
Potential Uses of Wild Germplasms of Grain Legumes for Crop Improvement


Abstract
:1. Introduction
2. A Brief History of the Domestication of Major Grain Legumes
3. Plant Domestication: More Than a Syndrome
4. Revisiting the Genetic Diversities and Potentials of Wild Relatives of Crop Plants
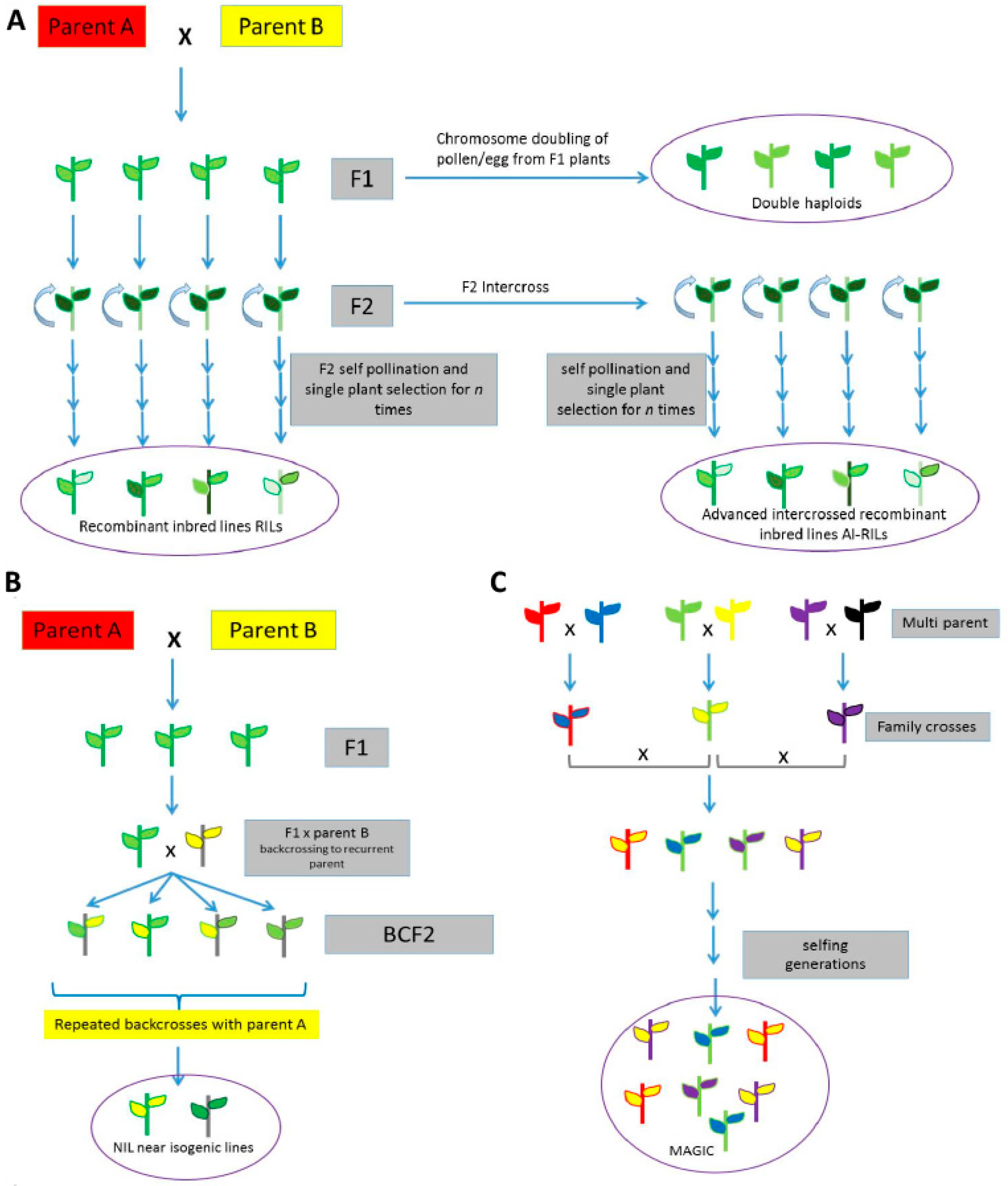
5. Traditional and Sequencing-Based Genetic Mapping Using Wild Relatives
6. Use of Genetic Diversity in Wild Relatives to Improve Grain Legume Performance
7. Conclusions and Perspectives
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| QTL | Quantitative trait locus |
| InDel | Insertion/deletion |
| CNV | Copy number variation |
| PAV | Present/absent variation |
| cM | Centimorgan |
| A-BC | Advanced backcross population |
| RIL | Recombinant inbred line |
| NIL | Near-isogenic line |
| NAM | Nested association mapping |
| MAGIC | Multi-parent advanced generation inter-cross |
| LD | Linkage disequilibrium |
| GWAS | Genome-wide association studies |
| SNP | Single nucleotide polymorphism |
| DArT | Diversity Arrays Technology |
| RAD | Restriction site-associated DNA marker |
| RRL | Reduced-representation library |
| CRoPS | Complexity reduction of polymorphic sequence |
| GBS | Genotyping-by-sequencing |
| IBL | Inbred backcross line |
| MATE | Multidrug and toxic compound extrusion |
| SRAP | Sequence-related amplified polymorphism |
| TRAP | Target region amplification polymorphism |
| SRR | Simple sequence repeat |
| RAPD | Random amplified polymorphic DNA |
| AFLP | Amplified fragment length polymorphism |
Appendix A
| Gene | A DNA sequence that determines the appearance of hereditary characteristics in living organisms |
| Allele | Each alternative form of a gene, occupying the same position in each pair of homologous chromosomes |
| Fst (fixation index) | Measure of population differentiation due to the genetic structure |
| Phenotype | A set of inherited characteristics that is dependent on both the genes and the environment |
| Genotype | A set of genes that are characteristic of each organism or individual |
| QTL (quantitative trait locus) | A locus on the chromosome that is associated with the quantitative variation of a trait |
| Association mapping | A germplasm-based approach to characterize QTLs or variations by exploiting the historic linkage disequilibrium to associate phenotypes with the underlying genotypes |
| Joint-linkage association mapping | A family-based approach to characterize QTLs or variations that are shared across families. It differs in power and scope from the characterization of QTLs based on bi-parental populations. |
| Marker-assisted selection | A set of tools that use gene markers to select, in a precise manner, the plants with the genetic potential to produce the desired trait for breeding |
| Genomics-assisted breeding | A set of genomics tools (using high-throughput approaches) to select in a targeted manner the plants with the genetic potential to produce the desired trait for breeding |
| Introgression | The gene flow from one genetic background (individual) to another gene pool (individual) by backcrossing with one of its parent |
| Backcross | Crossing of offspring lines with one of the original parental line |
| Linkage drag | Offspring with undesirable genetic background inherited from one of the parental lines |
| SNP (Single Nucleotide Polymorphism) | Single-nucleotide variations on the DNA sequence within a population or between paired chromosomes |
References
- Acquaah, G. Principles of Plant Genetics and Breeding; John Wiley & Sons: Oxford, UK, 2009. [Google Scholar]
- FAO. How to Feed the World 2050. Available online: http://www.fao.org/wsfs/forum2050/wsfs-forum/en/ (accessed on 29 January 2017).
- Lobell, D.B.; Schlenker, W.; Costa-Roberts, J. Climate trends and global crop production since 1980. Science 2011, 333, 616–620. [Google Scholar] [CrossRef] [PubMed]
- Tanksley, S.D.; McCouch, S.R. Seed banks and molecular maps: Unlocking genetic potential from the wild. Science 1997, 277, 1063–1066. [Google Scholar] [CrossRef] [PubMed]
- Dwivedi, S.L.; Upadhyaya, H.D.; Stalker, H.T.; Blair, M.W.; Bertioli, D.J.; Nielen, S.; Ortiz, R. Enhancing crop gene pools with beneficial traits using wild relatives. Plant Breed. Rev. 2008, 30, 179–230. [Google Scholar]
- Harlan, J.R. Genetic resources in wild relatives of crops. Crop Sci. 1976, 16, 329–333. [Google Scholar] [CrossRef]
- McCouch, S. Diversifying selection in plant breeding. PLoS Biol. 2004, 2, e347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, X.H.; Han, B. Natural variations and genome-wide association studies in crop plants. Annu. Rev. Plant Biol. 2014, 65, 531–551. [Google Scholar] [CrossRef] [PubMed]
- Li, M.-W.; Muñoz, N.B.; Wong, C.-F.; Wong, F.-L.; Wong, K.-S.; Wong, J.W.-H.; Qi, X.; Li, K.-P.; Ng, M.-S.; Lam, H.-M. QTLs regulating the contents of antioxidants, phenolics, and flavonoids in soybean seeds share a common genomic region. Front. Plant Sci. 2016, 7, 854. [Google Scholar] [CrossRef] [PubMed]
- Asif, M.; Rooney, L.W.; Ali, R.; Riaz, M.N. Application and opportunities of pulses in food system: A review. Crit. Rev. Food Sci. Nutr. 2013, 53, 1168–1179. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.J.; Nelson, R.L. Methodology for creating alloplasmic soybean lines by using Glycine tomentella as a maternal parent. Plant Breed. 2014, 133, 624–631. [Google Scholar] [CrossRef]
- Zamir, D. Improving plant breeding with exotic genetic libraries. Nat. Rev. Genet. 2001, 2, 983–989. [Google Scholar] [CrossRef] [PubMed]
- Foyer, C.H.; Lam, H.-M.; Nguyen, H.T.; Siddique, K.H.; Varshney, R.K.; Colmer, T.D.; Cowling, W.; Bramley, H.; Mori, T.A.; Hodgson, J.M.; et al. Neglecting legumes has compromised human health and sustainable food production. Nat. Plants 2016, 2, 16112. [Google Scholar] [CrossRef]
- Smýkal, P.; Coyne, C.J.; Ambrose, M.J.; Maxted, N.; Schaefer, H.; Blair, M.W.; Berger, J.; Greene, S.L.; Nelson, M.N.; Besharat, N. Legume crops phylogeny and genetic diversity for science and breeding. Crit. Rev. Plant Sci. 2015, 34, 43–104. [Google Scholar] [CrossRef] [Green Version]
- Moose, S.P.; Mumm, R.H. Molecular plant breeding as the foundation for 21st century crop improvement. Plant Physiol. 2008, 147, 969–977. [Google Scholar] [CrossRef] [PubMed]
- Nutritional Benefits of Pulses. Available online: http://www.fao.org/fileadmin/user_upload/pulses-2016/docs/factsheets/Nutrition_EN_PRINT.pdf (accessed on 29 January 2017).
- Kislev, M.E.; Bar-Yosef, O. The legumes: The earliest domesticated plants in the near east? Curr. Anthropol. 1988, 29, 175–179. [Google Scholar] [CrossRef] [Green Version]
- Zohary, D.; Hopf, M. Domestication of pulses in the old world: Legumes were companions of wheat and barley when agriculture began in the Near East. Science 1973, 182, 887–894. [Google Scholar] [CrossRef] [PubMed]
- Harlan, J.R.; de Wet, J.M. Toward a rational classification of cultivated plants. Taxon 1971, 20, 509–517. [Google Scholar] [CrossRef]
- Hymowitz, T. On the domestication of the soybean. Econ. Bot. 1970, 24, 408–421. [Google Scholar] [CrossRef]
- Soybeans (Glycine max)—The Plant History of the Marvelous Soybean. Available online: http://archaeology.about.com/od/Domesticated-Plants/fl/Soybeans-Glycine-max-The-Plant-History-of-the-Marvelous-Soybean.htm (accessed on 18 January 2017).
- Smith, K. Soybeans-History and Future. Available online: http://www.soymeal.org/FactSheets/HistorySoybeanUse.pdf (accessed on 29 January 2017).
- Singh, A.; Simpson, C. Biosystematics and genetic resources. In The Groundnut Crop; Springer: Dordrecht, The Netherlands, 1994; pp. 96–137. [Google Scholar]
- Bonavia, D. Los Gavilanes: Mar, Desierto y oásis en la Historia del Hombre: Precerámico Peruano; Instituto Arqueológico Alemán: Lima, Perú, 1982; pp. 449–490. [Google Scholar]
- Krapovickas, A.G. WC Taxonomía del género arachis (Leguminosae). Bonplandia 1994, 8, 1–4. [Google Scholar]
- Dillehay, T.D.; Rossen, J.; Andres, T.C.; Williams, D.E. Preceramic adoption of peanut, squash, and cotton in Northern Peru. Science 2007, 316, 1890–1893. [Google Scholar] [CrossRef] [PubMed]
- Grabiele, M.; Chalup, L.; Robledo, G.; Seijo, G. Genetic and geographic origin of domesticated peanut as evidenced by 5S rDNA and chloroplast DNA sequences. Plant Syst. Evol. 2012, 298, 1151–1165. [Google Scholar] [CrossRef]
- Bertioli, D.J.; Cannon, S.B.; Froenicke, L.; Huang, G.; Farmer, A.D.; Cannon, E.K.S.; Liu, X.; Gao, D.; Clevenger, J.; Dash, S.; et al. The genome sequences of Arachis duranensis and Arachis ipaensis, the diploid ancestors of cultivated peanut. Nat. Genet. 2016, 48, 438–446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Janila, P.; Variath, M.T.; Pandey, M.K.; Desmae, H.; Motagi, B.N.; Okori, P.; Manohar, S.S.; Rathnakumar, A.L.; Radhakrishnan, T.; Liao, B.; et al. Genomic tools in groundnut breeding program: Status and perspectives. Front. Plant Sci. 2016, 7, 289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daniel, Z.; Maria, H. Domestication of Plants in the Old World; Oxford University Press: New York, NY, USA, 2000. [Google Scholar]
- Ambrose, M. From Near East center of origin, the prized pea migrates throughout world. Divers. Arlingt. Then Wash. 1995, 11, 118–119. [Google Scholar]
- Smýkal, P.; Aubert, G.; Burstin, J.; Coyne, C.J.; Ellis, N.T.; Flavell, A.J.; Ford, R.; Hýbl, M.; Macas, J.; Neumann, P. Pea (Pisum sativum L.) in the genomic era. Agronomy 2012, 2, 74–115. [Google Scholar] [CrossRef]
- Pea (Pisum sativum L.) Domestication-the History of Peas and Humans. Available online: http://archaeology.about.com/od/Domesticated-Plants/fl/Pea-Pisum-sativum-L-Domestication-The-History-of-Peas-and-Humans.htm (accessed on 18 January 2017).
- Smýkal, P.; Kenicer, G.; Flavell, A.J.; Corander, J.; Kosterin, O.; Redden, R.J.; Ford, R.; Coyne, C.J.; Maxted, N.; Ambrose, M.J. Phylogeny, phylogeography and genetic diversity of the Pisum genus. Plant Genet. Resour. 2011, 9, 4–18. [Google Scholar] [CrossRef]
- Kerem, Z.; Lev-Yadun, S.; Gopher, A.; Weinberg, P.; Abbo, S. Chickpea domestication in the neolithic levant through the nutritional perspective. J. Archaeol. Sci. 2007, 34, 1289–1293. [Google Scholar] [CrossRef]
- Shahal, A.; Inbar, Z.; Efrat, S.; Simcha, L.-Y.; Zohar, K.; Avi, G. Wild lentil and chickpea harvest in Israel: Bearing on the origins of Near Eastern farming. J. Archaeol. Sci. 2008, 35, 3172–3177. [Google Scholar]
- Warkentin, T.; Banniza, S.; Vandenberg, A. CDC frontier kabuli chickpea. Can. J. Plant Sci. 2005, 85, 909–910. [Google Scholar] [CrossRef]
- Delgado-Salinas, A.; Bibler, R.; Lavin, M. Phylogeny of the genus Phaseolus (Leguminosae): A recent diversification in an ancient landscape. Syst. Bot. 2006, 31, 779–791. [Google Scholar] [CrossRef]
- Delgado-Salinas, A.; Turley, T.; Richman, A.; Lavin, M. Phylogenetic analysis of the cultivated and wild species of Phaseolus (Fabaceae). Syst. Bot. 1999, 438–460. [Google Scholar] [CrossRef]
- Freytag, G.F.; Debouck, D.G. Phaseolus costaricensis, a new wild bean species (Phaseolinae, Leguminosae) from Costa Rica and Panama, Central America. Novon 1996, 6, 157–163. [Google Scholar] [CrossRef]
- Schmutz, J.; McClean, P.E.; Mamidi, S.; Wu, G.A.; Cannon, S.B.; Grimwood, J.; Jenkins, J.; Shu, S.; Song, Q.; Chavarro, C.; et al. A reference genome for common bean and genome-wide analysis of dual domestications. Nat. Genet. 2014, 46, 707–713. [Google Scholar] [CrossRef] [PubMed]
- Bitocchi, E.; Nanni, L.; Bellucci, E.; Rossi, M.; Giardini, A.; Zeuli, P.S.; Logozzo, G.; Stougaard, J.; McClean, P.; Attene, G. Mesoamerican origin of the common bean (Phaseolus vulgaris L.) is revealed by sequence data. Proc. Natl. Acad. Sci. USA 2012, 109, E788–E796. [Google Scholar] [CrossRef] [PubMed]
- Bellucci, E.; Bitocchi, E.; Rau, D.; Rodriguez, M.; Biagetti, E.; Giardini, A.; Attene, G.; Nanni, L.; Papa, R. Genomics of origin, domestication and evolution of Phaseolus vulgaris. In Genomics of Plant Genetic Resources; Springer: Dordrecht, The Netherlands, 2014; pp. 483–507. [Google Scholar]
- Ljuština, M.; Mikić, A. Archaeological evidence for the domestication of lentil (Lens culinaris) and its distribution in Europe. J. Lentil Res. 2010, 4, 26–29. [Google Scholar]
- Sonnante, G.; Hammer, K.; Pignone, D. From the cradle of agriculture a handful of lentils: History of domestication. Rend. Lincei 2009, 20, 21–37. [Google Scholar] [CrossRef]
- D’Andrea, A.C.; Kahlheber, S.; Logan, A.L.; Watson, D.J. Early domesticated cowpea (Vigna unguiculata) from Central Ghana. Antiquity 2007, 81, 686–698. [Google Scholar] [CrossRef]
- Singh, B. Advances in Cowpea Research; IITA-JIRCAS: Ibadan, Nigeria, 1997. [Google Scholar]
- Perrino, P.; Laghetti, G.; Zeuli, P.S.; Monti, L. Diversification of cowpea in the mediterranean and other centres of cultivation. Genet. Resour. Crop Evol. 1993, 40, 121–132. [Google Scholar] [CrossRef]
- Kurlovich, B.S. Lupins: Geography, Classification, Genetic Resources and Breeding; Bogouslav Kourlovitch: Saint Petersburg, Russia, 2002. [Google Scholar]
- Fuller, D.; Korisettar, R.; Venkatasubbaiah, P.; Jones, M.K. Early plant domestications in Southern India: Some preliminary archaeobotanical results. Veg. Hist. Archaeobot. 2004, 13, 115–129. [Google Scholar] [CrossRef]
- Fuller, D.Q.; Harvey, E.L. The archaeobotany of indian pulses: Identification, processing and evidence for cultivation. Environ. Archaeol. 2006, 11, 219–246. [Google Scholar] [CrossRef]
- Gepts, P.; Papa, R. Evolution during domestication. eLS 2002. [Google Scholar] [CrossRef]
- Fuller, D.Q.; Denham, T.; Arroyo-Kalin, M.; Lucas, L.; Stevens, C.J.; Qin, L.; Allaby, R.G.; Purugganan, M.D. Convergent evolution and parallelism in plant domestication revealed by an expanding archaeological record. Proc. Natl. Acad. Sci. USA 2014, 111, 6147–6152. [Google Scholar] [CrossRef] [PubMed]
- Abbo, S.; van-Oss, R.P.; Gopher, A.; Saranga, Y.; Ofner, I.; Peleg, Z. Plant domestication versus crop evolution: A conceptual framework for cereals and grain legumes. Trends Plant Sci. 2014, 19, 351–360. [Google Scholar] [CrossRef] [PubMed]
- Fuller, D.Q. Contrasting patterns in crop domestication and domestication rates: Recent archaeobotanical insights from the old world. Ann. Bot. 2007, 100, 903–924. [Google Scholar] [CrossRef] [PubMed]
- Kluyver, T.A.; Charles, M.; Jones, G.; Rees, M.; Osborne, C.P. Did greater burial depth increase the seed size of domesticated legumes? J. Exp. Bot. 2013, 64, 4101–4108. [Google Scholar] [CrossRef] [PubMed]
- Abbo, S.; Lev-Yadun, S.; Gopher, A. The “human mind” as a common denominator in plant domestication. J. Exp. Bot. 2014, 65, 1917–1920. [Google Scholar] [CrossRef] [PubMed]
- Zeder, M.A. The origins of agriculture in the Near East. Curr. Anthropol. 2011, 52, S221–S235. [Google Scholar] [CrossRef]
- Larson, G.; Piperno, D.R.; Allaby, R.G.; Purugganan, M.D.; Andersson, L.; Arroyo-Kalin, M.; Barton, L.; Vigueira, C.C.; Denham, T.; Dobney, K. Current perspectives and the future of domestication studies. Proc. Natl. Acad. Sci. USA 2014, 111, 6139–6146. [Google Scholar] [CrossRef] [PubMed]
- Meyer, R.S.; DuVal, A.E.; Jensen, H.R. Patterns and processes in crop domestication: An historical review and quantitative analysis of 203 global food crops. New Phytol. 2012, 196, 29–48. [Google Scholar] [CrossRef] [PubMed]
- Kaul, S.; Koo, H.L.; Jenkins, J.; Rizzo, M.; Rooney, T.; Tallon, L.J.; Feldblyum, T.; Nierman, W.; Benito, M.I.; Lin, X.Y.; et al. Analysis of the genome sequence of the flowering plant Arabidopsis thaliana. Nature 2000, 408, 796–815. [Google Scholar]
- International Rice Genome Sequencing Project. The map-based sequence of the rice genome. Nature 2005, 436, 793–800. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Hu, S.; Wang, J.; Wong, G.K.; Li, S.; Liu, B.; Deng, Y.; Dai, L.; Zhou, Y.; Zhang, X.; et al. A draft sequence of the rice genome (Oryza sativa L. ssp. Indica). Science 2002, 296, 79–92. [Google Scholar] [CrossRef] [PubMed]
- Hayden, E.C. Technology: The $1000 genome. Nature 2014, 507, 294–295. [Google Scholar] [CrossRef] [PubMed]
- Schmutz, J.; Cannon, S.B.; Schlueter, J.; Ma, J.; Mitros, T.; Nelson, W.; Hyten, D.L.; Song, Q.; Thelen, J.J.; Cheng, J.; et al. Genome sequence of the palaeopolyploid soybean. Nature 2010, 463, 178–183. [Google Scholar] [CrossRef] [PubMed]
- Qi, X.; Li, M.-W.; Xie, M.; Liu, X.; Ni, M.; Shao, G.; Song, C.; Yim, A.K.-Y.; Tao, Y.; Wong, F.-L.; et al. Identification of a novel salt tolerance gene in wild soybean by whole-genome sequencing. Nat. Commun. 2014, 5, 4340. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.-H.; Zhou, G.; Ma, J.; Jiang, W.; Jin, L.-G.; Zhang, Z.; Guo, Y.; Zhang, J.; Sui, Y.; Zheng, L.; et al. De novo assembly of soybean wild relatives for pan-genome analysis of diversity and agronomic traits. Nat. Biotechnol. 2014, 32, 1045–1052. [Google Scholar] [CrossRef] [PubMed]
- Jain, M.; Misra, G.; Patel, R.K.; Priya, P.; Jhanwar, S.; Khan, A.W.; Shah, N.; Singh, V.K.; Garg, R.; Jeena, G. A draft genome sequence of the pulse crop chickpea (Cicer arietinum L.). Plant J. 2013, 74, 715–729. [Google Scholar] [CrossRef] [PubMed]
- Varshney, R.K.; Song, C.; Saxena, R.K.; Azam, S.; Yu, S.; Sharpe, A.G.; Cannon, S.; Baek, J.; Rosen, B.D.; Tar’an, B. Draft genome sequence of chickpea (Cicer arietinum) provides a resource for trait improvement. Nat. Biotechnol. 2013, 31, 240–246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parween, S.; Nawaz, K.; Roy, R.; Pole, A.K.; Suresh, B.V.; Misra, G.; Jain, M.; Yadav, G.; Parida, S.K.; Tyagi, A.K. An advanced draft genome assembly of a desi type chickpea (Cicer arietinum L.). Sci. Rep. 2015, 5, 12806. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Nawaz, K.; Parween, S.; Roy, R.; Sahu, K.; Pole, A.K.; Khandal, H.; Srivastava, R.; Parida, S.K.; Chattopadhyay, D. Draft genome sequence of Cicer reticulatum L., the wild progenitor of chickpea provides a resource for agronomic trait improvement. DNA Res. 2016. [Google Scholar] [CrossRef] [PubMed]
- Vlasova, A.; Capella-Gutiérrez, S.; Rendón-Anaya, M.; Hernández-Oñate, M.; Minoche, A.E.; Erb, I.; Câmara, F.; Prieto-Barja, P.; Corvelo, A.; Sanseverino, W. Genome and transcriptome analysis of the Mesoamerican common bean and the role of gene duplications in establishing tissue and temporal specialization of genes. Genome Biol. 2016, 17, 32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Timko, M.P.; Rushton, P.J.; Laudeman, T.W.; Bokowiec, M.T.; Chipumuro, E.; Cheung, F.; Town, C.D.; Chen, X. Sequencing and analysis of the gene-rich space of cowpea. BMC Genom. 2008, 9, 103. [Google Scholar] [CrossRef] [PubMed]
- Hane, J.K.; Ming, Y.; Kamphuis, L.G.; Nelson, M.N.; Garg, G.; Atkins, C.A.; Bayer, P.E.; Bravo, A.; Bringans, S.; Cannon, S. A comprehensive draft genome sequence for lupin (Lupinus angustifolius), an emerging health food: Insights into plant-microbe interactions and legume evolution. Plant Biotechnol. J. 2016. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Tao, Y.; Zheng, Z.; Zhang, Q.; Zhou, G.; Sweetingham, M.W.; Howieson, J.G.; Li, C. Draft genome sequence, and a sequence-defined genetic linkage map of the legume crop species Lupinus angustifolius L. PLoS ONE 2013, 8, e64799. [Google Scholar] [CrossRef] [PubMed]
- Varshney, R.K.; Chen, W.; Li, Y.; Bharti, A.K.; Saxena, R.K.; Schlueter, J.A.; Donoghue, M.T.; Azam, S.; Fan, G.; Whaley, A.M. Draft genome sequence of pigeonpea (Cajanus cajan), an orphan legume crop of resource-poor farmers. Nat. Biotechnol. 2012, 30, 83–89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, N.K.; Gupta, D.K.; Jayaswal, P.K.; Mahato, A.K.; Dutta, S.; Singh, S.; Bhutani, S.; Dogra, V.; Singh, B.P.; Kumawat, G. The first draft of the pigeonpea genome sequence. J. Plant Biochem. Biotechnol. 2012, 21, 98–112. [Google Scholar] [CrossRef] [PubMed]
- Lam, H.M.; Xu, X.; Liu, X.; Chen, W.; Yang, G.; Wong, F.L.; Li, M.W.; He, W.; Qin, N.; Wang, B.; et al. Resequencing of 31 wild and cultivated soybean genomes identifies patterns of genetic diversity and selection. Nat. Genet. 2010, 42, 1053–1059. [Google Scholar] [CrossRef] [PubMed]
- Chung, W.H.; Jeong, N.; Kim, J.; Lee, W.K.; Lee, Y.G.; Lee, S.H.; Yoon, W.; Kim, J.H.; Choi, I.Y.; Choi, H.K.; et al. Population structure and domestication revealed by high-depth resequencing of korean cultivated and wild soybean genomes. DNA Res. 2014, 21, 153–167. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Jiang, Y.; Wang, Z.; Gou, Z.; Lyu, J.; Li, W.; Yu, Y.; Shu, L.; Zhao, Y.; Ma, Y.; et al. Resequencing 302 wild and cultivated accessions identifies genes related to domestication and improvement in soybean. Nat. Biotechnol. 2015, 33, 408–414. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, M.; Rau, D.; Bitocchi, E.; Bellucci, E.; Biagetti, E.; Carboni, A.; Gepts, P.; Nanni, L.; Papa, R.; Attene, G. Landscape genetics, adaptive diversity and population structure in Phaseolus vulgaris. New Phytol. 2016, 209, 1781–1794. [Google Scholar] [CrossRef] [PubMed]
- Moreno, M.-T.; Cubero, J. Variation in Cicer arietinum L. Euphytica 1978, 27, 465–485. [Google Scholar] [CrossRef]
- Bajaj, D.; Das, S.; Badoni, S.; Kumar, V.; Singh, M.; Bansal, K.C.; Tyagi, A.K.; Parida, S.K. Genome-wide high-throughput SNP discovery and genotyping for understanding natural (functional) allelic diversity and domestication patterns in wild chickpea. Sci. Rep. 2015, 5, 12468. [Google Scholar] [CrossRef] [PubMed]
- Kochert, G.; Halward, T.; Branch, W.; Simpson, C. RFLP variability in peanut (Arachis hypogaea L.) cultivars and wild species. Theor. Appl. Genet. 1991, 81, 565–570. [Google Scholar] [CrossRef] [PubMed]
- Varshney, R.; Penmetsa, R.; Dutta, S.; Kulwal, P.; Saxena, R.; Datta, S.; Sharma, T.; Rosen, B.; Carrasquilla-Garcia, N.; Farmer, A. Pigeonpea genomics initiative (PGI): An international effort to improve crop productivity of pigeonpea (Cajanus cajan L.). Mol. Breed. 2010, 26, 393–408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kassa, M.T.; Penmetsa, R.V.; Carrasquilla-Garcia, N.; Sarma, B.K.; Datta, S.; Upadhyaya, H.D.; Varshney, R.K.; von Wettberg, E.J.; Cook, D.R. Genetic patterns of domestication in pigeonpea (Cajanus cajan (L.) Millsp.) and wild cajanus relatives. PLoS ONE 2012, 7, e39563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lentil Genome Pre-Release. Available online: http://knowpulse.usask.ca/portal/lentil-genome (accessed on 18 January 2017).
- Plant and Fungi Data Integration. Available online: https://urgi.versailles.inra.fr/Data/Genome/Genome-data-access (accessed on 18 January 2017).
- The Cowpea Genomics Initiative. Available online: http://cowpeagenomics.med.virginia.edu/ (accessed on 18 January 2017).
- Simon, M.; Benko-Iseppon, A.-M.; Resende, L.; Winter, P.; Kahl, G. Genetic diversity and phylogenetic relationships in Vigna Savi germplasm revealed by DNA amplification fingerprinting. Genome 2007, 50, 538–547. [Google Scholar] [PubMed]
- Nkongolo, K. Genetic characterization of malawian cowpea (Vigna unguiculata (L.) walp) landraces: Diversity and gene flow among accessions. Euphytica 2003, 129, 219–228. [Google Scholar] [CrossRef]
- Ghalmi, N.; Malice, M.; Jacquemin, J.-M.; Ounane, S.-M.; Mekliche, L.; Baudoin, J.-P. Morphological and molecular diversity within algerian cowpea (Vigna unguiculata (L.) Walp.) landraces. Genet. Resour. Crop Evol. 2010, 57, 371–386. [Google Scholar] [CrossRef]
- Wang, M.L.; Barkley, N.A.; Gillaspie, G.A.; Pederson, G.A. Phylogenetic relationships and genetic diversity of the USDA Vigna germplasm collection revealed by gene-derived markers and sequencing. Genet. Res. 2008, 90, 467–480. [Google Scholar] [CrossRef] [PubMed]
- Wamalwa, E.N.; Muoma, J.; Wekesa, C. Genetic diversity of cowpea (Vigna unguiculata (L.) Walp.) accession in Kenya gene bank based on simple sequence repeat markers. Int. J. Genom. 2016, 2016, 8956412. [Google Scholar] [CrossRef] [PubMed]
- Xiong, H.; Shi, A.; Mou, B.; Qin, J.; Motes, D.; Lu, W.; Ma, J.; Weng, Y.; Yang, W.; Wu, D. Genetic diversity and population structure of cowpea (Vigna unguiculata L. Walp). PLoS ONE 2016, 11, e0160941. [Google Scholar] [CrossRef] [PubMed]
- Coulibaly, S.; Pasquet, R.; Papa, R.; Gepts, P. AFLP analysis of the phenetic organization and genetic diversity of Vigna unguiculata L. Walp. Reveals extensive gene flow between wild and domesticated types. Theor. Appl. Genet. 2002, 104, 358–366. [Google Scholar] [CrossRef] [PubMed]
- Michelle, C.-A. Análisis de la Variabilidad Genética entre treinta accesiones de tarwi (Lupinus mutabilis Sweet) usando marcadores moleculares ISSR. Sci. Agropecu. 2016, 6, 17–30. [Google Scholar]
- Atchison, G.W.; Nevado, B.; Eastwood, R.J.; Contreras-Ortiz, N.; Reynel, C.; Madriñán, S.; Filatov, D.A.; Hughes, C.E. Lost crops of the Incas: Origins of domestication of the Andean pulse crop tarwi, Lupinus mutabilis. Am. J. Bot. 2016, 103, 1592–1606. [Google Scholar] [CrossRef] [PubMed]
- Iqbal, M.J.; Mamidi, S.; Ahsan, R.; Kianian, S.F.; Coyne, C.J.; Hamama, A.A.; Narina, S.S.; Bhardwaj, H.L. Population structure and linkage disequilibrium in Lupinus albus L. Germplasm and its implication for association mapping. Theor. Appl. Genet. 2012, 125, 517–530. [Google Scholar] [CrossRef] [PubMed]
- Wolko, B.; Clements, J.C.; Naganowska, B.; Nelson, M.N.; Yang, H. Wild Crop Relatives: Genomic and Breeding Resources. Legume Crops and Forages; Kole, C., Ed.; Springer: Heidelberg, Germany, 2011; p. 321. [Google Scholar]
- Sudheesh, S.; Sawbridge, T.I.; Cogan, N.O.; Kennedy, P.; Forster, J.W.; Kaur, S. De novo assembly and characterisation of the field pea transcriptome using RNA-seq. BMC Genom. 2015, 16, 611. [Google Scholar] [CrossRef] [PubMed]
- Tayeh, N.; Aubert, G.; Pilet-Nayel, M.-L.; Lejeune-Hénaut, I.; Warkentin, T.D.; Burstin, J. Genomic tools in pea breeding programs: Status and perspectives. Front. Plant Sci. 2015, 6, 1037. [Google Scholar] [CrossRef] [PubMed]
- Collard, B.C.Y.; Mackill, D.J. Marker-assisted selection: An approach for precision plant breeding in the twenty-first century. Philos. Trans. R. Soc. B Biol. Sci. 2008, 363, 557–572. [Google Scholar] [CrossRef] [PubMed]
- Desta, Z.A.; Ortiz, R. Genomic selection: Genome-wide prediction in plant improvement. Trends Plant Sci. 2014, 19, 592–601. [Google Scholar] [CrossRef] [PubMed]
- Tanksley, S.; Nelson, J. Advanced backcross QTL analysis: A method for the simultaneous discovery and transfer of valuable QTL from unadapted germplasm into elite breeding lines. Theor. Appl. Genet. 1996, 92, 191–203. [Google Scholar] [CrossRef] [PubMed]
- Alonso-Blanco, C.; Koornneef, M.; Stam, P. The use of recombinant inbred lines (RILs) for genetic mapping. In Arabidopsis Protocols; Martinez-Zapater, J., Salinas, J., Eds.; Humana Press: Totowa, NJ, USA, 1998; pp. 137–146. [Google Scholar]
- Kooke, R.; Wijnker, E.; Keurentjes, J.J. Backcross populations and near isogenic lines. Methods Mol. Biol. 2012, 871, 3–16. [Google Scholar] [PubMed]
- Yu, J.M.; Holland, J.B.; McMullen, M.D.; Buckler, E.S. Genetic design and statistical power of nested association mapping in maize. Genetics 2008, 178, 539–551. [Google Scholar] [CrossRef] [PubMed]
- Bandillo, N.; Raghavan, C.; Muyco, P.A.; Sevilla, M.A.L.; Lobina, I.T.; Dilla-Ermita, C.J.; Tung, C.-W.; McCouch, S.; Thomson, M.; Mauleon, R. Multi-parent advanced generation inter-cross (MAGIC) populations in rice: Progress and potential for genetics research and breeding. Rice 2013, 6, 11. [Google Scholar] [CrossRef] [PubMed]
- Acquaah, G. Principles of Plant Genetics and Breeding, 2nd ed.; John Wiley & Sons: Oxford, UK, 2012. [Google Scholar]
- Hufford, M.B.; Xu, X.; van Heerwaarden, J.; Pyhajarvi, T.; Chia, J.M.; Cartwright, R.A.; Elshire, R.J.; Glaubitz, J.C.; Guill, K.E.; Kaeppler, S.M.; et al. Comparative population genomics of maize domestication and improvement. Nat. Genet. 2012, 44, 808–811. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.H.; Kurata, N.; Wei, X.H.; Wang, Z.X.; Wang, A.; Zhao, Q.; Zhao, Y.; Liu, K.Y.; Lu, H.Y.; Li, W.J.; et al. A map of rice genome variation reveals the origin of cultivated rice. Nature 2012, 490, 497–501. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Liu, X.; Ge, S.; Jensen, J.D.; Hu, F.; Li, X.; Dong, Y.; Gutenkunst, R.N.; Fang, L.; Huang, L.; et al. Resequencing 50 accessions of cultivated and wild rice yields markers for identifying agronomically important genes. Nat. Biotechnol. 2012, 30, 105–111. [Google Scholar] [CrossRef] [PubMed]
- Hu, Z.B.; Zhang, D.; Zhang, G.Z.; Kan, G.Z.; Hong, D.L.; Yu, D.Y. Association mapping of yield-related traits and SSR markers in wild soybean (Glycine soja Sieb. and Zucc.). Breed. Sci. 2014, 63, 441–449. [Google Scholar] [CrossRef] [PubMed]
- Jaccoud, D.; Peng, K.M.; Feinstein, D.; Kilian, A. Diversity arrays: A solid state technology for sequence information independent genotyping. Nucleic Acids Res. 2001, 29, E25. [Google Scholar] [CrossRef] [PubMed]
- Miller, M.R.; Dunham, J.P.; Amores, A.; Cresko, W.A.; Johnson, E.A. Rapid and cost-effective polymorphism identification and genotyping using restriction site associated DNA (RAD) markers. Genome Res. 2007, 17, 240–248. [Google Scholar] [CrossRef] [PubMed]
- Pratap, A.; Kumar, J. Alien gene transfer in crop plants: An introduction. In Alien Gene Transfer in Crop Plants, Volume 1: Innovations, Methods and Risk Assessment; Pratap, A., Kumar, J., Eds.; Springer: New York, NY, USA, 2014; pp. 1–23. [Google Scholar]
- Xie, W.; Feng, Q.; Yu, H.; Huang, X.; Zhao, Q.; Xing, Y.; Yu, S.; Han, B.; Zhang, Q. Parent-independent genotyping for constructing an ultrahigh-density linkage map based on population sequencing. Proc. Natl. Acad. Sci. USA 2010, 107, 10578–10583. [Google Scholar] [CrossRef] [PubMed]
- Pandey, M.K.; Roorkiwal, M.; Singh, V.K.; Ramalingam, A.; Kudapa, H.; Thudi, M.; Chitikineni, A.; Rathore, A.; Varshney, R.K. Emerging genomic tools for legume breeding: Current status and future prospects. Front. Plant Sci. 2016, 7, 455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, X.H.; Feng, Q.; Qian, Q.; Zhao, Q.; Wang, L.; Wang, A.H.; Guan, J.P.; Fan, D.L.; Weng, Q.J.; Huang, T.; et al. High-throughput genotyping by whole-genome resequencing. Genome Res. 2009, 19, 1068–1076. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.H.; Xie, W.B.; Wang, J.; Xing, Y.Z.; Xu, C.G.; Li, X.H.; Xiao, J.H.; Zhang, Q.F. Gains in QTL detection using an ultra-high density SNP map based on population sequencing relative to traditional RFLP/SSR markers. PLoS ONE 2011, 6, e17595. [Google Scholar]
- Iquira, E.; Humira, S.; François, B. Association mapping of QTLs for sclerotinia stem rot resistance in a collection of soybean plant introductions using a genotyping by sequencing (GBS) approach. BMC Plant Biol. 2015, 15, 5. [Google Scholar] [CrossRef] [PubMed]
- Manavalan, L.P.; Prince, S.J.; Musket, T.A.; Chaky, J.; Deshmukh, R.; Vuong, T.D.; Song, L.; Cregan, P.B.; Nelson, J.C.; Shannon, J.G.; et al. Identification of novel QTL governing root architectural traits in an interspecific soybean population. PLoS ONE 2015, 10, e0120490. [Google Scholar] [CrossRef] [PubMed]
- Muñoz, N.; Qi, X.; Li, M.; Xie, M.; Gao, Y.; Cheung, M.; Wong, F.; Lam, H. Improvement in nitrogen fixation capacity could be part of the domestication process in soybean. Heredity 2016, 117, 84–93. [Google Scholar] [CrossRef] [PubMed]
- Mkwaila, W.; Terpstra, K.A.; Ender, M.; Kelly, J.D. Identification of QTL for agronomic traits and resistance to white mold in wild and landrace germplasm of common bean. Plant Breed. 2011, 130, 665–672. [Google Scholar] [CrossRef]
- Mamidi, S.; Miklas, P.N.; Trapp, J.; Felicetti, E.; Grimwood, J.; Schmutz, J.; Lee, R.; McClean, P.E. Sequence-based introgression mapping identifies candidate white mold tolerance genes in common bean. Plant Genome 2016, 9. [Google Scholar] [CrossRef] [PubMed]
- Blair, M.W.; Iriarte, G.; Beebe, S. QLT analysis of yield traits in an advanced backcross population derived from a cultivated Andean× wild common bean (Phaseolus vulgaris L.) cross. Theor. Appl. Genet. 2006, 112, 1149–1163. [Google Scholar] [CrossRef] [PubMed]
- Blair, M.W.; Izquierdo, P. Use of the advanced backcross-QTL method to transfer seed mineral accumulation nutrition traits from wild to Andean cultivated common beans. Theor. Appl. Genet. 2012, 125, 1015–1031. [Google Scholar] [CrossRef] [PubMed]
- O’Rourke, J.A.; Iniguez, L.P.; Fu, F.; Bucciarelli, B.; Miller, S.S.; Jackson, S.A.; McClean, P.E.; Li, J.; Dai, X.; Zhao, P.X. An RNA-seq based gene expression atlas of the common bean. BMC Genom. 2014, 15, 866. [Google Scholar] [CrossRef] [PubMed]
- Crampton, M.; Sripathi, V.R.; Hossain, K.; Kalavacharla, V. Analyses of methylomes derived from Meso-American common bean (Phaseolus vulgaris L.) using MeDIP-seq and whole genome sodium bisulfite-sequencing. Front. Plant Sci. 2016, 7. [Google Scholar] [CrossRef] [PubMed]
- Ariani, A.; y Teran, J.C.B.M.; Gepts, P. Genome-wide identification of SNPs and copy number variation in common bean (Phaseolus vulgaris L.) using genotyping-by-sequencing (GBS). Mol. Breed. 2016, 36. [Google Scholar] [CrossRef]
- Perseguini, J.M.K.C.; Oblessuc, P.R.; Rosa, J.R.B.F.; Gomes, K.A.; Chiorato, A.F.; Carbonell, S.A.M.; Garcia, A.A.F.; Vianello, R.P.; Benchimol-Reis, L.L. Genome-wide association studies of anthracnose and angular leaf spot resistance in common bean (Phaseolus vulgaris L.). PLoS ONE 2016, 11, e0150506. [Google Scholar] [CrossRef] [PubMed]
- Schröder, S.; Mamidi, S.; Lee, R.; McKain, M.R.; McClean, P.E.; Osorno, J.M. Optimization of genotyping by sequencing (GBS) data in common bean (Phaseolus vulgaris L.). Mol. Breed. 2016, 36. [Google Scholar] [CrossRef]
- Wang, D.; Graef, G.; Procopiuk, A.; Diers, B. Identification of putative QTL that underlie yield in interspecific soybean backcross populations. Theor. Appl. Genet. 2004, 108, 458–467. [Google Scholar] [CrossRef] [PubMed]
- Winter, S.M.; Shelp, B.J.; Anderson, T.R.; Welacky, T.W.; Rajcan, I. QTL associated with horizontal resistance to soybean cyst nematode in Glycine soja PI464925B. Theor. Appl. Genet. 2007, 114, 461–472. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Li, C.; Davis, E.L.; Wang, J.; Griffin, J.D.; Kofsky, J.; Song, B.-H. Genome-wide association study of resistance to soybean Cyst nematode (Heterodera glycines) HG type 2.5.7 in wild soybean (Glycine soja). Front. Plant Sci. 2016, 7, 1214. [Google Scholar] [CrossRef] [PubMed]
- Bajaj, D.; Das, S.; Upadhyaya, H.D.; Ranjan, R.; Badoni, S.; Kumar, V.; Tripathi, S.; Gowda, C.L.; Sharma, S.; Singh, S. A genome-wide combinatorial strategy dissects complex genetic architecture of seed coat color in chickpec. Front. Plant Sci. 2015, 6, 979. [Google Scholar] [CrossRef] [PubMed]
- Upadhyaya, H.D.; Bajaj, D.; Das, S.; Saxena, M.S.; Badoni, S.; Kumar, V.; Tripathi, S.; Gowda, C.; Sharma, S.; Tyagi, A.K. A genome-scale integrated approach aids in genetic dissection of complex flowering time trait in chickpea. Plant Mol. Biol. 2015, 89, 403–420. [Google Scholar] [CrossRef] [PubMed]
- Saxena, M.S.; Bajaj, D.; Das, S.; Kujur, A.; Kumar, V.; Singh, M.; Bansal, K.C.; Tyagi, A.K.; Parida, S.K. An integrated genomic approach for rapid delineation of candidate genes regulating agro-morphological traits in chickpea. DNA Res. 2014, 21, 695–710. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, R.; Singh, M.; Bajaj, D.; Parida, S.K. A high-resolution InDel (insertion–deletion) markers-anchored consensus genetic map identifies major QTLs governing pod number and seed yield in chickpea. Front. Plant Sci. 2016, 7, 1362. [Google Scholar] [CrossRef] [PubMed]
- Das, S.; Singh, M.; Srivastava, R.; Bajaj, D.; Saxena, M.S.; Rana, J.C.; Bansal, K.C.; Tyagi, A.K.; Parida, S.K. MQTL-seq delineates functionally relevant candidate gene harbouring a major QTL regulating pod number in chickpea. DNA Res. 2016, 23, 53–65. [Google Scholar] [CrossRef] [PubMed]
- Leal-Bertioli, S.C.; José, A.C.; Alves-Freitas, D.M.; Moretzsohn, M.C.; Guimarães, P.M.; Nielen, S.; Vidigal, B.S.; Pereira, R.W.; Pike, J.; Fávero, A.P. Identification of candidate genome regions controlling disease resistance in Arachis. BMC Plant Biol. 2009, 9, 112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leal-Bertioli, S.C.; Moretzsohn, M.C.; Roberts, P.A.; Ballén-Taborda, C.; Borba, T.C.; Valdisser, P.A.; Vianello, R.P.; Araújo, A.C.G.; Guimarães, P.M.; Bertioli, D.J. Genetic mapping of resistance to Meloidogyne arenaria in Arachis stenosperma: A new source of nematode resistance for peanut. G3 2016, 6, 377–390. [Google Scholar] [CrossRef] [PubMed]
- Fonceka, D.; Tossim, H.A.; Rivallan, R.; Vignes, H.; Faye, I.; Ndoye, O.; Moretzsohn, M.C.; Bertioli, D.J.; Glaszmann, J.C.; Courtois, B.; et al. Fostered and left behind alleles in peanut: Interspecific QTL mapping reveals footprints of domestication and useful natural variation for breeding. BMC Plant Biol. 2012, 12, 26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burow, M.D.; Simpson, C.E.; Paterson, A.H.; Starr, J.L. Identification of peanut (Arachis hypogaea L.) RAPD markers diagnostic of root-knot nematode (Meloidogyne arenaria (Neal) Chitwood) resistance. Mol. Breed. 1996, 2, 369–379. [Google Scholar] [CrossRef]
- Fonceka, D.; Tossim, H.-A.; Rivallan, R.; Vignes, H.; Lacut, E.; de Bellis, F.; Faye, I.; Ndoye, O.; Leal-Bertioli, S.C.; Valls, J.F. Construction of chromosome segment substitution lines in peanut (Arachis hypogaea L.) using a wild synthetic and QTL mapping for plant morphology. PLoS ONE 2012, 7, e48642. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fondevilla, S.; Satovic, Z.; Rubiales, D.; Moreno, M.T.; Torres, A.M. Mapping of quantitative trait loci for resistance to Mycosphaerella pinodes in Pisum sativum subsp. syriacum. Mol. Breed. 2008, 21, 439–454. [Google Scholar] [CrossRef]
- Weeden, N.F. Genetic changes accompanying the domestication of Pisum sativum: Is there a common genetic basis to the “domestication syndrome” for legumes? Ann. Bot. 2007, 100, 1017–1025. [Google Scholar] [CrossRef] [PubMed]
- Saxena, K.; Ariyanayagam, R.; Reddy, L. Genetics of a high-selfing trait in pigeonpea. Euphytica 1992, 59, 125–127. [Google Scholar] [CrossRef]
- Saxena, K.; Kumar, R.; Rao, P. Pigeonpea nutrition and its improvement. J. Crop Prod. 2002, 5, 227–260. [Google Scholar] [CrossRef]
- Saxena, K.; Kumar, R.; Latha, K.M.; Dalvi, V. Commercial pigeonpea hybrids are just a few steps away. Indian J. Pulses Res. 2006, 19, 7–16. [Google Scholar]
- Saxena, R.K.; Cui, X.; Thakur, V.; Walter, B.; Close, T.J.; Varshney, R.K. Single feature polymorphisms (SFPS) for drought tolerance in pigeonpea (Cajanus spp.). Funct. Integr. Genom. 2011, 11, 651–657. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tullu, A.; Bett, K.; Banniza, S.; Vail, S.; Vandenberg, A. Widening the genetic base of cultivated lentil through hybridization of Lens culinaris “Eston” and L. ervoides accession IG 72815. Can. J. Plant Sci. 2013, 93, 1037–1047. [Google Scholar] [CrossRef]
- Andargie, M.; Knudsen, J.T.; Pasquet, R.S.; Gowda, B.S.; Muluvi, G.M.; Timko, M.P. Mapping of quantitative trait loci for floral scent compounds in cowpea (Vigna unguiculata L.). Plant Breed. 2014, 133, 92–100. [Google Scholar] [CrossRef]
- Andargie, M.; Pasquet, R.S.; Gowda, B.S.; Muluvi, G.M.; Timko, M.P. Construction of a SSR-based genetic map and identification of QTL for domestication traits using recombinant inbred lines from a cross between wild and cultivated cowpea (V. unguiculata (L.) Walp.). Mol. Breed. 2011, 28, 413–420. [Google Scholar] [CrossRef]
- Andargie, M.; Pasquet, R.S.; Muluvi, G.M.; Timko, M.P. Quantitative trait loci analysis of flowering time related traits identified in recombinant inbred lines of cowpea (Vigna unguiculata). Genome 2013, 56, 289–294. [Google Scholar] [CrossRef] [PubMed]
- Kongjaimun, A.; Kaga, A.; Tomooka, N.; Somta, P.; Shimizu, T.; Shu, Y.; Isemura, T.; Vaughan, D.A.; Srinives, P. An SSR-based linkage map of yardlong bean (Vigna unguiculata (L.) Walp. subsp. unguiculata Sesquipedalis Group) and QTL analysis of pod length. Genome 2012, 55, 81–92. [Google Scholar] [PubMed]
- Kongjaimun, A.; Kaga, A.; Tomooka, N.; Somta, P.; Vaughan, D.A.; Srinives, P. The genetics of domestication of yardlong bean, Vigna unguiculata (L.) Walp. ssp. unguiculata cv.-gr. sesquipedalis. Ann. Bot. 2012, 109, 1185–1200. [Google Scholar] [PubMed]
- Kongjaimun, A.; Somta, P.; Tomooka, N.; Kaga, A.; Vaughan, D.A.; Srinives, P. QTL mapping of pod tenderness and total soluble solid in yardlong bean [Vigna unguiculata (L.) Walp. subsp. unguiculata cv.-gr. sesquipedalis]. Euphytica 2013, 189, 217–223. [Google Scholar] [CrossRef]
- Varshney, R.K. Exciting journey of 10 years from genomes to fields and markets: Some success stories of genomics-assisted breeding in chickpea, pigeonpea and groundnut. Plant Sci. 2016, 242, 98–107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, R.; Sharma, P.; Varshney, R.K.; Sharma, S.; Singh, N. Chickpea improvement: Role of wild species and genetic markers. Biotechnol. Genet. Eng. Rev. 2008, 25, 267–314. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, R.; Bajaj, D.; Malik, A.; Singh, M.; Parida, S.K. Transcriptome landscape of perennial wild Cicer microphyllum uncovers functionally relevant molecular TAGs regulating agronomic traits in chickpea. Sci. Rep. 2016, 6. [Google Scholar] [CrossRef] [PubMed]
- Kujur, A.; Upadhyaya, H.D.; Bajaj, D.; Gowda, C.; Sharma, S.; Tyagi, A.K.; Parida, S.K. Identification of candidate genes and natural allelic variants for QTLs governing plant height in chickpea. Sci. Res. 2016, 6, 27968. [Google Scholar] [CrossRef] [PubMed]
- Bajaj, D.; Srivastava, R.; Nath, M.; Tripathi, S.; Bharadwaj, C.; Upadhyaya, H.D.; Tyagi, A.K.; Parida, S.K. Ecotilling-based association mapping efficiently delineates functionally relevant natural allelic variants of candidate genes governing agronomic traits in chickpea. Front. Plant Sci. 2016, 7, 450. [Google Scholar] [CrossRef] [PubMed]
- Kudapa, H.; Azam, S.; Sharpe, A.G.; Taran, B.; Li, R.; Deonovic, B.; Cameron, C.; Farmer, A.D.; Cannon, S.B.; Varshney, R.K. Comprehensive transcriptome assembly of chickpea (Cicer arietinum L.) using sanger and next generation sequencing platforms: Development and applications. PLoS ONE 2014, 9, e86039. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bertioli, D.J.; Seijo, G.; Freitas, F.O.; Valls, J.F.; Leal-Bertioli, S.C.; Moretzsohn, M.C. An overview of peanut and its wild relatives. Plant Genet. Resour. 2011, 9, 134–149. [Google Scholar] [CrossRef] [Green Version]
- Hong, Y.; Pandey, M.K.; Liu, Y.; Chen, X.; Liu, H.; Varshney, R.K.; Liang, X.; Huang, S. Identification and evaluation of single-nucleotide polymorphisms in allotetraploid peanut (Arachis hypogaea L.) based on amplicon sequencing combined with high resolution melting (HRM) analysis. Front. Plant Sci. 2015, 6, 1068. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, L.; Redden, R.J.; Zong, X.; Berger, J.; Bennett, S.J. Ecogeographic analysis of pea collection sites from China to determine potential sites with abiotic stresses. Genet. Resour. Crop Evol. 2013, 60, 1801–1815. [Google Scholar] [CrossRef]
- Boutet, G.; Alves Carvalho, S.; Falque, M.; Peterlongo, P.; Lhuillier, E.; Bouchez, O.; Lavaud, C.; Pilet-Nayel, M.-L.; Rivière, N.; Baranger, A. SNP discovery and genetic mapping using genotyping by sequencing of whole genome genomic DNA from a pea RIL population. BMC Genom. 2016, 17, 121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharpe, A.G.; Ramsay, L.; Sanderson, L.-A.; Fedoruk, M.J.; Clarke, W.E.; Li, R.; Kagale, S.; Vijayan, P.; Vandenberg, A.; Bett, K.E. Ancient orphan crop joins modern era: Gene-based SNP discovery and mapping in lentil. BMC Genom. 2013, 14, 192. [Google Scholar] [CrossRef] [PubMed]
- Saxena, K. Genetic improvement of pigeonpea—A review. Trop. Plant Biol. 2008, 1, 159–178. [Google Scholar] [CrossRef]
- Saxena, R.K.; Saxena, K.; Varshney, R.K. Application of SSR markers for molecular characterization of hybrid parents and purity assessment of ICPH 2438 hybrid of pigeonpea [Cajanus cajan (L.) Millspaugh]. Mol. Breed. 2010, 26, 371–380. [Google Scholar] [CrossRef]
- Bohra, A.; Jha, R.; Singh, I.P.; Pandey, G.; Pareek, S.; Basu, P.S.; Chaturvedi, S.K.; Singh, N.P. Novel CMS lines in pigeonpea [Cajanus cajan (L.) Millspaugh] derived from cytoplasmic substitutions, and their effective restoration and deployment in hybrid breeding. Crop J. 2017, 5, 89–94. [Google Scholar] [CrossRef]
- Saxena, K.; Sultana, R.; Mallikarjuna, N.; Saxena, R.; Kumar, R.; Sawargaonkar, S.; Varshney, R. Male-sterility systems in pigeonpea and their role in enhancing yield. Plant Breed. 2010, 129, 125–134. [Google Scholar] [CrossRef]
- Pazhamala, L.; Saxena, R.K.; Singh, V.K.; Sameerkumar, C.; Kumar, V.; Sinha, P.; Patel, K.; Obala, J.; Kaoneka, S.R.; Tongoona, P. Genomics-assisted breeding for boosting crop improvement in pigeonpea (Cajanus cajan). Front. Plant Sci. 2015, 6, 50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mallikarjuna, N.; Jadhav, D.; Reddy, P. Introgression of Cajanus platycarpus genome into cultivated pigeonpea, C. cajan. Euphytica 2006, 149, 161–167. [Google Scholar] [CrossRef] [Green Version]
- Saxena, K.; Singh, L.; Reddy, M.; Singh, U.; Lateef, S.; Sharma, S.; Remanandan, P. Intra species variation in Atylosia scarabaeoides (L.) Benth., a wild relative of pigeonpea (Cajanus cajan (L.) Millsp.). Euphytica 1990, 49, 185–191. [Google Scholar]
- Boukar, O.; Fatokun, C.A.; Huynh, B.-L.; Roberts, P.A.; Close, T.J. Genomic tools in cowpea breeding programs: Status and perspectives. Front. Plant Sci. 2016, 7, 757. [Google Scholar] [CrossRef] [PubMed]
- Fatokun, C.A.; Singh, B.B. Interspecific hybridization between Vigna pubescens and V. unquiculata (L.) Walp through embryo rescue. Plant Cell Tissue Organ Cult. 1987, 9, 229–233. [Google Scholar] [CrossRef]
- Souleymane, A.; Aken’Ova, M.; Fatokun, C.; Alabi, O. Screening for resistance to cowpea aphid (Aphis craccivora koch) in wild and cultivated cowpea (Vigna unguiculata L. Walp.) accessions. Int. J. Sci. Environ. Technol. 2013, 2, 611–621. [Google Scholar]
- HarvEST. Available online: http://harvest.ucr.edu/ (accessed on 18 January 2017).
- Wyrwa, K.; Książkiewicz, M.; Szczepaniak, A.; Susek, K.; Podkowiński, J.; Naganowska, B. Integration of Lupinus angustifolius L. (narrow-leafed lupin) genome maps and comparative mapping within legumes. Chromosome Res. 2016, 24, 355–358. [Google Scholar] [CrossRef] [PubMed]
- Parra-González, L.B.; Aravena-Abarzúa, G.A.; Navarro-Navarro, C.S.; Udall, J.; Maughan, J.; Peterson, L.M.; Salvo-Garrido, H.E.; Maureira-Butler, I.J. Yellow lupin (Lupinus luteus L.) transcriptome sequencing: Molecular marker development and comparative studies. BMC Genom. 2012, 13. [Google Scholar] [CrossRef]
- Gao, L.-L.; Hane, J.K.; Kamphuis, L.G.; Foley, R.; Shi, B.-J.; Atkins, C.A.; Singh, K.B. Development of genomic resources for the narrow-leafed lupin (Lupinus angustifolius): Construction of a bacterial artificial chromosome (BAC) library and BAC-end sequencing. BMC Genom. 2011, 12, 521. [Google Scholar] [CrossRef] [PubMed]
- Tian, L.; Peel, G.J.; Lei, Z.; Aziz, N.; Dai, X.; He, J.; Watson, B.; Zhao, P.X.; Sumner, L.W.; Dixon, R.A. Transcript and proteomic analysis of developing white lupin (Lupinus albus L.) roots. BMC Plant Biol. 2009, 9, 1. [Google Scholar] [CrossRef] [PubMed]
- Lulsdorf, M.M.; Ferrie, A.; Slater, S.M.H.; Yuan, H.Y. Methods and Role of Embryo Rescue Technique in Alien Gene Transfer. In Alien Gene Transfer in Crop Plants, Volume 1: Innovations, Methods and Risk Assessment; Pratap, A., Kumar, J., Eds.; Springer: New York, NY, USA, 2014. [Google Scholar]
- Hajjar, R.; Hodgkin, T. The use of wild relatives in crop improvement: A survey of developments over the last 20 years. Euphytica 2007, 156, 1–13. [Google Scholar] [CrossRef]
- Warschefsky, E.; Penmetsa, R.V.; Cook, D.R.; von Wettberg, E.J. Back to the wilds: Tapping evolutionary adaptations for resilient crops through systematic hybridization with crop wild relatives. Am. J. Bot. 2014, 101, 1791–1800. [Google Scholar] [CrossRef] [PubMed]
- Hawkes, J. The importance of wild germplasm in plant breeding. Euphytica 1977, 26, 615–621. [Google Scholar] [CrossRef]
- Zhang, H.; Mittal, N.; Leamy, L.J.; Barazani, O.; Song, B.H. Back into the wild-apply untapped genetic diversity of wild relatives for crop improvement. Evol. Appl. 2017, 10, 5–24. [Google Scholar] [CrossRef] [PubMed]
- Upadhyaya, H.D.; Dwivedi, S.L.; Ambrose, M.; Ellis, N.; Berger, J.; Smýkal, P.; Debouck, D.; Duc, G.; Dumet, D.; Flavell, A. Legume genetic resources: Management, diversity assessment, and utilization in crop improvement. Euphytica 2011, 180, 27–47. [Google Scholar] [CrossRef]
- Kilian, B.; Graner, A. NGS technologies for analyzing germplasm diversity in genebanks. Brief. Funct. Genom. 2012, 11, 38–50. [Google Scholar] [CrossRef] [PubMed]
- Van Treuren, R.; van Hintum, T.J. Next-generation genebanking: Plant genetic resources management and utilization in the sequencing era. Plant Genet. Resour. 2014, 12, 298–307. [Google Scholar] [CrossRef]
- Ma, C.; Zhang, H.H.; Wang, X. Machine learning for big data analytics in plants. Trends Plant Sci. 2014, 19, 798–808. [Google Scholar] [CrossRef] [PubMed]
- Maxted, N.; Dulloo, E.; V Ford-Lloyd, B.; Iriondo, J.M.; Jarvis, A. Gap analysis: A tool for complementary genetic conservation assessment. Divers. Distrib. 2008, 14, 1018–1030. [Google Scholar] [CrossRef]
- Ramírez-Villegas, J.; Khoury, C.; Jarvis, A.; Debouck, D.G.; Guarino, L. A gap analysis methodology for collecting crop genepools: A case study with Phaseolus beans. PLoS ONE 2010, 5, e13497. [Google Scholar] [CrossRef] [PubMed]
- Dempewolf, H.; Eastwood, R.J.; Guarino, L.; Khoury, C.K.; Müller, J.V.; Toll, J. Adapting agriculture to climate change: A global initiative to collect, conserve, and use crop wild relatives. Agroecol. Sust. Food 2014, 38, 369–377. [Google Scholar] [CrossRef]
- Castañeda-Álvarez, N.P.; Khoury, C.K.; Achicanoy, H.A.; Bernau, V.; Dempewolf, H.; Eastwood, R.J.; Guarino, L.; Harker, R.H.; Jarvis, A.; Maxted, N. Global conservation priorities for crop wild relatives. Nat. Plants 2016, 2, 16022. [Google Scholar] [CrossRef]


{kind=link}
| Crop | Genus | Species | Germplasm | Assembled Genome Size (Mb) | Predicted Genome Size (Mb) | Available Database(s) * | Reference |
|---|---|---|---|---|---|---|---|
| Soybean | Glycine | max | Cultivated (William 82) | 950 | 1115 | 1, 2 | [65] |
| Soybean | Glycine | soja | Wild | 868 | 1170 | [66] | |
| Soybean | Glycine | soja | Wild | 813–985 | 889–1118 | [67] | |
| Peanut | Arachis | duranensis | Wild | 1211 | 1250 | 3 | [28] |
| Peanut | Arachis | ipaensis | Wild | 1512 | 1560 | [28] | |
| Pea | Pisum | sativum | N/D | N/D | 4685 # | 4, 5 | [13] |
| Chickpea | Cicer | arietinum | Cultivated (desi-type) | 520 | 740 | 6, 4, 7 | [68] |
| Chickpea | Cicer | arietinum | Cultivated (kabuli-type) | 532 | 738 | [69] | |
| Chickpea | Cicer | arietinum | Cultivated (desi-type) | 511 | 740 | [70] | |
| Chickpea | Cicer | reticulatum | Wild | 416 | 817 | [71] | |
| Common bean | Phaseolus | vulgaris | Landrace | 473 | 587 | 6, 4, 8, 2 | [41] |
| Common bean | Phaseolus | vulgaris | Breeding lines | 550 | 587 | [72] | |
| Lentil | Lens | culinaris | N/D | N/D | 4032 # | 4 | [13] |
| Cowpea | Vigna | unguiculata | Cultivated (IT97K-499-35) | Not complete | 620 | 9 | [73] |
| Lupin | Lupinus | angustifolius | Cultivated (Tanjil) | 609 | 951 | 6, 10, 11 | [74] |
| Lupin | Lupinus | angustifolius | Cultivated (Tanjil) | 598 | 1153 | [75] | |
| Pigeonpea | Cajanus | cajan | Cultivated (Asha) | 606 | 833 | 6 | [76] |
| Pigeonpea | Cajanus | cajan | Cultivated (Asha) | 511 | 858 | [77] |
| Grain Legume | Trait/s | Population Strategy | Genotyping/Mapping Strategy | QTL/Gene | Reference |
|---|---|---|---|---|---|
| Soybean | Salt tolerance, seed anthocyanin content, pod and seed number per plant, growth period, seed coat color, pod color, trailing growth, leaf length/width ratio, nodule number per plant with cultivated incompatible rhizobia strain | RILs (cultivated × wild) | GBS-WGR | CHX cation anti-transporter for salt tolerance, QTLs for the others traits | [66] |
| Soybean | Nodule fresh weight per plant, root fresh weight, total plant fresh weight and ureides (μmol per plant) | RILs (cultivated × wild) | GBS-WGR | QTLs | [124] |
| Soybean | Antioxidants, phenolics, and flavonoids in seeds | RILs (cultivated × wild) | GBS-WGR | MATE transporters | [9] |
| Soybean | Sclerotinia stem rot resistance | 101 lines | GBS-association mapping | QTL | [122] |
| Soybean | Root traits (tap root length and lateral root number) and shoot length | BC2F5 (cultivated × wild) | SSR and SNP markers | QTLs | [123] |
| Soybean | Oil content, flower color, seed coat color, pubescence form and reported domestication-related QTLs | 302 wild and cultivated accessions | GBS-association mapping | GWAS signals associated | [80] |
| Soybean | Yield, height and maturity | BC2F4 (cultivated × wild) | SSR markers | QTLs | [134] |
| Soybean | Soybean cyst nematode resistance | RILs (cultivated × wild) | SSR markers | QTLs | [135] |
| Soybean | Soybean cyst nematode resistance | 235 wild soybean accessions | GBS-GWAS | QTLs | [136] |
| Common bean | White mold resistance | NILs source of resistance from Andean genotype Jatu Rong | InDel, SCAR, SNP and phaseolin markers | QTLs | [126] |
| Common bean | Seed weight, seed size, days to flowering, yield, plant height | BC2F3:5 (cultivated × wild) | Microsatellite, SCAR, and phaseolin markers | QTLs | [127] |
| Common bean | White mold resistance | BC2F3 (cultivated × wild) BC1F4:5 (landrace × cultivated) | SSR, SRAP, TRAP markers | QLTs | [125] |
| Common bean | Seed weight, seed mineral accumulation: iron concentration (ppm), zinc concentration (ppm), iron content (mg/seed), zinc content (mg/seed) | BC2F3:5 (cultivated × wild) | Microsatellite markers | QTLs | [128] and cited herein |
| Chickpea | Seed coat color | Germplasm collection (93 cultivated desi and kabuli and 79 wild) and RILs (landrace × landrace) | GBS-WGR-association and QTL mapping | MATE transporter | [137] |
| Chickpea | Flowering time | Germplasm collection (92 cultivated desi and kabuli including beaded, landraces and wild) | Genome-wide GBS- and candidate gene-based genotyping | Eight potential known/candidate flowering time-regulating genes and QTLs | [138] |
| Chickpea | 100-seed weight, pod and branch number/plant and plant hairiness | RILs (cultivated × wild) | SSR and SNP polymorphism marker-based | QTLs and seed weight regulating ABI3VP1 transcription factor | [139] |
| Chickpea | Pod number and seed yield per plant | Two F5 mapping populations (cultivated × wild) | GBS-WGR SNP InDel markers | QTLs | [140,141] |
| Chickpea | 100-seed weight | RILs (cultivated × landrace) | GBS-WGR | QTLs | [137] |
| Peanut | Root-knot nematodes resistance, drought-related traits and agronomic/domestication traits | RILs (wild × wild) | SNP markers and integrated consensus map from Shirasawa et al. 2013 [142] | QTLs | [143] |
| Peanut | Water availability, flowering precocity, seed and pod number, length and size, and pod maturity | 87 BC3F1 and 55 BC2F2 [cultivated × wild amphidiploid (A. ipaensis × A. duranensis)] | SSR markers | QTLs | [144] |
| Peanut | Root-knot nematode resistance | BC4F2 population (cultivated × wilds) | RAPD markers | Resistance associated to markers | [145] |
| Peanut | Plant growth habit, height of the main stem, plant spread and flower color | Chromosome segment substitution lines (CSSLs). [wild synthetic allotetraploid (A. ipaensis × A. duranensis) × cultivated] | SSR markers | QTLs | [146] |
| Peanut | Late leaf spot resistance | F2 (A. duranensis × A. stenosperma) | Microsatellite, AFLP and legume anchor markers | QTLs | [142] |
| Pea | Mycosphaerella pinodes resistance | RILs (cultivated × wild) | RAPD, Sequence-tagged site and expressed sequence tag markers | QTLs | [147] |
| Pea | Seed weight, root/shoot ratio, flowering response, pod dehiscence, seed dormancy, plant height, basal branching | Set of five recombinant inbred populations: wild × cultivated F12; cultivated × wild F6; cultivated × primitive BC1F4; primitive × wild F4; cultivated × landrace F5 | Morphological Markers, allozyme variation and RAPD | QTLs and genes | [148] |
| Pigeonpea | Cleistogamous line | F2 (cultivated × wild) | N/A | Gene | [149] |
| Pigeonpea | High protein | Cultivated × wild | N/A | Traditional breeding (pedigree method) | [150] |
| Pigeonpea | Male sterility lines | BCn (cultivated × wild) | N/A | Traditional breeding | [151] |
| Pigeonpea | Drought tolerance and pod borer insect resistance | F2 (wild × cultivated) | Single feature polymorphisms (SFPs) | Genes | [152] |
| Lentil | Anthracnose resistance, seed yield, biomass, straw yield, seed weight, harvest index, podding ability and stand at maturity | RILs (wild × cultivated) | N/A | N/A potential material for breeding | [153] |
| Cowpea | Floral scent compounds, seed size, pod fiber layer thickness, seed weight, time of flower opening, days to flower | RILs (cultivated × wild) | SSR | QTLs | [154,155,156] |
| Yardlong bean (Vigna unguiculata ssp. unguiculata cv.-gr. sesquipedalis) | Pod length | BC1F1 (cultivated × wild) | SSR | QTLs | [157] |
| Yardlong bean | Domestication related traits | BC1F1 (cultivated × wild) | SSR | QTLs | [158] |
| Yardlong bean | Pod tenderness | BC1F1 and F2 (cultivated × wild) | SSR | QTLs | [159] |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Muñoz, N.; Liu, A.; Kan, L.; Li, M.-W.; Lam, H.-M. Potential Uses of Wild Germplasms of Grain Legumes for Crop Improvement. Int. J. Mol. Sci. 2017, 18, 328. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms18020328
Muñoz N, Liu A, Kan L, Li M-W, Lam H-M. Potential Uses of Wild Germplasms of Grain Legumes for Crop Improvement. International Journal of Molecular Sciences. 2017; 18(2):328. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms18020328
Chicago/Turabian StyleMuñoz, Nacira, Ailin Liu, Leo Kan, Man-Wah Li, and Hon-Ming Lam. 2017. "Potential Uses of Wild Germplasms of Grain Legumes for Crop Improvement" International Journal of Molecular Sciences 18, no. 2: 328. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms18020328