
Charcot Marie Tooth 2B Peripheral Sensory Neuropathy: How Rab7 Mutations Impact NGF Signaling?

Abstract
:1. Introduction
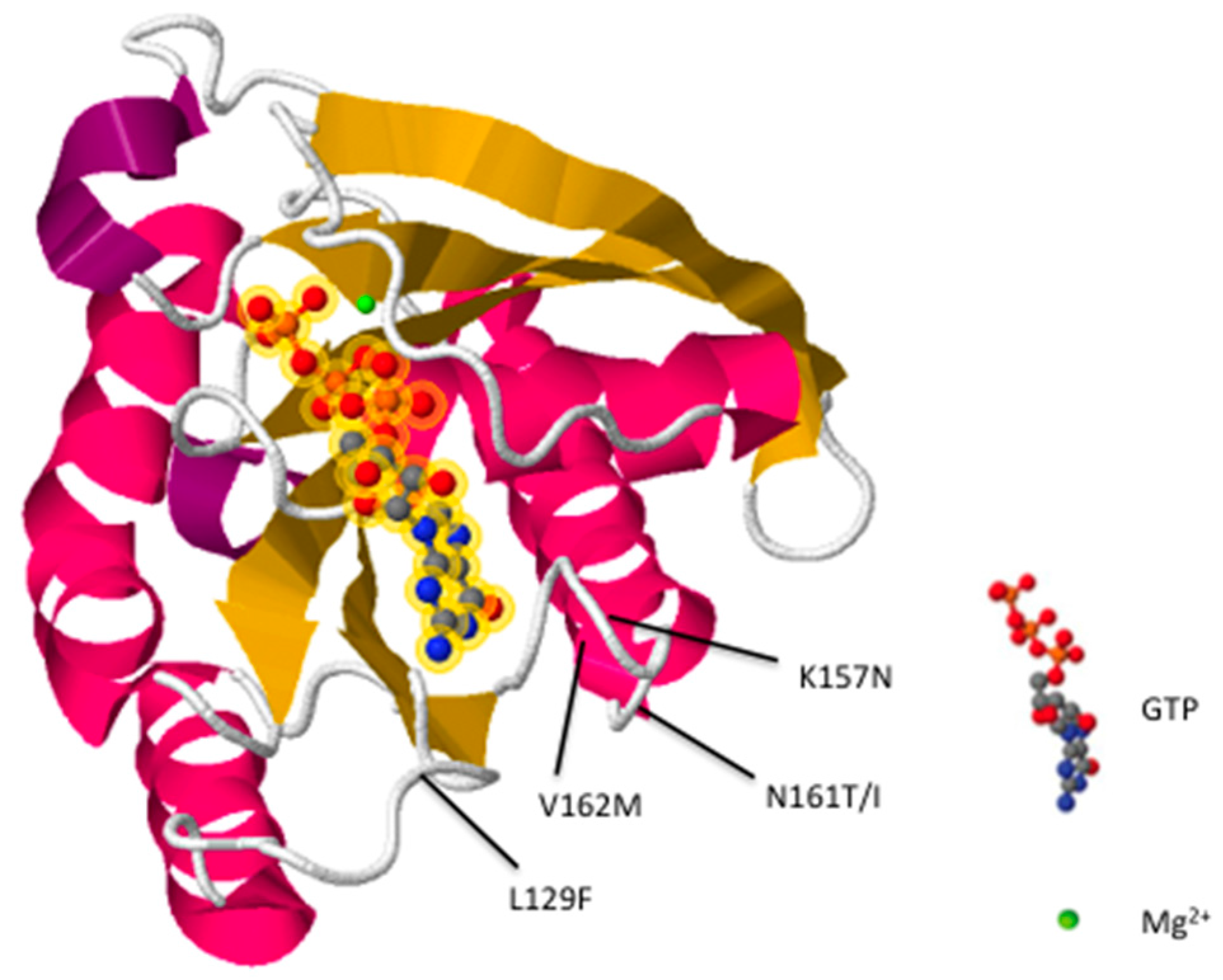
2. Rab7 Mutations Are Associated with CMT2B
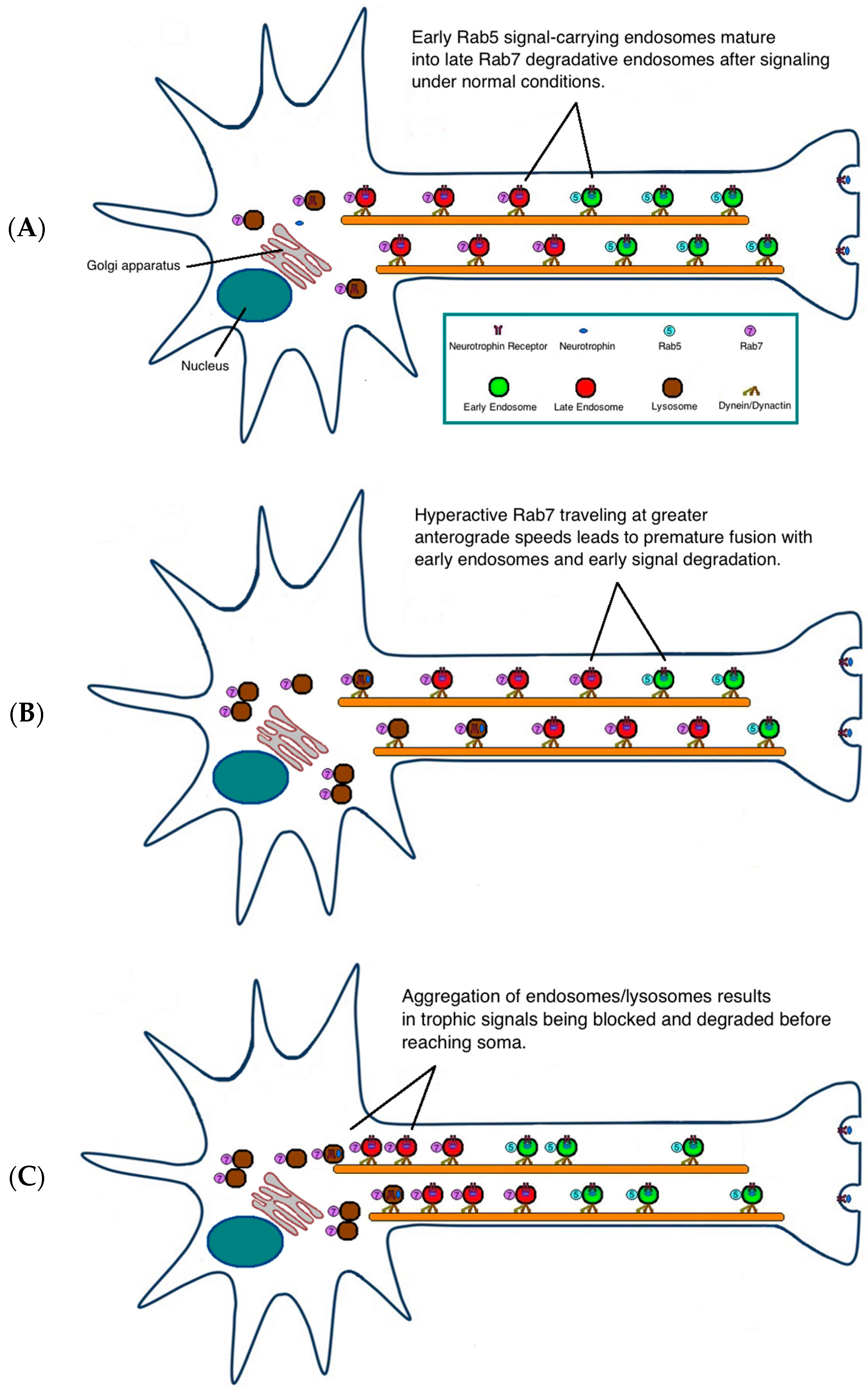
3. Possible Pathogenic Mechanisms
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Barisic, N.; Claeys, K.G.; Sirotković-Skerlev, M.; Löfgren, A.; Nelis, E.; de Jonghe, P.; Timmerman, V. Charcot-Marie-Tooth disease: A clinico-genetic confrontation. Ann. Hum. Genet. 2008, 72, 416–441. [Google Scholar] [CrossRef] [PubMed]
- Auer-Grumbach, M. Hereditary sensory neuropathies. Drugs Today 2004, 40, 385–394. [Google Scholar] [CrossRef] [PubMed]
- Auer-Grumbach, M.; De Jonghe, P.; Verhoeven, K.; Timmerman, V.; Wagner, K.; Hartung, H.P.; Nicholson, G.A. Autosomal dominant inherited neuropathies with prominent sensory loss and mutilations: A review. Arch. Neurol. 2003, 60, 329–334. [Google Scholar] [CrossRef] [PubMed]
- Auer–Grumbach, M.; De Jonghe, P.; Wagner, K.; Verhoeven, K.; Hartung, H.P.; Timmerman, V. Phenotype-genotype correlations in a CMT2B family with refined 3q13-q22 locus. Neurology 2000, 55, 1552–1557. [Google Scholar] [CrossRef] [PubMed]
- Auer-Grumbach, M.; Mauko, B.; Auer-Grumbach, P.; Pieber, T.R. Molecular genetics of hereditary sensory neuropathies. Neuromol. Med. 2006, 8, 147–158. [Google Scholar] [CrossRef]
- Auer-Grumbach, M.; Wagner, K.; Timmerman, V.; De Jonghe, P.; Hartung, H.P. Ulcero-mutilating neuropathy in an Austrian kinship without linkage to hereditary motor and sensory neuropathy IIB and hereditary sensory neuropathy I loci. Neurology 2000, 54, 45–52. [Google Scholar] [CrossRef] [PubMed]
- Gutmann, L.; Shy, M. Update on Charcot-Marie-Tooth disease. Curr. Opin. Neurol. 2015, 5, 462–467. [Google Scholar] [CrossRef] [PubMed]
- Mathis, S.; Goizet, C.; Tazir, M.; Magdelaine, C.; Lia, A.S.; Magy, L.; Vallat, J.M. Charcot-Marie-Tooth diseases: An update and some new proposals for the classification. J. Med. Genet. 2015. [Google Scholar] [CrossRef] [PubMed]
- Ekins, S.; Litterman, N.K.; Arnold, R.J.; Burgess, R.W.; Freundlich, J.S.; Gray, S.J.; Higgins, J.J.; Langley, B.; Willis, D.E.; Notterpek, L.; et al. A brief review of recent Charcot-Marie-Tooth research and priorities. F1000 Res. 2015, 4, 53. [Google Scholar] [CrossRef] [PubMed]
- Chartier-Harlin, M.C.; Kachergus, J.; Roumier, C.; Mouroux, V.; Douay, X.; Lincoln, S.; Levecque, C.; Larvor, L.; Andrieux, J.; Hulihan, M.; et al. α-synuclein locus duplication as a cause of familial Parkinson’s disease. Lancet 2004, 364, 1167–1169. [Google Scholar] [CrossRef]
- Harding, A.E.; Thomas, P.K. The clinical features of hereditary motor and sensory neuropathy types I and II. Brain 1980, 103, 259–280. [Google Scholar] [CrossRef] [PubMed]
- Bird, T.D. Charcot-Marie-Tooth Neuropathy Type 2; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Gemignani, F.; Marbini, A. Charcot-Marie-Tooth disease (CMT): Distinctive phenotypic and genotypic features in CMT type 2. J. Neurol. Sci. 2001, 184, 1–9. [Google Scholar] [CrossRef]
- Kwon, J.M.; Elliott, J.L.; Yee, W.C.; Ivanovich, J.; Scavarda, N.J.; Moolsintong, P.J.; Goodfellow, P.J. Assignment of a second Charcot-Marie-Tooth type II locus to chromosome 3q. Am. J. Hum. Genet. 1995, 57, 853–858. [Google Scholar] [PubMed]
- Lus, G.; Nelis, E.; Jordanova, A.; Löfgren, A.; Cavallaro, T.; Ammendola, A.; Melone, M.A.B.; Rizzuto, N.; Timmerman, V.; Cotrufo, R.; et al. Charcot-Marie-Tooth disease with giant axons: A clinicopathological and genetic entity. Neurology 2003, 61, 988–990. [Google Scholar] [CrossRef] [PubMed]
- Marrosu, M.G.; Vaccargiu, S.; Marrosu, G.; Vannelli, A.; Cianchetti, C.; Muntoni, F. Charcot-Marie-Tooth disease type 2 associated with mutation of the myelin protein zero gene. Neurology 1998, 50, 1397–1401. [Google Scholar] [CrossRef] [PubMed]
- Meggouh, F.; Bienfait, H.M.; Weterman, M.A.; de Visser, M.; Baas, F. Charcot-Marie-Tooth disease due to a de novo mutation of the Rab7 gene. Neurology 2006, 67, 1476–1478. [Google Scholar] [CrossRef] [PubMed]
- Mersiyanova, I.V.; Perepelov, A.V.; Polyakov, A.V.; Sitnikov, V.F.; Dadali, E.L.; Oparin, R.B.; Petrin, A.N.; Evgrafov, O.V. A new variant of Charcot-Marie-Tooth disease type 2 is probably the result of a mutation in the neurofilament-light gene. Am. J. Hum. Genet. 2000, 67, 37–46. [Google Scholar] [CrossRef] [PubMed]
- Mathis, S.; Magy, L.; Vallat, J.M. Therapeutic options in Charcot-Marie-Tooth diseases. Expert Rev. Neurother. 2015, 15, 355–366. [Google Scholar] [CrossRef] [PubMed]
- Jerath, N.U.; Shy, M.E. Hereditary motor and sensory neuropathies: Understanding molecular pathogenesis could lead to future treatment strategies. Biochim. Biophys. Acta 2015, 1852, 667–678. [Google Scholar] [CrossRef] [PubMed]
- Brennan, K.M.; Bai, Y.; Shy, M.E. Demyelinating CMT—What’s known, what’s new and what’s in store? Neurosci. Lett. 2015, 596, 14–26. [Google Scholar] [CrossRef] [PubMed]
- Fridman, V.; Bundy, B.; Reilly, M.M.; Pareyson, D.; Bacon, C.; Burns, J.; Day, J.; Feely, S.; Finkel, R.S.; Grider, T.; et al. CMT subtypes and disease burden in patients enrolled in the Inherited Neuropathies Consortium natural history study: A cross-sectional analysis. J. Neurol. Neurosurg. Psychiatry 2015, 86, 873–878. [Google Scholar] [CrossRef] [PubMed]
- Bennett, C.L.; Chance, P.F. Molecular pathogenesis of hereditary motor, sensory and autonomic neuropathies. Curr. Opin. Neurol. 2001, 14, 621–627. [Google Scholar] [CrossRef] [PubMed]
- Vance, J.M.; Speer, M.C.; Stajich, J.M.; West, S.; Wolpert, C.; Gaskell, P.; Lennon, F.; Tim, R.M.; Rozear, M.; Othmane, K.B. Misclassification and linkage of hereditary sensory and autonomic neuropathy type 1 as Charcot-Marie-Tooth disease, type 2B. Am. J. Hum. Genet. 1996, 59, 258–262. [Google Scholar] [PubMed]
- Verpoorten, N.; De Jonghe, P.; Timmerman, V. Disease mechanisms in hereditary sensory and autonomic neuropathies. Neurobiol. Dis. 2006, 21, 247–255. [Google Scholar] [CrossRef] [PubMed]
- Skre, H. Genetic and clinical aspects of Charcot-Marie-Tooth’s disease. Clin. Genet. 1974, 6, 98–118. [Google Scholar] [CrossRef] [PubMed]
- Reilly, M.M. Classification and diagnosis of the inherited neuropathies. Ann. Indian Acad. Neurol. 2009, 12, 80–88. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Han, C.; Liu, W.; Wang, P.; Zhang, X. A novel Rab7 mutation in a Chinese family with Charcot-Marie-Tooth type 2B disease. Gene 2014, 534, 431–434. [Google Scholar] [CrossRef] [PubMed]
- Dawkins, J.L.; Hulme, D.J.; Brahmbhatt, S.B.; Auer-Grumbach, M.; Nicholson, G.A. Mutations in SPTLC1, encoding serine palmitoyltransferase, long chain base subunit-1, cause hereditary sensory neuropathy type I. Nat. Genet. 2001, 27, 309–312. [Google Scholar] [CrossRef] [PubMed]
- Bucci, C.; Thomsen, P.; Nicoziani, P.; McCarthy, J.; van Deurs, B. Rab7: A key to lysosome biogenesis. Mol. Biol. Cell 2000, 11, 467–480. [Google Scholar] [CrossRef] [PubMed]
- Cai, H.; Reinisch, K.; Ferro-Novick, S. Coats, tethers, Rabs, and SNAREs work together to mediate the intracellular destination of a transport vesicle. Dev. Cell. 2007, 12, 671–682. [Google Scholar] [CrossRef] [PubMed]
- Grosshans, B.L.; Ortiz, D.; Novick, P. Rabs and their effectors: Achieving specificity in membrane traffic. Proc. Natl. Acad. Sci. USA 2006, 103, 11821–11827. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Kenan, R.F.B.; Osakada, Y.; Xu, W.; Sinit, R.S.; Chen, L.; Zhao, X.; Chen, J.Y.; Cui, B.; Wu, C. Defective axonal transport of Rab7 GTPase results in dysregulated trophic signaling. J. Neurosci. 2013, 33, 7451–7462. [Google Scholar] [CrossRef] [PubMed]
- Houlden, H.; King, R.H.; Muddle, J.R.; Warner, T.T.; Reilly, M.M.; Orrell, R.W.; Ginsberg, L. A novel Rab7 mutation associated with ulcero-mutilating neuropathy. Ann. Neurol. 2004, 56, 586–590. [Google Scholar] [CrossRef] [PubMed]
- Verhoeven, K.; De Jonghe, P.; Coen, K.; Verpoorten, N.; Auer-Grumbach, M.; Kwon, J.M.; FitzPatrick, D.; Schmedding, E.; De Vriendt, E.; Jacobs, A.; et al. Mutations in the small GTPase late endosomal protein Rab7 cause Charcot-Marie-Tooth type 2B neuropathy. Am. J. Hum. Genet. 2003, 72, 722–727. [Google Scholar] [CrossRef] [PubMed]
- Cogli, L.; Piro, F.; Bucci, C. Rab7 and the CMT2B disease. Biochem. Soc. Trans. 2009, 37, 1027–1031. [Google Scholar] [CrossRef] [PubMed]
- McCray, B.A.; Skordalakes, E.; Taylor, J.P. Disease mutations in Rab7 result in unregulated nucleotide exchange and inappropriate activation. Hum. Mol. Genet. 2010, 19, 1033–1047. [Google Scholar] [CrossRef] [PubMed]
- Cogli, L.; Progida, C.; Lecci, R.; Bramato, R.; Krüttgen, A.; Bucci, C. CMT2B-associated Rab7 mutants inhibit Neurite outgrowth. Acta Neuropathol. 2010, 120, 491–501. [Google Scholar] [CrossRef] [PubMed]
- Spinosa, M.R.; Progida, C.; De Luca, A.; Colucci, A.M.R.; Alifano, P.; Bucci, C. Functional characterization of Rab7 mutant proteins associated with Charcot-Marie-Tooth type 2B disease. J. Neurosci. 2008, 28, 1640–1648. [Google Scholar] [CrossRef] [PubMed]
- Cogli, L.; Progida, C.; Thomas, C.L.; Spencer-Dene, B.; Donno, C.; Schiavo, G.; Bucci, C. Charcot-Marie-Tooth type 2B disease-causing Rab7a mutant proteins show altered interaction with the neuronal intermediate filament peripherin. Acta Neuropathol. 2013, 125, 257–272. [Google Scholar] [CrossRef] [PubMed]
- Cherry, S.; Jin, E.J.; Özel, M.N.; Lu, Z.; Agi, E.; Wang, D.; Jung, W.H.; Epstein, D.; Meinertzhagen, I.A.; Chan, C.C.; et al. Charcot-Marie-Tooth 2B mutations in Rab7 cause dosage-dependent neurodegeneration due to partial loss of function. Elife 2013, 2, e01064. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.; Wang, T.; Loh, E.; Hong, W.; Song, H. Structural basis for recruitment of RILP by small GTPase Rab7. EMBO J. 2005, 24, 1491–1501. [Google Scholar] [CrossRef] [PubMed]
- Ponomareva, O.Y.; Eliceiri, K.W.; Halloran, M.C. Charcot-Marie-Tooth 2b associated Rab7 mutations cause axon growth and guidance defects during vertebrate sensory neuron development. Neural Dev. 2016, 11, 2. [Google Scholar] [CrossRef] [PubMed]
- Saxena, S.; Bucci, C.; Weis, J.; Kruttgen, A. The small GTPase Rab7 controls the endosomal trafficking and neuritogenic signaling of the nerve growth factor receptor TrkA. J. Neurosci. 2005, 25, 10930–10940. [Google Scholar] [CrossRef] [PubMed]
- BasuRay, S.; Mukherjee, S.; Romero, E.; Wilson, M.C.; Wandinger-Ness, A. Rab7 mutants associated with Charcot-Marie-Tooth disease exhibit enhanced NGF-stimulated signaling. PLoS ONE 2010, 5, e15351. [Google Scholar] [CrossRef] [PubMed]
- Cogli, L.; Progida, C.; Bramato, R.; Bucci, C. Vimentin phosphorylation and assembly are regulated by the small GTPase Rab7a. Biochim. Biophys. Acta 2013, 1833, 1283–1293. [Google Scholar] [CrossRef] [PubMed]
- Mizuno, K.; Kitamura, A.; Sasaki, T. Rabring7, a novel Rab7 target protein with a RING finger motif. Mol. Biol. Cell 2003, 14, 3741–3752. [Google Scholar] [CrossRef] [PubMed]
- Taub, N.; Teis, D.; Ebner, H.L.; Hess, M.W.; Huber, L.A. Late endosomal traffic of the epidermal growth factor receptor ensures spatial and temporal fidelity of mitogen-activated protein kinase signaling. Mol. Biol. Cell 2007, 18, 4698–4710. [Google Scholar] [CrossRef] [PubMed]
- Bottger, G.; Nagelkerken, B.; van der Sluijs, P. Rab4 and Rab7 define distinct nonoverlapping endosomal compartments. J. Biol. Chem. 1996, 271, 29191–29197. [Google Scholar] [PubMed]
- Seals, D.F.; Eitzen, G.; Margolis, N.; Wickner, W.T.; Price, A. A Ypt/Rab effector complex containing the Sec1 homolog Vps33p is required for homotypic vacuole fusion. Proc. Natl. Acad. Sci. USA 2000, 97, 9402–9407. [Google Scholar] [CrossRef] [PubMed]
- Wurmser, A.E.; Sato, T.K.; Emr, S.D. New component of the vacuolar class C-Vps complex couples nucleotide exchange on the Ypt7 GTPase to SNARE-dependent docking and fusion. J. Cell Biol. 2000, 151, 551–562. [Google Scholar] [CrossRef] [PubMed]
- Rink, J.; Ghigo, E.; Kalaidzidis, Y.; Zerial, M. Rab conversion as a mechanism of progression from early to late endosomes. Cell 2005, 122, 735–749. [Google Scholar] [CrossRef] [PubMed]
- Russell, M.R.; Nickerson, D.P.; Odorizzi, G. Molecular mechanisms of late endosome morphology, identity and sorting. Curr. Opin. Cell. Biol. 2006, 18, 422–428. [Google Scholar] [CrossRef] [PubMed]
- Cantalupo, G.; Alifano, P.; Roberti, V.; Bruni, C.B.; Bucci, C. Rab-interacting lysosomal protein (RILP): The Rab7 effector required for transport to lysosomes. EMBO J. 2001, 20, 683–693. [Google Scholar] [CrossRef] [PubMed]
- Jordens, I.; Fernandez-Borja, M.; Marsman, M.; Dusseljee, S.; Janssen, L.; Calafat, J.; Janssen, H.; Wubbolts, R.; Neefjes, J. The Rab7 effector protein RILP controls lysosomal transport by inducing the recruitment of dynein-dynactin motors. Curr. Biol. 2001, 11, 1680–1685. [Google Scholar] [CrossRef]
- Progida, C.; Malerød, L.; Stuffers, S.; Brech, A.; Bucci, C.; Stenmark, H. RILP is required for the proper morphology and function of late endosomes. J. Cell Sci. 2007, 120, 3729–3737. [Google Scholar] [CrossRef] [PubMed]
- Johansson, M.; Rocha, N.; Zwart, W.; Jordens, I.; Janssen, L.; Kuijl, C.; Olkkonen, V.M.; Neefjes, J. Activation of endosomal dynein motors by stepwise assembly of Rab7-RILP-p150Glued, ORP1L, and the receptor betalll spectrin. J. Cell Biol. 2007, 176, 459–471. [Google Scholar] [CrossRef] [PubMed]
- Nakada-Tsukui, K.; Saito-Nakano, Y.; Ali, V.; Nozaki, T. A retromerlike complex is a novel Rab7 effector that is involved in the transport of the virulence factor cysteine protease in the enteric protozoan parasite Entamoeba histolytica. Mol. Biol. Cell 2005, 16, 5294–5303. [Google Scholar] [CrossRef] [PubMed]
- Rojas, R.; van Vlijmen, T.; Mardones, G.A.; Prabhu, Y.; Rojas, A.L.; Mohammed, S.; Heck, A.J.; Raposo, G.; van der Sluijs, P.; Bonifacino, J.S. Regulation of retromer recruitment to endosomes by sequential action of Rab5 and Rab7. J. Cell Biol. 2008, 183, 513–526. [Google Scholar] [CrossRef] [PubMed]
- Sakane, A.; Hatakeyama, S.; Sasaki, T. Involvement of Rabring7 in EGF receptor degradation as an E3 ligase. Biochem. Biophys. Res. Commun. 2007, 357, 1058–1064. [Google Scholar] [CrossRef] [PubMed]
- Janssens, K.; Goethals, S.; Atkinson, D.; Ermanoska, B.; Fransen, E.; Jordanova, A.; Auer-Grumbach, M.; Asselbergh, B.; Timmerman, V. Human Rab7 mutation mimics features of Charcot-Marie-Tooth neuropathy type 2B in Drosophila. Neurobiol. Dis. 2014, 65, 211–219. [Google Scholar] [CrossRef] [PubMed]
- Seaman, M.N.; Harbour, M.E.; Tattersall, D.; Read, E.; Bright, N. Membrane recruitment of the cargo-selective retromer subcomplex is catalysed by the small GTPase Rab7 and inhibited by the Rab-GAP TBC1D5. J. Cell Sci. 2009, 122, 2371–2382. [Google Scholar] [CrossRef] [PubMed]
- Harrison, M.S.; Hung, C.S.; Liu, T.T.; Christiano, R.; Walther, T.C.; Burd, C.G. A mechanism for retromer endosomal coat complex assembly with cargo. Proc. Natl. Acad. Sci. USA 2014, 111, 267–272. [Google Scholar] [CrossRef] [PubMed]
- BasuRay, S.; Mukherjee, S.; Romero, E.G.; Seaman, M.N.; Wandinger-Ness, A. Rab7 mutants associated with Charcot-Marie-Tooth disease cause delayed growth factor receptor transport and altered endosomal and nuclear signaling. J. Biol. Chem. 2013, 288, 1135–1149. [Google Scholar] [CrossRef] [PubMed]
- Choudhury, A.; Dominguez, M.; Puri, V.; Sharma, D.K.; Narita, K.; Wheatley, C.L.; Marks, D.L.; Pagano, R.E. Rab proteins mediate Golgi transport of caveola-internalized glycosphingolipids and correct lipid trafficking in Niemann-Pick C cells. J. Clin. Investig. 2002, 109, 1541–1550. [Google Scholar] [CrossRef] [PubMed]
- Beilina, A.; Rudenko, I.N.; Kaganovich, A.; Civiero, L.; Chau, H.; Kalia, S.K.; Kalia, L.V.; Lobbestael, E.; Chia, R.; Ndukwe, K.; et al. Unbiased screen for interactors of leucine-rich repeat kinase 2 supports a common pathway for sporadic and familial Parkinson disease. Proc. Natl. Acad. Sci. USA 2014, 111, 2626–2631. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Satake, W.; Nakabayashi, Y.; Mizuta, I.; Hirota, Y.; Ito, C.; Kubo, M.; Kawaguchi, T.; Tsunoda, T.; Watanabe, M.; Takeda, A.; et al. Genome-wide association study identifies common variants at four loci as genetic risk factors for Parkinson’s disease. Nat. Genet. 2009, 41, 1303–1307. [Google Scholar] [CrossRef] [PubMed]
- Simon-Sanchez, J.; Schulte, C.; Bras, J.M.; Sharma, M.; Gibbs, J.R.; Berg, D.; Paisan-Ruiz, C.; Lichtner, P.; Scholz, S.W.; Hernandez, D.G.; et al. Genome-wide association study reveals genetic risk underlying Parkinson’s disease. Nat. Genet. 2009, 41, 1308–1312. [Google Scholar] [CrossRef] [PubMed]
- MacLeod, D.A.; Rhinn, H.; Kuwahara, T.; Zolin, A.; Di Paolo, G.; McCabe, B.D.; Marder, K.S.; Honig, L.S.; Clark, L.N.; Small, S.A.; et al. RAB7L1 interacts with LRRK2 to modify intraneuronal protein sorting and Parkinson’s disease risk. Neuron 2013, 77, 425–439. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Effector | Function | CMT2B Rab7 | Reference |
|---|---|---|---|
| HOPS Complex | Tethering (regulates endosomal membrane fusion)/GEF | Possible decreased interaction. CMT2B mutants can exchange GTP in GEF-independent manner. | [30,37,49,50,51,52,53] |
| RILP/ORP1L | Recruit and activate dynein-dynactin motor complex. Regulate late endosome/lysosome organization and transport | Increased interaction shown in L129F and V162M mutants. | [37,54,55,56,57] |
| Vps13C | Vacuolar protein sorting-associated protein | Increased interaction shown in L129F and V162M mutants. | [37] |
| Retromer Core Complex | Regulates retrograde transport from endosomes to trans-Golgi network. | Possible increased interaction. Rab7 binds in nucleotide dependent manner. | [58,59] |
| Rabring7 | Ubiquitin ligase that regulates EGFR degradation | Possible increased interaction. Rab7 binds in nucleotide dependent manner. | [45,47,48,60] |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, H.; Wu, C. Charcot Marie Tooth 2B Peripheral Sensory Neuropathy: How Rab7 Mutations Impact NGF Signaling? Int. J. Mol. Sci. 2017, 18, 324. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms18020324
Liu H, Wu C. Charcot Marie Tooth 2B Peripheral Sensory Neuropathy: How Rab7 Mutations Impact NGF Signaling? International Journal of Molecular Sciences. 2017; 18(2):324. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms18020324
Chicago/Turabian StyleLiu, Harry, and Chengbiao Wu. 2017. "Charcot Marie Tooth 2B Peripheral Sensory Neuropathy: How Rab7 Mutations Impact NGF Signaling?" International Journal of Molecular Sciences 18, no. 2: 324. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms18020324