scholar] [CrossRef] [PubMed]
- Zourlidou, A.; Gidalevitz, T.; Kristiansen, M.; Landles, C.; Woodman, B.; Wells, D.J.; Latchman, D.S.; de Belleroche, J.; Tabrizi, S.J.; Morimoto, R.I.; et al. Hsp27 overexpression in the R6/2 mouse model of Huntington’s disease: Chronic neurodegeneration does not induce Hsp27 activation. Hum. Mol. Genet. 2007, 16, 1078–1090. [Google Scholar] [CrossRef] [PubMed]
- Das, N.; Levine, R.L.; Orr, W.C.; Sohal, R.S. Selectivity of protein oxidative damage during aging in Drosophila melanogaster. Biochem. J. 2001, 360, 209–216. [Google Scholar] [CrossRef] [PubMed]
- Browne, S.E.; Bowling, A.C.; Mac Garvey, U.; Baik, M.J.; Berger, S.C.; Muqit, M.M.; Bird, E.D.; Beal, M.F. Oxidative damage and metabolic dysfunction in Huntington’s disease: Selective vulnerability of the basal ganglia. Ann. Neurol. 1997, 41, 646–653. [Google Scholar] [CrossRef] [PubMed]
- Browne, S.E.; Ferrante, R.J.; Beal, M.F. Oxidative stress in Huntington’s disease. Brain Pathol. 1999, 9, 147–163. [Google Scholar] [CrossRef] [PubMed]
- Wyttenbach, A.; Sauvageot, O.; Carmichael, J.; Diaz-Latoud, C.; Arrigo, A.P.; Rubinsztein, D.C. Hock protein 27 prevents cellular polyglutamine toxicity and suppresses the increase of reactive oxygen species caused by huntingtin. Hum. Mol. Genet. 2002, 11, 137–1151. [Google Scholar] [CrossRef]
- Chen, C.M.; Wu, Y.R.; Cheng, M.L.; Liu, J.L.; Lee, Y.M.; Lee, P.W.; Soong, B.W.; Chiu, D.T. Increased oxidative damage and mitochondrial abnormalities in the peripheral blood of Huntington’s disease patients. Biochem. Biophys. Res. Commun. 2007, 359, 335–340. [Google Scholar] [CrossRef] [PubMed]
- Perluigi, M.; Poon, H.F.; Maragos, W.; Pierce, W.M.; Klein, J.B.; Calabrese, V.; Cini, C.; de Marco, C.; Butterfield, D.A. Proteomic analysis of protein expression and oxidative modification in R6/2 transgenic mice: A model of Huntington disease. Mol. Cell. Proteom. 2005, 4, 1849–1861. [Google Scholar] [CrossRef] [PubMed]
- Karpuj, M.V.; Garren, H.; Slunt, H.; Price, D.L.; Gusella, J.; Becher, M.W.; Steinman, L. Transglutaminase aggregates huntingtin into nonamyloidogenic polymers, and its enzymatic activity increases in Huntington’s disease. Proc. Natl. Acad. Sci. USA. 1999, 96, 7388–7393. [Google Scholar] [CrossRef] [PubMed]
- Lesort, M.; Chun, W.; Johnson, G.V.W.; Ferrante, R.J. Tissue transglutaminase is increased in Huntington’s disease brain. J. Neurochem. 1999, 73, 2018–2027. [Google Scholar] [PubMed]
- Karpuj, M.V.; Becher, M.W.; Springer, J.E.; Chabas, D.; Youssef, S.; Pedotti, R.; Mitchell, D.; Steinman, L. Prolonged survival and decreased abnormal movements in transgenic model of Huntington disease, with administration of the transglutaminase inhibitor cystamine. Nat. Med. 2002, 8, 143–149. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.Y.; Marekov, L.; Bubber, P.; Browne, S.E.; Stavrovskaya, I.; Lee, J.; Steinert, P.M.; Blass, J.P.; Beal, M.F.; Gibson, G.E.; et al. Mitochondrial aconitase is a transglutaminase 2 substrate: Transglutamination is a probable mechanism contributing to high-molecular-weight aggregates of aconitase and loss of aconitase activity in Huntington disease brain. Neurochem. Res. 2005, 30, 1245–1255. [Google Scholar] [CrossRef] [PubMed]
- Maiuri, L.; Luciani, A.; Giardino, I.; Raia, V.; Villella, V.R.; D’Apolito, M.; Pettoello-Mantovani, M.; Guido, S.; Ciacci, C.; Cimmino, M.; et al. Tissue transglutaminase activation modulates inflammation in cystic fibrosis via PPARgamma down-regulation. J. Immunol. 2008, 180, 7697–7705. [Google Scholar] [CrossRef] [PubMed]
- Naseri, N.N.; Bonica, J.; Xu, H.; Park, L.C.; Arjomand, J.; Chen, Z.; Gibson, G.E. Novel Metabolic Abnormalities in the Tricarboxylic Acid Cycle in Peripheral Cells From Huntington’s Disease Patients. PLoS ONE 2016, 11, e0160384. [Google Scholar] [CrossRef] [PubMed]
- Wright, D.J.; Renoir, T.; Smith, Z.M.; Frazier, A.E.; Francis, P.S.; Thorburn, D.R.; McGee, S.L.; Hannan, A.J.; Gray, L.J. N-Acetylcysteine improves mitochondrial function and ameliorates behavioral deficits in the R6/1 mouse model of Huntington’s disease. Transl. Psychiatry 2015, 5, e492. [Google Scholar] [CrossRef] [PubMed]
- Ferrante, R.J.; Andreassen, O.A.; Dedeoglu, A.; Ferrante, K.L.; Jenkins, B.G.; Hersch, S.M.; Beal, M.F. Therapeutic effects of coenzyme Q10 and remacemide in transgenic mouse models of Huntington’s disease. J. Neurosci. 2002, 22, 1592–1599. [Google Scholar] [PubMed]
- Yang, L.; Calingasan, N.Y.; Wille, E.J.; Cormier, K.; Smith, K.; Ferrante, R.J.; Beal, M.F. Combination therapy with coenzyme Q10 and creatine produces additive neuroprotective effects in models of Parkinson’s and Huntington’s diseases. J. Neurochem. 2009, 109, 1427–1439. [Google Scholar] [CrossRef] [PubMed]
- Klivenyi, P.; Ferrante, R.J.; Gardian, G.; Browne, S.; Chabrier, P.E.; Beal, M.F. Increased survival and neuroprotective effects of BN82451 in a transgenic mouse model of Huntington’s disease. J. Neurochem. 2003, 86, 267–272. [Google Scholar] [CrossRef] [PubMed]
- Stack, C.; Ho, D.; Wille, E.; Calingasan, N.Y.; Williams, C.; Liby, K.; Sporn, M.; Dumont, M.; Beal, M.F. Triterpenoids CDDO-ethyl amide and CDDO-trifluoroethyl amide improve the behavioral phenotype and brain pathology in a transgenic mouse model of Huntington’s disease. Free Radic. Biol. Med. 2010, 49, 147–158. [Google Scholar] [CrossRef] [PubMed]
- Sawa, A.; Wiegand, G.W.; Cooper, J.; Margolis, R.L.; Sharp, A.H.; Lawler, J.F., Jr.; Greenamyre, J.T.; Snyder, S.H.; Ross, C.A. Increased apoptosis of Huntington disease lymphoblasts associated with repeat length-dependent mitochondrial depolarization. Nat. Med. 1999, 5, 1194–1198. [Google Scholar] [PubMed]
- Varani, K.; Abbracchio, M.P.; Cannella, M.; Cislaghi, G.; Giallonardo, P.; Mariotti, C.; Cattabriga, E.; Cattabeni, F.; Borea, P.A.; Squitieri, F.; et al. Aberrant A2A receptor function in peripheral blood cells in Huntington’s disease. FASEB. J. 2003, 17, 2148–2150. [Google Scholar] [CrossRef] [PubMed]
- Maglione, V.; Giallonardo, P.; Cannella, M.; Martino, T.; Frati, L.; Squitieri, F.T. Adenosine A(2A) receptor dysfunction correlates with age at onset anticipation in blood platelets of subjects with Huntington’s disease. Am. J. Med. Genet. Part B Neuropsychiatr. Genet. 2005, 139, 101–105. [Google Scholar] [CrossRef] [PubMed]
- Underwood, B.R.; Broadhurst, D.; Dunn, W.B.; Ellis, D.I.; Michell, A.W.; Vacher, C.; Mosedale, D.E.; Kell, D.B.; Barker, R.A.; Grainger, D.J.; et al. Huntington disease patients and transgenic mice have similar pro-catabolic serum metabolite profiles. Brain 2006, 129, 877–886. [Google Scholar] [CrossRef] [PubMed]
- Hersch, S.M.; Gevorkian, S.; Marder, K.; Moskowitz, C.; Feigin, A.; Cox, M.; Como, P.; Zimmerman, C.; Lin, M.; Zhang, L.; et al. Creatine in Huntington disease is safe, tolerable, bioavailable in brain and reduces serum 8OH2’dG. Neurology 2006, 66, 250–252. [Google Scholar] [CrossRef] [PubMed]
- Battista, N.; Bari, M.; Tarditi, A.; Mariotti, C.; Bachoud-Lévi, A.C.; Zuccato, C.; Finazzi-Agrò, A.; Genitrini, S.; Peschanski, M.; Di Donato, S.; et al. Severe deficiency of the fatty acid amide hydrolase (FAAH) activity segregates with the Huntington’s disease mutation in peripheral lymphocytes. Neurobiol. Dis. 2007, 27, 108–116. [Google Scholar] [CrossRef] [PubMed]
- Mochel, F.; Charles, P.; Seguin, F.; Barritault, J.; Coussieu, C.; Perin, L.; Le Bouc, Y.; Gervais, C.; Carcelain, G.; Vassault, A.; et al. Early energy deficit in Huntington disease: Identification of a plasma biomarker traceable during disease progression. PLoS ONE 2007, 2, e647. [Google Scholar] [CrossRef] [PubMed]
- Chang, K.H.; Chen, Y.C.; Wu, Y.R.; Lee, W.F.; Chen, C.M. Downregulation of genes involved in metabolism and oxidative stress in the peripheral leukocytes of Huntington’s disease patients. PLoS ONE 2012, 7, e46492. [Google Scholar] [CrossRef] [PubMed]
- Chang, K.H.; Wu, Y.R.; Chen, Y.C.; Chen, C.M. Plasma Inflammatory biomarkers for Huntington’s disease patients and mouse model. Brain Behav. Immun. 2015, 44, 121–127. [Google Scholar] [CrossRef] [PubMed]
- Cheng, M.L.; Chang, K.H.; Wu, Y.R.; Chen, C.M. Metabolic disturbances in plasma as biomarkers for Huntington’s disease. J. Nutr. Biochem. 2016, 31, 38–44. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Chopra, V.; Chopra, R.; Locascio, J.J.; Liao, Z.; Ding, H.; Zheng, B.; Matson, W.R.; Ferrante, R.J.; Rosas, H.D.; et al. Transcriptional modulator H2A histone family, member Y (H2AFY) marks Huntington disease activity in man and mouse. Proc. Natl. Acad. Sci. USA 2011, 108, 17141–17146. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | Controls (n = 25) | PreHD Carriers (n = 6) | HD Patients (n = 19) | HD1 Patients (n = 7) | HD2 Patients (n = 12) |
|---|---|---|---|---|---|
| Gender (male/female) | 15/10 | 4/2 | 13/6 | 5/2 | 8/4 |
| Age (years) | 48.1 ± 2.4 (30–76) | 46.5 ± 7.8 (21–69) | 48.7 ± 2.6 (24–67) | 51.4 ± 5.5 (24–67) | 47.3 ± 2.6 (25–59) |
| Age at symptom onset (years) | 44.4 ± 2.6 (15–62) | 43.7 ± 5.9 (15–62) | 44.8 ± 2.5 (24–56) | ||
| Disease duration (years) | 4.5 ± 0.9 (1–15) | 7.9 ± 1.7 (3–15) | 2.5 ± 0.3 (1–4) | ||
| Expanded CAG repeat No | 40.2 ± 0.7 (38–43) | 44.9 ± 1.3 (40–62) | 46.7 ± 2.7 (41–62) | 43.8 ± 1.2 (40–56) | |
| UHDRS | |||||
| Motor score | 0 | 30.8 ± 4.7 (9–69) | 50.3 ± 7.3 (24–69) | 19.4 ± 2.9 (9–37) | |
| Independence scale | 100 | 81.1 ± 4.6 (40–100) | 60.0 ± 6.5 (40–80) | 93.3 ± 1.9 (80–100) | |
| Functional capacity | 13 | 9.9 ± 0.7 (3–13) | 6.4 ± 1.0 (3–10) | 12.0 ± 0.25 (11–13) |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, C.-M.; Wu, Y.-R.; Chang, K.-H. Altered Aconitase 2 Activity in Huntington’s Disease Peripheral Blood Cells and Mouse Model Striatum. Int. J. Mol. Sci. 2017, 18, 2480. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms18112480
Chen C-M, Wu Y-R, Chang K-H. Altered Aconitase 2 Activity in Huntington’s Disease Peripheral Blood Cells and Mouse Model Striatum. International Journal of Molecular Sciences. 2017; 18(11):2480. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms18112480
Chicago/Turabian StyleChen, Chiung-Mei, Yih-Ru Wu, and Kuo-Hsuan Chang. 2017. "Altered Aconitase 2 Activity in Huntington’s Disease Peripheral Blood Cells and Mouse Model Striatum" International Journal of Molecular Sciences 18, no. 11: 2480. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms18112480