
Redox Proteomics and Platelet Activation: Understanding the Redox Proteome to Improve Platelet Quality for Transfusion

Abstract
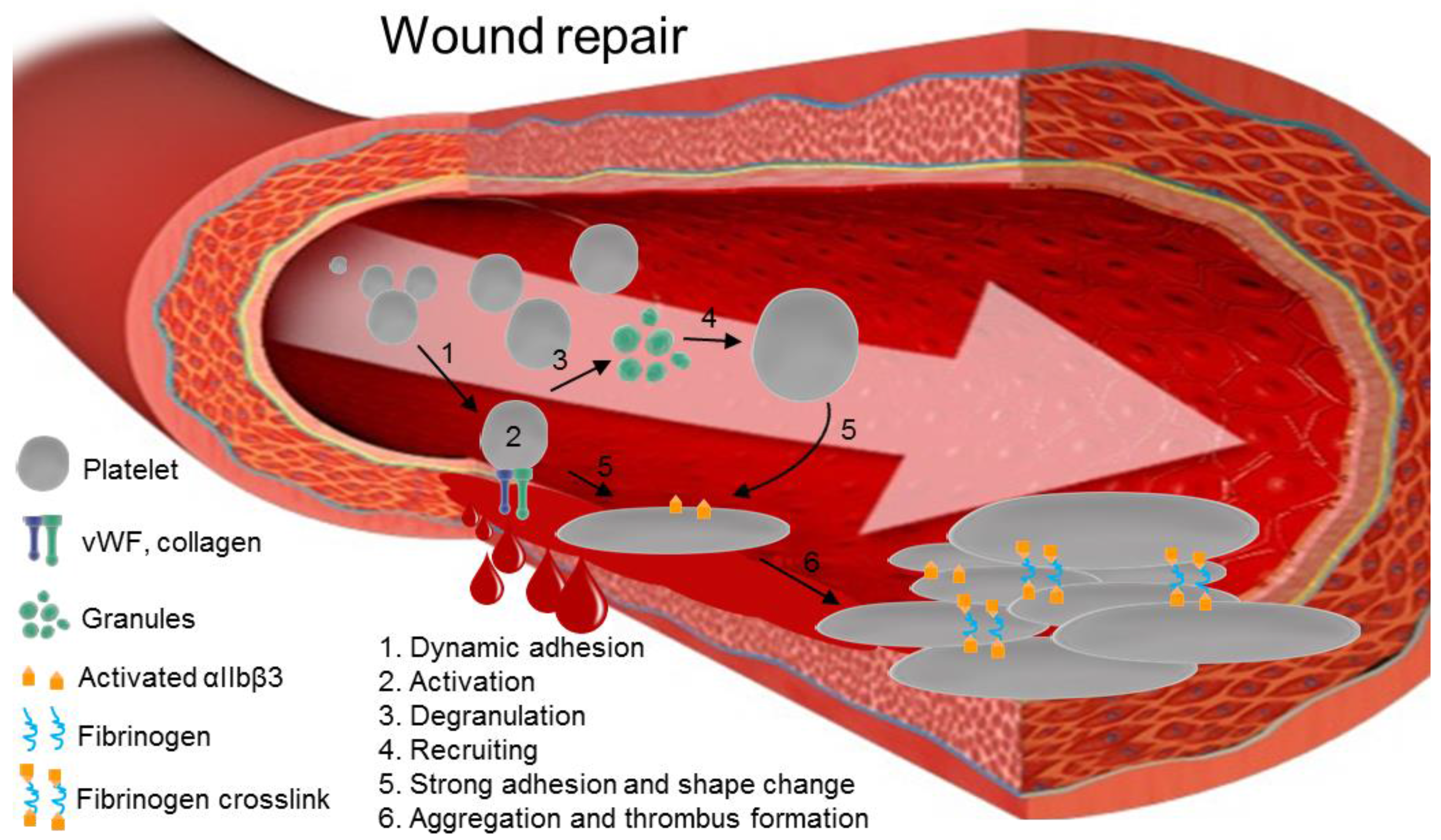
:1. Introduction
2. Alterations and Oxidation of Platelet Proteins
3. The Role of ROS in Platelet Function
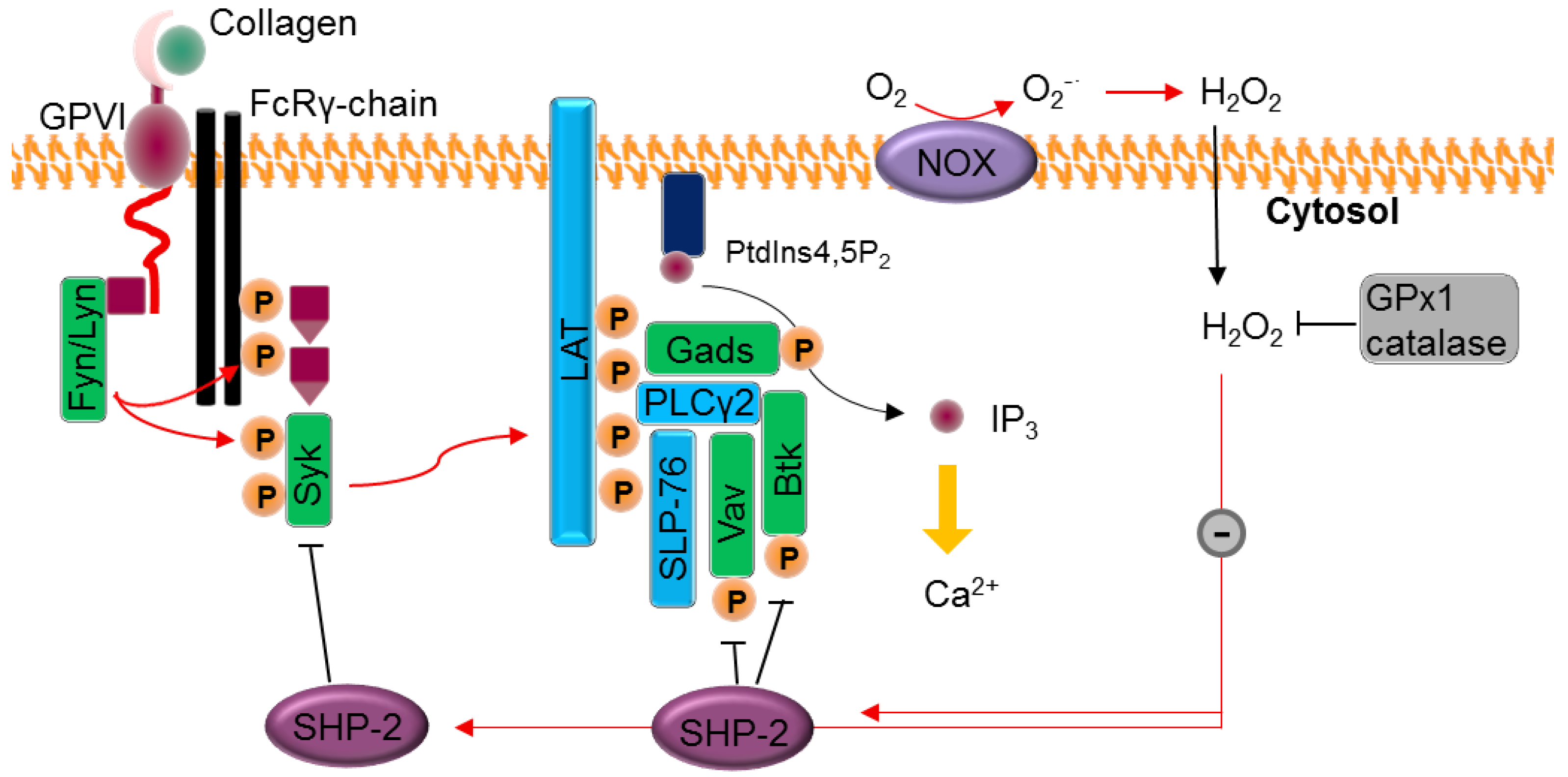
3.1. Platelet Activation Pathways Stimulate ROS Production
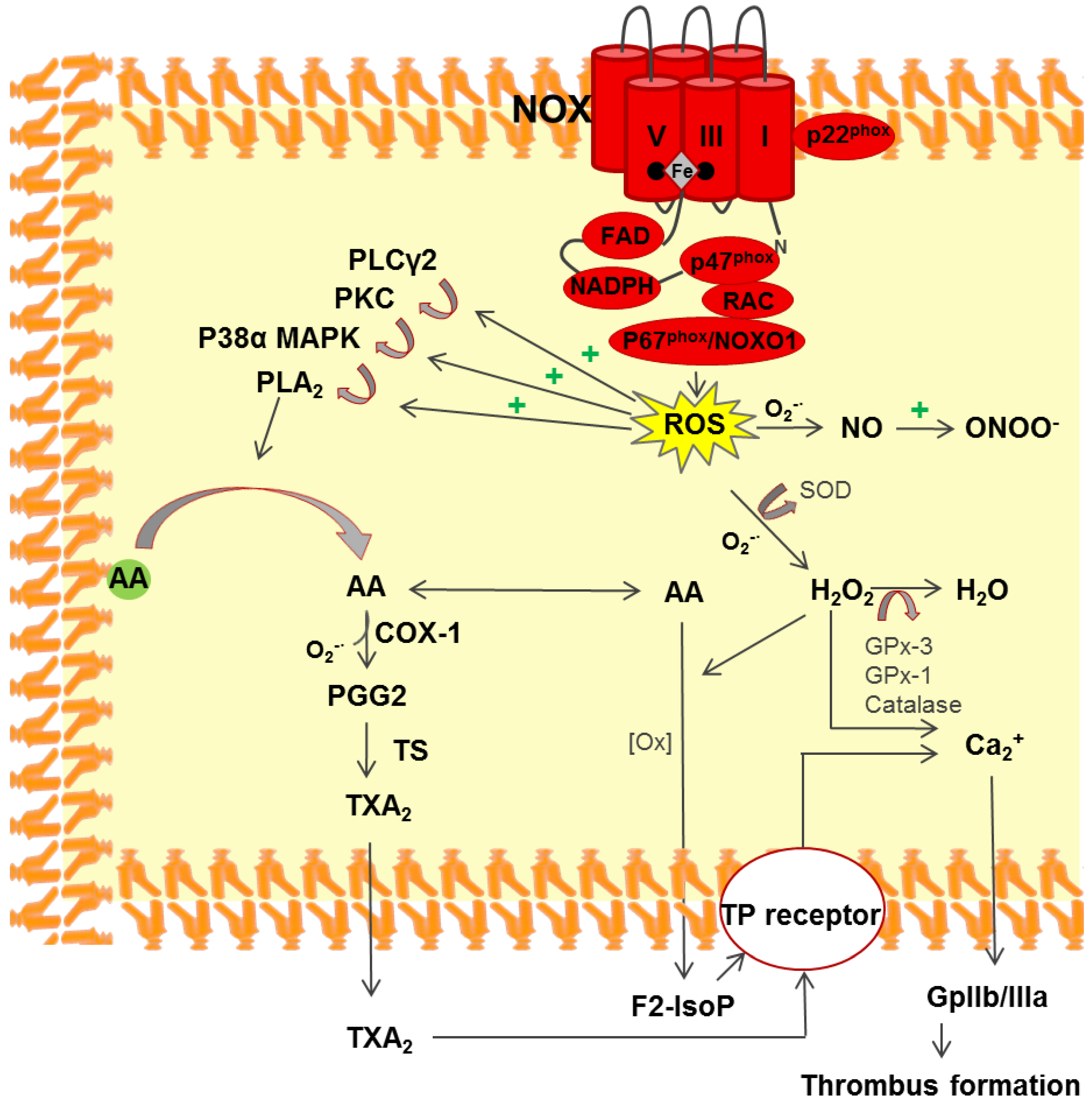
3.2. NADPH Oxidase
3.3. Platelet Activation via ROS
4. Role of Redox Cysteine in Platelets
4.1. The Involvement of Cysteine in ROS Sensing and Defense
4.2. Cysteine-Mediated Redox Signaling
5. ROS-Phosphorylation Crosstalk
6. The Overlap of Redox Proteomics and Transfusion
6.1. The Impact of Antioxidants on PI-Treated PCs
6.2. Cold Storage and Cryopreservation
6.3. Whole Blood Inactivation and Cold Storage
7. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| 2D-GE | Two dimensional-gel electrophoresis |
| F-actin | Actin filaments |
| AA | Ascorbic acid |
| CCI | Corrected count increment |
| COX1 | Cyclooxygenase 1 |
| CyPA | Cyclophilin A |
| DCF | 2′,7′-dichlorofluorescein |
| F2-IsoP | F2 isoprostane |
| GP-Ib-IX | Glycoprotein Ib-IX |
| GPVI | Glycoprotein VI |
| H2DCFDA | 2′,7′-dichlorodihydrofluorescein diacetate |
| IP3 | Isoprostane 3 |
| LAT | Linker of activated T-cells |
| mPT | Mitochondria permeability transition |
| NOX | NADPH oxidase |
| PC | Platelet concentrate |
| PDI | Protein disulfide isomerase |
| PGG2 | Prostaglandin G2 |
| PI | Pathogen inactivation |
| PIT | Pathogen inactivation technology |
| PLA2 | Phospholipase A2 |
| PLCg-2 | Phospholipase Cg 2 |
| PrxII | Peroxiredoxin-II |
| PTM | Post-translational modification |
| oxPTM | Oxidative PTM |
| RBC | Red blood cell |
| ROS | Reactive oxygen species |
| RT | Room temperature |
| SOD | Superoxide dismutase |
| TRAF4 | TNF receptor associated factor 4 |
| TXA2 | Thromboxane A2 |
References
- Garraud, O. Editorial: Platelets as immune cells in physiology and immunopathology. Front. Immunol. 2015, 6, 274. [Google Scholar] [CrossRef] [PubMed]
- Rondina, M.T.; Weyrich, A.S.; Zimmerman, G.A. Platelets as cellular effectors of inflammation in vascular diseases. Circ. Res. 2013, 112, 1506–1519. [Google Scholar] [CrossRef] [PubMed]
- Holbro, A.; Infanti, L.; Sigle, J.; Buser, A. Platelet transfusion: Basic aspects. Swiss Med. Wkly. 2013, 143, 13885. [Google Scholar] [CrossRef] [PubMed]
- Hoffmeister, K.M.; Felbinger, T.W.; Falet, H.; Denis, C.V.; Bergmeier, W.; Mayadas, T.N.; von Andrian, U.H.; Wagner, D.D.; Stossel, T.P.; Hartwig, J.H. The clearance mechanism of chilled blood platelets. Cell 2003, 112, 87–97. [Google Scholar] [CrossRef]
- Salunkhe, V.; van der Meer, P.F.; de Korte, D.; Seghatchian, J.; Gutiérrez, L. Development of blood transfusion product pathogen reduction treatments: A review of methods, current applications and demands. Transfus. Apher. Sci. 2015, 52, 19–34. [Google Scholar] [CrossRef] [PubMed]
- Kaiser-Guignard, J.; Canellini, G.; Lion, N.; Abonnenc, M.; Osselaer, J.-C.; Tissot, J.-D. The clinical and biological impact of new pathogen inactivation technologies on platelet concentrates. Blood Rev. 2014, 28, 235–241. [Google Scholar] [CrossRef] [PubMed]
- Prowse, C.V. Component pathogen inactivation: A critical review. Vox Sang. 2013, 104, 183–199. [Google Scholar] [CrossRef] [PubMed]
- Abonnenc, M.; Sonego, G.; Kaiser-Guignard, J.; Crettaz, D.; Prudent, M.; Tissot, J.-D.; Lion, N. In vitro evaluation of pathogen-inactivated buffy coat-derived platelet concentrates during storage: Psoralen-based photochemical treatment step-by-step. Blood Transfus. 2015, 13, 255–264. [Google Scholar] [PubMed]
- Ignatova, A.A.; Karpova, O.V.; Trakhtman, P.E.; Rumiantsev, S.A.; Panteleev, M.A. Functional characteristics and clinical effectiveness of platelet concentrates treated with riboflavin and ultraviolet light in plasma and in platelet additive solution. Vox Sang. 2016, 110, 244–252. [Google Scholar] [CrossRef] [PubMed]
- Janetzko, K.; Hinz, K.; Marschner, S.; Goodrich, R.; Kluter, H. Evaluation of different preparation procedures of pathogen reduction technology mirasol(R)-treated platelets collected by plateletpheresis. Transfus. Med. Hemother. 2009, 36, 309–315. [Google Scholar] [CrossRef] [PubMed]
- Maurer-Spurej, E.; Larsen, R.; Labrie, A.; Heaton, A.; Chipperfield, K. Microparticle content of platelet concentrates is predicted by donor microparticles and is altered by production methods and stress. Transfus. Apher. Sci. 2016, 55, 35–43. [Google Scholar] [CrossRef] [PubMed]
- Kerkhoffs, J.L.H. Evaluation of platelet transfusion clinical trials—Response to Corash & Sherman. Br. J. Haematol. 2011, 153, 531–532. [Google Scholar]
- Kerkhoffs, J.L.H.; van Putten, W.L.J.; Novotny, V.M.J.; Boekhorst, P.; Schipperus, M.R.; Zwaginga, J.J.; van Pampus, L.C.M.; de Greef, G.E.; Luten, M.; Huijgens, P.C.; et al. Clinical effectiveness of leucoreduced, pooled donor platelet concentrates, stored in plasma or additive solution with and without pathogen reduction. Br. J. Haematol. 2010, 150, 209–217. [Google Scholar] [CrossRef] [PubMed]
- Corash, L.; Sherman, C.D. Evaluation of platelet transfusion clinical trials. Br. J. Haematol. 2011, 153, 529–531. [Google Scholar] [CrossRef] [PubMed]
- Cazenave, J.P.; Follea, G.; Bardiaux, L.; Boiron, J.M.; Lafeuillade, B.; Debost, M.; Lioure, B.; Harousseau, J.L.; Tabrizi, R.; Cahn, J.Y.; et al. A randomized controlled clinical trial evaluating the performance and safety of platelets treated with MIRASOL pathogen reduction technology. Transfusion 2010, 50, 2362–2375. [Google Scholar] [CrossRef] [PubMed]
- Lozano, M.; Knutson, F.; Tardivel, R.; Cid, J.; Maymo, R.M.; Lof, H.; Roddie, H.; Pelly, J.; Docherty, A.; Sherman, C.; et al. A multi-centre study of therapeutic efficacy and safety of platelet components treated with amotosalen and ultraviolet A pathogen inactivation stored for 6 or 7 d prior to transfusion. Br. J. Haematol. 2011, 153, 393–401. [Google Scholar] [CrossRef] [PubMed]
- Heddle, N.M.; Ness, P.M. Consort and clinical trial reporting: Room for improvement. Transfusion 2016, 56, 781–783. [Google Scholar] [CrossRef] [PubMed]
- Cook, R.J.; Heddle, N.M. Clinical trials evaluating pathogen-reduced platelet products: Methodologic issues and recommendations. Transfusion 2013, 53, 1843–1855. [Google Scholar] [CrossRef] [PubMed]
- Feys, H.B.; van Aelst, B.; Devreese, K.; Devloo, R.; Coene, J.; Vandekerckhove, P.; Compernolle, V. Oxygen removal during pathogen inactivation with riboflavin and UV light preserves protein function in plasma for transfusion. Vox Sang. 2014, 106, 307–315. [Google Scholar] [CrossRef] [PubMed]
- Johnson, L.; Marks, D. Treatment of platelet concentrates with the mirasol pathogen inactivation system modulates platelet oxidative stress and NF-κB activation. Transfus. Med. Hemother. 2015, 42, 167–173. [Google Scholar] [CrossRef] [PubMed]
- Kitamura, N.; Kohtani, S.; Nakagaki, R. Molecular aspects of furocoumarin reactions: Photophysics, photochemistry, photobiology, and structural analysis. J. Photochem. Photobiol. C Photochem. Rev. 2005, 6, 168–185. [Google Scholar] [CrossRef]
- Cardoso, D.R.; Libardi, S.H.; Skibsted, L.H. Riboflavin as a photosensitizer. Effects on human health and food quality. Food Funct. 2012, 3, 487–502. [Google Scholar] [CrossRef] [PubMed]
- Herrling, T.; Jung, K.; Fuchs, J. Measurements of UV-generated free radicals/reactive oxygen species (ROS) in skin. Spectrochim. Acta A Mol. Biomol. Spectrosc. 2006, 63, 840–845. [Google Scholar] [CrossRef] [PubMed]
- Abonnenc, M.; Sonego, G.; Crettaz, D.; Aliotta, A.; Prudent, M.; Tissot, J.-D.; Lion, N. In vitro study of platelet function confirms the contribution of the ultraviolet B (UVB) radiation in the lesions observed in riboflavin/UVB-treated platelet concentrates. Transfusion 2015, 55, 2219–2230. [Google Scholar] [CrossRef] [PubMed]
- Freedman, J.E. Oxidative stress and platelets. Arterioscler. Thromb. Vasc. Biol. 2008, 28, s11–s16. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Zehnder, J.L. Oxidative stress and immune thrombocytopenia. Consult. Hematol. 2013, 50, e1–e4. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Bae, H.Y.; Kim, S.Y. Clinical marker of platelet hyperreactivity in diabetes mellitus. Diabetes Metab. J. 2013, 37, 423–428. [Google Scholar] [CrossRef] [PubMed]
- Pignatelli, P.; Pulcinelli, F.M.; Lenti, L.; Gazzaniga, P.P.; Violi, F. Hydrogen peroxide is involved in collagen-induced platelet activation. Blood 1998, 91, 484–490. [Google Scholar] [PubMed]
- Chakrabarti, A.; Halder, S.; Karmakar, S. Erythrocyte and platelet proteomics in hematological disorders. Proteomics Clin. Appl. 2016, 10, 403–414. [Google Scholar] [CrossRef] [PubMed]
- Circu, M.L.; Aw, T.Y. Reactive oxygen species, cellular redox systems and apoptosis. Free Radic. Biol. Med. 2010, 48, 749–762. [Google Scholar] [CrossRef] [PubMed]
- Begonja, A.J.; Gambaryan, S.; Geiger, J.; Aktas, B.; Pozgajova, M.; Nieswandt, B.; Walter, U. Platelet NAD(P)H-oxidase–generated ROS production regulates αIIbβ3-integrin activation independent of the NO/cGMP pathway. Blood 2005, 106, 2757–2760. [Google Scholar] [CrossRef] [PubMed]
- Krötz, F.; Sohn, H.Y.; Gloe, T.; Zahler, S.; Riexinger, T.; Schiele, T.M.; Becker, B.F.; Theisen, K.; Klauss, V.; Pohl, U. NAD(P)H oxidase-dependent platelet superoxide anion release increases platelet recruitment. Blood 2002, 100, 917. [Google Scholar] [CrossRef] [PubMed]
- Go, Y.-M.; Jones, D.P. The redox proteome. J. Biol. Chem. 2013, 288, 26512–26520. [Google Scholar] [CrossRef] [PubMed]
- Singh, H.; Chaudhary, R.; Ray, V. Evaluation of platelet storage lesions in platelet concentrates stored for seven days. Indian J. Med. Res. 2003, 118, 243–246. [Google Scholar] [PubMed]
- Thiele, T.; Iuga, C.; Janetzky, S.; Schwertz, H.; Gesell Salazar, M.; Furll, B.; Volker, U.; Greinacher, A.; Steil, L. Early storage lesions in apheresis platelets are induced by the activation of the integrin αIIbβ(3) and focal adhesion signaling pathways. J. Proteom. 2012, 76, 297–315. [Google Scholar] [CrossRef] [PubMed]
- Schubert, P.; Devine, D.V. Towards targeting platelet storage lesion-related signaling pathways. Blood Transfus. 2010, 8, 69–72. [Google Scholar]
- Prudent, M.; D’Alessandro, A.; Cazenave, J.P.; Devine, D.V.; Gachet, C.; Greinacher, A.; Lion, N.; Schubert, P.; Steil, L.; Thiele, T.; et al. Proteome changes in platelets after pathogen inactivation—An interlaboratory consensus. Transfus. Med. Rev. 2014, 28, 72–83. [Google Scholar] [CrossRef] [PubMed]
- Schubert, P.; Culibrk, B.; Karwal, S.; Goodrich, R.P.; Devine, D.V. Protein translation occurs in platelet concentrates despite riboflavin/UV light pathogen inactivation treatment. Proteomics Clin. Appl. 2016, 10, 839–850. [Google Scholar] [CrossRef] [PubMed]
- Harrison, P.; Briggs, C. Chapter 27—Platelet Counting. In Platelets, 3rd ed.; Michelson, A.D., Ed.; Elsevier: London, UK, 2013; pp. 547–557. [Google Scholar]
- Landry, P.; Plante, I.; Ouellet, D.L.; Perron, M.P.; Rousseau, G.; Provost, P. Existence of a microRNA pathway in anucleate platelets. Nat. Struct. Mol. Biol. 2009, 16, 961–966. [Google Scholar] [CrossRef] [PubMed]
- Burkhart, J.M.; Vaudel, M.; Gambaryan, S.; Radau, S.; Walter, U.; Martens, L.; Geiger, J.; Sickmann, A.; Zahedi, R.P. The first comprehensive and quantitative analysis of human platelet protein composition allows the comparative analysis of structural and functional pathways. Blood 2012, 120, e73. [Google Scholar] [CrossRef] [PubMed]
- Marrocco, C.; D’Alessandro, A.; Girelli, G.; Zolla, L. Proteomic analysis of platelets treated with γ irradiation versus a commercial photochemical pathogen reduction technology. Transfusion 2013, 53, 1808–1820. [Google Scholar] [CrossRef] [PubMed]
- Prudent, M.; Crettaz, D.; Delobel, J.; Tissot, J.-D.; Lion, N. Proteomic analysis of Intercept-treated platelets. J. Proteom. 2012, 76, 316–328. [Google Scholar] [CrossRef] [PubMed]
- Hechler, B.; Ohlmann, P.; Chafey, P.; Ravanat, C.; Eckly, A.; Maurer, E.; Mangin, P.; Isola, H.; Cazenave, J.P.; Gachet, C. Preserved functional and biochemical characteristics of platelet components prepared with amotosalen and ultraviolet A for pathogen inactivation. Transfusion 2013, 53, 1187–1200. [Google Scholar] [CrossRef] [PubMed]
- Thiele, T.; Sablewski, A.; Iuga, C.; Bakchoul, T.; Bente, A.; Gorg, S.; Volker, U.; Greinacher, A.; Steil, L. Profiling alterations in platelets induced by amotosalen/UVA pathogen reduction and γ irradiation—A LC-ESI-MS/MS-based proteomics approach. Blood Transfus. 2012, 10, 63–70. [Google Scholar]
- Madian, A.G.; Regnier, F.E. Detection of oxidized polypeptides. U.S. Patent 9,134,318, 15 September 2015. [Google Scholar]
- Suzuki, Y.J.; Carini, M.; Butterfield, D.A. Protein carbonylation. Antioxid. Redox Signal. 2010, 12, 323–325. [Google Scholar] [CrossRef] [PubMed]
- Prudent, M.; Sonego, G.; Abonnenc, M.; Tissot, J.-D.; Lion, N. LC-MS/MS Analysis and comparison of oxidative damages on peptides induced by pathogen reduction technologies for platelets. J. Am. Soc. Mass Spectrom. 2014, 25, 651–661. [Google Scholar] [CrossRef] [PubMed]
- Cattaruzza, M.; Hecker, M. Protein carbonylation and decarboylation: A new twist to the complex response of vascular cells to oxidative stress. Circ. Res. 2008, 102, 273–274. [Google Scholar] [CrossRef] [PubMed]
- Wright, A.; Bubb, W.A.; Hawkins, C.L.; Davies, M.J. Singlet oxygen-mediated protein oxidation: Evidence for the formation of reactive side chain peroxides on tyrosine residues. Photochem. Photobiol. 2002, 76, 35–46. [Google Scholar] [CrossRef]
- Abonnenc, M.; Crettaz, D.; Marvin, L.; Grund, B.; Sonego, G.; Bardyn, M.; Tissot, J.-D.; Prudent, M.; Rochat, B.; Lion, N. Metabolomic profiling highlights oxidative damages in platelet concentrates treated for pathogen inactivation and shows protective role of urate. Metabolomics 2016, 12, 188. [Google Scholar] [CrossRef]
- Dalle-Donne, I.; Aldini, G.; Carini, M.; Colombo, R.; Rossi, R.; Milzani, A. Protein carbonylation, cellular dysfunction, and disease progression. J. Cell. Mol. Med. 2006, 10, 389–406. [Google Scholar] [CrossRef] [PubMed]
- Reid, S.; Johnson, L.; Woodland, N.; Marks, D.C. Pathogen reduction treatment of buffy coat platelet concentrates in additive solution induces proapoptotic signaling. Transfusion 2012, 52, 2094–2103. [Google Scholar] [CrossRef] [PubMed]
- Wong, C.M.; Cheema, A.K.; Zhang, L.; Suzuki, Y.J. Protein carbonylation as a novel mechanism in redox signaling. Circ. Res. 2008, 102, 310–318. [Google Scholar] [CrossRef] [PubMed]
- Májek, P.; Reicheltová, Z.; Štikarová, J.; Suttnar, J.; Sobotková, A.; Dyr, J.E. Proteome changes in platelets activated by arachidonic acid, collagen, and thrombin. Proteome Sci. 2010, 8, 1–13. [Google Scholar]
- Alexandru, N.; Constantin, A.; Popov, D. Carbonylation of platelet proteins occurs as consequence of oxidative stress and thrombin activation, and is stimulated by ageing and type 2 diabetes. Clin. Chem. Lab. Med. 2008, 46, 528–536. [Google Scholar] [CrossRef] [PubMed]
- Bakdash, N.; Williams, M.S. Spatially distinct production of reactive oxygen species regulates platelet activation. Free Radic. Biol. Med. 2008, 45, 158–166. [Google Scholar] [CrossRef] [PubMed]
- Carrim, N.; Arthur, J.F.; Hamilton, J.R.; Gardiner, E.E.; Andrews, R.K.; Moran, N.; Berndt, M.C.; Metharom, P. Thrombin-induced reactive oxygen species generation in platelets: A novel role for protease-activated receptor 4 and GPIbα. Redox Biol. 2015, 6, 640–647. [Google Scholar] [CrossRef] [PubMed]
- Carrim, N.; Walsh, T.G.; Consonni, A.; Torti, M.; Berndt, M.C.; Metharom, P. Role of focal adhesion tyrosine kinases in GPVI-dependent platelet activation and reactive oxygen species formation. PLoS ONE 2014, 9, e113679. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Soe, N.N.; Sowden, M.; Xu, Y.; Modjeski, K.; Baskaran, P.; Kim, Y.; Smolock, E.M.; Morrell, C.N.; Berk, B.C. Cyclophilin A is an important mediator of platelet function by regulating integrin αIIbβ3 bidirectional signalling. Thromb. Haemost. 2014, 111, 873–882. [Google Scholar] [CrossRef] [PubMed]
- Soe, N.N.; Sowden, M.; Baskaran, P.; Smolock, E.M.; Kim, Y.; Nigro, P.; Berk, B.C. Cyclophilin A is required for angiotensin II-induced p47phox translocation to caveolae in vascular smooth muscle cells. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 2147–2153. [Google Scholar] [CrossRef] [PubMed]
- Caccese, D.; Praticò, D.; Ghiselli, A.; Natoli, S.; Pignatelli, P.; Sanguigni, V.; Iuliano, L.; Violi, F. Superoxide anion and hydroxyl radical release by collagen-induced platelet aggregation—Role of arachidonic acid metabolism. Thromb. Haemost. 2000, 83, 485–490. [Google Scholar] [PubMed]
- Arthur, J.F.; Gardiner, E.E.; Kenny, D.; Andrews, R.K.; Berndt, M.C. Platelet receptor redox regulation. Platelets 2008, 19, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Arthur, J.F.; Shen, Y.; Gardiner, E.E.; Coleman, L.; Murphy, D.; Kenny, D.; Andrews, R.K.; Berndt, M.C. TNF receptor-associated factor 4 (TRAF4) is a novel binding partner of glycoprotein Ib and glycoprotein VI in human platelets. J. Thromb. Haemost. 2011, 9, 163–172. [Google Scholar] [CrossRef] [PubMed]
- Arthur, J.F.; Qiao, J.; Shen, Y.; Davis, A.K.; Dunne, E.; Berndt, M.C.; Gardiner, E.E.; Andrews, R.K. ITAM receptor-mediated generation of reactive oxygen species in human platelets occurs via Syk-dependent and Syk-independent pathways. J. Thromb. Haemost. 2012, 10, 1133–1141. [Google Scholar] [CrossRef] [PubMed]
- Korbecki, J.; Baranowska-Bosiacka, I.; Gutowska, I.; Chlubek, D. The effect of reactive oxygen species on the synthesis of prostanoids from arachidonic acid. J. Physiol. Pharmacol. 2013, 64, 409–421. [Google Scholar] [PubMed]
- Morel, A.; Miller, E.; Bijak, M.; Saluk, J. The increased level of COX-dependent arachidonic acid metabolism in blood platelets from secondary progressive multiple sclerosis patients. Mol. Cell. Biochem. 2016, 420, 85–94. [Google Scholar] [CrossRef] [PubMed]
- Lambeth, J.D. NOX enzymes and the biology of reactive oxygen. Nat. Rev. Immunol. 2004, 4, 181–189. [Google Scholar] [CrossRef] [PubMed]
- Violi, F.; Pignatelli, P. Platelet NOX, a novel target for anti-thrombotic treatment. Thromb. Haemost. 2014, 111, 817–823. [Google Scholar] [CrossRef] [PubMed]
- Bedard, K.; Krause, K.-H. The NOX Family of ROS-generating NADPH oxidases: Physiology and pathophysiology. Physiol. Rev. 2007, 87, 245–313. [Google Scholar] [CrossRef] [PubMed]
- Jiang, F.; Zhang, Y.; Dusting, G.J. NADPH oxidase-mediated redox signaling: roles in cellular stress response, stress tolerance, and tissue repair. Pharmacol. Rev. 2011, 63, 218–242. [Google Scholar] [CrossRef] [PubMed]
- Finazzi-Agrò, A.; Menichelli, A.; Persiani, M.; Biancini, G.; del Principe, D. Hydrogen peroxide release from human blood platelets. Biochim. Biophys. Acta 1982, 718, 21–25. [Google Scholar] [CrossRef]
- Delaney, M.K.; Kim, K.; Estevez, B.; Xu, Z.; Stojanovic-Terpo, A.; Shen, B.; Ushio-Fukai, M.; Cho, J.; Du, X. Differential roles of the NADPH-oxidase 1 and 2 in platelet activation and thrombosis. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 846–854. [Google Scholar] [CrossRef] [PubMed]
- Walsh, T.G.; Berndt, M.C.; Carrim, N.; Cowman, J.; Kenny, D.; Metharom, P. The role of NOX1 and NOX2 in GPVI-dependent platelet activation and thrombus formation. Redox Biol. 2014, 2, 178–186. [Google Scholar] [CrossRef] [PubMed]
- Pignatelli, P.; Carnevale, R.; Di Santo, S.; Bartimoccia, S.; Sanguigni, V.; Lenti, L.; Finocchi, A.; Mendolicchio, L.; Soresina, A.R.; Plebani, A.; et al. Inherited human gp91phox deficiency is associated with impaired isoprostane formation and platelet dysfunction. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 423–434. [Google Scholar] [CrossRef] [PubMed]
- Iuliano, L.; Praticò, D.; Ghiselli, A.; Bonavita, M.S.; Violi, F. Superoxide dismutase triggers activation of “primed” platelets. Arch. Biochem. Biophys. 1991, 289, 180–183. [Google Scholar] [CrossRef]
- Yun, S.-H.; Sim, E.-H.; Goh, R.-Y.; Park, J.-I.; Han, J.-Y. Platelet activation: The mechanisms and potential biomarkers. BioMed Res. Int. 2016, 2016, 9060143. [Google Scholar] [CrossRef] [PubMed]
- Sangkuhl, K.; Shuldiner, A.R.; Klein, T.E.; Altman, R.B. Platelet aggregation pathway. Pharmacogenet. Genom. 2011, 21, 516–521. [Google Scholar] [CrossRef] [PubMed]
- Dayal, S.; Wilson, K.M.; Motto, D.G.; Miller, F.J.; Chauhan, A.K.; Lentz, S.R. Hydrogen peroxide promotes aging-related platelet hyperactivation and thrombosis. Circulation 2013, 127, 1308–1316. [Google Scholar] [CrossRef] [PubMed]
- Jang, J.Y.; Min, J.H.; Chae, Y.H.; Baek, J.Y.; Wang, S.B.; Park, S.J.; Oh, G.T.; Lee, S.-H.; Ho, Y.-S.; Chang, T.-S. Reactive oxygen species play a critical role in collagen-induced platelet activation via SHP-2 oxidation. Antioxid. Redox Signal. 2014, 20, 2528–2540. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Du, J.; Zhao, L.; Wang, X.; Zhang, Y.; Yan, R.; Dai, J.; Liu, G.; Zhang, F.; Dai, K. The role of intraplatelet reactive oxygen species in the regulation of platelet glycoprotein Ibα ectodomain shedding. Thromb. Res. 2013, 132, 696–701. [Google Scholar] [CrossRef] [PubMed]
- Brill, A.; Chauhan, A.K.; Canault, M.; Walsh, M.T.; Bergmeier, W.; Wagner, D.D. Oxidative stress activates ADAM17/TACE and induces its target receptor shedding in platelets in a p38-dependent fashion. Cardiovasc. Res. 2009, 84, 137–144. [Google Scholar] [CrossRef] [PubMed]
- Choo, H.-J.; Saafir, T.B.; Mkumba, L.; Wagner, M.B.; Jobe, S.M. Mitochondrial calcium and reactive oxygen species regulate agonist-initiated platelet phosphatidylserine exposure. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 2946–2955. [Google Scholar] [CrossRef] [PubMed]
- McStay, G.P.; Clarke, S.J.; Halestrap, A.P. Role of critical thiol groups on the matrix surface of the adenine nucleotide translocase in the mechanism of the mitochondrial permeability transition pore. Biochem. J. 2002, 367, 541–548. [Google Scholar] [CrossRef] [PubMed]
- Essex, D.W. The role of thiols and disulfides in platelet function. Antioxid. Redox Signal. 2004, 6, 736–746. [Google Scholar] [CrossRef] [PubMed]
- Yan, B.; Smith, J.W. A redox site involved in integrin activation. J. Biol. Chem. 2000, 275, 39964–39972. [Google Scholar] [CrossRef] [PubMed]
- Verhaar, R.; Dekkers, D.W.; de Cuyper, I.M.; Ginsberg, M.H.; de Korte, D.; Verhoeven, A.J. UV-C irradiation disrupts platelet surface disulfide bonds and activates the platelet integrin αIIbβ3. Blood 2008, 112, 4935–4939. [Google Scholar] [CrossRef] [PubMed]
- Mailloux, R.J.; Jin, X.; Willmore, W.G. Redox regulation of mitochondrial function with emphasis on cysteine oxidation reactions. Redox Biol. 2014, 2, 123–139. [Google Scholar] [CrossRef] [PubMed]
- Chung, H.S.; Wang, S.-B.; Venkatraman, V.; Murray, C.I.; van Eyk, J.E. Cysteine oxidative post-translational modifications: Emerging regulation in the cardiovascular system. Circ. Res. 2013, 112, 382–392. [Google Scholar] [CrossRef] [PubMed]
- Farah, M.E.; Amberg, D.C. Conserved actin cysteine residues are oxidative stress sensors that can regulate cell death in yeast. Mol. Biol. Cell 2007, 18, 1359–1365. [Google Scholar] [CrossRef] [PubMed]
- Van der Meer, P.F.; Bontekoe, I.J.; Daal, B.B.; de Korte, D. Riboflavin and UV light treatment of platelets: A protective effect of platelet additive solution? Transfusion 2015, 55, 1900–1908. [Google Scholar] [CrossRef] [PubMed]
- Johnson, L.; Winter, K.M.; Reid, S.; Hartkopf-Theis, T.; Marschner, S.; Goodrich, R.P.; Marks, D.C. The effect of pathogen reduction technology (Mirasol) on platelet quality when treated in additive solution with low plasma carryover. Vox Sang. 2011, 101, 208–214. [Google Scholar] [CrossRef] [PubMed]
- Carballal, S.; Radi, R.; Kirk, M.C.; Barnes, S.; Freeman, B.A.; Alvarez, B. Sulfenic acid formation in human serum albumin by hydrogen peroxide and peroxynitrite. Biochemistry 2003, 42, 9906–9914. [Google Scholar] [CrossRef] [PubMed]
- Jang, J.Y.; Wang, S.B.; Min, J.H.; Chae, Y.H.; Baek, J.Y.; Yu, D.-Y.; Chang, T.-S. Peroxiredoxin II is an antioxidant enzyme that negatively regulates collagen-stimulated platelet function. J. Biol. Chem. 2015, 290, 11432–11442. [Google Scholar] [CrossRef] [PubMed]
- Gupta, V.; Carroll, K.S. Sulfenic acid chemistry, detection and cellular lifetime. Biochim. Biophys. Acta 2014, 1840, 847–875. [Google Scholar] [CrossRef] [PubMed]
- Paulsen, C.E.; Carroll, K.S. Cysteine-mediated redox signaling: Chemistry, biology, and tools for discovery. Chem. Rev. 2013, 113, 4633–4679. [Google Scholar] [CrossRef] [PubMed]
- Chiarugi, P.; Cirri, P. Redox regulation of protein tyrosine phosphatases during receptor tyrosine kinase signal transduction. Trends Biochem. Sci. 2003, 28, 509–514. [Google Scholar] [CrossRef]
- Brennan, J.P.; Bardswell, S.C.; Burgoyne, J.R.; Fuller, W.; Schröder, E.; Wait, R.; Begum, S.; Kentish, J.C.; Eaton, P. Oxidant-induced activation of type I protein kinase A is mediated by RI subunit interprotein disulfide bond formation. J. Biol. Chem. 2006, 281, 21827–21836. [Google Scholar] [CrossRef] [PubMed]
- Fiaschi, T.; Cozzi, G.; Raugei, G.; Formigli, L.; Ramponi, G.; Chiarugi, P. Redox regulation of β-actin during integrin-mediated cell adhesion. J. Biol. Chem. 2006, 281, 22983–22991. [Google Scholar] [CrossRef] [PubMed]
- Chiarugi, P.; Pani, G.; Giannoni, E.; Taddei, L.; Colavitti, R.; Raugei, G.; Symons, M.; Borrello, S.; Galeotti, T.; Ramponi, G. Reactive oxygen species as essential mediators of cell adhesion: The oxidative inhibition of a FAK tyrosine phosphatase is required for cell adhesion. J. Cell Biol. 2003, 161, 933–944. [Google Scholar] [CrossRef] [PubMed]
- Giannoni, E.; Buricchi, F.; Raugei, G.; Ramponi, G.; Chiarugi, P. Intracellular reactive oxygen species activate Src tyrosine kinase during cell adhesion and anchorage-dependent cell growth. Mol. Cell. Biol. 2005, 25, 6391–6403. [Google Scholar] [CrossRef] [PubMed]
- Messens, J.; Collet, J.-F. Thiol-disulfide exchange in signaling: Disulfide bonds as a switch. Antioxid. Redox Signal. 2013, 18, 1594–1596. [Google Scholar] [CrossRef] [PubMed]
- Furie, B.; Flaumenhaft, R. Thiol isomerases in thrombus formation. Circ. Res. 2014, 114, 1162–1173. [Google Scholar] [CrossRef] [PubMed]
- Cho, J.; Kennedy, D.R.; Lin, L.; Huang, M.; Merrill-Skoloff, G.; Furie, B.C.; Furie, B. Protein disulfide isomerase capture during thrombus formation in vivo depends on the presence of β3 integrins. Blood 2012, 120, 647–655. [Google Scholar] [CrossRef] [PubMed]
- Janiszewski, M.; Lopes, L.R.; Carmo, A.O.; Pedro, M.A.; Brandes, R.P.; Santos, C.X.C.; Laurindo, F.R.M. Regulation of NAD(P)H oxidase by associated protein disulfide isomerase in vascular smooth muscle cells. J. Biol. Chem. 2005, 280, 40813–40819. [Google Scholar] [CrossRef] [PubMed]
- Sefton, B.M. Overview of protein phosphorylation. Curr. Protoc. Cell Biol. 2001. [Google Scholar] [CrossRef]
- Immler, D.; Gremm, D.; Kirsch, D.; Spengler, B.; Presek, P.; Meyer, H.E. Identification of phosphorylated proteins from thrombin-activated human platelets isolated by two-dimensional gel electrophoresis by electrospray ionization-tandem mass spectrometry (ESI-MS/MS) and liquid chromatography-electrospray ionization-mass spectrometry (LC-ESI-MS). Electrophoresis 1998, 19, 1015–1023. [Google Scholar] [PubMed]
- Zahedi, R.P.; Lewandrowski, U.; Wiesner, J.; Wortelkamp, S.; Moebius, J.; Schütz, C.; Walter, U.; Gambaryan, S.; Sickmann, A. Phosphoproteome of resting human platelets. J. Proteome Res. 2008, 7, 526–534. [Google Scholar] [CrossRef] [PubMed]
- Zimman, A.; Titz, B.; Komisopoulou, E.; Biswas, S.; Graeber, T.G.; Podrez, E.A. Phosphoproteomic analysis of platelets activated by pro-thrombotic oxidized phospholipids and thrombin. PLoS ONE 2014, 9, e84488. [Google Scholar] [CrossRef] [PubMed]
- Van Marwijk Kooy, M.; Akkerman, J.W.N.; van Asbeck, S.; Borghuis, L.; van Prooijen, H.C. UVB radiation exposes fibrinogen binding sites on platelets by activating protein kinase C via reactive oxygen species. Br. J. Haematol. 1993, 83, 253–258. [Google Scholar] [CrossRef] [PubMed]
- Schubert, P.; Culibrk, B.; Coupland, D.; Scammell, K.; Gyongyossy-Issa, M.; Devine, D.V. Riboflavin and ultraviolet light treatment potentiates vasodilator-stimulated phosphoprotein Ser-239 phosphorylation in platelet concentrates during storage. Transfusion 2012, 52, 397–408. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, U.R.; Geiger, J.; Walter, U.; Eigenthaler, M. Flow cytometry analysis of intracellular VASP phosphorylation for the assessment of activating and inhibitory signal transduction pathways in human platelets definition and detection of ticlopidine/clopidogrel effects. Thromb. Haemost. 1999, 82, 1145–1152. [Google Scholar] [PubMed]
- Fedor, M.; Simonova, R.; Fedorova, J.; Skornova, I.; Duraj, L.; Samos, M.; Stasko, J.; Kovar, F.; Mokan, M.; Kubisz, P. Role of VASP phosphorylation assay in monitoring the antiplatelet therapy. Acta Med. Martiniana 2013, 13, 21–26. [Google Scholar]
- Schubert, P.; Coupland, D.; Culibrk, B.; Goodrich, R.P.; Devine, D.V. Riboflavin and ultraviolet light treatment of platelets triggers p38MAPK signaling: Inhibition significantly improves in vitro platelet quality after pathogen reduction treatment. Transfusion 2013, 53, 3164–3173. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Schubert, P.; Culibrk, B.; Devine, D.V. p38MAPK is involved in apoptosis development in apheresis platelet concentrates after riboflavin and ultraviolet light treatment. Transfusion 2015, 55, 848–857. [Google Scholar] [CrossRef] [PubMed]
- Paulsen, C.E.; Truong, T.H.; Garcia, F.J.; Homann, A.; Gupta, V.; Leonard, S.E.; Carroll, K.S. Peroxide-dependent sulfenylation of the EGFR catalytic site enhances kinase activity. Nat. Chem. Biol. 2012, 8, 57–64. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Gupta, V.; Carroll, K.S.; Liebler, D.C. Site-specific mapping and quantification of protein S-sulfenylation in cells. Nat. Commun. 2014, 5, 4776. [Google Scholar] [CrossRef] [PubMed]
- Lion, N.; Tissot, J.-D.; Prudent, M. Is proteomics still knockin’ on the hematological door? Proteomics Clin. Appl. 2016, 10, 765–766. [Google Scholar] [CrossRef] [PubMed]
- Spinella, P.C.; Pidcoke, H.F.; Strandenes, G.; Hervig, T.; Fisher, A.; Jenkins, D.; Yazer, M.; Stubbs, J.; Murdock, A.; Sailliol, A.; et al. Whole blood for hemostatic resuscitation of major bleeding. Transfusion 2016, 56, 190–202. [Google Scholar] [CrossRef] [PubMed]
- Johnson, L.; Coorey, C.P.; Marks, D.C. The hemostatic activity of cryopreserved platelets is mediated by phosphatidylserine-expressing platelets and platelet microparticles. Transfusion 2014, 54, 1917–1926. [Google Scholar] [CrossRef] [PubMed]
- Johnson, L.; Tan, S.; Wood, B.; Davis, A.; Marks, D.C. Refrigeration and cryopreservation of platelets differentially affect platelet metabolism and function: A comparison with conventional platelet storage conditions. Transfusion 2016, 56, 1807–1818. [Google Scholar] [CrossRef] [PubMed]
- Cap, A.P. Platelet storage: A license to chill. Transfusion 2016, 56, 13–16. [Google Scholar] [CrossRef] [PubMed]
- Abonnenc, M.; Crettaz, D.; Tacchini, P.; Di Vincenzo, L.; Sonego, G.; Prudent, M.; Tissot, J.-D.; Lion, N. Antioxidant power as a quality control marker for completeness of amotosalen and ultraviolet A photochemical treatments in platelet concentrates and plasma units. Transfusion 2016, 56, 1819–1827. [Google Scholar] [CrossRef] [PubMed]
- Tacchini, P.; Lesch, A.; Neequaye, A.; Lagger, G.; Liu, J.; Cortés-Salazar, F.; Girault, H.H. Electrochemical pseudo-titration of water-soluble antioxidants. Electroanalysis 2013, 25, 922–930. [Google Scholar] [CrossRef]
- Olas, B.; Wachowicz, B.; Nowak, P.; Kedzierska, M.; Tomczak, A.; Stochmal, A.; Oleszek, W.; Jeziorski, A.; Piekarski, J. Studies on antioxidant properties of polyphenol-rich extract from berries of Aronia melanocarpa in blood platelets. J. Physiol. Pharmacol. 2008, 59, 823–835. [Google Scholar] [PubMed]
- Manasa, K.; Vani, R. Influence of oxidative stress on stored platelets. Adv. Hematol. 2016, 2016, 4091461. [Google Scholar] [CrossRef] [PubMed]
- Olas, B.; Wachowicz, B. Resveratrol and vitamin C as antioxidants in blood platelets. Thromb. Res. 2002, 106, 143–148. [Google Scholar] [CrossRef]
- Sobotková, A.; Mášová-Chrastinová, L.; Suttnar, J.; Štikarová, J.; Májek, P.; Reicheltová, Z.; Kotlín, R.; Weisel, J.W.; Malý, M.; Dyr, J.E. Antioxidants change platelet responses to various stimulating events. Free Radic. Biol. Med. 2009, 47, 1707–1714. [Google Scholar] [CrossRef] [PubMed]
- Shen, M.Y.; Hsiao, G.; Liu, C.L.; Fong, T.H.; Lin, K.H.; Chou, D.S.; Sheu, J.R. Inhibitory mechanisms of resveratrol in platelet activation: Pivotal roles of p38 MAPK and NO/cyclic GMP. Br. J. Haematol. 2007, 139, 475–485. [Google Scholar] [CrossRef] [PubMed]
- The Effect of Vitamin E and β carotene on the incidence of lung cancer and other cancers in male smokers. N. Engl. J. Med. 1994, 330, 1029–1035.
- Acker, J.P.; Marks, D.C.; Sheffield, W.P. Quality assessment of established and emerging blood components for transfusion. J. Blood Transfus. 2016, 2016, 4860284. [Google Scholar] [CrossRef] [PubMed]
- Gerber, B.; Alberio, L.; Rochat, S.; Stenner, F.; Manz, M.G.; Buser, A.; Schanz, U.; Stussi, G. Safety and efficacy of cryopreserved autologous platelet concentrates in. Transfusion 2016, 56, 2426–2437. [Google Scholar] [CrossRef] [PubMed]
- Barnard, M.R.; MacGregor, H.; Ragno, G.; Pivacek, L.E.; Khuri, S.F.; Michelson, A.D.; Valeri, C.R. Fresh, liquid-preserved, and cryopreserved platelets: Adhesive surface receptors and membrane procoagulant activity. Transfusion 1999, 39, 880–888. [Google Scholar] [CrossRef] [PubMed]
- Italiano, J.E.; Mairuhu, A.T.; Flaumenhaft, R. Clinical relevance of microparticles from platelets and megakaryocytes. Curr. Opin. Hematol. 2010, 17, 578–584. [Google Scholar] [CrossRef] [PubMed]
- Roos, M.A.; Gennero, L.; Denysenko, T.; Reguzzi, S.; Cavallo, G.; Pescarmona, G.P.; Ponzetto, A. Microparticles in physiological and in pathological conditions. Cell Biochem. Funct. 2010, 28, 539–548. [Google Scholar] [CrossRef] [PubMed]
- Holbrook, L.-M.; Watkins, N.A.; Simmonds, A.D.; Jones, C.I.; Ouwehand, W.H.; Gibbins, J.M. Platelets release novel thiol isomerase enzymes which are recruited to the cell surface following activation. Br. J. Haematol. 2010, 148, 627–637. [Google Scholar] [CrossRef] [PubMed]
- Essex, D.W.; Li, M. Protein disulphide isomerase mediates platelet aggregation and secretion. Br. J. Haematol. 1999, 104, 448–454. [Google Scholar] [CrossRef] [PubMed]
- Reinhardt, C.; von Brühl, M.-L.; Manukyan, D.; Grahl, L.; Lorenz, M.; Altmann, B.; Dlugai, S.; Hess, S.; Konrad, I.; Orschiedt, L.; et al. Protein disulfide isomerase acts as an injury response signal that enhances fibrin generation via tissue factor activation. J. Clin. Investig. 2008, 118, 1110–1122. [Google Scholar] [CrossRef] [PubMed]
- Valeri, C.R.; Macgregor, H.; Ragno, G. Correlation between in vitro aggregation and thromboxane A2 production in fresh, liquid-preserved, and cryopreserved human platelets: Effect of agonists, pH, and plasma and saline resuspension. Transfusion 2005, 45, 596–603. [Google Scholar] [CrossRef] [PubMed]
- Rasongles, P.; Angelini-Tibert, M.F.; Simon, P.; Currie, C.; Isola, H.; Kientz, D.; Slaedts, M.; Jacquet, M.; Sundin, D.; Lin, L.; et al. Transfusion of platelet components prepared with photochemical pathogen inactivation treatment during a Chikungunya virus epidemic in Ile de La Réunion. Transfusion 2009, 49, 1083–1091. [Google Scholar] [CrossRef] [PubMed]
- Picker, S.M. Current methods for the reduction of blood-borne pathogens: A comprehensive literature review. Blood Transfus. 2013, 11, 343–348. [Google Scholar] [PubMed]
- Henschler, R.; Seifried, E.; Mufti, N. Development of the S-303 pathogen inactivation technology for red blood cell concentrates. Transfus. Med. Hemother. 2011, 38, 33–42. [Google Scholar] [CrossRef] [PubMed]
- Mufti, N.A.; Erickson, A.C.; North, A.K.; Hanson, D.; Sawyer, L.; Corash, L.M.; Lin, L. Treatment of whole blood (WB) and red blood cells (RBC) with S-303 inactivates pathogens and retains in vitro quality of stored RBC. Biologicals 2010, 38, 14–19. [Google Scholar] [CrossRef] [PubMed]
- Schubert, P.; Culibrk, B.; Karwal, S.; Serrano, K.; Levin, E.; Bu, D.; Bhakta, V.; Sheffield, W.P.; Goodrich, R.P.; Devine, D.V. Whole blood treated with riboflavin and ultraviolet light: Quality assessment of all blood components produced by the buffy coat method. Transfusion 2015, 55, 815–823. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Name | Formulae | Oxidation State |
|---|---|---|
| Cysteine | R–SH | −II |
| Disulfide bridge | R–S–S–R | −I |
| Glutathionylation | R–S–S–G | −I |
| Sulfenic acid | R–SOH | 0 |
| Sulfinic acid | R–SO2H | +II |
| Sulfonic acid | R–SO3H | +IV |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sonego, G.; Abonnenc, M.; Tissot, J.-D.; Prudent, M.; Lion, N. Redox Proteomics and Platelet Activation: Understanding the Redox Proteome to Improve Platelet Quality for Transfusion. Int. J. Mol. Sci. 2017, 18, 387. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms18020387
Sonego G, Abonnenc M, Tissot J-D, Prudent M, Lion N. Redox Proteomics and Platelet Activation: Understanding the Redox Proteome to Improve Platelet Quality for Transfusion. International Journal of Molecular Sciences. 2017; 18(2):387. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms18020387
Chicago/Turabian StyleSonego, Giona, Mélanie Abonnenc, Jean-Daniel Tissot, Michel Prudent, and Niels Lion. 2017. "Redox Proteomics and Platelet Activation: Understanding the Redox Proteome to Improve Platelet Quality for Transfusion" International Journal of Molecular Sciences 18, no. 2: 387. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms18020387