Proteinase-Activated Receptor 2 May Drive Cancer Progression by Facilitating TGF-β Signaling

 , , and
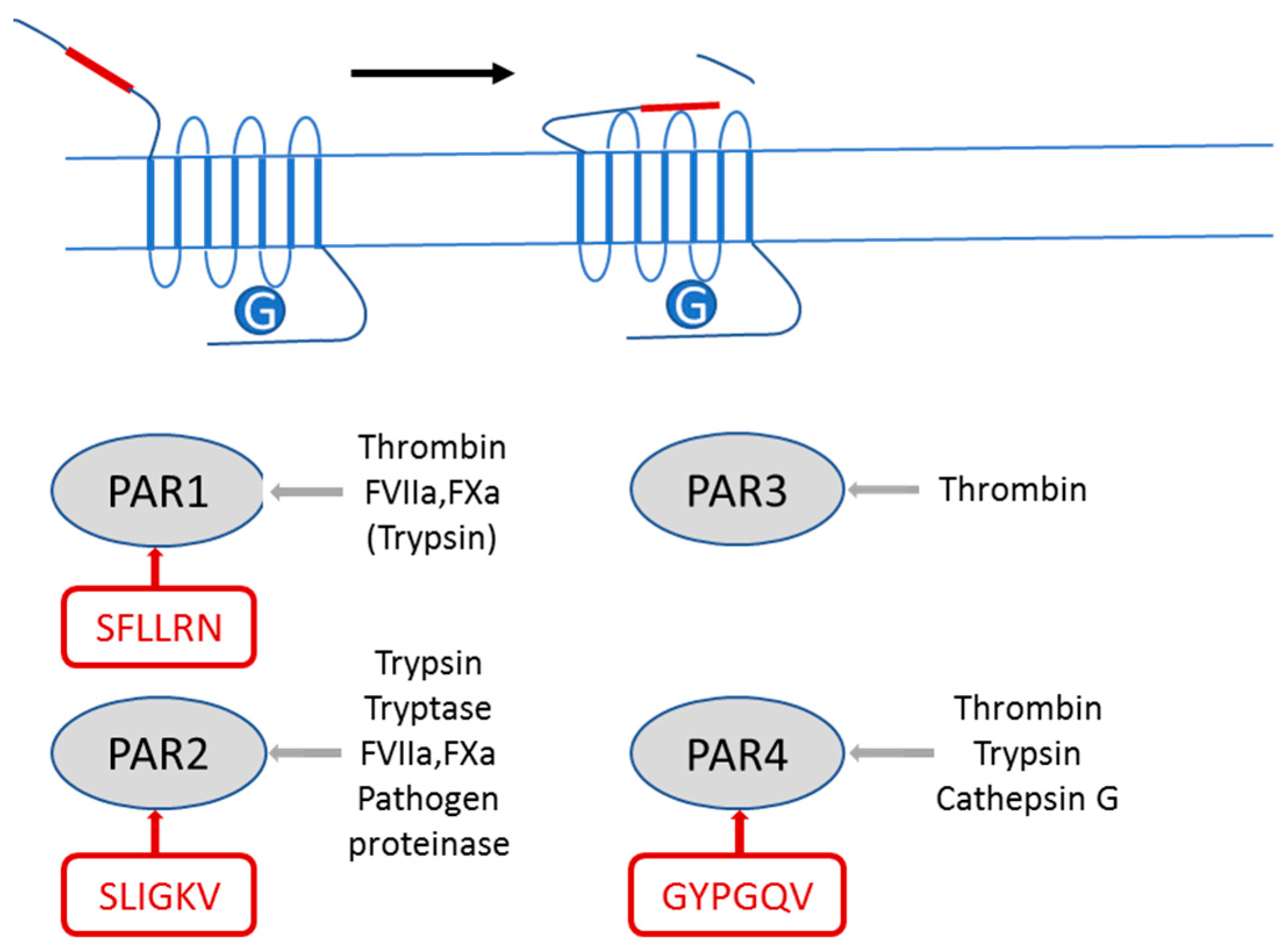
, , and 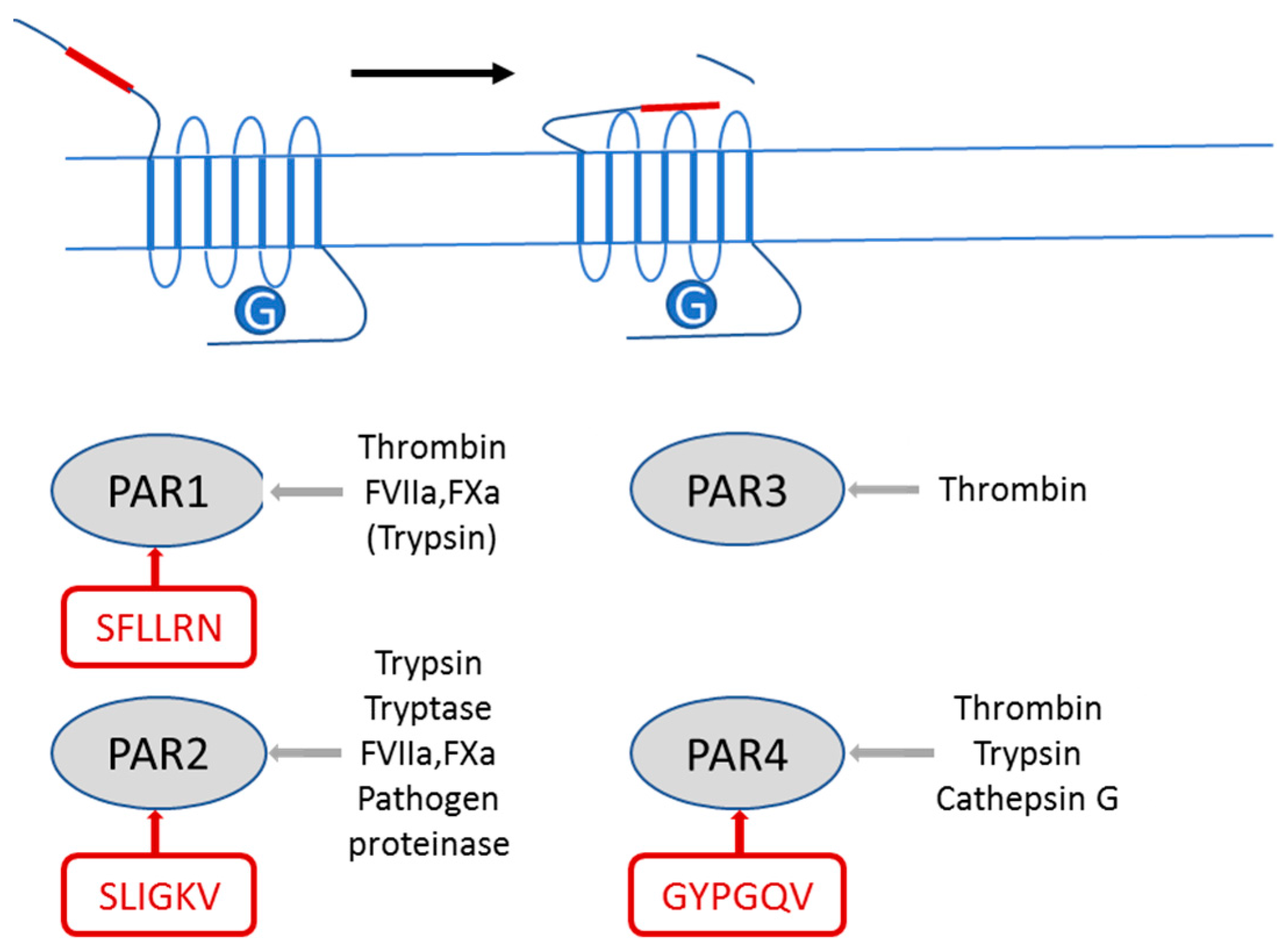{kind=link}
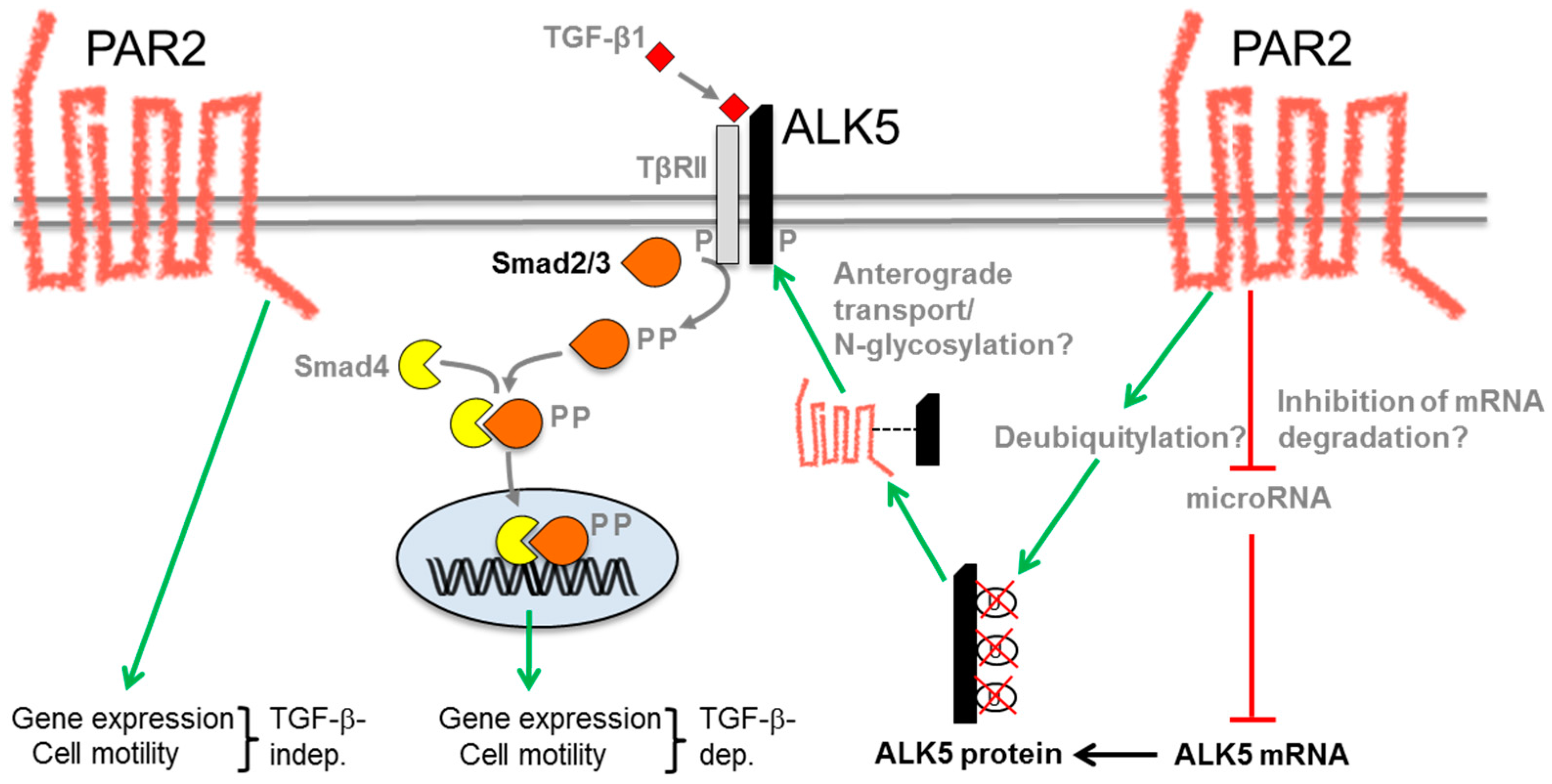
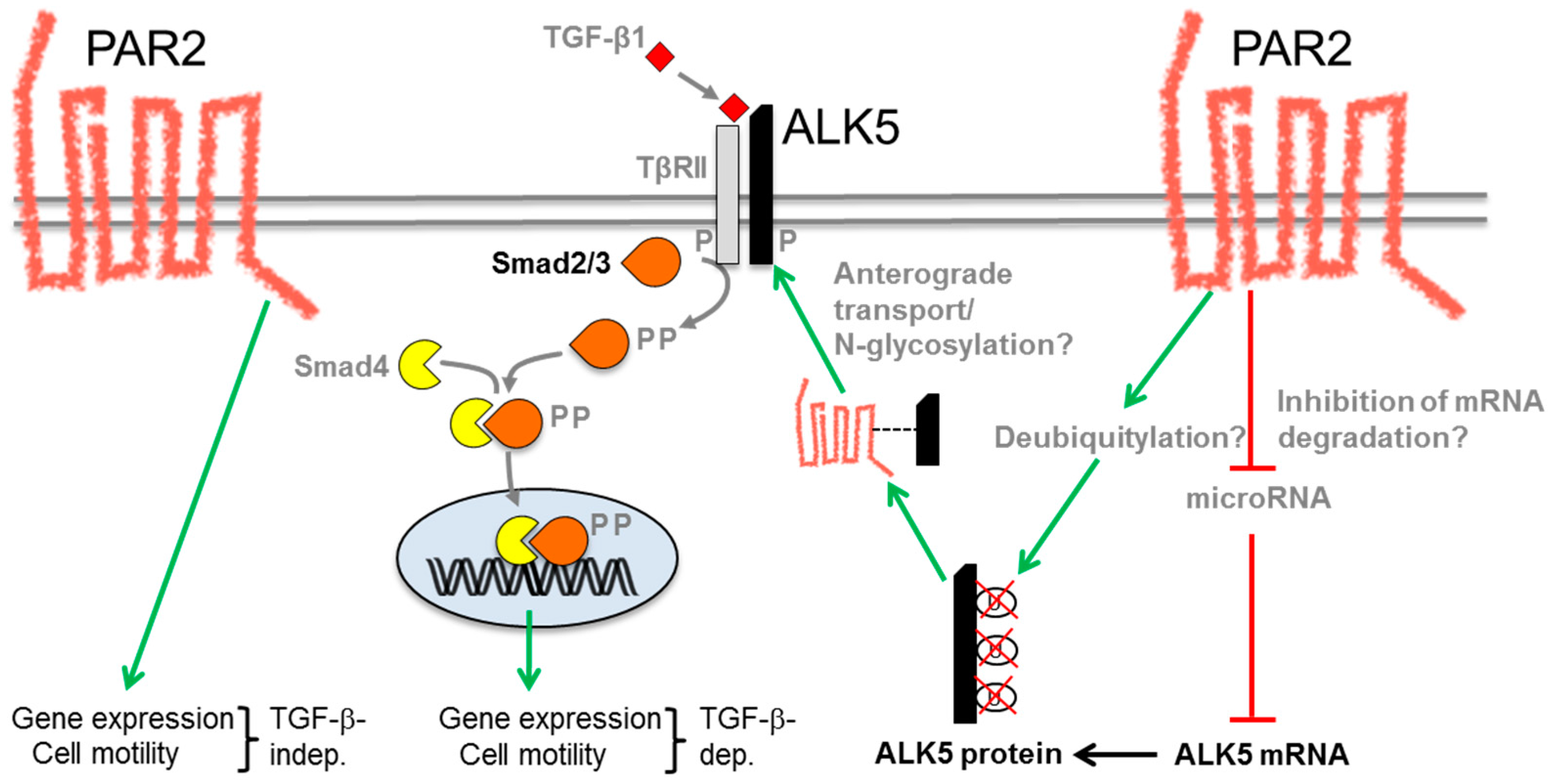{kind=link}
Abstract
:1. Introduction
2. PAR2 and TGF-β
2.1. PAR2 Signaling
2.2. The Role of PAR2 in Pancreatic and Liver Cancer
2.3. TGF-β Signaling
2.4. The Role of TGF-β in Pancreatic and Liver Cancer
3. Evidence for PAR and TGF-β/ALK5 Signaling Crosstalk
4. PAR2 and Its Requirement for TGF-β1-Mediated Cellular Responses and TGF-β/ALK5-Induced Signaling
4.1. TGF-β/ALK5-Induced Signaling
4.2. Cell Migration and Invasion
4.3. Epithelial–Mesenchymal Transition (EMT) and EMT-Associated Alterations Relevant for Therapy
5. PAR2 Signaling Functions Required for Promoting TGF-β Signaling
6. Molecular Mechanism of the PAR2–TGF-β Interaction
7. Potential Implications of PAR2 Inhibition in Anti-Cancer Therapy
Conflicts of Interest
References
- Hollenberg, M.D.; Mihara, K.; Polley, D.; Suen, J.Y.; Han, A.; Fairlie, D.P.; Ramachandran, R. Biased signalling and proteinase-activated receptors (PARs): Targeting inflammatory disease. Br. J. Pharmacol. 2014, 171, 1180–1194. [Google Scholar] [CrossRef] [PubMed]
- Gieseler, F.; Ungefroren, H.; Settmacher, U.; Hollenberg, M.D.; Kaufmann, R. Proteinase-activated receptors (PARs)—Focus on receptor-receptor-interactions and their physiological and pathophysiological impact. Cell Commun. Signal. 2013, 11, 86. [Google Scholar] [CrossRef] [PubMed]
- Nichols, H.L.; Saffeddine, M.; Theriot, B.S.; Hegde, A.; Polley, D.; El-Mays, T.; Vliagoftis, H.; Hollenberg, M.D.; Wilson, E.H.; Walker, J.K.; et al. β-Arrestin-2 mediates the proinflammatory effects of proteinase-activated receptor-2 in the airway. Proc. Natl. Acad. Sci. USA 2012, 109, 16660–16665. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Gao, J.; Zhao, T.; Wei, L.; Wu, W.; Bai, Y.; Zou, D.; Li, Z. Proteinase-activated receptor 2 mediates thermal hyperalgesia and is upregulated in a rat model of chronic pancreatitis. Pancreas 2011, 40, 300–307. [Google Scholar] [CrossRef] [PubMed]
- Knight, V.; Tchongue, J.; Lourensz, D.; Tipping, P.; Sievert, W. Protease-activated receptor 2 promotes experimental liver fibrosis in mice and activates human hepatic stellate cells. Hepatology 2012, 55, 879–887. [Google Scholar] [CrossRef] [PubMed]
- Kaufmann, R.; Oettel, C.; Horn, A.; Halbhuber, K.J.; Eitner, A.; Krieg, R.; Katenkamp, K.; Henklein, P.; Westermann, M.; Böhmer, F.D.; et al. Met receptor tyrosine kinase transactivation is involved in proteinase-activated receptor-2-mediated hepatocellular carcinoma cell invasion. Carcinogenesis 2009, 30, 1487–1496. [Google Scholar] [CrossRef] [PubMed]
- Shi, X.; Gangadharan, B.; Brass, L.F.; Ruf, W.; Mueller, B.M. Protease-activated receptors (PAR1 and PAR2) contribute to tumor cell motility and metastasis. Mol. Cancer Res. 2004, 2, 395–402. [Google Scholar] [PubMed]
- Versteeg, H.H.; Schaffner, F.; Kerver, M.; Ellies, L.G.; Andrade-Gordon, P.; Mueller, B.M.; Ruf, W. Protease-activated receptor (PAR) 2, but not PAR1, signaling promotes the development of mammary adenocarcinoma in polyoma middle T mice. Cancer Res. 2008, 68, 7219–7227. [Google Scholar] [CrossRef] [PubMed]
- Iwaki, K.; Shibata, K.; Ohta, M.; Endo, Y.; Uchida, H.; Tominaga, M.; Okunaga, R.; Kai, S.; Kitano, S. A small interfering RNA targeting proteinase-activated receptor-2 is effective in suppression of tumor growth in a Panc1 xenograft model. Int. J. Cancer 2008, 122, 658–663. [Google Scholar] [CrossRef] [PubMed]
- Segal, L.; Katz, L.S.; Lupu-Meiri, M.; Shapira, H.; Sandbank, J.; Gershengorn, M.C.; Oron, Y. Proteinase-activated receptors differentially modulate in vitro invasion of human pancreatic adenocarcinoma PANC1 cells in correlation with changes in the expression of CDC42 protein. Pancreas 2014, 43, 103–108. [Google Scholar] [CrossRef] [PubMed]
- Fujimoto, D.; Hirono, Y.; Goi, T.; Katayama, K.; Hirose, K.; Yamaguchi, A. Expression of protease activated receptor-2 (PAR-2) in gastric cancer. J. Surg. Oncol. 2006, 93, 139–144. [Google Scholar] [CrossRef] [PubMed]
- Olejar, T.; Vetvicka, D.; Zadinova, M.; Pouckova, P.; Kukal, J.; Jezek, P.; Matej, R. Dual role of host Par2 in a murine model of spontaneous metastatic B16 melanoma. Anticancer Res. 2014, 34, 3511–3515. [Google Scholar] [PubMed]
- Shi, K.; Queiroz, K.C.; Roelofs, J.J.; van Noesel, C.J.; Richel, D.J.; Spek, C.A. Protease-activated receptor 2 suppresses lymphangiogenesis and subsequent lymph node metastasis in a murine pancreatic cancer model. J. Pathol. 2014, 234, 398–409. [Google Scholar] [CrossRef] [PubMed]
- Gamperl, H.; Plattfaut, C.; Freund, A.; Quecke, T.; Theophil, F.; Gieseler, F. Extracellular vesicles from malignant effusions induce tumor cell migration: Inhibitory effect of LMWH tinzaparin. Cell Biol. Int. 2016, 40, 1050–1061. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, O.; Egami, H.; Ishiko, T.; Ishikawa, S.; Kamohara, H.; Hidaka, H.; Mita, S.; Ogawa, M. Expression of proteinase-activated receptor-2 in human pancreatic cancer: A possible relation to cancer invasion and induction of fibrosis. Int. J. Oncol. 2003, 22, 295–300. [Google Scholar] [CrossRef] [PubMed]
- Ungefroren, H.; Sebens, S.; Seidl, D.; Lehnert, H.; Hass, R. Interaction of tumor cells with the microenvironment. Cell Commun. Signal. 2011, 9, 18. [Google Scholar] [CrossRef] [PubMed]
- Pickup, M.; Novitskiy, S.; Moses, H.L. The roles of TGFβ in the tumour microenvironment. Nat. Rev. Cancer 2013, 13, 788–799. [Google Scholar] [CrossRef] [PubMed]
- Clouston, H.W.; Davenport, A.; Gregson, H.; Shaker, H.; Duff, S.; Kirwan, C.C. PO-51—Expression of proteins of the tissue factor thrombin pathway is upregulated in the stroma and epithelium of colorectal cancer. Thromb. Res. 2016, 140, S195. [Google Scholar] [CrossRef]
- Mußbach, F.; Ungefroren, H.; Günther, B.; Katenkamp, K.; Henklein, P.; Westermann, M.; Settmacher, U.; Lenk, L.; Sebens, S.; Müller, J.P.; et al. Proteinase-activated receptor 2 (PAR2) in hepatic stellate cells—Evidence for a role in hepatocellular carcinoma growth in vivo. Mol. Cancer 2016, 15, 54. [Google Scholar] [CrossRef] [PubMed]
- Shaker, H.; Bundred, N.J.; Albadry, H.; Nicholson, S.; Castle, J.; Lumsden, L.J.; Pritchard, S.; Landberg, G.; Kirwan, C.C. PO-21—Stromal fibroblasts in preinvasive breast cancer (ductal carcinoma in situ, DCIS) demonstrate a cancer-like procoagulant phenotypic switch that may facilitate invasion. Thromb. Res. 2016, 140, S184. [Google Scholar] [CrossRef]
- Yang, G.; Yang, X. Smad4-mediated TGF-beta signaling in tumorigenesis. Int. J. Biol. Sci. 2010, 6, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Miyazono, K.; Ehata, S.; Koinuma, D. Tumour-promoting functions of transforming growth factor-β in progression of cancer. Ups. J. Med. Sci. 2012, 117, 143–152. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.E. Non-Smad pathways in TGF-beta signaling. Cell Res. 2009, 19, 128–139. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.K.; Pardoux, C.; Hall, M.C.; Lee, P.S.; Warburton, D.; Qing, J.; Smith, S.M.; Derynck, R. TGF-beta activates Erk MAP kinase signalling through direct phosphorylation of ShcA. EMBO J. 2007, 26, 3957–3967. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Chen, J.K.; Nagai, K.; Plieth, D.; Tan, M.; Lee, T.C.; Threadgill, D.W.; Neilson, E.G.; Harris, R.C. EGFR signaling promotes TGFb-dependent renal fibrosis. J. Am. Soc. Nephrol. 2012, 23, 215–224. [Google Scholar] [CrossRef] [PubMed]
- Uchiyama-Tanaka, Y.; Matsubara, H.; Mori, Y.; Kosaki, A.; Kishimoto, N.; Amano, K.; Higashiyama, S.; Iwasaka, T. Involvement of HB-EGF and EGF receptor transactivation in TGF-beta-mediated fibronectin expression in mesangial cells. Kidney Int. 2002, 62, 799–808. [Google Scholar] [CrossRef] [PubMed]
- Midgley, A.C.; Rogers, M.; Hallett, M.B.; Clayton, A.; Bowen, T.; Phillips, A.O.; Steadman, R. Transforming growth factor-β1 (TGF-β1)-stimulated fibroblast to myofibroblast differentiation is mediated by hyaluronan (HA)-facilitated epidermal growth factor receptor (EGFR) and CD44 co-localization in lipid rafts. J. Biol. Chem. 2013, 288, 14824–14838. [Google Scholar] [CrossRef] [PubMed]
- Jechlinger, M.; Sommer, A.; Moriggl, R.; Seither, P.; Kraut, N.; Capodiecci, P.; Donovan, M.; Cordon-Cardo, C.; Beug, H.; Grünert, S. Autocrine PDGFR signaling promotes mammary cancer metastasis. J. Clin. Investig. 2006, 116, 1561–1570. [Google Scholar] [CrossRef] [PubMed]
- Ferrari, G.; Pintucci, G.; Seghezzi, G.; Hyman, K.; Galloway, A.C.; Mignatti, P. VEGF, a prosurvival factor, acts in concert with TGF-beta1 to induce endothelial cell apoptosis. Proc. Natl. Acad. Sci. USA 2006, 103, 17260–17265. [Google Scholar] [CrossRef] [PubMed]
- Jarad, M.; Kuczynski, E.A.; Morrison, J.; Viloria-Petit, A.M.; Coomber, B.L. Release of endothelial cell associated VEGFR2 during TGF-β modulated angiogenesis in vitro. BMC Cell Biol. 2017, 18, 10. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y. Hepatocyte growth factor in kidney fibrosis: Therapeutic potential and mechanisms of action. Am. J. Physiol. Ren. Physiol. 2004, 287, F7–F16. [Google Scholar] [CrossRef] [PubMed]
- Gallo, S.; Sala, V.; Gatti, S.; Crepaldi, T. Cellular and molecular mechanisms of HGF/Met in the cardiovascular system. Clin. Sci. 2015, 129, 1173–1193. [Google Scholar] [CrossRef] [PubMed]
- Nlandu Khodo, S.; Neelisetty, S.; Woodbury, L.; Green, E.; Harris, R.C.; Zent, R.; Gewin, L. Deleting the TGF-β receptor in proximal tubules impairs HGF signaling. Am. J. Physiol. Ren. Physiol. 2016, 310, F499–F510. [Google Scholar] [CrossRef] [PubMed]
- Mizuno, S.; Matsumoto, K.; Kurosawa, T.; Mizuno-Horikawa, Y.; Nakamura, T. Reciprocal balance of hepatocyte growth factor and transforming growth factor-β1 in renal fibrosis in mice. Kidney Int. 2000, 57, 937–948. [Google Scholar] [CrossRef] [PubMed]
- Neuzillet, C.; de Gramont, A.; Tijeras-Raballand, A.; de Mestier, L.; Cros, J.; Faivre, S.; Raymond, E. Perspectives of TGF-β inhibition in pancreatic and hepatocellular carcinomas. Oncotarget 2014, 5, 78–94. [Google Scholar] [CrossRef] [PubMed]
- Amundadottir, L.T. Pancreatic Cancer Genetics. Int. J. Biol. Sci. 2016, 12, 314–325. [Google Scholar] [CrossRef] [PubMed]
- Jones, S.; Zhang, X.; Parsons, D.W.; Lin, J.C.; Leary, R.J.; Angenendt, P.; Mankoo, P.; Carter, H.; Kamiyama, H.; Jimeno, A.; et al. Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science 2008, 321, 1801–1806. [Google Scholar] [CrossRef] [PubMed]
- Ijichi, H.; Chytil, A.; Gorska, A.E.; Aakre, M.E.; Fujitani, Y.; Fujitani, S.; Wright, C.V.; Moses, H.L. Aggressive pancreatic ductal adenocarcinoma in mice caused by pancreas-specific blockade of transforming growth factor-beta signaling in cooperation with active Kras expression. Genes Dev. 2006, 20, 3147–3160. [Google Scholar] [CrossRef] [PubMed]
- Schniewind, B.; Groth, S.; Sebens Muerkoster, S.; Sipos, B.; Schafer, H.; Kalthoff, H.; Ungefroren, H. Dissecting the role of TGF-beta type I receptor/ALK5 in pancreatic ductal adenocarcinoma: Smad activation is crucial for both the tumor suppressive and prometastatic function. Oncogene 2007, 26, 4850–4862. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Yu, N.; Lee, C. Vicious cycle of TGF-β signaling in tumor progression and metastasis. Am. J. Clin. Exp. Urol. 2014, 2, 149–155. [Google Scholar] [PubMed]
- Breuhahn, K.; Longerich, T.; Schirmacher, P. Dysregulation of growth factor beta signalling in hepatocellular carcinoma. Oncogene 2006, 25, 3787–3800. [Google Scholar] [CrossRef] [PubMed]
- Abou-Shady, M.; Baer, H.U.; Friess, H.; Berberat, P.; Zimmermann, A.; Graber, H.; Gold, L.I.; Korc, M.; Büchler, M.W. Transforming growth factor betas and their signaling receptors in human hepatocellular carcinoma. Am. J. Surg. 1999, 177, 209–215. [Google Scholar] [CrossRef]
- De Bleser, P.J.; Niki, T.; Rogiers, V.; Geerts, A. Transforming growth factor-beta gene expression in normal and fibrotic rat liver. J. Hepatol. 1997, 26, 886–893. [Google Scholar] [CrossRef]
- George, J.; Roulot, D.; Koteliansky, V.E.; Bissell, D.M. In vivo inhibition of rat stellate cell activation by soluble transforming growth factor beta type II receptor: A potential new therapy for hepatic fibrosis. Proc. Natl. Acad. Sci. USA 1999, 96, 12719–12724. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Hu, H.; Yin, J.Q. Therapeutic strategies against TGF-beta signaling pathway in hepatic fibrosis. Liver Int. 2006, 26, 8–22. [Google Scholar] [CrossRef] [PubMed]
- Presser, L.D.; McRae, S.; Waris, G. Activation of TGF-β1 promoter by hepatitis C virus-induced AP-1 and Sp1: Role of TGF-β1 in hepatic stellate cell activation and invasion. PLoS ONE 2013, 8, e56367. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Hui, A.; Albanis, E.; Arthur, M.; O’Byrne, S.; Blaner, W.; Mukherjee, P.; Friedman, S.; Eng, F. Human hepatic stellate cell lines, LX-1 and LX-2: New tools for analysis of hepatic fibrosis. Gut 2005, 54, 142–151. [Google Scholar] [CrossRef] [PubMed]
- Saito, A.; Osuga, Y.; Yoshino, O.; Takamura, M.; Hirata, T.; Hirota, Y.; Koga, K.; Harada, M.; Takemura, Y.; Yano, T.; et al. TGF-β1 induces proteinase-activated receptor 2 (PAR2) expression in endometriotic stromal cells and stimulates PAR2 activation-induced secretion of IL-6. Hum. Reprod. 2011, 26, 1892–1898. [Google Scholar] [CrossRef] [PubMed]
- Chung, H.; Ramachandran, R.; Hollenberg, M.D.; Muruve, D.A. Proteinase-activated receptor-2 transactivation of epidermal growth factor receptor and transforming growth factor-β receptor signaling pathways contributes to renal fibrosis. J. Biol. Chem. 2013, 288, 37319–37331. [Google Scholar] [CrossRef] [PubMed]
- Zeeh, F.; Witte, D.; Gädeken, T.; Rauch, B.H.; Grage-Griebenow, E.; Leinung, N.; Fromm, S.J.; Stölting, S.; Mihara, K.; Kaufmann, R.; et al. Proteinase-activated receptor 2 promotes TGF-β-dependent cell motility in pancreatic cancer cells by sustaining expression of the TGF-β type I receptor ALK5. Oncotarget 2016, 7, 41095–41109. [Google Scholar] [CrossRef] [PubMed]
- Ungefroren, H.; Lenschow, W.; Chen, W.B.; Faendrich, F.; Kalthoff, H. Regulation of biglycan gene expression by transforming growth factor-beta requires MKK6-p38 mitogen-activated protein Kinase signaling downstream of Smad signaling. J. Biol. Chem. 2003, 278, 11041–11049. [Google Scholar] [CrossRef] [PubMed]
- Ungefroren, H.; Witte, D.; Fiedler, C.; Gädeken, T.; Kaufmann, R.; Lehnert, H.; Gieseler, F.; Rauch, B.H. The role of ERK activation in PAR2 agonist and TGF-beta1-induced cell migration. Int. J. Mol. Sci. 2017. under review. [Google Scholar]
- Ungefroren, H.; University Hospital Schleswig-Holstein, Lübeck, Germany. Unpublished work. 2017.
- Ungefroren, H.; Witte, D.; Mihara, K.; Rauch, B.H.; Henklein, P.; Johren, O.; Bonni, S.; Settmacher, U.; Lehnert, H.; Hollenberg, M.D.; et al. TGF-β1/ALK5-mediated cell migration is dependent on the protein PAR2 but not on PAR2-stimulated Gq-calcium signaling. Mol. Pharmacol. 2017, 92, 519–532. [Google Scholar] [CrossRef] [PubMed]
- Jaber, M.; Maoz, M.; Kancharla, A.; Agranovich, D.; Peretz, T.; Grisaru-Granovsky, S.; Uziely, B.; Bar-Shavit, R. Protease-activated-receptor-2 affects protease-activated-receptor-1-driven breast cancer. Cell. Mol. Life Sci. 2014, 71, 2517–2533. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, M.R.; McIntosh, K.A.; Pediani, J.D.; Robben, J.; Cooke, A.E.; Nilsson, M.; Gould, G.W.; Mundell, S.; Milligan, G.; Plevin, R. Novel role for proteinase-activated receptor 2 (PAR2) in membrane trafficking of proteinase-activated receptor 4 (PAR4). J. Biol. Chem. 2012, 287, 16656–16669. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lima, L.G.; Monteiro, R.Q. Activation of blood coagulation in cancer: Implications for tumour progression. Biosci. Rep. 2013, 33, e00064. [Google Scholar] [CrossRef] [PubMed]
- Tang, S.C.; Chen, Y.C. Novel therapeutic targets for pancreatic cancer. World J. Gastroenterol. 2014, 20, 10825–10844. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Elsner, T.; Botella, L.M.; Velasco, B.; Corbí, A.; Attisano, L.; Bernabéu, C. Synergistic cooperation between hypoxia and transforming growth factor-beta pathways on human vascular endothelial growth factor gene expression. J. Biol. Chem. 2001, 276, 38527–38535. [Google Scholar] [CrossRef] [PubMed]
- Ramachandran, R.; Noorbakhsh, F.; Defea, K.; Hollenberg, M.D. Targeting proteinase-activated receptors: Therapeutic potential and challenges. Nat. Rev. Drug Discov. 2012, 11, 69–86. [Google Scholar] [CrossRef] [PubMed]
- Ellenrieder, V.; Hendler, S.F.; Boeck, W.; Seufferlein, T.; Menke, A.; Ruhland, C.; Adler, G.; Gress, T.M. Transforming growth factor beta1 treatment leads to an epithelial-mesenchymal transdifferentiation of pancreatic cancer cells requiring extracellular signal-regulated kinase 2 activation. Cancer Res. 2001, 61, 4222–4228. [Google Scholar] [PubMed]
- Yau, M.K.; Liu, L.; Suen, J.Y.; Lim, J.; Lohman, R.J.; Jiang, Y.; Cotterell, A.J.; Barry, G.D.; Mak, J.Y.; Vesey, D.A.; et al. PAR2 Modulators Derived from GB88. ACS Med. Chem. Lett. 2016, 7, 1179–1184. [Google Scholar] [CrossRef] [PubMed]
- Cheng, R.K.Y.; Fiez-Vandal, C.; Schlenker, O.; Edman, K.; Aggeler, B.; Brown, D.G.; Brown, G.A.; Cooke, R.M.; Dumelin, C.E.; Doré, A.S.; et al. Structural insight into allosteric modulation of protease-activated receptor 2. Nature 2017, 545, 112–115. [Google Scholar] [CrossRef] [PubMed]
- Calone, I.; Souchelnytskyi, S. Inhibition of TGFβ signaling and its implication in anticancer treatments. Exp. Oncol. 2012, 34, 9–16. [Google Scholar] [PubMed]
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ungefroren, H.; Witte, D.; Rauch, B.H.; Settmacher, U.; Lehnert, H.; Gieseler, F.; Kaufmann, R. Proteinase-Activated Receptor 2 May Drive Cancer Progression by Facilitating TGF-β Signaling. Int. J. Mol. Sci. 2017, 18, 2494. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms18112494
Ungefroren H, Witte D, Rauch BH, Settmacher U, Lehnert H, Gieseler F, Kaufmann R. Proteinase-Activated Receptor 2 May Drive Cancer Progression by Facilitating TGF-β Signaling. International Journal of Molecular Sciences. 2017; 18(11):2494. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms18112494
Chicago/Turabian StyleUngefroren, Hendrik, David Witte, Bernhard H. Rauch, Utz Settmacher, Hendrik Lehnert, Frank Gieseler, and Roland Kaufmann. 2017. "Proteinase-Activated Receptor 2 May Drive Cancer Progression by Facilitating TGF-β Signaling" International Journal of Molecular Sciences 18, no. 11: 2494. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms18112494