Predictive and Experimental Approaches for Elucidating Protein–Protein Interactions and Quaternary Structures


Abstract
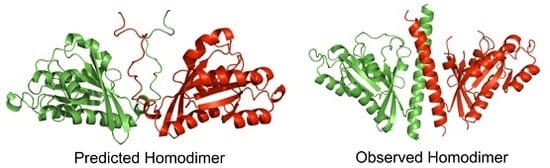
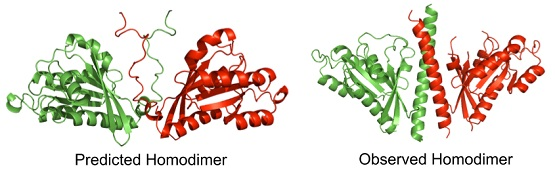
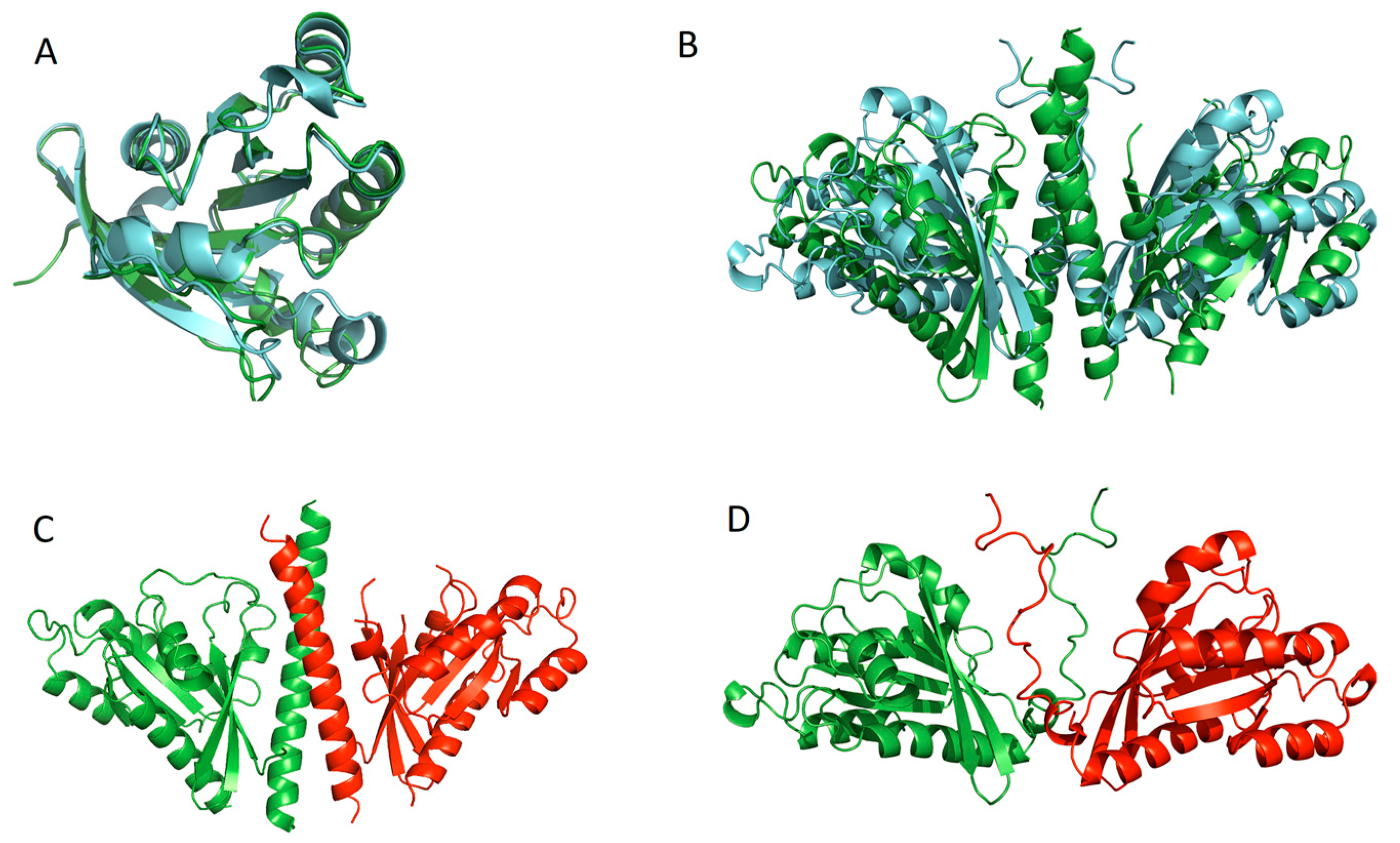
:
1. Introduction
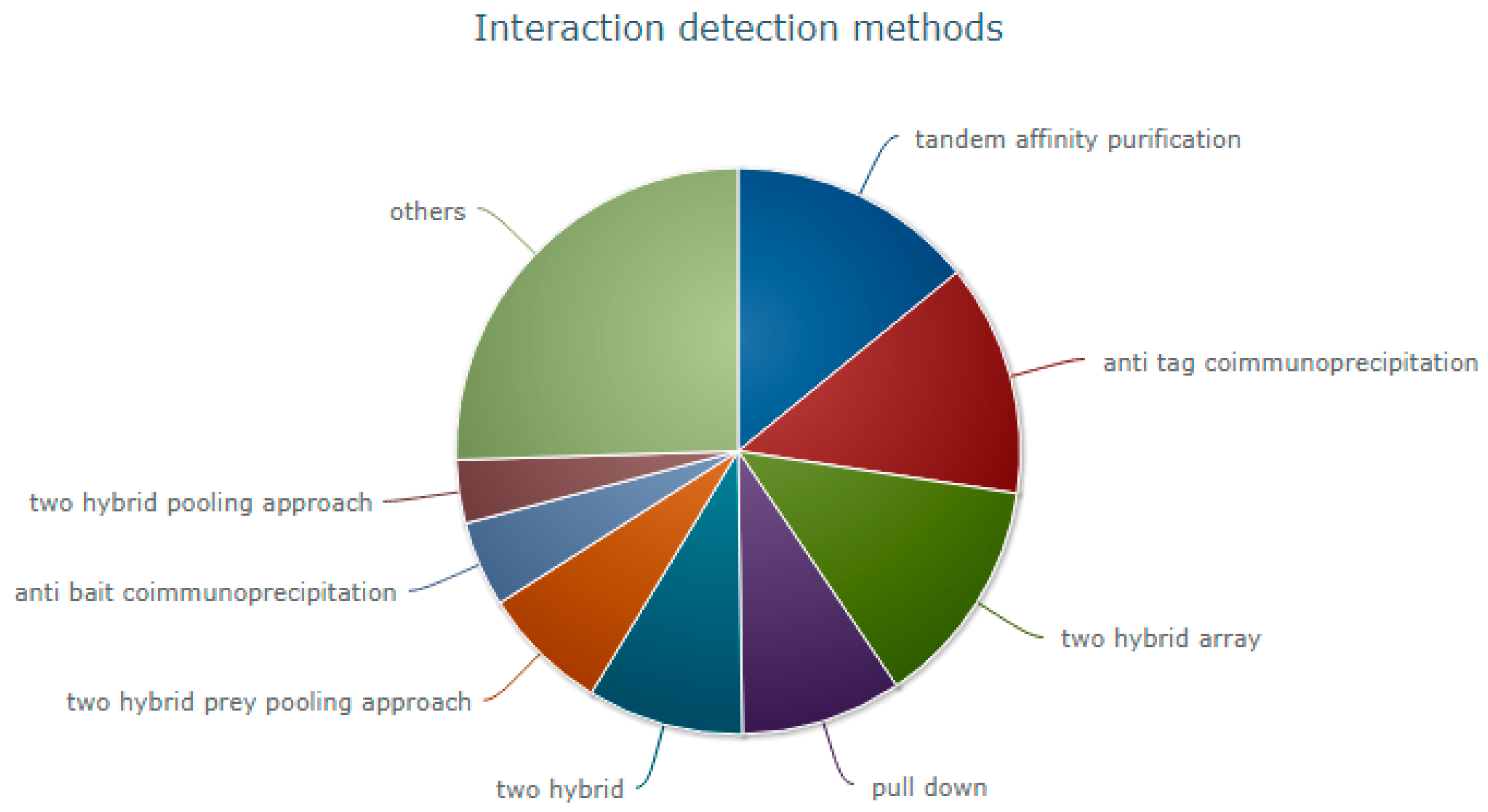
2. Experimental Methods for Determining Protein–Protein Interactions (PPIs)
Sparse Experimental Data on Prediction of Protein Interactions and Modelling of Their Complexes
3. In Silico Methods for Modelling Protein–Protein Complexes
3.1. Computational Methods for Protein–Protein Interaction Modelling
3.2. Quality Assessment of Quaternary Structure Predictions
4. CASP, CAPRI and CAMEO: Driving the Development of in Silico Quaternary Structure Prediction Methods and Their Fusion with In Vitro Data
4.1. CASP
4.2. CAMEO
4.3. CAPRI
5. Conclusions
Author Contributions
Conflicts of Interest
Abbreviations
| NMR | Nuclear magnetic resonance |
| SAXS | Small-angle X-ray scattering |
| PPI | Protein–protein interaction |
| TAP | Tandem affinity purification |
| Co-IP | Co-immunoprecipitation |
| CD | Circular Dichroism |
| ECs | Evolutionary couplings |
| GPU | Graphics processing unit |
| NOE | Nuclear Overhauser effect |
| RDC | Residual dipolar coupling |
| Cryo-EM | Cryo-electron microscopy |
| PDB | Protein Data Bank |
| RMSD | Root mean square deviation |
| CASP | Critical Assessment of techniques for Structure Prediction |
| CAPRI | Critical assessment of prediction of interactions |
| CAMEO | Continuous automated model evaluation |
| LDDT | Local distance difference test on all atoms |
| 3DZD | 3D Zernike descriptor |
| FFT | Fast Fourier transform |
References
- Levinthal’s Paradox. Available online: http://web.archive.org/web/20110523080407/http://www-miller.ch.cam.ac.uk/levinthal/levinthal.html (accessed on 25 May 2015).
- RCSB PDB—Holdings Report. Available online: https://www.rcsb.org/pdb/statistics/holdings.do (accessed on 22 November 2017).
- Emwas, A.-H.M. The strengths and weaknesses of NMR spectroscopy and mass spectrometry with particular focus on metabolomics research. Methods Mol. Biol. 2015, 1277, 161–193. [Google Scholar] [CrossRef] [PubMed]
- Kikhney, A.G.; Svergun, D.I. A practical guide to small angle X-ray scattering (SAXS) of flexible and intrinsically disordered proteins. FEBS Lett. 2015, 589, 2570–2577. [Google Scholar] [CrossRef] [PubMed]
- Szklarczyk, D.; Jensen, L.J. Protein-Protein Interaction Databases; Meyerkord, C.L., Fu, H., Eds.; Methods in Molecular Biology; Springer: New York, NY, USA, 2015; ISBN 978-1-4939-2424-0. [Google Scholar]
- Ehrenberger, T.; Cantley, L.C.; Yaffe, M.B. Computational Prediction of Protein-Protein Interaction; Meyerkord, C.L., Fu, H., Eds.; Springer: New York, NY, USA, 2015; ISBN 978-1-4939-2424-0. [Google Scholar]
- Moult, J.; Fidelis, K.; Kryshtafovych, A.; Schwede, T.; Tramontano, A. Critical Assessment of Methods of Protein Structure Prediction (CASP)—Round XII. Proteins Struct. Funct. Bioinform. 2017. [Google Scholar] [CrossRef] [PubMed]
- Janin, J. Welcome to CAPRI: A critical assessment of PRedicted interactions. Proteins Struct. Funct. Genet. 2002, 47, 257–257. [Google Scholar] [CrossRef]
- Haas, J.; Roth, S.; Arnold, K.; Kiefer, F.; Schmidt, T.; Bordoli, L.; Schwede, T. The protein model portal—A comprehensive resource for protein structure and model information. Database 2013, 2013. [Google Scholar] [CrossRef] [PubMed]
- IntAct. Available online: https://www.ebi.ac.uk/intact/ (accessed on 4 October 2017).
- Günzl, A.; Schimanski, B. Tandem Affinity Purification of Proteins. In Current Protocols in Protein Science; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2001; ISBN 978-0-471-14086-3. [Google Scholar]
- Puig, O.; Caspary, F.; Rigaut, G.; Rutz, B.; Bouveret, E.; Bragado-Nilsson, E.; Wilm, M.; Seraphin, B. The tandem affinity purification (TAP) method: A general procedure of protein complex purification. Methods 2001, 24, 218–229. [Google Scholar] [CrossRef] [PubMed]
- Bauch, A.; Superti-Furga, G. Charting protein complexes, signaling pathways, and networks in the immune system. Immunol. Rev. 2006, 210, 187–207. [Google Scholar] [CrossRef] [PubMed]
- Gavin, A.-C.; Bosche, M.; Krause, R.; Grandi, P.; Marzioch, M.; Bauer, A.; Schultz, J.; Rick, J.M.; Michon, A.-M.; Cruciat, C.-M.; et al. Functional organization of the yeast proteome by systematic analysis of protein complexes. Nature 2002, 415, 141–147. [Google Scholar] [CrossRef] [PubMed]
- Lee, C. Coimmunoprecipitation Assay. In Circadian Rhythms: Methods and Protocols; Rosato, E., Ed.; Humana Press: Totowa, NJ, USA, 2007; pp. 401–406. ISBN 978-1-59745-257-1. [Google Scholar]
- Ren, L.; Emery, D.; Kaboord, B.; Chang, E.; Qoronfleh, M.W. Improved immunomatrix methods to detect protein–protein interactions. J. Biochem. Biophys. Methods 2003, 57, 143–157. [Google Scholar] [CrossRef]
- Phizicky, E.M.; Fields, S. Protein–protein interactions: Methods for detection and analysis. Microbiol. Rev. 1995, 59, 94–123. [Google Scholar] [PubMed]
- Fields, S.; Song, O. A novel genetic system to detect protein–protein interactions. Nature 1989, 340, 245–246. [Google Scholar] [CrossRef] [PubMed]
- Estojak, J.; Brent, R.; Golemis, E.A. Correlation of two-hybrid affinity data with in vitro measurements. Mol. Cell. Biol. 1995, 15, 5820–5829. [Google Scholar] [CrossRef] [PubMed]
- Deane, C.M.; Salwinski, L.; Xenarios, I.; Eisenberg, D. Protein interactions: Two methods for assessment of the reliability of high throughput observations. Mol. Cell. Proteom. 2002, 1, 349–356. [Google Scholar] [CrossRef]
- Semple, J.I.; Sanderson, C.M.; Campbell, R.D. The jury is out on “guilt by association” trials. Brief. Funct. Genomic. 2002, 1, 40–52. [Google Scholar] [CrossRef]
- Louche, A.; Salcedo, S.P.; Bigot, S. Protein–protein interactions: Pull-down assays. Methods Mol. Biol. 2017, 1615, 247–255. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.N.; Goodrich, J.A. Protein–protein interaction assays: Eliminating false positive interactions. Nat. Methods 2006, 3, 135–139. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Snyder, M. Protein chip technology. Curr. Opin. Chem. Biol. 2003, 7, 55–63. [Google Scholar] [CrossRef]
- Zhu, H.; Klemic, J.F.; Chang, S.; Bertone, P.; Casamayor, A.; Klemic, K.G.; Smith, D.; Gerstein, M.; Reed, M.A.; Snyder, M. Analysis of yeast protein kinases using protein chips. Nat. Genet. 2000, 26, 283–289. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Xu, D. Computational analyses of high-throughput protein–protein interaction data. Curr. Protein Pept. Sci. 2003, 4, 159–181. [Google Scholar] [CrossRef] [PubMed]
- O’Connell, M.R.; Gamsjaeger, R.; Mackay, J.P. The structural analysis of protein–protein interactions by NMR spectroscopy. Proteomics 2009, 9, 5224–5232. [Google Scholar] [CrossRef] [PubMed]
- Gao, G.; Williams, J.G.; Campbell, S.L. Protein–protein Interaction analysis by nuclear magnetic resonance spectroscopy. In Protein–Protein Interactions: Methods and Applications; Fu, H., Ed.; Humana Press: Totowa, NJ, USA, 2004; pp. 79–91. ISBN 978-1-59259-762-8. [Google Scholar]
- Hermjakob, H.; Montecchi-Palazzi, L.; Bader, G.; Wojcik, J.; Salwinski, L.; Ceol, A.; Moore, S.; Orchard, S.; Sarkans, U.; von Mering, C.; et al. The HUPO PSI’s molecular interaction format—A community standard for the representation of protein interaction data. Nat. Biotechnol. 2004, 22, 177–183. [Google Scholar] [CrossRef] [PubMed]
- Narayan, P.; Orte, A.; Clarke, R.W.; Bolognesi, B.; Hook, S.; Ganzinger, K.A.; Meehan, S.; Wilson, M.R.; Dobson, C.M.; Klenerman, D. The extracellular chaperone clusterin sequesters oligomeric forms of the amyloid-β(1–40) peptide. Nat. Struct. Mol. Biol. 2011, 19, 79–83. [Google Scholar] [CrossRef] [PubMed]
- Heegaard, N.H. Affinity in electrophoresis. Electrophoresis 2009, 30, S229–S239. [Google Scholar] [CrossRef] [PubMed]
- Orchard, S.; Hermjakob, H.; Julian, R.K.J.; Runte, K.; Sherman, D.; Wojcik, J.; Zhu, W.; Apweiler, R. Common interchange standards for proteomics data: Public availability of tools and schema. Proteomics 2004, 4, 490–491. [Google Scholar] [CrossRef] [PubMed]
- Manzano, C.; Contreras-Martel, C.; El Mortaji, L.; Izore, T.; Fenel, D.; Vernet, T.; Schoehn, G.; di Guilmi, A.M.; Dessen, A. Sortase-mediated pilus fiber biogenesis in Streptococcus pneumoniae. Structure 2008, 16, 1838–1848. [Google Scholar] [CrossRef] [PubMed]
- Rogers, K.R. Principles of affinity-based biosensors. Mol. Biotechnol. 2000, 14, 109–129. [Google Scholar] [CrossRef]
- Wallace, B.A.; Janes, R.W. Synchrotron radiation circular dichroism spectroscopy of proteins: Secondary structure, fold recognition and structural genomics. Curr. Opin. Chem. Biol. 2001, 5, 567–571. [Google Scholar] [CrossRef]
- Jelesarov, I.; Bosshard, H.R. Isothermal titration calorimetry and differential scanning calorimetry as complementary tools to investigate the energetics of biomolecular recognition. J. Mol. Recognit. 1999, 12, 3–18. [Google Scholar] [CrossRef]
- Murata, K.; Mitsuoka, K.; Hirai, T.; Walz, T.; Agre, P.; Heymann, J.B.; Engel, A.; Fujiyoshi, Y. Structural determinants of water permeation through aquaporin-1. Nature 2000, 407, 599–605. [Google Scholar] [CrossRef] [PubMed]
- Honke, K.; Kotani, N. The enzyme-mediated activation of radical source reaction: A new approach to identify partners of a given molecule in membrane microdomains. J. Neurochem. 2011, 116, 690–695. [Google Scholar] [CrossRef] [PubMed]
- Van Liempd, S.; Morrison, D.; Sysmans, L.; Nelis, P.; Mortishire-Smith, R. Development and validation of a higher-throughput equilibrium dialysis assay for plasma protein binding. J. Lab. Autom. 2011, 16, 56–67. [Google Scholar] [CrossRef] [PubMed]
- Muchowski, P.J.; Schaffar, G.; Sittler, A.; Wanker, E.E.; Hayer-Hartl, M.K.; Hartl, F.U. Hsp70 and hsp40 chaperones can inhibit self-assembly of polyglutamine proteins into amyloid-like fibrils. Proc. Natl. Acad. Sci. USA 2000, 97, 7841–7846. [Google Scholar] [CrossRef] [PubMed]
- Demirdoven, N.; Cheatum, C.M.; Chung, H.S.; Khalil, M.; Knoester, J.; Tokmakoff, A. Two-dimensional infrared spectroscopy of antiparallel β-sheet secondary structure. J. Am. Chem. Soc. 2004, 126, 7981–7990. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prakasam, A.K.; Maruthamuthu, V.; Leckband, D.E. Similarities between heterophilic and homophilic cadherin adhesion. Proc. Natl. Acad. Sci. USA 2006, 103, 15434–15439. [Google Scholar] [CrossRef] [PubMed]
- Leavitt, S.; Freire, E. Direct measurement of protein binding energetics by isothermal titration calorimetry. Curr. Opin. Struct. Biol. 2001, 11, 560–566. [Google Scholar] [CrossRef]
- Murphy, R.M. Static and dynamic light scattering of biological macromolecules: What can we learn? Curr. Opin. Biotechnol. 1997, 8, 25–30. [Google Scholar] [CrossRef]
- Badr, C.E. Bioluminescence imaging: Basics and practical limitations. Methods Mol. Biol. 2014, 1098, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Duhr, S.; Braun, D. Why molecules move along a temperature gradient. Proc. Natl. Acad. Sci. USA 2006, 103, 19678–19682. [Google Scholar] [CrossRef] [PubMed]
- Chatake, T.; Tanaka, I.; Umino, H.; Arai, S.; Niimura, N. The hydration structure of a Z-DNA hexameric duplex determined by a neutron diffraction technique. Acta Crystallogr. D Biol. Crystallogr. 2005, 61, 1088–1098. [Google Scholar] [CrossRef] [PubMed]
- Hanson, B.L. Getting protein solvent structures down cold. Proc. Natl. Acad. Sci. USA 2004, 101, 16393–16394. [Google Scholar] [CrossRef] [PubMed]
- Pellecchia, M.; Sem, D.S.; Wuthrich, K. NMR in drug discovery. Nat. Rev. Drug Discov. 2002, 1, 211–219. [Google Scholar] [CrossRef] [PubMed]
- Rammensee, S.; Slotta, U.; Scheibel, T.; Bausch, A.R. Assembly mechanism of recombinant spider silk proteins. Proc. Natl. Acad. Sci. USA 2008, 105, 6590–6595. [Google Scholar] [CrossRef] [PubMed]
- Udenfriend, S.; Gerber, L.D.; Brink, L.; Spector, S. Scintillation proximity radioimmunoassay utilizing 125I-labeled ligands. Proc. Natl. Acad. Sci. USA 1985, 82, 8672–8676. [Google Scholar] [CrossRef] [PubMed]
- Kranz, J.K.; Clemente, J.C. Binding techniques to study the allosteric energy cycle. Methods Mol. Biol. 2012, 796, 3–17. [Google Scholar] [CrossRef] [PubMed]
- Cotruvo, J.A.J.; Stubbe, J. NrdI, a flavodoxin involved in maintenance of the diferric-tyrosyl radical cofactor in Escherichia coli class Ib ribonucleotide reductase. Proc. Natl. Acad. Sci. USA 2008, 105, 14383–14388. [Google Scholar] [CrossRef] [PubMed]
- Lowe, T.M.; Eddy, S.R. A computational screen for methylation guide snoRNAs in yeast. Science 1999, 283, 1168–1171. [Google Scholar] [CrossRef] [PubMed]
- Modesti, M.; Ristic, D.; van der Heijden, T.; Dekker, C.; van Mameren, J.; Peterman, E.J.G.; Wuite, G.J.L.; Kanaar, R.; Wyman, C. Fluorescent human RAD51 reveals multiple nucleation sites and filament segments tightly associated along a single DNA molecule. Structure 2007, 15, 599–609. [Google Scholar] [CrossRef] [PubMed]
- Unger, V.M. Electron cryomicroscopy methods. Curr. Opin. Struct. Biol. 2001, 11, 548–554. [Google Scholar] [CrossRef]
- Tanabe, Y.; Fujita, E.; Momoi, T. FOXP2 promotes the nuclear translocation of POT1, but FOXP2(R553H), mutation related to speech-language disorder, partially prevents it. Biochem. Biophys. Res. Commun. 2011, 410, 593–596. [Google Scholar] [CrossRef] [PubMed]
- Denhardt, D.T. Mechanism of action of antisense RNA. Sometime inhibition of transcription, processing, transport, or translation. Ann. N. Y. Acad. Sci. 1992, 660, 70–76. [Google Scholar] [CrossRef] [PubMed]
- Chiu, Y.-L.; Rana, T.M. RNAi in human cells: Basic structural and functional features of small interfering RNA. Mol. Cell 2002, 10, 549–561. [Google Scholar] [CrossRef]
- Karimova, G.; Pidoux, J.; Ullmann, A.; Ladant, D. A bacterial two-hybrid system based on a reconstituted signal transduction pathway. Proc. Natl. Acad. Sci. USA 1998, 95, 5752–5756. [Google Scholar] [CrossRef] [PubMed]
- Rossi, F.; Charlton, C.A.; Blau, H.M. Monitoring protein–protein interactions in intact eukaryotic cells by β-galactosidase complementation. Proc. Natl. Acad. Sci. USA 1997, 94, 8405–8410. [Google Scholar] [CrossRef] [PubMed]
- Galarneau, A.; Primeau, M.; Trudeau, L.-E.; Michnick, S.W. β-lactamase protein fragment complementation assays as in vivo and in vitro sensors of protein protein interactions. Nat. Biotechnol. 2002, 20, 619–622. [Google Scholar] [CrossRef] [PubMed]
- Hu, C.-D.; Chinenov, Y.; Kerppola, T.K. Visualization of interactions among bZIP and Rel family proteins in living cells using bimolecular fluorescence complementation. Mol. Cell 2002, 9, 789–798. [Google Scholar] [CrossRef]
- Remy, I.; Michnick, S.W. Clonal selection and in vivo quantitation of protein interactions with protein-fragment complementation assays. Proc. Natl. Acad. Sci. USA 1999, 96, 5394–5399. [Google Scholar] [CrossRef] [PubMed]
- Lemmens, I.; Eyckerman, S.; Zabeau, L.; Catteeuw, D.; Vertenten, E.; Verschueren, K.; Huylebroeck, D.; Vandekerckhove, J.; Tavernier, J. Heteromeric MAPPIT: A novel strategy to study modification-dependent protein–protein interactions in mammalian cells. Nucleic Acids Res. 2003, 31, e75. [Google Scholar] [CrossRef] [PubMed]
- Stefan, E.; Aquin, S.; Berger, N.; Landry, C.R.; Nyfeler, B.; Bouvier, M.; Michnick, S.W. Quantification of dynamic protein complexes using Renilla luciferase fragment complementation applied to protein kinase A activities in vivo. Proc. Natl. Acad. Sci. USA 2007, 104, 16916–16921. [Google Scholar] [CrossRef] [PubMed]
- Hubsman, M.; Yudkovsky, G.; Aronheim, A. A novel approach for the identification of protein–protein interaction with integral membrane proteins. Nucleic Acids Res. 2001, 29, E18. [Google Scholar] [CrossRef] [PubMed]
- Kato, N.; Jones, J. The split luciferase complementation assay. Methods Mol. Biol. 2010, 655, 359–376. [Google Scholar] [CrossRef] [PubMed]
- Russ, W.P.; Engelman, D.M. TOXCAT: A measure of transmembrane helix association in a biological membrane. Proc. Natl. Acad. Sci. USA 1999, 96, 863–868. [Google Scholar] [CrossRef] [PubMed]
- Dyer, K.N.; Hammel, M.; Rambo, R.P.; Tsutakawa, S.E.; Rodic, I.; Classen, S.; Tainer, J.A.; Hura, G.L. High-throughput SAXS for the characterization of biomolecules in solution: A practical approach. Methods Mol. Biol. 2014, 1091, 245–258. [Google Scholar] [CrossRef] [PubMed]
- Jiménez-García, B.; Pons, C.; Svergun, D.I.; Bernadó, P.; Fernández-Recio, J. pyDockSAXS: Protein–protein complex structure by SAXS and computational docking. Nucleic Acids Res. 2015. [Google Scholar] [CrossRef] [PubMed]
- Skou, S.; Gillilan, R.E.; Ando, N. Synchrotron-based small-angle X-ray scattering of proteins in solution. Nat. Protoc. 2014, 9, 1727–1739. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Huang, Y.J.; Hopf, T.A.; Sander, C.; Marks, D.S.; Montelione, G.T. Protein structure determination by combining sparse NMR data with evolutionary couplings. Nat. Meth. 2015, 12, 751–754. [Google Scholar] [CrossRef] [PubMed]
- Latek, D.; Ekonomiuk, D.; Kolinski, A. Protein structure prediction: Combining de novo modeling with sparse experimental data. J. Comput. Chem. 2007, 28, 1668–1676. [Google Scholar] [CrossRef] [PubMed]
- Robinson, P.J.; Trnka, M.J.; Pellarin, R.; Greenberg, C.H.; Bushnell, D.A.; Davis, R.; Burlingame, A.L.; Sali, A.; Kornberg, R.D. Molecular architecture of the yeast Mediator complex. eLife 2015, 4. [Google Scholar] [CrossRef] [PubMed]
- Tang, X.; Bruce, J.E. Chemical cross-linking for protein–protein interaction studies. In Mass Spectrometry of Proteins and Peptides: Methods and Protocols; Lipton, M.S., Paša-Tolic, L., Eds.; Humana Press: Totowa, NJ, USA, 2009; pp. 283–293. ISBN 978-1-59745-493-3. [Google Scholar]
- Kluger, R.; Alagic, A. Chemical cross-linking and protein–protein interactions—A review with illustrative protocols. Bioorg. Chem. 2004, 32, 451–472. [Google Scholar] [CrossRef] [PubMed]
- Nogales, E. The development of cryo-EM into a mainstream structural biology technique. Nat. Methods 2016, 13, 24–27. [Google Scholar] [CrossRef] [PubMed]
- Bai, X.; McMullan, G.; Scheres, S.H. How cryo-EM is revolutionizing structural biology. Trends Biochem. Sci. 2015, 40, 49–57. [Google Scholar] [CrossRef] [PubMed]
- Statistics: EMDataBank. Available online: http://www.emdatabank.org/statistics.html (accessed on 10 October 2017).
- Skiniotis, G. A snapshot of cryo-EM. Protein Sci. 2017, 26, 5–7. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.-W.; Wang, J.-W. How cryo-electron microscopy and X-ray crystallography complement each other. Protein Sci. 2017, 26, 32–39. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Tan, D.; Woolford, J.L.; Dong, M.-Q.; Gao, N. Atomic modeling of the ITS2 ribosome assembly subcomplex from cryo-EM together with mass spectrometry-identified protein–protein crosslinks. Protein Sci. 2017, 26, 103–112. [Google Scholar] [CrossRef] [PubMed]
- Gadkari, R.A.; Srinivasan, N. Prediction of protein–protein interactions in dengue virus coat proteins guided by low resolution cryoEM structures. BMC Struct. Biol. 2010, 10, 17. [Google Scholar] [CrossRef] [PubMed]
- Gadkari, R.A.; Varughese, D.; Srinivasan, N. Recognition of interaction interface residues in low-resolution structures of protein assemblies solely from the positions of Cα atoms. PLoS ONE 2009, 4. [Google Scholar] [CrossRef] [PubMed]
- Bernstein, F.C.; Koetzle, T.F.; Williams, G.J.; Meyer, E.F.; Brice, M.D.; Rodgers, J.R.; Kennard, O.; Shimanouchi, T.; Tasumi, M. The protein data bank. FEBS J. 1977, 80, 319–324. [Google Scholar]
- Gray, J.J.; Moughon, S.; Wang, C.; Schueler-Furman, O.; Kuhlman, B.; Rohl, C.A.; Baker, D. Protein–protein docking with simultaneous optimization of rigid-body displacement and side-chain conformations. J. Mol. Biol. 2003, 331, 281–299. [Google Scholar] [CrossRef]
- Chen, R.; Li, L.; Weng, Z. ZDOCK: An initial-stage protein-docking algorithm. Proteins Struct. Funct. Bioinform. 2003, 52, 80–87. [Google Scholar] [CrossRef] [PubMed]
- Pierce, B.G.; Wiehe, K.; Hwang, H.; Kim, B.-H.; Vreven, T.; Weng, Z. ZDOCK server: Interactive docking prediction of protein–protein complexes and symmetric multimers. Bioinformatics 2014, 30, 1771–1773. [Google Scholar] [CrossRef] [PubMed]
- Pierce, B.; Weng, Z. ZRANK: Reranking protein docking predictions with an optimized energy function. Proteins Struct. Funct. Bioinform. 2007, 67, 1078–1086. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Schueler-Furman, O.; Andre, I.; London, N.; Fleishman, S.J.; Bradley, P.; Qian, B.; Baker, D. RosettaDock in CAPRI rounds 6–12. Proteins Struct. Funct. Bioinform. 2007, 69, 758–763. [Google Scholar] [CrossRef] [PubMed]
- Sønderby, P.; Rinnan, Å.; Madsen, J.J.; Harris, P.; Bukrinski, J.T.; Peters, G.H.J. Small-angle X-ray scattering data in combination with RosettaDock improves the docking energy landscape. J. Chem. Inf. Model. 2017, 57, 2463–2475. [Google Scholar] [CrossRef] [PubMed]
- Katchalski-Katzir, E.; Shariv, I.; Eisenstein, M.; Friesem, A.A.; Aflalo, C.; Vakser, I.A. Molecular surface recognition: Determination of geometric fit between proteins and their ligands by correlation techniques. Proc. Natl. Acad. Sci. USA 1992, 89, 2195–2199. [Google Scholar] [CrossRef] [PubMed]
- Tovchigrechko, A.; Vakser, I.A. GRAMM-X public web server for protein–protein docking. Nucleic Acids Res. 2006, 34, W310–W314. [Google Scholar] [CrossRef] [PubMed]
- Macindoe, G.; Mavridis, L.; Venkatraman, V.; Devignes, M.-D.; Ritchie, D.W. HexServer: An FFT-based protein docking server powered by graphics processors. Nucleic Acids Res. 2010, 38, W445–W449. [Google Scholar] [CrossRef] [PubMed]
- Ohue, M.; Shimoda, T.; Suzuki, S.; Matsuzaki, Y.; Ishida, T.; Akiyama, Y. MEGADOCK 4.0: An ultra–high-performance protein–protein docking software for heterogeneous supercomputers. Bioinformatics 2014, 30, 3281–3283. [Google Scholar] [CrossRef] [PubMed]
- Garzon, J.I.; Lopéz-Blanco, J.R.; Pons, C.; Kovacs, J.; Abagyan, R.; Fernandez-Recio, J.; Chacon, P. FRODOCK: A new approach for fast rotational protein–protein docking. Bioinformatics 2009, 25, 2544–2551. [Google Scholar] [CrossRef] [PubMed]
- Tobi, D. Designing coarse grained-and atom based-potentials for protein–protein docking. BMC Struct. Biol. 2010, 10, 40. [Google Scholar] [CrossRef] [PubMed]
- Moal, I.H.; Torchala, M.; Bates, P.A.; Fernández-Recio, J. The scoring of poses in protein–protein docking: current capabilities and future directions. BMC Bioinform. 2013, 14, 286. [Google Scholar] [CrossRef] [PubMed]
- Pierce, B.; Tong, W.; Weng, Z. M-ZDOCK: A grid-based approach for Cn symmetric multimer docking. Bioinformatics 2005, 21, 1472–1478. [Google Scholar] [CrossRef] [PubMed]
- Kozakov, D.; Hall, D.R.; Xia, B.; Porter, K.A.; Padhorny, D.; Yueh, C.; Beglov, D.; Vajda, S. The ClusPro web server for protein–protein docking. Nat. Protoc. 2017, 12, 255–278. [Google Scholar] [CrossRef] [PubMed]
- Comeau, S.R.; Gatchell, D.W.; Vajda, S.; Camacho, C.J. ClusPro: An automated docking and discrimination method for the prediction of protein complexes. Bioinformatics 2004, 20, 45–50. [Google Scholar] [CrossRef] [PubMed]
- Yueh, C.; Hall, D.R.; Xia, B.; Padhorny, D.; Kozakov, D.; Vajda, S. ClusPro-DC: Dimer classification by the CLUSPRO server for protein–protein docking. J. Mol. Biol. 2017, 429, 372–381. [Google Scholar] [CrossRef] [PubMed]
- Xia, B.; Mamonov, A.; Leysen, S.; Allen, K.N.; Strelkov, S.V.; Paschalidis, I.C.; Vajda, S.; Kozakov, D. Accounting for observed small angle X-ray scattering profile in the protein–protein docking server cluspro. J. Comput. Chem. 2015, 36, 1568–1572. [Google Scholar] [CrossRef] [PubMed]
- Duhovny, D.; Nussinov, R.; Wolfson, H.J. Efficient unbound docking of rigid molecules. In International Workshop on Algorithms in Bioinformatics; Springer: New York, NY, USA, 2002; pp. 185–200. [Google Scholar]
- Esquivel-Rodríguez, J.; Yang, Y.D.; Kihara, D. Multi-LZerD: Multiple protein docking for asymmetric complexes. Proteins Struct. Funct. Bioinform. 2012. [Google Scholar] [CrossRef] [PubMed]
- Schneidman-Duhovny, D.; Inbar, Y.; Polak, V.; Shatsky, M.; Halperin, I.; Benyamini, H.; Barzilai, A.; Dror, O.; Haspel, N.; Nussinov, R. Taking geometry to its edge: Fast unbound rigid (and hinge-bent) docking. Proteins Struct. Funct. Bioinform. 2003, 52, 107–112. [Google Scholar] [CrossRef] [PubMed]
- Peterson, L.X.; Kim, H.; Esquivel-Rodriguez, J.; Roy, A.; Han, X.; Shin, W.-H.; Zhang, J.; Terashi, G.; Lee, M.; Kihara, D. Human and server docking prediction for CAPRI round 30–35 using LZerD with combined scoring functions: Scoring LZerD CAPRI Docking Predictions. Proteins Struct. Funct. Bioinform. 2017, 85, 513–527. [Google Scholar] [CrossRef] [PubMed]
- Peterson, L.X.; Shin, W.-H.; Kim, H.; Kihara, D. Improved performance in CAPRI round 37 using LZerD docking and template-based modeling with combined scoring functions. Proteins Struct. Funct. Bioinform. 2017. [Google Scholar] [CrossRef] [PubMed]
- Pierce, B.G.; Hourai, Y.; Weng, Z. Accelerating protein docking in ZDOCK using an advanced 3D convolution library. PLoS ONE 2011, 6, e24657. [Google Scholar] [CrossRef] [PubMed]
- Venkatraman, V.; Yang, Y.D.; Sael, L.; Kihara, D. Protein–protein docking using region-based 3D Zernike descriptors. BMC Bioinform. 2009, 10, 407. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y. TM-align: A protein structure alignment algorithm based on the TM-score. Nucleic Acids Res. 2005, 33, 2302–2309. [Google Scholar] [CrossRef] [PubMed]
- Maghrabi, A.H.A.; McGuffin, L.J. ModFOLD6: An accurate web server for the global and local quality estimation of 3D protein models. Nucleic Acids Res. 2017, 45, W416–W421. [Google Scholar] [CrossRef] [PubMed]
- Uziela, K.; Shu, N.; Wallner, B.; Elofsson, A. ProQ3: Improved model quality assessments using Rosetta energy terms. Sci. Rep. 2016, 6. [Google Scholar] [CrossRef] [PubMed]
- Uziela, K.; Hurtado, D.M.; Shu, N.; Wallner, B.; Elofsson, A. ProQ3D: Improved model quality assessments using deep learning. Bioinformatics 2017, 33, 1578–1580. [Google Scholar] [CrossRef] [PubMed]
- Elofsson, A.; Joo, K.; Keasar, C.; Lee, J.; Maghrabi, A.H.A.; Manavalan, B.; McGuffin, L.J.; Ménendez Hurtado, D.; Mirabello, C.; Pilstål, R.; et al. Methods for estimation of model accuracy in CASP12. Proteins Struct. Funct. Bioinform. 2017. [Google Scholar] [CrossRef] [PubMed]
- Shimoda, T.; Ishida, T.; Suzuki, S.; Ohue, M.; Akiyama, Y. MEGADOCK-GPU: Acceleration of—Docking Calculation on GPUs. In Proceedings of the International Conference on Bioinformatics, Computational Biology and Biomedical Informatics; BCB’13; ACM: New York, NY, USA, 2013; pp. 883:883–883:889. [Google Scholar]
- Lensink, M.F.; Velankar, S.; Kryshtafovych, A.; Huang, S.-Y.; Schneidman-Duhovny, D.; Sali, A.; Segura, J.; Fernandez-Fuentes, N.; Viswanath, S.; Elber, R.; et al. Prediction of homo- and hetero-protein complexes by protein docking and template-based modeling: A CASP-CAPRI experiment. Proteins Struct. Funct. Bioinform. 2016. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, S.; Zhang, Y. MM-align: A quick algorithm for aligning multiple-chain protein complex structures using iterative dynamic programming. Nucleic Acids Res. 2009, 37, e83. [Google Scholar] [CrossRef] [PubMed]
- McGuffin, L.J.; Atkins, J.D.; Salehe, B.R.; Shuid, A.N.; Roche, D.B. IntFOLD: An integrated server for modelling protein structures and functions from amino acid sequences. Nucleic Acids Res. 2015. [Google Scholar] [CrossRef] [PubMed]
- McGuffin, L.J.; Roche, D.B. Automated tertiary structure prediction with accurate local model quality assessment using the intfold-ts method. Proteins Struct. Funct. Bioinform. 2011, 79, 137–146. [Google Scholar] [CrossRef] [PubMed]
- Roche, D.B.; Buenavista, M.T.; Tetchner, S.J.; McGuffin, L.J. The IntFOLD server: An integrated web resource for protein fold recognition, 3D model quality assessment, intrinsic disorder prediction, domain prediction and ligand binding site prediction. Nucleic Acids Res. 2011, 39, W171–W176. [Google Scholar] [CrossRef] [PubMed]
- McGuffin, L.J.; Shuid, A.N.; Kempster, R.; Maghrabi, A.H.A.; Nealon, J.O.; Salehe, B.R.; Atkins, J.D.; Roche, D.B. Accurate template-based modeling in CASP12 using the IntFOLD4-TS, ModFOLD6, and ReFOLD methods. Proteins Struct. Funct. Bioinform. 2017. [Google Scholar] [CrossRef] [PubMed]
- Shuid, A.N.; Kempster, R.; McGuffin, L.J. ReFOLD: A server for the refinement of 3D protein models guided by accurate quality estimates. Nucleic Acids Res. 2017, 45, W422–W428. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharya, D.; Cheng, J. i3Drefine software for protein 3D structure refinement and its assessment in CASP10. PLoS ONE 2013, 8, e69648. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Huang, C.; Zheng, G.; Bohm, E.; Bhatele, A.; Phillips, J.C.; Yu, H.; Kale, L.V. Scalable molecular dynamics with NAMD on the IBM Blue Gene/L system. IBM J. Res. Dev. 2008, 52, 177–188. [Google Scholar] [CrossRef]
- Roche, D.B.; Buenavista, M.T.; McGuffin, L.J. Assessing the quality of modelled 3D protein structures using the ModFOLD server. Methods Mol. Biol. 2014, 1137, 83–103. [Google Scholar] [CrossRef] [PubMed]
- McGuffin, L.J.; Buenavista, M.T.; Roche, D.B. The ModFOLD4 server for the quality assessment of 3D protein models. Nucleic Acids Res. 2013, 41, W368–W372. [Google Scholar] [CrossRef] [PubMed]
- Huwe, P.J.; Xu, Q.; Shapovalov, M.V.; Modi, V.; Andrake, M.D.; Dunbrack, R.L. Biological function derived from predicted structures in CASP11: CASP11 biological function prediction. Proteins Struct. Funct. Bioinform. 2016, 84, 370–391. [Google Scholar] [CrossRef] [PubMed]
- Kryshtafovych, A.; Monastyrskyy, B.; Fidelis, K. CASP11 statistics and the prediction center evaluation system: Prediction center in CASP11. Proteins Struct. Funct. Bioinform. 2016, 84, 15–19. [Google Scholar] [CrossRef] [PubMed]
- Kryshtafovych, A.; Moult, J.; Baslé, A.; Burgin, A.; Craig, T.K.; Edwards, R.A.; Fass, D.; Hartmann, M.D.; Korycinski, M.; Lewis, R.J.; et al. Some of the most interesting CASP11 targets through the eyes of their authors: CASP11 target highlights. Proteins Struct. Funct. Bioinform. 2016, 84, 34–50. [Google Scholar] [CrossRef] [PubMed]
- Kryshtafovych, A.; Moult, J.; Bales, P.; Bazan, J.F.; Biasini, M.; Burgin, A.; Chen, C.; Cochran, F.V.; Craig, T.K.; Das, R.; et al. Challenging the state of the art in protein structure prediction: Highlights of experimental target structures for the 10th Critical Assessment of Techniques for Protein Structure Prediction Experiment CASP10: CASP10 Target Highlights. Proteins Struct. Funct. Bioinform. 2014, 82, 26–42. [Google Scholar] [CrossRef] [PubMed]
- Moult, J.; Fidelis, K.; Kryshtafovych, A.; Schwede, T.; Tramontano, A. Critical assessment of methods of protein structure prediction: Progress and new directions in round XI. Proteins 2016, 84, 4–14. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
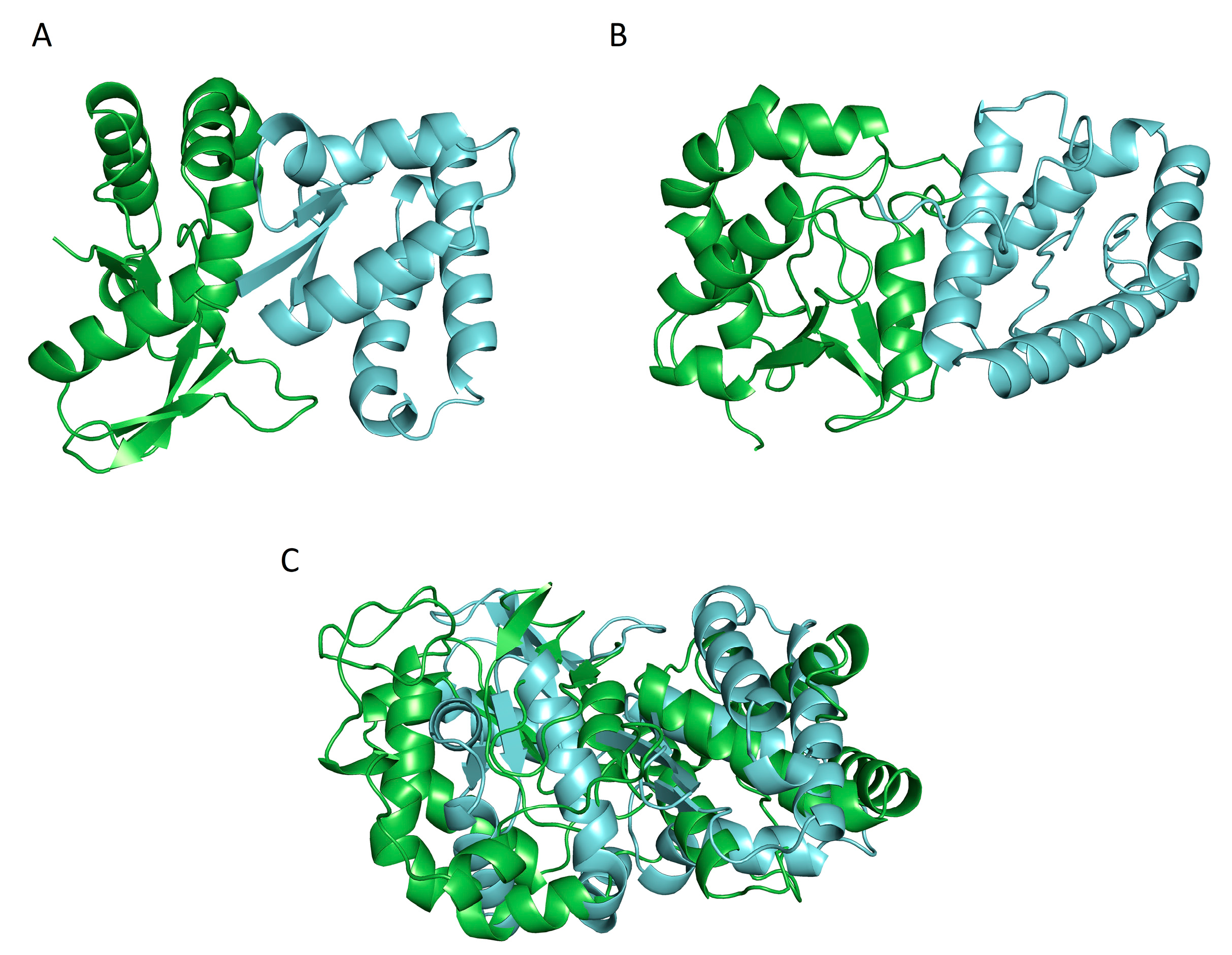{kind=link}
| Interaction Detection Method | Current Available Techniques | Reference |
|---|---|---|
| Biochemical | Affinity technology | [29] |
| Aggregation assay | [30] | |
| Chromatography technology | [29] | |
| Cosedimentation | [29] | |
| Cross-linking study | [29] | |
| Electrophoretic mobility-based method | [31] | |
| Enzymatic study | [29] | |
| Footprinting | [29] | |
| Nucleotide exchange assay | [32] | |
| Polymerization | [33] | |
| Probe interaction assay | [29] | |
| Biophysical | Biosensor | [34] |
| Circular dichroism | [35] | |
| Mass spectrometry | [29] | |
| Differential scanning calorimetry | [36] | |
| Electron diffraction | [37] | |
| Electron resonance | [29] | |
| Enzyme-mediated activation of radical sources | [38] | |
| Equilibrium dialysis | [39] | |
| Filter trap assay | [40] | |
| Fluorescence technology | [30] | |
| Infrared spectroscopy | [41] | |
| Intermolecular force | [42] | |
| Isothermal titration calorimetry | [43] | |
| Light scattering | [44] | |
| Luminescence technology | [45] | |
| Microscale thermophoresis | [46] | |
| Molecular sieving | [17] | |
| Neutron diffraction | [47] | |
| Neutron fibre diffraction | [48] | |
| Nuclear magnetic resonance | [49] | |
| Rheology measurement | [50] | |
| Scintillation proximity assay | [51] | |
| Small angle neutron scattering | [35] | |
| Thermal shift binding | [52] | |
| Ultraviolet- visible spectroscopy | [53] | |
| X-ray crystallography | [29] | |
| Genetic interference | Chemical RNA modification plus base | [54] |
| Random spore analysis | [29] | |
| Synthetic genetic analysis | [29] | |
| Imaging techniques | Atomic force microscopy | [55] |
| Confocal microscopy | [29] | |
| Electron microscopy | [56] | |
| Fluorescence microscopy | [29] | |
| Light microscopy | [29] | |
| Super-resolution microscopy | [29] | |
| X-ray tomography | [29] | |
| Phenotype-based detection assay | Nuclear translocation assay | [57] |
| Post-transcriptional interference | Antisense oligonucleotides | [29] |
| Antisense RNA | [58] | |
| RNA interference | [59] | |
| Protein complementation assays | Adenylate cyclase complementation | [60] |
| β-galactosidase complementation | [61] | |
| β lactamase complementation | [62] | |
| Bimolecular fluorescence complementation | [63] | |
| Dihydrofolate reductase reconstruction | [64] | |
| Mammalian protein–protein interaction trap | [65] | |
| Protein kinase A complementation | [66] | |
| Reverse ras recruitment system | [67] | |
| Split luciferase complementation | [68] | |
| Tox-R dimerization assay | [69] | |
| Transcriptional complementation assay | [29] |
| Name | Method | URL | Reference |
|---|---|---|---|
| RosettaDock | RosettaDock is a Monte Carlo (MC) based multi-scale docking algorithm. | https://www.rosettacommons.org/ | [87] |
| ZDOCK | FFT used to perform a 3D search of the spatial degrees of freedom between two molecules., utilizes a pairwise statistical potential in the scoring function. | http://zdock.umassmed.edu/ | [110] |
| GRAMM-X | The best surface match between molecules is determined by correlation technique using FFT, uses a smoothed Lennard-Jones potential on a fine grid during the global search FFT stage. | http://vakser.compbio.ku.edu/resources/gramm/ | [94] |
| HexServer | Uses a closed-form 6D spherical polar FFT correlation expression from which arbitrary multi-dimensional multi-property multi-resolution FFT correlations may be generated. | http://hexserver.loria.fr/ | [95] |
| MEGADOCK | MEGADOCK uses a Katchalski-Katzir algorithm and searches probable docking structures in a grid-based 3D space using FFT. MEGADOCK employs a scoring function in which only shape complementarity and electrostatics are considered. The method is set up to perform massive numbers of calculations that are run on parallel computing systems. | http://www.bi.cs.titech.ac.jp/megadock/ | [96] |
| FRODOCK | FRODOCK projects the interaction terms of a potential protein complex into 3D grid-based potentials using spherical harmonics approximations to accelerate the search; this is itself an extension of the FFT alogrithm. | http://frodock.chaconlab.org/ | [97] |
| M-ZDOCK | A grid-based FFT approach generates symmetrical multimers which are searched for the highest quality structure rather than creating predicted structures with ZDOCK and filtering for adjacent symmetrical structures. | http://zdock.umassmed.edu/m-zdock/ | [100] |
| ClusPro | The ClusPro docking algorithm evaluates multiple presumed complexes, retaining a pre-set number with encouraging surface complementarities, next a filtering method is applied to this set of structures, selecting those with good electrostatic and DE free energies for further clustering. | https://cluspro.bu.edu/home.php | [101] |
| Patchdock | Two molecules have their surfaces divided into patches based on the surface shape. The surface of the proteins is calculated, a segmentation algorithm for detection of geometric patches is applied and the patches are filtered, so that only patches with residues involved in binding are retained. A surface patch matching procedure applies geometric hashing and pose clustering matching techniques to match the patches previously detected. | https://bioinfo3d.cs.tau.ac.il/PatchDock/ | [105] |
| LZerD | Uses the 3DZD a rotational invariant mathematical surface representation of proteins to generate predictions. | http://www.kiharalab.org/proteindocking/lzerd.php | [111] |
| Multi-LZerD | Uses pairwise docking predictions from LZerD, these are then combined using a genetic algorithm and several scoring methods are used. | http://kiharalab.org/proteindocking/multilzerd.php | [106] |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nealon, J.O.; Philomina, L.S.; McGuffin, L.J. Predictive and Experimental Approaches for Elucidating Protein–Protein Interactions and Quaternary Structures. Int. J. Mol. Sci. 2017, 18, 2623. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms18122623
Nealon JO, Philomina LS, McGuffin LJ. Predictive and Experimental Approaches for Elucidating Protein–Protein Interactions and Quaternary Structures. International Journal of Molecular Sciences. 2017; 18(12):2623. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms18122623
Chicago/Turabian StyleNealon, John Oliver, Limcy Seby Philomina, and Liam James McGuffin. 2017. "Predictive and Experimental Approaches for Elucidating Protein–Protein Interactions and Quaternary Structures" International Journal of Molecular Sciences 18, no. 12: 2623. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms18122623