Phylodynamic and Genetic Diversity of Canine Parvovirus Type 2c in Taiwan

 ,
, 
Abstract
:1. Introduction
2. Results
2.1. Sequence Comparison and Amino Acid Sequence Analysis
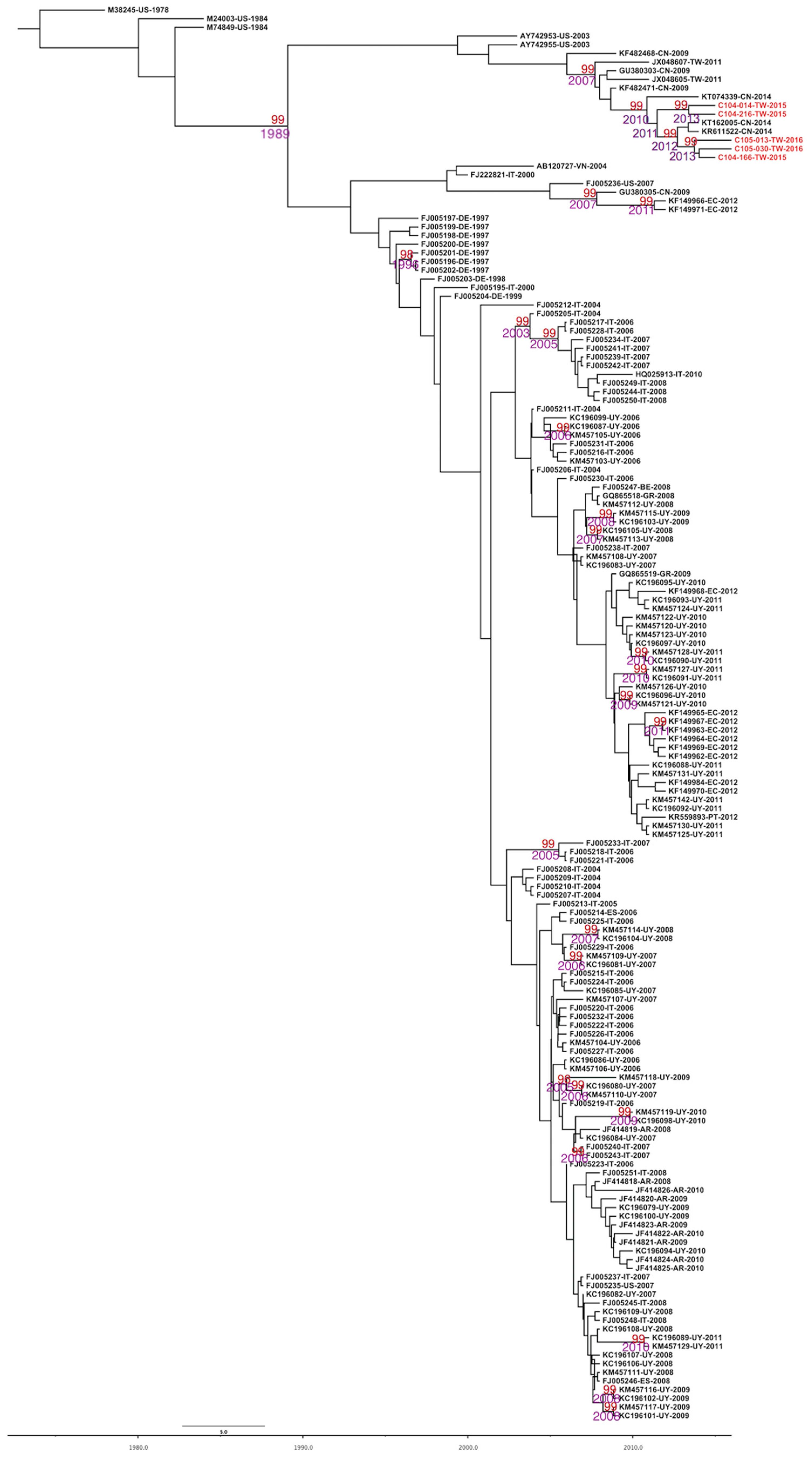
2.2. Evolutionary Rates and the Most Recent Common Ancestor of CPV-2 and CPV-2c
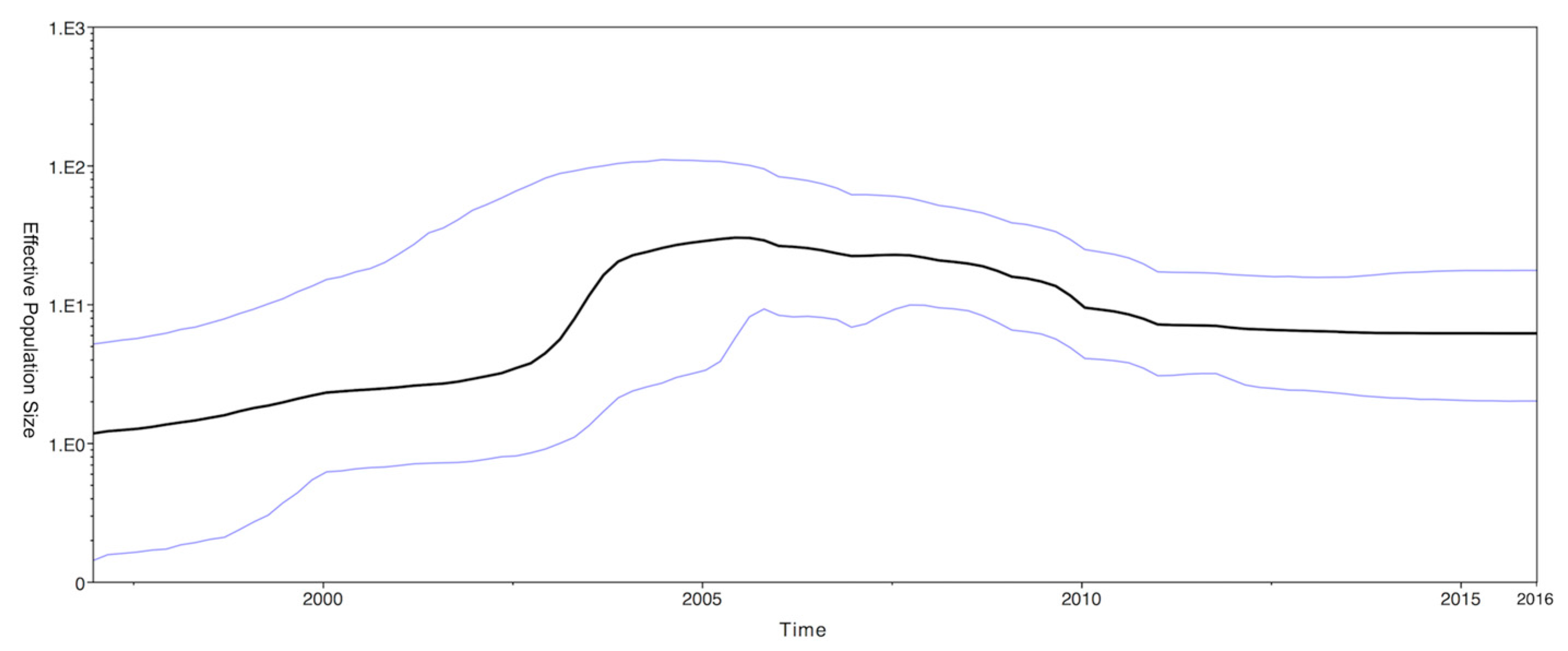
2.3. Phylodynamics of CPV-2c
2.4. Selection Pressures in the CPV-2 VP2 Protein
3. Discussion
4. Materials and Methods
4.1. Ethics Statement and Study Design
4.2. Complete VP2 Gene Amplification and Sequencing
4.3. Phylodynamic Analysis
4.4. Selection Pressure of CPV2 VP2 Protein Genes
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Shackelton, L.A.; Parrish, C.R.; Truyen, U.; Holmes, E.C. High rate of viral evolution associated with the emergence of carnivore parvovirus. Proc. Natl. Acad. Sci. USA 2005, 102, 379–384. [Google Scholar] [CrossRef] [PubMed]
- Martella, V.; Decaro, N.; Buonavoglia, C. Evolution of CPV-2 and implication for antigenic/genetic characterization. Virus Genes 2006, 33, 11–13. [Google Scholar] [CrossRef] [PubMed]
- Lopez de Turiso, J.A.; Cortes, E.; Ranz, A.; Garcia, J.; Sanz, A.; Vela, C.; Casal, J.I. Fine mapping of canine parvovirus b cell epitopes. J. Gen. Virol. 1991, 72 Pt 10, 2445–2456. [Google Scholar] [CrossRef] [PubMed]
- Chang, S.F.; Sgro, J.Y.; Parrish, C.R. Multiple amino acids in the capsid structure of canine parvovirus coordinately determine the canine host range and specific antigenic and hemagglutination properties. J. Virol. 1992, 66, 6858–6867. [Google Scholar] [PubMed]
- Palermo, L.M.; Hueffer, K.; Parrish, C.R. Residues in the apical domain of the feline and canine transferrin receptors control host-specific binding and cell infection of canine and feline parvoviruses. J. Virol. 2003, 77, 8915–8923. [Google Scholar] [CrossRef] [PubMed]
- Parker, J.S.; Parrish, C.R. Canine parvovirus host range is determined by the specific conformation of an additional region of the capsid. J. Virol. 1997, 71, 9214–9222. [Google Scholar] [PubMed]
- Buonavoglia, C.; Martella, V.; Pratelli, A.; Tempesta, M.; Cavalli, A.; Buonavoglia, D.; Bozzo, G.; Elia, G.; Decaro, N.; Carmichael, L. Evidence for evolution of canine parvovirus type 2 in Italy. J. Gen. Virol. 2001, 82, 3021–3025. [Google Scholar] [CrossRef] [PubMed]
- Decaro, N.; Buonavoglia, C. Canine parvovirus: A review of epidemiological and diagnostic aspects, with emphasis on type 2c. Vet. Microbiol. 2012, 155, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Decaro, N.; Desario, C.; Lucente, M.S.; Amorisco, F.; Campolo, M.; Elia, G.; Cavalli, A.; Martella, V.; Buonavoglia, C. Specific identification of feline panleukopenia virus and its rapid differentiation from canine parvoviruses using minor groove binder probes. J. Virol. Methods 2008, 147, 67–71. [Google Scholar] [CrossRef] [PubMed]
- Decaro, N.; Desario, C.; Addie, D.D.; Martella, V.; Vieira, M.J.; Elia, G.; Zicola, A.; Davis, C.; Thompson, G.; Thiry, E.; et al. The study molecular epidemiology of canine parvovirus, europe. Emerg. Infect. Dis. 2007, 13, 1222–1224. [Google Scholar] [CrossRef] [PubMed]
- Decaro, N.; Elia, G.; Martella, V.; Campolo, M.; Desario, C.; Camero, M.; Cirone, F.; Lorusso, E.; Lucente, M.S.; Narcisi, D.; et al. Characterisation of the canine parvovirus type 2 variants using minor groove binder probe technology. J. Virol. Methods 2006, 133, 92–99. [Google Scholar] [CrossRef] [PubMed]
- Kapil, S.; Cooper, E.; Lamm, C.; Murray, B.; Rezabek, G.; Johnston, L., 3rd; Campbell, G.; Johnson, B. Canine parvovirus types 2c and 2b circulating in north american dogs in 2006 and 2007. J. Clin. Microbiol. 2007, 45, 4044–4047. [Google Scholar] [CrossRef] [PubMed]
- Touihri, L.; Bouzid, I.; Daoud, R.; Desario, C.; El Goulli, A.F.; Decaro, N.; Ghorbel, A.; Buonavoglia, C.; Bahloul, C. Molecular characterization of canine parvovirus-2 variants circulating in tunisia. Virus Genes 2009, 38, 249–258. [Google Scholar] [CrossRef] [PubMed]
- Gallo Calderon, M.; Wilda, M.; Boado, L.; Keller, L.; Malirat, V.; Iglesias, M.; Mattion, N.; La Torre, J. Study of canine parvovirus evolution: Comparative analysis of full-length VP2 gene sequences from argentina and international field strains. Virus Genes 2012, 44, 32–39. [Google Scholar] [CrossRef] [PubMed]
- Muz, D.; Oguzoglu, T.C.; Timurkan, M.O.; Akin, H. Characterization of the partial VP2 gene region of canine parvoviruses in domestic cats from turkey. Virus Genes 2012, 44, 301–308. [Google Scholar] [CrossRef] [PubMed]
- Miranda, C.; Parrish, C.R.; Thompson, G. Epidemiological evolution of canine parvovirus in the portuguese domestic dog population. Vet. Microbiol. 2016, 183, 37–42. [Google Scholar] [CrossRef] [PubMed]
- Miranda, C.; Thompson, G. Canine parvovirus: The worldwide occurrence of antigenic variants. J. Gen. Virol. 2016, 97, 2043–2057. [Google Scholar] [CrossRef] [PubMed]
- Chiang, S.Y.; Wu, H.Y.; Chiou, M.T.; Chang, M.C.; Lin, C.N. Identification of a novel canine parvovirus type 2c in Taiwan. Virol. J. 2016, 13, 160. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.N.; Chien, C.H.; Chiou, M.T.; Chueh, L.L.; Hung, M.Y.; Hsu, H.S. Genetic characterization of type 2a canine parvoviruses from Taiwan reveals the emergence of an ILE324 mutation in VP2. Virol. J. 2014, 11, 39. [Google Scholar] [CrossRef] [PubMed]
- Calderon, M.G.; Romanutti, C.; A, D.A.; Keller, L.; Mattion, N.; La Torre, J. Evolution of canine parvovirus in argentina between years 2003 and 2010: CPV2C has become the predominant variant affecting the domestic dog population. Virus Res. 2011, 157, 106–110. [Google Scholar] [CrossRef] [PubMed]
- Chang, W.L.; Chang, A.C.; Pan, M.J. Antigenic types of canine parvoviruses prevailing in Taiwan. Vet. Rec. 1996, 138, 447. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.C.; Chen, W.D.; Lin, S.L.; Chan, J.P.; Wong, M.L. Phylogenetic analysis of canine parvovirus VP2 gene in Taiwan. Virus Genes 2005, 31, 171–174. [Google Scholar] [CrossRef] [PubMed]
- Chou, S.J.; Lin, H.T.; Wu, J.T.; Yang, W.C.; Chan, K.W. Genotyping of canine parvovirus type 2 VP2 gene in southern Taiwan in 2011. Taiwan Vet. J. 2013, 39, 81–92. [Google Scholar]
- Kumar, S.; Stecher, G.; Tamura, K. Mega7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [PubMed]
- Silva, S.P.; Silva, L.; Rodrigues, E.D.L.; Cardoso, J.F.; Tavares, F.N.; Souza, W.M.; Santos, C.M.P.; Martins, F.M.S.; Jesus, I.S.; Brito, T.C.; et al. Full-length genomic and molecular characterization of canine parvovirus in dogs from north of brazil. Genet. Mol. Res. 2017, 16. [Google Scholar] [CrossRef] [PubMed]
- Hoelzer, K.; Shackelton, L.A.; Parrish, C.R.; Holmes, E.C. Phylogenetic analysis reveals the emergence, evolution and dispersal of carnivore parvoviruses. J. Gen. Virol. 2008, 89, 2280–2289. [Google Scholar] [CrossRef] [PubMed]
- Pereira, C.A.; Leal, E.S.; Durigon, E.L. Selective regimen shift and demographic growth increase associated with the emergence of high-fitness variants of canine parvovirus. Infect. Genet. Evol. 2007, 7, 399–409. [Google Scholar] [CrossRef] [PubMed]
- Duffy, S.; Shackelton, L.A.; Holmes, E.C. Rates of evolutionary change in viruses: Patterns and determinants. Nat. Rev. Genet. 2008, 9, 267–276. [Google Scholar] [CrossRef] [PubMed]
- Aldaz, J.; Garcia-Diaz, J.; Calleros, L.; Sosa, K.; Iraola, G.; Marandino, A.; Hernandez, M.; Panzera, Y.; Perez, R. High local genetic diversity of canine parvovirus from ecuador. Vet. Microbiol. 2013, 166, 214–219. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, M.; Tohya, Y.; Miyazawa, T.; Mochizuki, M.; Phung, H.T.; Nguyen, N.H.; Huynh, L.M.; Nguyen, L.T.; Nguyen, P.N.; Nguyen, P.V.; et al. A novel antigenic variant of canine parvovirus from a vietnamese dog. Arch. Virol. 2004, 149, 2261–2269. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Yang, S.; Zhang, W.; Zhang, T.; Xie, Z.; Feng, H.; Wang, S.; Xia, X. Phylogenetic analysis of the VP2 gene of canine parvoviruses circulating in china. Virus Genes 2010, 40, 397–402. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Wang, J.; Jiang, Y.; Cheng, Y.; Lin, P.; Zhu, H.; Han, G.; Yi, L.; Zhang, S.; Guo, L.; et al. Typing of canine parvovirus strains circulating in North-East China. Transbound. Emerg. Dis. 2015, 64, 495–503. [Google Scholar] [CrossRef] [PubMed]
- Han, S.C.; Guo, H.C.; Sun, S.Q.; Shu, L.; Wei, Y.Q.; Sun, D.H.; Cao, S.Z.; Peng, G.N.; Liu, X.T. Full-length genomic characterizations of two canine parvoviruses prevalent in northwest china. Arch. Microbiol. 2015, 197, 621–626. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Guo, H.C.; Wei, Y.Q.; Shu, L.; Wang, J.; Li, J.S.; Cao, S.Z.; Sun, S.Q. Phylogenetic analysis of canine parvovirus isolates from Sichuan and Gansu provinces of china in 2011. Transbound. Emerg. Dis. 2015, 62, 91–95. [Google Scholar] [CrossRef] [PubMed]
- Mukhopadhyay, H.K.; Matta, S.L.; Amsaveni, S.; Antony, P.X.; Thanislass, J.; Pillai, R.M. Phylogenetic analysis of canine parvovirus partial VP2 gene in India. Virus Genes 2013, 48, 89–95. [Google Scholar] [CrossRef] [PubMed]
- Perez, R.; Bianchi, P.; Calleros, L.; Francia, L.; Hernandez, M.; Maya, L.; Panzera, Y.; Sosa, K.; Zoller, S. Recent spreading of a divergent canine parvovirus type 2a (CPV-2a) strain in a CPV-2c homogenous population. Vet. Microbiol. 2012, 155, 214–219. [Google Scholar] [CrossRef] [PubMed]
- Perez, R.; Calleros, L.; Marandino, A.; Sarute, N.; Iraola, G.; Grecco, S.; Blanc, H.; Vignuzzi, M.; Isakov, O.; Shomron, N.; et al. Phylogenetic and genome-wide deep-sequencing analyses of canine parvovirus reveal co-infection with field variants and emergence of a recent recombinant strain. PLoS ONE 2014, 9, e111779. [Google Scholar] [CrossRef] [PubMed]
- Agbandje, M.; McKenna, R.; Rossmann, M.G.; Strassheim, M.L.; Parrish, C.R. Structure determination of feline Panleukopenia virus empty particles. Proteins 1993, 16, 155–171. [Google Scholar] [CrossRef] [PubMed]
- Tsao, J.; Chapman, M.S.; Agbandje, M.; Keller, W.; Smith, K.; Wu, H.; Luo, M.; Smith, T.J.; Rossmann, M.G.; Compans, R.W.; et al. The three-dimensional structure of canine parvovirus and its functional implications. Science 1991, 251, 1456–1464. [Google Scholar] [CrossRef] [PubMed]
- Zhong, Z.; Liang, L.; Zhao, J.; Xu, X.; Cao, X.; Liu, X.; Zhou, Z.; Ren, Z.; Shen, L.; Geng, Y.; et al. First isolation of new canine parvovirus 2a from Tibetan mastiff and global analysis of the full-length VP2 gene of canine parvoviruses 2 in china. Int. J. Mol. Sci. 2014, 15, 12166–12187. [Google Scholar] [CrossRef] [PubMed]
- Yi, L.; Tong, M.; Cheng, Y.; Song, W.; Cheng, S. Phylogenetic analysis of canine parvovirus VP2 gene in china. Transbound. Emerg. Dis. 2014, 63, e262–e269. [Google Scholar] [CrossRef] [PubMed]
- Geng, Y.; Guo, D.; Li, C.; Wang, E.; Wei, S.; Wang, Z.; Yao, S.; Zhao, X.; Su, M.; Wang, X.; et al. Co-circulation of the rare CPV-2c with unique Gln370Arg substitution, new CPV-2b with unique Thr440Ala substitution and new CPV-2a with high prevalence and variation in Heilongjiang province, northeast china. PLoS ONE 2015, 10, e0137288. [Google Scholar] [CrossRef] [PubMed]
- Jeoung, S.Y.; Ahn, S.J.; Kim, D. Genetic analysis of VP2 gene of canine parvovirus isolates in Korea. J. Vet. Med. Sci. 2008, 70, 719–722. [Google Scholar] [CrossRef] [PubMed]
- Yoon, S.H.; Jeong, W.; Kim, H.J.; An, D.J. Molecular insights into the phylogeny of canine parvovirus 2 (CPV-2) with emphasis on Korean isolates: A bayesian approach. Arch. Virol. 2009, 154, 1353–1360. [Google Scholar] [CrossRef] [PubMed]
- Phromnoi, S.; Sirinarumitr, K.; Sirinarumitr, T. Sequence analysis of VP2 gene of canine parvovirus isolates in thailand. Virus Genes 2010, 41, 23–29. [Google Scholar] [CrossRef] [PubMed]
- Soma, T.; Taharaguchi, S.; Ohinata, T.; Ishii, H.; Hara, M. Analysis of the VP2 protein gene of canine parvovirus strains from affected dogs in japan. Res. Vet. Sci. 2013, 94, 368–371. [Google Scholar] [CrossRef] [PubMed]
- Mittal, M.; Chakravarti, S.; Mohapatra, J.K.; Chug, P.K.; Dubey, R.; Narwal, P.S.; Kumar, A.; Churamani, C.P.; Kanwar, N.S. Molecular typing of canine parvovirus strains circulating from 2008–2012 in an organized kennel in India reveals the possibility of vaccination failure. Infect. Genet. Evol. 2014. [Google Scholar] [CrossRef] [PubMed]
- Csagola, A.; Varga, S.; Lorincz, M.; Tuboly, T. Analysis of the full-length VP2 protein of canine parvoviruses circulating in Hungary. Arch. Virol. 2014, 159, 2441–2444. [Google Scholar] [CrossRef] [PubMed]
- Hueffer, K.; Parrish, C.R. Parvovirus host range, cell tropism and evolution. Curr. Opin. Microbiol. 2003, 6, 392–398. [Google Scholar] [CrossRef]
- Guo, L.; Yang, S.L.; Chen, S.J.; Zhang, Z.; Wang, C.; Hou, R.; Ren, Y.; Wen, X.; Cao, S.; Guo, W.; et al. Identification of canine parvovirus with the Q370R point mutation in the VP2 gene from a giant panda (Ailuropoda melanoleuca). Virol. J. 2013, 10, 163. [Google Scholar] [CrossRef] [PubMed]
- Simpson, A.A.; Chandrasekar, V.; Hebert, B.; Sullivan, G.M.; Rossmann, M.G.; Parrish, C.R. Host range and variability of calcium binding by surface loops in the capsids of canine and feline parvoviruses. J. Mol. Biol. 2000, 300, 597–610. [Google Scholar] [CrossRef] [PubMed]
- Notredame, C.; Higgins, D.G.; Heringa, J. T-coffee: A novel method for fast and accurate multiple sequence alignment. J. Mol. Biol. 2000, 302, 205–217. [Google Scholar] [CrossRef] [PubMed]
- Guindon, S.; Dufayard, J.F.; Lefort, V.; Anisimova, M.; Hordijk, W.; Gascuel, O. New algorithms and methods to estimate maximum-likelihood phylogenies: Assessing the performance of phyml 3.0. Syst. Biol. 2010, 59, 307–321. [Google Scholar] [CrossRef] [PubMed]
- Rambaut, A.; Lam, T.T.; Max Carvalho, L.; Pybus, O.G. Exploring the temporal structure of heterochronous sequences using tempest (formerly Path-O-Gen). Virus Evol. 2016, 2, vew007. [Google Scholar] [CrossRef] [PubMed]
- Drummond, A.J.; Suchard, M.A.; Xie, D.; Rambaut, A. Bayesian phylogenetics with beauti and the beast 1.7. Mol. Biol. Evol. 2012, 29, 1969–1973. [Google Scholar] [CrossRef] [PubMed]
- Shapiro, B.; Rambaut, A.; Drummond, A.J. Choosing appropriate substitution models for the phylogenetic analysis of protein-coding sequences. Mol. Biol. Evol. 2006, 23, 7–9. [Google Scholar] [CrossRef] [PubMed]
- Baele, G.; Lemey, P.; Bedford, T.; Rambaut, A.; Suchard, M.A.; Alekseyenko, A.V. Improving the accuracy of demographic and molecular clock model comparison while accommodating phylogenetic uncertainty. Mol. Biol. Evol. 2012, 29, 2157–2167. [Google Scholar] [CrossRef] [PubMed]
- Murrell, B.; Moola, S.; Mabona, A.; Weighill, T.; Sheward, D.; Kosakovsky Pond, S.L.; Scheffler, K. Fubar: A fast, unconstrained bayesian approximation for inferring selection. Mol. Biol. Evol. 2013, 30, 1196–1205. [Google Scholar] [CrossRef] [PubMed]
- Poon, A.F.; Frost, S.D.; Pond, S.L. Detecting signatures of selection from DNA sequences using datamonkey. Methods Mol. Biol. 2009, 537, 163–183. [Google Scholar] [PubMed]
- Delport, W.; Poon, A.F.; Frost, S.D.; Kosakovsky Pond, S.L. Datamonkey 2010: A suite of phylogenetic analysis tools for evolutionary biology. Bioinformatics 2010, 26, 2455–2457. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Country | Year | Amino Acid at Position | Accession Numbers | ||||||
|---|---|---|---|---|---|---|---|---|---|
| 5 | 267 | 324 | 370 | 383 | 410 | 440 | |||
| Taiwan | 2015 | A | Y a | I a | R a | R a | L a | T | KU244254 |
| 2015 | A | Y a | I a | R a | R a | P | T | KX421787 | |
| 2015 | G a | Y a | I a | R a | Q | P | T | KX421786 | |
| 2016 | G a | Y a | I a | R a | Q | P | T | KX421788, KX421789 | |
| China | 2009 | A | Y | I | Q | Q | P | T | GU380303 |
| 2009 | A | F | Y | Q | Q | P | T | GU380305 | |
| 2014 | G | Y | I | R | Q | P | T | KR611522, KT162005 | |
| Argentina | 2008–2009 | A | F | Y | Q | Q | P | T | JF414818–JF414820 |
| 2009–2010 | A | F | Y | Q | Q | P | A | JF414821–JF414825 | |
| 2010 | A | F | Y | Q | Q | P | T | JF414826 | |
| Belgium | 2008 | A | F | Y | Q | Q | P | T | FJ005247 |
| Ecuador | 2012 | A | F | Y | Q | Q | P | T | KF149962–KF149965, KF149967–KF149971, KF149984, |
| 2012 | A | F | Y | Q | Q | P | S | KF149966 | |
| Germany | 1997–1999 | A | F | Y | Q | Q | P | T | FJ005196–FJ005204 |
| Greece | 2008–2009 | A | F | Y | Q | Q | P | T | GQ865518, GQ865519 |
| Italy | 2000–2010 | A | F | Y | Q | Q | P | T | FJ222821, FJ005195, FJ005205–FJ005213, FJ005215–FJ005234, FJ005237–FJ005245, FJ005248–FJ005251, HQ025913 |
| India | 2014 | A | F | Y | Q | Q | P | T | KP071956 |
| Spain | 2006–2008 | A | F | Y | Q | Q | P | T | FJ005214, FJ005246 |
| Uruguay | 2006–2011 | A | F | Y | Q | Q | P | T | KC196079–KC196093, KC196080–KC196093, KC196095–KC196109, KC196111–KC196114, KM457103–KM457110, KM457115–KM457131, KM457142 |
| 2010 | A | F | Y | Q | Q | P | A | KC196094 | |
| USA | 2007 | A | F | Y | Q | Q | P | T | FJ005235 |
| 2007 | A | F | Y | Q | Q | P | A | FJ005236 | |
| Vietnam | 2004 | A | F | Y | Q | Q | P | T | AB120727 |
| TMRCA | Substitution Rates Sub/Site/Year (10−4) | Mean Relative Substitution Rate | SE of Mean | |
|---|---|---|---|---|
| VP2 gene | 4.586 (3.284~6.076) | |||
| CPV-2 | 1973 (1963–1978) a | |||
| CPV-2c | 1990 (1984–1996) | 6.071 (4.277~8.132) | ||
| CPV-2c Taiwanese strains | 2011 (2010–2013) | |||
| 1st + 2nd codon position | 0.4 (0.302~0.495) | 5.215 × 10−4 | ||
| 3rd codon position | 2.199 (2.011~2.396) | 1.043 × 10−3 |
| Positively Selected Sites | No. of Negatively Selected Sites | Mean dN/dS | |||||
|---|---|---|---|---|---|---|---|
| SLAC a | FEL a | FUBA b | SLAC a | FEL a | FUBAR b | ||
| CPV-2 VP2 gene | Non | Non | 426 | 8 | 46 | 31 | 0.108 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lin, Y.-C.; Chiang, S.-Y.; Wu, H.-Y.; Lin, J.-H.; Chiou, M.-T.; Liu, H.-F.; Lin, C.-N. Phylodynamic and Genetic Diversity of Canine Parvovirus Type 2c in Taiwan. Int. J. Mol. Sci. 2017, 18, 2703. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms18122703
Lin Y-C, Chiang S-Y, Wu H-Y, Lin J-H, Chiou M-T, Liu H-F, Lin C-N. Phylodynamic and Genetic Diversity of Canine Parvovirus Type 2c in Taiwan. International Journal of Molecular Sciences. 2017; 18(12):2703. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms18122703
Chicago/Turabian StyleLin, Yung-Cheng, Shu-Yun Chiang, Hung-Yi Wu, Jih-Hui Lin, Ming-Tang Chiou, Hsin-Fu Liu, and Chao-Nan Lin. 2017. "Phylodynamic and Genetic Diversity of Canine Parvovirus Type 2c in Taiwan" International Journal of Molecular Sciences 18, no. 12: 2703. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms18122703