
Integrating Pharmacoproteomics into Early-Phase Clinical Development: State-of-the-Art, Challenges, and Recommendations


Abstract
:1. Introduction and Background
2. History
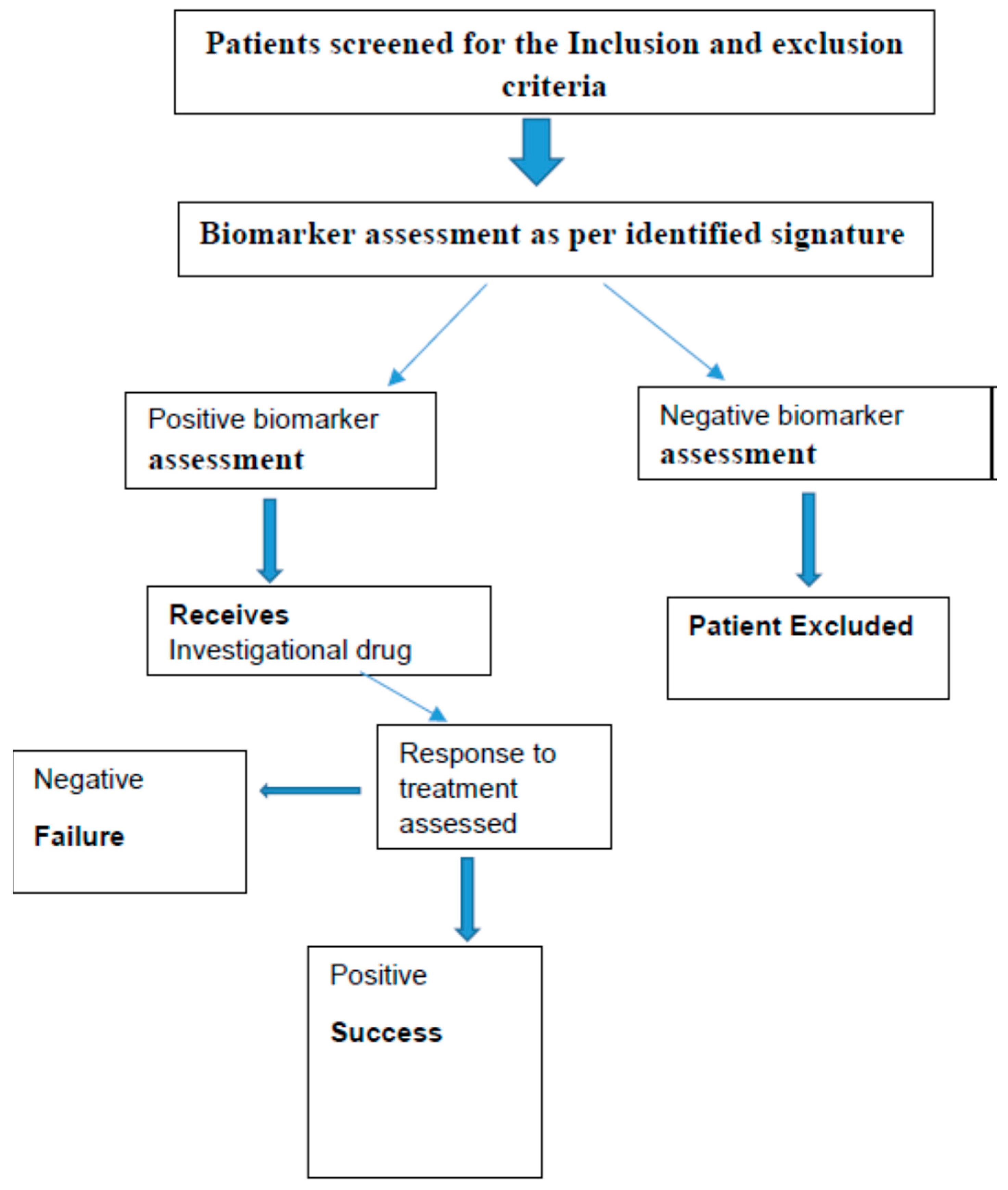
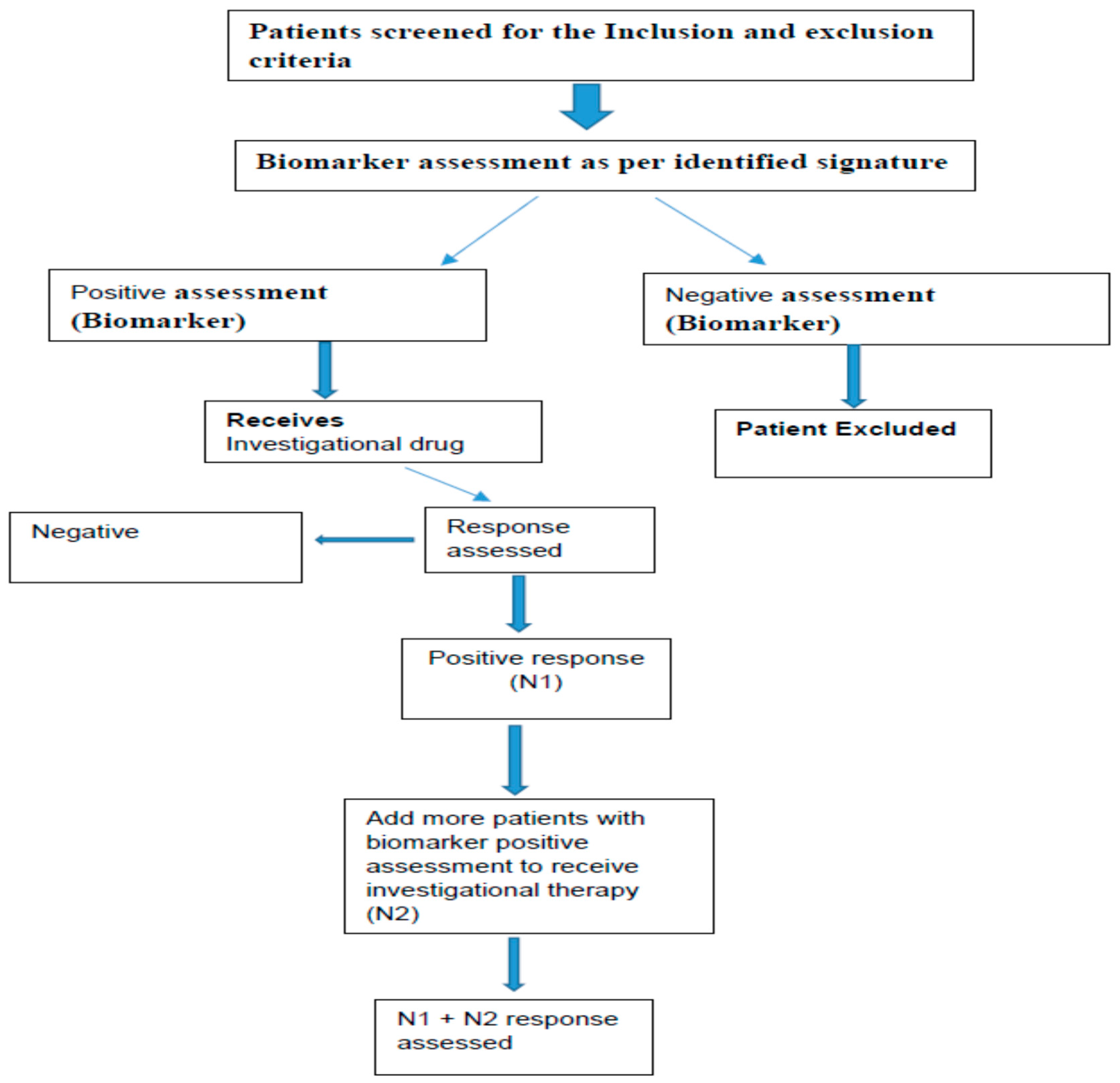
3. Pharmacoproteomics in Early Phase Development
4. Clinicaltrials.gov and PubMed Analyses
4.1. Purpose
4.2. Methodology
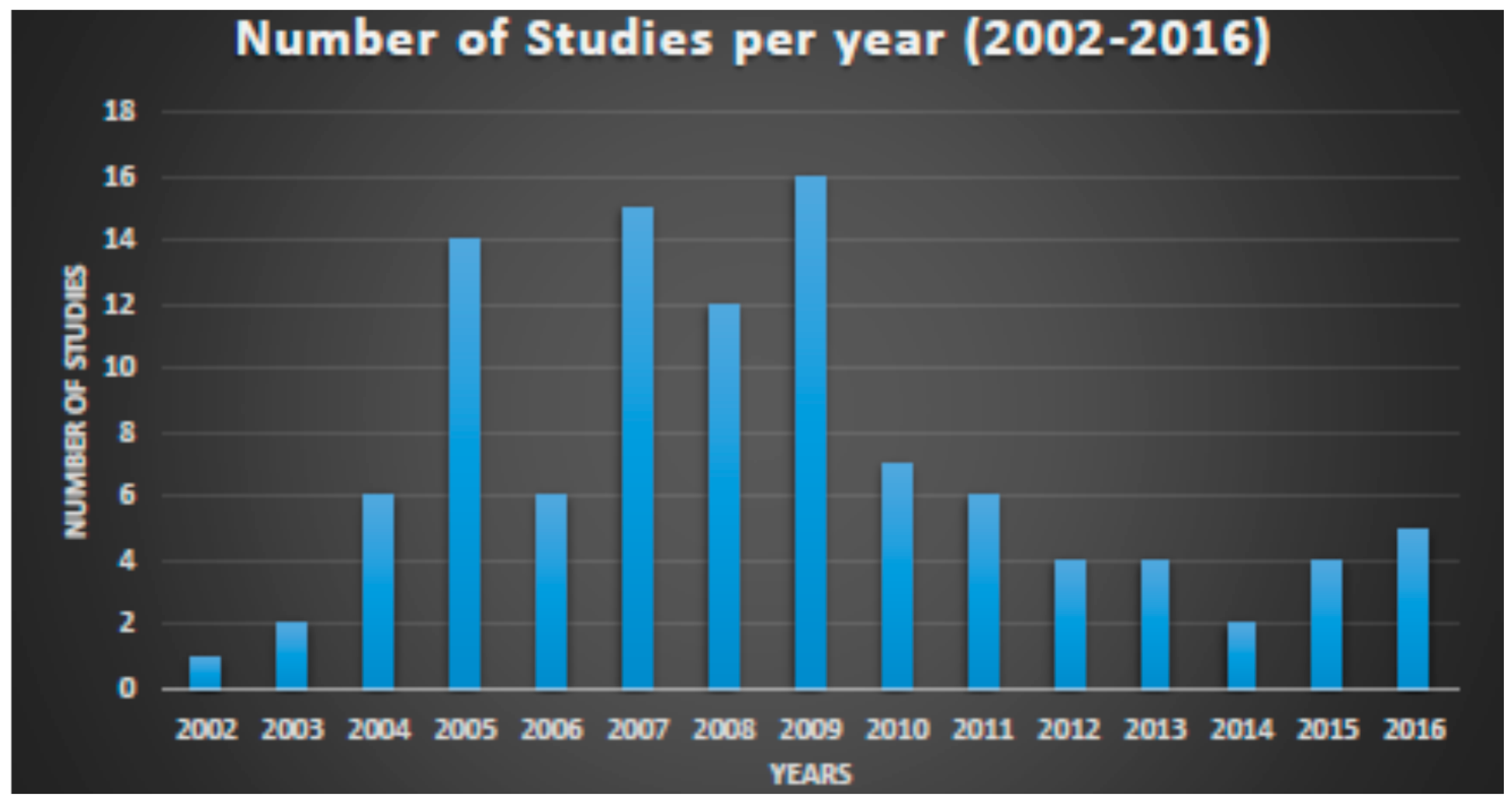
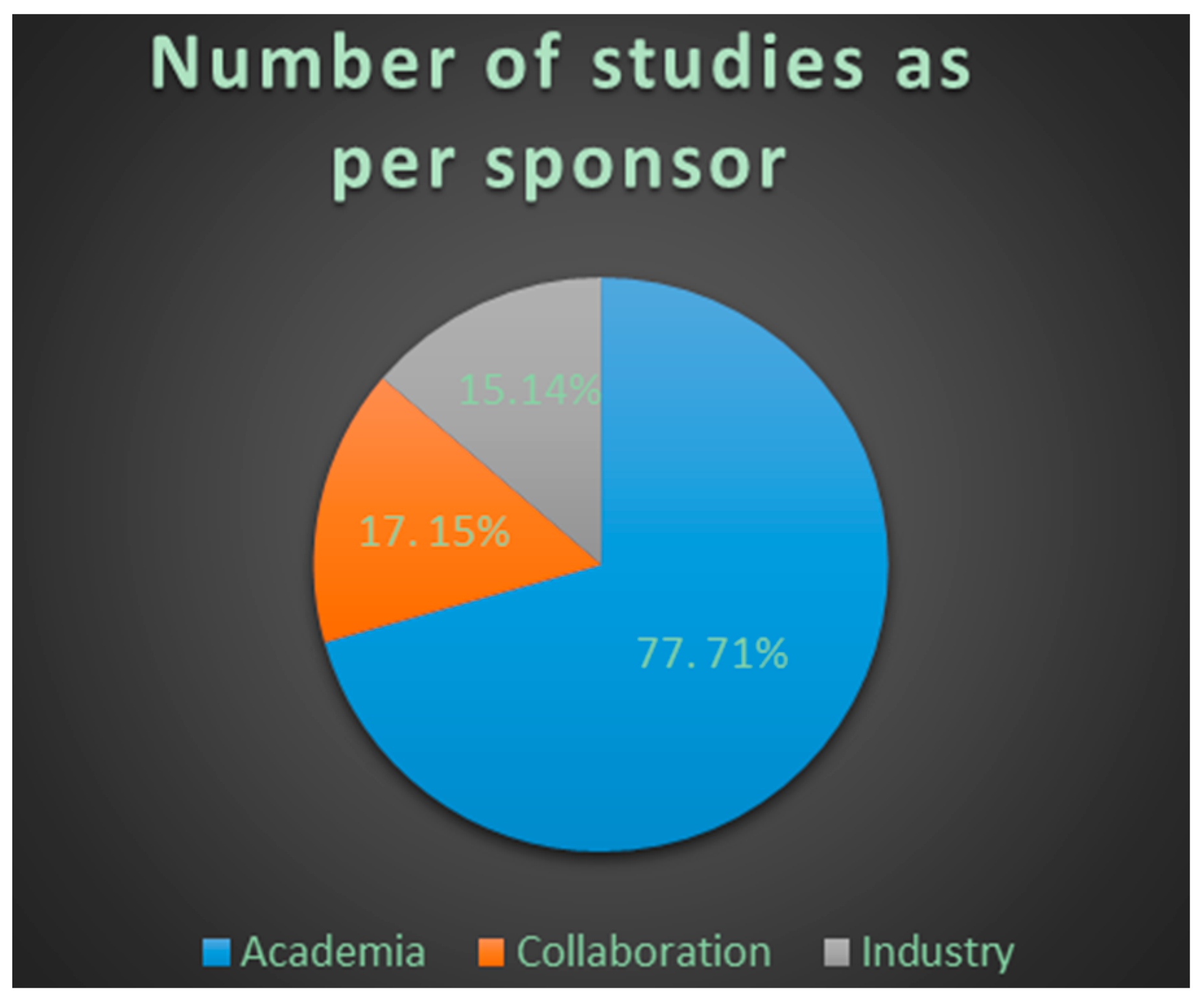
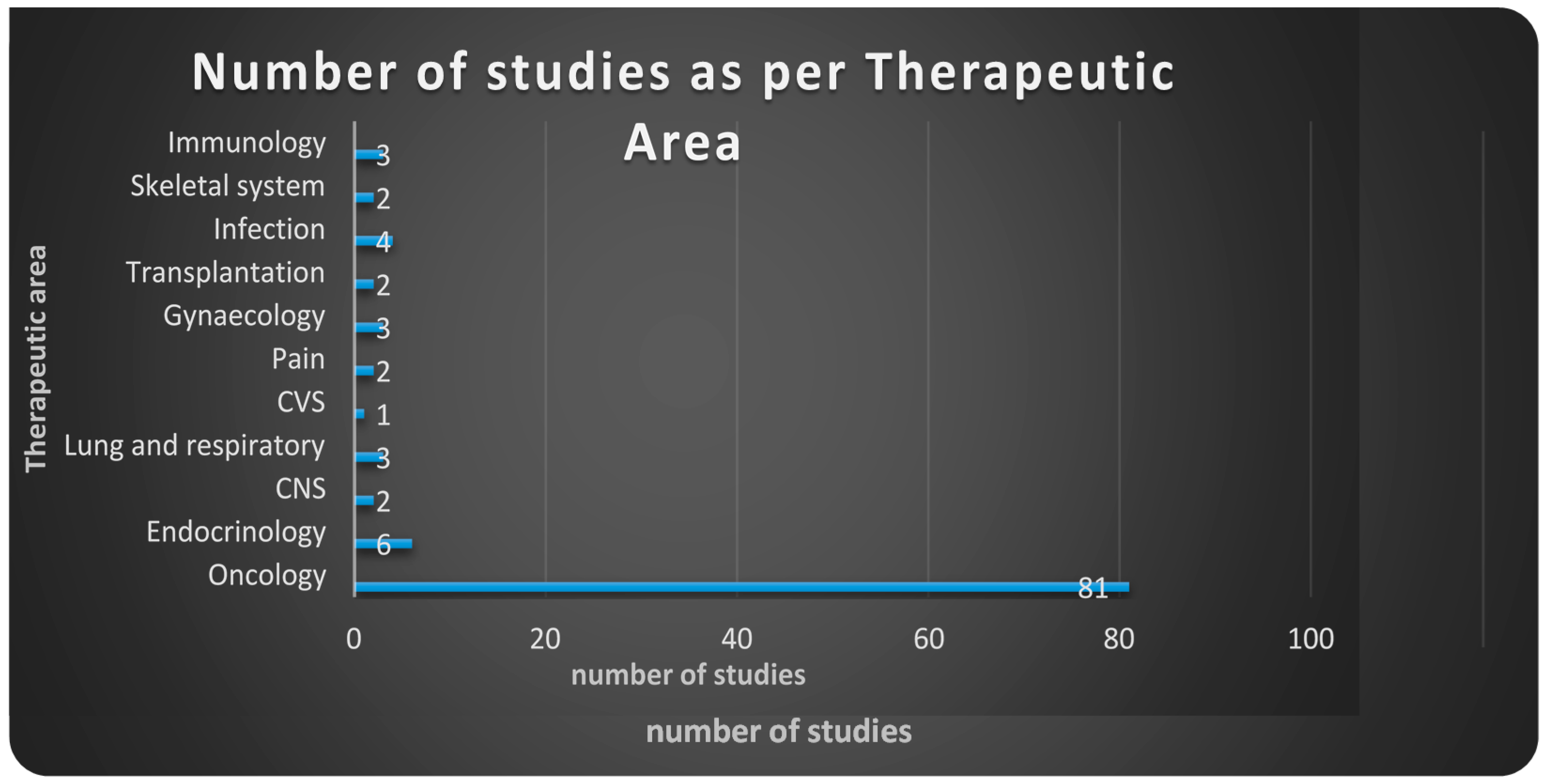
4.3. Results
4.4. Conclusions
4.5. Limitations
4.6. Challenges
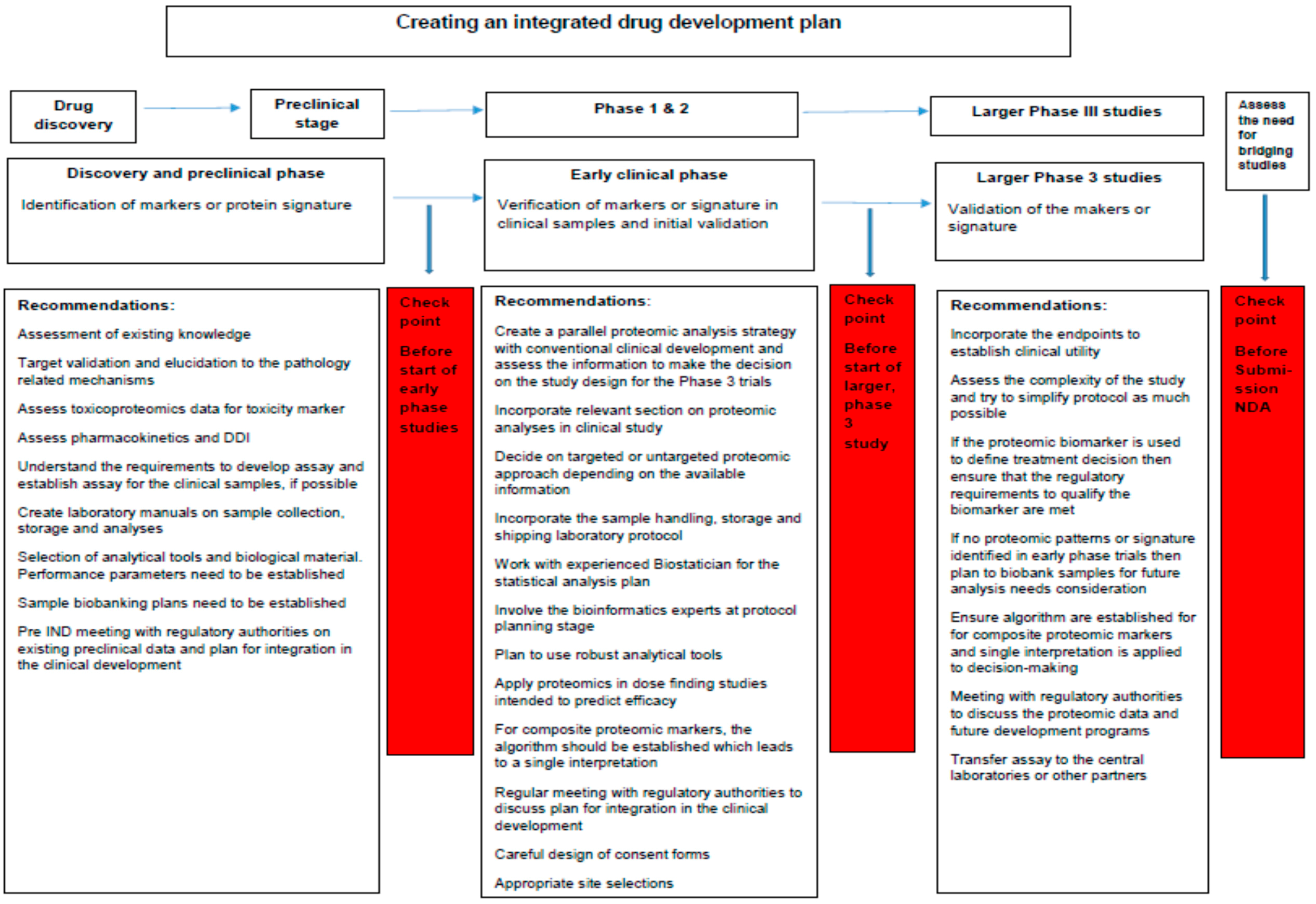
4.7. Recommendations
5. Conclusions
5.1. Box 1: Analytic Approaches Used in Proteomic and Pharmacoproteomic Studies
5.1.1. 2D Gel Electrophoresis (2DE) [91]
5.1.2. Mass Spectrometry [92]
5.1.3. ELISA and Immunohistochemistry
5.1.4. Multidirectional Protein Identification Technology (MudPIT) [92,93]
5.1.5. Isobaric Tags for Relative and Absolute Quantification (iTRAQ) [52]
5.1.6. Stable Isotopic Labeling by Amino Acids in Cell Culture (SILAC) [48,49,78]
5.1.7. Isotope Coded Affinity Tags (ICAT) [50,51]
5.1.8. Functional Proteomic Analyses [12]
5.1.9. Activity-Based Protein Profiling [96,103,104]
5.1.10. Reverse Phase Protein Arrays (RPPA) [107,108]
Supplementary Materials
Author Contributions
Conflicts of Interest
References
- Woodcock, J. The PCAST report on pharmaceutical innovation: Implications for the FDA. Clin. Pharmacol. Ther. 2013, 94, 297–300. [Google Scholar] [CrossRef] [PubMed]
- Food and Drug Administration (FDA). Innovation or Stagnation: Challenge and Opportunity on the Critical Path to New Medical Products. Available online: http://www.fda.gov/oc/initiatives/criticalpath/whitepaper.html (accessed on 12 February 2017).
- Pammolli, F.; Magazzini, L.; Riccaboni, M. The productivity crisis in pharmaceutical R&D. Nat. Rev. Drug Discov. 2011, 10, 428–438. [Google Scholar] [PubMed]
- Munos, B. Lessons from 60 years of pharmaceutical innovation. Nat. Rev. Drug Discov. 2009, 8, 959–968. [Google Scholar] [CrossRef] [PubMed]
- Walsh, B.T.; Seidman, S.N.; Sysko, R.; Gould, M. Placebo response in studies of major depression: Variable, substantial, and growing. JAMA 2002, 287, 1840–1847. [Google Scholar] [CrossRef] [PubMed]
- Scannell, J.W.; Blanckley, A.; Boldon, H.; Warrington, B. Diagnosing the decline in pharmaceutical R&D efficiency. Nat. Rev. Drug Discov. 2012, 11, 191–200. [Google Scholar] [PubMed]
- Paul, S.M.; Mytelka, D.S.; Dunwiddie, C.T.; Persinger, C.C.; Munos, B.H.; Lindborg, S.R.; Schacht, A.L. How to improve R&D productivity: The pharmaceutical industry’s grand challenge. Nat. Rev. Drug Discov. 2010, 9, 203–214. [Google Scholar] [PubMed]
- IOM. Evolution of Translational Omics: Lessons Learned and the Path Forward; The National Academies Press: Washington, DC, USA, 2012. [Google Scholar]
- Collins, F.S. Reengineering translational science: The time is right. Sci. Transl. Med. 2011, 3, 90cm17. [Google Scholar] [CrossRef] [PubMed]
- Wishart, D.S. Emerging applications of metabolomics in drug discovery and precision medicine. Nat. Rev. Drug Discov. 2016, 15, 473–484. [Google Scholar] [CrossRef] [PubMed]
- Boja, E.S.; Kinsinger, C.R.; Rodriguez, H.; Srinivas, P. Integration of omics sciences to advance biology and medicine. Clin. Proteom. 2014, 11, 45. [Google Scholar] [CrossRef]
- Russell, C.; Rahman, A.; Mohammed, A.R. Application of genomics, proteomics and metabolomics in drug discovery, development and clinic. Ther. Deliv. 2013, 4, 395–413. [Google Scholar] [CrossRef] [PubMed]
- McShane, L.M.; Cavenagh, M.M.; Lively, T.G.; Eberhard, D.A.; Bigbee, W.L.; Williams, P.M.; Mesirov, J.P.; Polley, M.Y.; Kim, K.Y.; Tricoli, J.V.; et al. Criteria for the use of omics-based predictors in clinical trials: Explanation and elaboration. BMC Med. 2013, 11, 220. [Google Scholar] [CrossRef] [PubMed]
- Burt, T.; Nandal, S. Pharmacometabolomics in Early-Phase Clinical Development. Clin. Transl. Sci. 2016. [Google Scholar] [CrossRef] [PubMed]
- Burt, T.; Dhillon, S. Pharmacogenomics in early-phase clinical development. Pharmacogenomics 2013, 14, 1085–1097. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Button, K.S.; Ioannidis, J.P.; Mokrysz, C.; Nosek, B.A.; Flint, J.; Robinson, E.S.; Munafo, M.R. Power failure: Why small sample size undermines the reliability of neuroscience. Nat. Rev. Neurosci. 2013, 14, 365–376. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.S.; Pinto, S.M.; Getnet, D.; Nirujogi, R.S.; Manda, S.S.; Chaerkady, R.; Madugundu, A.K.; Kelkar, D.S.; Isserlin, R.; Jain, S.; et al. A draft map of the human proteome. Nature 2014, 509, 575–581. [Google Scholar] [CrossRef] [PubMed]
- Wasinger, V.C.; Cordwell, S.J.; Cerpa-Poljak, A.; Yan, J.X.; Gooley, A.A.; Wilkins, M.R.; Duncan, M.W.; Harris, R.; Williams, K.L.; Humphery-Smith, I. Progress with gene-product mapping of the Mollicutes: Mycoplasma genitalium. Electrophoresis 1995, 16, 1090–1094. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, S. The role of proteomics in toxicology: Identification of biomarkers of toxicity by protein expression analysis. Biomarkers 2002, 7, 269–290. [Google Scholar] [CrossRef] [PubMed]
- Meister, W. Pharmacogenomics/pharmacoproteomics Europe. Pharmacogenomics 2002, 3, 449–452. [Google Scholar] [CrossRef] [PubMed]
- Moellering, R.E.; Cravatt, B.F. How chemoproteomics can enable drug discovery and development. Chem. Biol. 2012, 19, 11–22. [Google Scholar] [CrossRef] [PubMed]
- Rabilloud, T.; Lescuyer, P. Proteomics in mechanistic toxicology: History, concepts, achievements, caveats, and potential. Proteomics 2015, 15, 1051–1074. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Chan, D.W. Proteomic cancer biomarkers from discovery to approval: It’s worth the effort. Expert Rev. Proteom. 2014, 11, 135–136. [Google Scholar] [CrossRef] [PubMed]
- Hartley, H. Origin of the word “protein”. Nature 1951, 168, 244. [Google Scholar] [CrossRef] [PubMed]
- Thomson, J.J. Bakerian Lecture: Rays of Positive Electricity. Proc. R. Soc. Lond. Ser. A 1913, 89, 1–20. [Google Scholar] [CrossRef]
- Coons, A.H.; Creech, H.J.; Jones, R.N. Immunological Properties of an Antibody Containing a Fluorescent Group. Exp. Biol. Med. 1941, 47, 200–202. [Google Scholar] [CrossRef]
- Horning, M.G.; Knox, K.L.; Dalgliesh, C.E.; Horning, E.C. Gas-liquid chromatographic study and estimation of several urinary aromatic acids. Anal. Biochem. 1966, 17, 244–257. [Google Scholar] [CrossRef]
- Ryhage, R.; Stenhagen, E. Mass spectrometry in lipid research. J. Lipid Res. 1960, 1, 361–390. [Google Scholar] [PubMed]
- Engvall, E.; Perlmann, P. Enzyme-linked immunosorbent assay (ELISA). Quantitative assay of immunoglobulin G. Immunochemistry 1971, 8, 871–874. [Google Scholar] [CrossRef]
- Van Weemen, B.K.; Schuurs, A.H. Immunoassay using antigen-enzyme conjugates. FEBS Lett. 1971, 15, 232–236. [Google Scholar] [CrossRef]
- O’Farrell, P.H. High resolution two-dimensional electrophoresis of proteins. J. Biol. Chem. 1975, 250, 4007–4021. [Google Scholar] [PubMed]
- Klose, J. Protein mapping by combined isoelectric focusing and electrophoresis of mouse tissues. A novel approach to testing for induced point mutations in mammals. Humangenetik 1975, 26, 231–243. [Google Scholar] [PubMed]
- Scheele, G.A. Two-dimensional gel analysis of soluble proteins. Charaterization of guinea pig exocrine pancreatic proteins. J. Biol. Chem. 1975, 250, 5375–5385. [Google Scholar] [PubMed]
- Games, D.E.; Alcock, N.J.; van der Greef, J.; Nyssen, L.M.; Maarse, H.; Ten, M.C.; de Brauw, N. Analysis of pepper and capsicum oleoresins by high-performance liquid chromatography—Mass spectrometry and field desorption mass spectrometry. J. Chromatogr. A 1984, 294, 269–279. [Google Scholar] [CrossRef]
- Van der Greef, J.; Tas, A.C.; Bouwman, J.; Ten Noever de Brauw, M.C.; Schreurs, W.H.P. Evaluation of field-desorption and fast atom-bombardment mass spectrometric profiles by pattern recognition techniques. Anal. Chim. Acta 1983, 150, 45–52. [Google Scholar] [CrossRef]
- Bain, J.R.; Stevens, R.D.; Wenner, B.R.; Ilkayeva, O.; Muoio, D.M.; Newgard, C.B. Metabolomics applied to diabetes research: Moving from information to knowledge. Diabetes 2009, 58, 2429–2443. [Google Scholar] [CrossRef] [PubMed]
- Van der Greef, J.; Hankemeier, T.; McBurney, R.N. Metabolomics-based systems biology and personalized medicine: Moving towards n = 1 clinical trials? Pharmacogenomics 2006, 7, 1087–1094. [Google Scholar] [CrossRef] [PubMed]
- Anderson, N.L.; Edwards, J.J.; Giometti, C.S. High resolution two dimensional electrophoretic mapping of human proteins. In Proceedings of the Electrophoresis’79, Munich, Germany, October 15–17; Radola, B.J., Ed.; Walter de Gruyter: Berlin, Germany, 1980. [Google Scholar]
- Feuerstein, N.; Cooper, H.L. Rapid phosphorylation-dephosphorylation of specific proteins induced by phorbol ester in HL-60 cells. Further characterization of the phosphorylation of 17-kilodalton and 27-kilodalton proteins in myeloid leukemic cells and human monocytes. J. Biol. Chem. 1984, 259, 2782–2788. [Google Scholar] [PubMed]
- Zylber-Katz, E.; Glazer, R.I. Phospholipid- and Ca2+-dependent protein kinase activity and protein phosphorylation patterns in the differentiation of human promyelocytic leukemia cell line HL-60. Cancer Res. 1985, 45, 5159–5164. [Google Scholar] [PubMed]
- Aebersold, R.H.; Leavitt, J.; Saavedra, R.A.; Hood, L.E.; Kent, S.B. Internal amino acid sequence analysis of proteins separated by one- or two-dimensional gel electrophoresis after in situ protease digestion on nitrocellulose. Proc. Natl. Acad. Sci. USA 1987, 84, 6970–6974. [Google Scholar] [CrossRef] [PubMed]
- Aebersold, R.H.; Teplow, D.B.; Hood, L.E.; Kent, S.B. Electroblotting onto activated glass. High efficiency preparation of proteins from analytical sodium dodecyl sulfate-polyacrylamide gels for direct sequence analysis. J. Biol. Chem. 1986, 261, 4229–4238. [Google Scholar] [PubMed]
- Fenn, J.B.; Mann, M.; Meng, C.K.; Wong, S.F.; Whitehouse, C.M. Electrospray ionization for mass spectrometry of large biomolecules. Science 1989, 246, 64–71. [Google Scholar] [CrossRef] [PubMed]
- Karas, M.; Hillenkamp, F. Laser desorption ionization of proteins with molecular mass exceeding 10000 daltons. Anal. Chem. 1988, 60, 2299–2301. [Google Scholar] [CrossRef] [PubMed]
- Hutchens, T.W.; Yip, T.-T. New desorption strategies for the mass spectrometric analysis of macromolecules. Rapid Commun. Mass Spectrom. 1993, 7, 576–580. [Google Scholar] [CrossRef]
- Wilkins, M.R.; Pasquali, C.; Appel, R.D.; Ou, K.; Golaz, O.; Sanchez, J.C.; Yan, J.X.; Gooley, A.A.; Hughes, G.; Humphery-Smith, I.; et al. From proteins to proteomes: Large scale protein identification by two-dimensional electrophoresis and amino acid analysis. Biotechnology 1996, 14, 61–65. [Google Scholar] [CrossRef] [PubMed]
- Wilkins, M.R.; Sanchez, J.C.; Gooley, A.A.; Appel, R.D.; Humphery-Smith, I.; Hochstrasser, D.F.; Williams, K.L. Progress with proteome projects: Why all proteins expressed by a genome should be identified and how to do it. Biotechnol. Genet. Eng. Rev. 1996, 13, 19–50. [Google Scholar] [CrossRef] [PubMed]
- Gygi, S.P. Quantitative analysis of complex protein mixtures using isotope-coded affinity tags. Nat. Biotechnol. 1999, 17, 994–999. [Google Scholar] [CrossRef] [PubMed]
- Tunon, J.; Martin-Ventura, J.L.; Blanco-Colio, L.M.; Lorenzo, O.; Lopez, J.A.; Egido, J. Proteomic strategies in the search of new biomarkers in atherothrombosis. J. Am. Coll. Cardiol. 2010, 55, 2009–2016. [Google Scholar] [CrossRef] [PubMed]
- Ong, S.E. Stable isotope labeling by amino acids in cell culture, SILAC, as a simple and accurate approach to expression proteomics. Mol. Cell. Proteom. 2002, 1, 376–386. [Google Scholar] [CrossRef]
- Blagoev, B. A proteomics strategy to elucidate functional protein-protein interactions applied to EGF signaling. Nat. Biotechnol. 2003, 21, 315–318. [Google Scholar] [CrossRef] [PubMed]
- Ross, P.L.; Huang, Y.N.; Marchese, J.N.; Williamson, B.; Parker, K.; Hattan, S.; Khainovski, N.; Pillai, S.; Dey, S.; Daniels, S.; et al. Multiplexed protein quantitation in Saccharomyces cerevisiae using amine-reactive isobaric tagging reagents. Mol. Cell. Proteom. 2004, 3, 1154–1169. [Google Scholar] [CrossRef] [PubMed]
- Burt, T.; John, C.S.; Ruckle, J.L.; Vuong, L.T. Phase-0/microdosing studies using PET, AMS, and LC-MS/MS: A range of study methodologies and conduct considerations. Accelerating development of novel pharmaceuticals through safe testing in humans—A practical guide. Expert Opin. Drug Deliv. 2016, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Coleman, H.N.; Greenfield, W.W.; Stratton, S.L.; Vaughn, R.; Kieber, A.; Moerman-Herzog, A.M.; Spencer, H.J.; Hitt, W.C.; Quick, C.M.; Hutchins, L.F.; et al. Human papillomavirus type 16 viral load is decreased following a therapeutic vaccination. Cancer Immunol. Immunother. 2016, 65, 563–573. [Google Scholar] [CrossRef] [PubMed]
- Cheraghchi-Bashi, A.; Parker, C.A.; Curry, E.; Salazar, J.F.; Gungor, H.; Saleem, A.; Cunnea, P.; Rama, N.; Salinas, C.; Mills, G.B.; et al. A putative biomarker signature for clinically effective AKT inhibition: Correlation of in vitro, in vivo and clinical data identifies the importance of modulation of the mTORC1 pathway. Oncotarget 2015, 6, 41736–41749. [Google Scholar] [PubMed]
- Corcoran, R.B.; Atreya, C.E.; Falchook, G.S.; Kwak, E.L.; Ryan, D.P.; Bendell, J.C.; Hamid, O.; Messersmith, W.A.; Daud, A.; Kurzrock, R.; et al. Combined BRAF and MEK Inhibition With Dabrafenib and Trametinib in BRAF V600-Mutant Colorectal Cancer. J. Clin. Oncol. 2015, 33, 4023–4031. [Google Scholar] [CrossRef] [PubMed]
- Buscail, L.; Bournet, B.; Vernejoul, F.; Cambois, G.; Lulka, H.; Hanoun, N.; Dufresne, M.; Meulle, A.; Vignolle-Vidoni, A.; Ligat, L.; et al. First-in-man phase 1 clinical trial of gene therapy for advanced pancreatic cancer: Safety, biodistribution, and preliminary clinical findings. Mol. Ther. 2015, 23, 779–789. [Google Scholar] [CrossRef] [PubMed]
- Hare, B.J.; Haseltine, E.; Fleming, M.; Chelsky, D.; McIntosh, L.; Allard, R.; Botfield, M. A signature for immune response correlates with HCV treatment outcome in Caucasian subjects. J. Proteom. 2015, 116, 59–67. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.M.; Hays, J.L.; Annunziata, C.M.; Noonan, A.M.; Minasian, L.; Zujewski, J.A.; Yu, M.; Gordon, N.; Ji, J.; Sissung, T.M.; et al. Phase I/Ib study of olaparib and carboplatin in BRCA1 or BRCA2 mutation-associated breast or ovarian cancer with biomarker analyses. J. Natl. Cancer Inst. 2014, 106, dju089. [Google Scholar] [CrossRef] [PubMed]
- Cardin, D.B.; Goff, L.; Li, C.I.; Shyr, Y.; Winkler, C.; DeVore, R.; Schlabach, L.; Holloway, M.; McClanahan, P.; Meyer, K.; et al. Phase II trial of sorafenib and erlotinib in advanced pancreatic cancer. Cancer Med. 2014, 3, 572–579. [Google Scholar] [CrossRef] [PubMed]
- Maitland, M.L.; Levine, M.R.; Lacouture, M.E.; Wroblewski, K.E.; Chung, C.H.; Gordon, I.O.; Szeto, L.; Ratko, G.; Soltani, K.; Kozloff, M.F.; et al. Evaluation of a novel rash scale and a serum proteomic predictor in a randomized phase II trial of sequential or concurrent cetuximab and pemetrexed in previously treated non-small cell lung cancer. BMC Cancer 2014, 14, 5. [Google Scholar] [CrossRef] [PubMed]
- Templeton, A.J.; Dutoit, V.; Cathomas, R.; Rothermundt, C.; Bartschi, D.; Droge, C.; Gautschi, O.; Borner, M.; Fechter, E.; Stenner, F.; et al. Phase 2 trial of single-agent everolimus in chemotherapy-naive patients with castration-resistant prostate cancer (SAKK 08/08). Eur. Urol. 2013, 64, 150–158. [Google Scholar] [CrossRef] [PubMed]
- Azad, N.; Yu, M.; Davidson, B.; Choyke, P.; Chen, C.C.; Wood, B.J.; Venkatesan, A.; Henning, R.; Calvo, K.; Minasian, L.; et al. Translational predictive biomarker analysis of the phase 1b sorafenib and bevacizumab study expansion cohort. Mol. Cell. Proteom. 2013, 12, 1621–1631. [Google Scholar] [CrossRef] [PubMed]
- Stinchcombe, T.E.; Roder, J.; Peterman, A.H.; Grigorieva, J.; Lee, C.B.; Moore, D.T.; Socinski, M.A. A retrospective analysis of VeriStrat status on outcome of a randomized phase II trial of first-line therapy with gemcitabine, erlotinib, or the combination in elderly patients (age 70 years or older) with stage IIIB/IV non-small-cell lung cancer. J. Thorac. Oncol. 2013, 8, 443–451. [Google Scholar] [CrossRef] [PubMed]
- Akerley, W.; Boucher, K.; Rich, N.; Egbert, L.; Harker, G.; Bylund, J.; van Duren, T.; Reddy, C. A phase II study of bevacizumab and erlotinib as initial treatment for metastatic non-squamous, non-small cell lung cancer with serum proteomic evaluation. Lung Cancer 2013, 79, 307–311. [Google Scholar] [CrossRef] [PubMed]
- Chinnaiyan, P.; Chowdhary, S.; Potthast, L.; Prabhu, A.; Tsai, Y.Y.; Sarcar, B.; Kahali, S.; Brem, S.; Yu, H.M.; Rojiani, A.; et al. Phase I trial of vorinostat combined with bevacizumab and CPT-11 in recurrent glioblastoma. Neuro Oncol. 2012, 14, 93–100. [Google Scholar] [CrossRef] [PubMed]
- Jensen, A.D.; Nikoghosyan, A.; Hinke, A.; Debus, J.; Munter, M.W. Combined treatment of adenoid cystic carcinoma with cetuximab and IMRT plus C12 heavy ion boost: ACCEPT [ACC, Erbitux(R) and particle therapy]. BMC Cancer 2011, 11, 70. [Google Scholar] [CrossRef] [PubMed]
- Dalenc, F.; Doisneau-Sixou, S.F.; Allal, B.C.; Marsili, S.; Lauwers-Cances, V.; Chaoui, K.; Schiltz, O.; Monsarrat, B.; Filleron, T.; Renee, N.; et al. Tipifarnib plus tamoxifen in tamoxifen-resistant metastatic breast cancer: A negative phase II and screening of potential therapeutic markers by proteomic analysis. Clin. Cancer Res. 2010, 16, 1264–1271. [Google Scholar] [CrossRef] [PubMed]
- Tabernero, J.; Cervantes, A.; Rivera, F.; Martinelli, E.; Rojo, F.; von Heydebreck, A.; Macarulla, T.; Rodriguez-Braun, E.; Eugenia Vega-Villegas, M.; Senger, S.; et al. Pharmacogenomic and pharmacoproteomic studies of cetuximab in metastatic colorectal cancer: Biomarker analysis of a phase I dose-escalation study. J. Clin. Oncol. 2010, 28, 1181–1189. [Google Scholar] [CrossRef] [PubMed]
- Debucquoy, A.; Haustermans, K.; Daemen, A.; Aydin, S.; Libbrecht, L.; Gevaert, O.; de Moor, B.; Tejpar, S.; McBride, W.H.; Penninckx, F.; et al. Molecular response to cetuximab and efficacy of preoperative cetuximab-based chemoradiation in rectal cancer. J. Clin. Oncol. 2009, 27, 2751–2757. [Google Scholar] [CrossRef] [PubMed]
- Schilder, R.J.; Pathak, H.B.; Lokshin, A.E.; Holloway, R.W.; Alvarez, R.D.; Aghajanian, C.; Min, H.; Devarajan, K.; Ross, E.; Drescher, C.W.; et al. Phase II trial of single agent cetuximab in patients with persistent or recurrent epithelial ovarian or primary peritoneal carcinoma with the potential for dose escalation to rash. Gynecol. Oncol. 2009, 113, 21–27. [Google Scholar] [CrossRef] [PubMed]
- O’Byrne, K.J.; Danson, S.; Dunlop, D.; Botwood, N.; Taguchi, F.; Carbone, D.; Ranson, M. Combination therapy with gefitinib and rofecoxib in patients with platinum-pretreated relapsed non small-cell lung cancer. J. Clin. Oncol. 2007, 25, 3266–3273. [Google Scholar] [CrossRef] [PubMed]
- Posadas, E.M.; Kwitkowski, V.; Kotz, H.L.; Espina, V.; Minasian, L.; Tchabo, N.; Premkumar, A.; Hussain, M.M.; Chang, R.; Steinberg, S.M.; et al. A prospective analysis of imatinib-induced c-KIT modulation in ovarian cancer: A phase II clinical study with proteomic profiling. Cancer 2007, 110, 309–317. [Google Scholar] [CrossRef] [PubMed]
- Dragovich, T.; McCoy, S.; Fenoglio-Preiser, C.M.; Wang, J.; Benedetti, J.K.; Baker, A.F.; Hackett, C.B.; Urba, S.G.; Zaner, K.S.; Blanke, C.D.; et al. Phase II trial of erlotinib in gastroesophageal junction and gastric adenocarcinomas: SWOG 0127. J. Clin. Oncol. 2006, 24, 4922–4927. [Google Scholar] [CrossRef] [PubMed]
- Shusta, E.V. Blood-brain barrier genomics, proteomics, and new transporter discovery. NeuroRx 2005, 2, 151–161. [Google Scholar] [CrossRef] [PubMed]
- Boessen, R.; Heerspink, H.J.; de Zeeuw, D.; Grobbee, D.E.; Groenwold, R.H.; Roes, K.C. Improving clinical trial efficiency by biomarker-guided patient selection. Trials 2014, 15, 103. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, R.L.; Yang, S.N.; Taldone, T.; Chang, B.; Gerecitano, J.; Elenitoba-Johnson, K.; Shaknovich, R.; Tam, W.; Leonard, J.P.; Chiosis, G.; et al. Pharmacoproteomics identifies combinatorial therapy targets for diffuse large B cell lymphoma. J. Clin. Investig. 2015, 125, 4559–4571. [Google Scholar] [CrossRef] [PubMed]
- Food and Drug Administration (FDA). Draft Guidance: Guidance for Industry. Expedited Programs for Serious Conditions—Drugs and Biologics; Food and Drug Administration: Silver Spring, MD, USA, 2013.
- Riveros, C.; Dechartres, A.; Perrodeau, E.; Haneef, R.; Boutron, I.; Ravaud, P. Timing and completeness of trial results posted at ClinicalTrials.gov and published in journals. PLoS Med. 2013, 10, e1001566. [Google Scholar] [CrossRef] [PubMed]
- Diamandis, E.P. Cancer biomarkers: Can we turn recent failures into success? J. Natl. Cancer Inst. 2010, 102, 1462–1467. [Google Scholar] [CrossRef] [PubMed]
- Zhang, A.H.; Sun, H.; Yan, G.L.; Han, Y.; Wang, X.J. Serum proteomics in biomedical research: A systematic review. Appl. Biochem. Biotechnol. 2013, 170, 774–786. [Google Scholar] [CrossRef] [PubMed]
- Pavlou, M.P.; Diamandis, E.P.; Blasutig, I.M. The long journey of cancer biomarkers from the bench to the clinic. Clin. Chem. 2013, 59, 147–157. [Google Scholar] [CrossRef] [PubMed]
- D’Abramo, F.; Schildmann, J.; Vollmann, J. Research participants’ perceptions and views on consent for biobank research: A review of empirical data and ethical analysis. BMC Med. Ethics 2015, 16, 60. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Graber, A.; McBurney, R.N.; Balasubramanian, R. Sample size and statistical power considerations in high-dimensionality data settings: A comparative study of classification algorithms. BMC Bioinf. 2010, 11, 447. [Google Scholar] [CrossRef] [PubMed]
- Kummar, S.; Williams, P.M.; Lih, C.J.; Polley, E.C.; Chen, A.P.; Rubinstein, L.V.; Zhao, Y.; Simon, R.M.; Conley, B.A.; Doroshow, J.H. Application of molecular profiling in clinical trials for advanced metastatic cancers. J. Natl. Cancer Inst. 2015, 107, djv003. [Google Scholar] [CrossRef] [PubMed]
- Freidlin, B.; McShane, L.M.; Polley, M.-Y.C.; Korn, E.L. Randomized Phase II Trial Designs with Biomarkers. J. Clin. Oncol. 2012, 30, 3304–3309. [Google Scholar] [CrossRef] [PubMed]
- Edelman, M.J.; Schneider, C.P.; Tsai, C.M.; Kim, H.T.; Quoix, E.; Luft, A.V.; Kaleta, R.; Mukhopadhyay, P.; Trifan, O.C.; Whitaker, L.; et al. Randomized phase II study of ixabepilone or paclitaxel plus carboplatin in patients with non-small-cell lung cancer prospectively stratified by β-3 tubulin status. J. Clin. Oncol. 2013, 31, 1990–1996. [Google Scholar] [CrossRef] [PubMed]
- Seymour, L.; Ivy, S.P.; Sargent, D.; Spriggs, D.; Baker, L.; Rubinstein, L.; Ratain, M.J.; Le Blanc, M.; Stewart, D.; Crowley, J.; et al. The Design of Phase II Clinical Trials Testing Cancer Therapeutics: Consensus Recommendations from the Clinical Trial Design Task Force of the National Cancer Institute Investigational Drug Steering Committee. Clin. Cancer Res. 2010, 16, 1764–1769. [Google Scholar] [CrossRef] [PubMed]
- Freidlin, B.; Korn, E.L. Biomarker enrichment strategies: Matching trial design to biomarker credentials. Nat. Rev. Clin. Oncol. 2014, 11, 81–90. [Google Scholar] [CrossRef] [PubMed]
- Mandrekar, S.J.; Sargent, D.J. Clinical Trial Designs for Predictive Biomarker Validation: Theoretical Considerations and Practical Challenges. J. Clin. Oncol. 2009, 27, 4027–4034. [Google Scholar] [CrossRef] [PubMed]
- Burbaum, J.; Tobal, G.M. Proteomics in drug discovery. Curr. Opin. Chem. Biol. 2002, 6, 427–433. [Google Scholar] [CrossRef]
- McDonald, W.H.; Ohi, R.; Miyamoto, D.T.; Mitchison, T.J.; Yates III, J.R. Comparison of three directly coupled HPLC MS/MS strategies for identification of proteins from complex mixtures: Single-dimension LC-MS/MS, 2-phase MudPIT, and 3-phase MudPIT. Int. J. Mass Spectrom. 2002, 219, 245–251. [Google Scholar] [CrossRef]
- Washburn, M.P.; Wolters, D.; Yates, J.R. Large-scale analysis of the yeast proteome by multidimensional protein identification technology. Nat. Biotechnol. 2001, 19, 242–247. [Google Scholar] [CrossRef] [PubMed]
- Mann, M. Functional and quantitative proteomics using SILAC. Nat. Rev. Mol. Cell Biol. 2006, 7, 952–958. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Chevolot, Y.; Ataman-Önal, Y.; Choquet-Kastylevsky, G.; Souteyrand, E.; Laurenceau, E. Cancer biomarkers detection using 3D microstructured protein chip: Implementation of customized multiplex immunoassay. Sens. Actuators B Chem. 2012, 175, 22–28. [Google Scholar] [CrossRef]
- Wiedl, T.; Arni, S.; Roschitzki, B.; Grossmann, J.; Collaud, S.; Soltermann, A.; Hillinger, S.; Aebersold, R.; Weder, W. Activity-based proteomics: Identification of ABHD11 and ESD activities as potential biomarkers for human lung adenocarcinoma. J. Proteom. 2011, 74, 1884–1894. [Google Scholar] [CrossRef] [PubMed]
- Karas, M.; Bachmann, D.; Bahr, U.; Hillenkamp, F. Matrix-assisted ultraviolet laser desorption of non-volatile compounds. Int. J. Mass Spectrom. Ion Process. 1987, 78, 53–68. [Google Scholar] [CrossRef]
- Ho, C.S.; Lam, C.W.; Chan, M.H.; Cheung, R.C.; Law, L.K.; Lit, L.C.; Ng, K.F.; Suen, M.W.; Tai, H.L. Electrospray ionisation mass spectrometry: Principles and clinical applications. Clin. Biochem. 2003, 24, 3–12. [Google Scholar]
- Kim, H.J.; Kang, U.B.; Lee, H.; Jung, J.H.; Lee, S.T.; Yu, M.H.; Kim, H.; Lee, C. Profiling of differentially expressed proteins in stage IV colorectal cancers with good and poor outcomes. J. Proteom. 2012, 75, 2983–2997. [Google Scholar] [CrossRef] [PubMed]
- Hall, D.A.; Ptacek, J.; Snyder, M. Protein microarray technology. Mech. Ageing Dev. 2007, 128, 161–167. [Google Scholar] [CrossRef] [PubMed]
- Hoogenboom, H.R.; de Bruine, A.P.; Hufton, S.E.; Hoet, R.M.; Arends, J.W.; Roovers, R.C. Antibody phage display technology and its applications. Immunotechnology 1998, 4, 1–20. [Google Scholar] [CrossRef]
- He, M.; Taussig, M.J. Ribosome display: Cell-free protein display technology. Brief. Funct. Genom. Proteom. 2002, 1, 204–212. [Google Scholar] [CrossRef]
- Kramer, H.B.; Nicholson, B.; Kessler, B.M.; Altun, M. Detection of ubiquitin-proteasome enzymatic activities in cells: Application of activity-based probes to inhibitor development. Biochim. Biophys. Acta 2012, 1823, 2029–2037. [Google Scholar] [CrossRef] [PubMed]
- Fonovic, M.; Bogyo, M. Activity based probes for proteases: Applications to biomarker discovery, molecular imaging and drug screening. Curr. Pharm. Des. 2007, 13, 253–261. [Google Scholar] [CrossRef] [PubMed]
- Fonovic, M.; Bogyo, M. Activity-based probes as a tool for functional proteomic analysis of proteases. Expert Rev. Proteom. 2008, 5, 721–730. [Google Scholar] [CrossRef] [PubMed]
- Pichler, C.M.; Krysiak, J.; Breinbauer, R. Target identification of covalently binding drugs by activity-based protein profiling (ABPP). Bioorg. Med. Chem. 2016, 24, 3291–3303. [Google Scholar] [CrossRef] [PubMed]
- Paweletz, C.P.; Charboneau, L.; Bichsel, V.E.; Simone, N.L.; Chen, T.; Gillespie, J.W.; Emmert-Buck, M.R.; Roth, M.J.; Petricoin, E.F.; Liotta, L.A. Reverse phase protein microarrays which capture disease progression show activation of pro-survival pathways at the cancer invasion front. Oncogene 2001, 20, 1981–1989. [Google Scholar] [CrossRef] [PubMed]
- Boellner, S.; Becker, K.F. Reverse Phase Protein Arrays—Quantitative Assessment of Multiple Biomarkers in Biopsies for Clinical Use. Microarrays 2015, 4, 98–114. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Milestone | Year | Description | References |
|---|---|---|---|
| The word “protein” first used | 1838 | Swedish Chemist Jöns Jakob Berzelius | [24] |
| Mass spectrometer | 1913 | J.J. Thomson constructs the first mass spectrometer | [25] |
| Immunohistochemistry | 1941 | First description of the methodology | [26] |
| LC, GC, and MS in biology | 1960s | liquid (LC) and high-performance liquid chromatography (HPLC), gas chromatography (GC) and mass-spectrometry (MS) used to characterize physiologic and pathophysiologic states (quantitative) | [27,28] |
| Enzyme-linked immunosorbent assay (ELISA) | 1971 | First description of the methodology | [29] |
| [30] | |||
| Introduction of two-Dimensional gel | 1975 | The first protein studies that can be called proteomics began in 1975 with the introduction of the two-dimensional gel and mapping of the proteins from the bacterium Escherichia coli, guinea pig and mouse | [31] |
| [32] | |||
| [33] | |||
| Combining LC with MS = LCMS | 1980s | First interfaces for combining liquid chromatography with mass spectrometry (LC-MS) emerge | [34] |
| [35] | |||
| [36] | |||
| [37] | |||
| Attempt to catalog human proteins | 1980 | Attempt to catalog human proteins | [38] |
| 2-Dimentional electrophoresis (2DE) | 1984/5 | First studies to use 2DE for human protein separation | [39] |
| [40] | |||
| Development of microsequencing techniques for electroblotted proteins | 1986/7 | A major breakthrough was the development of microsequencing techniques for electroblotted proteins | [41] |
| [42] | |||
| Electrospray Ionization (ESI) and Matrix-assisted laser desorption/ionization (MALDI) | 1989 | ESI and MALDI first used to vaporize and ionize large molecules. This enabled transformation of proteins into the gas phase for MS analysis | [43] |
| [44] | |||
| Surface-enhanced laser desorption/ionization (SELDI) | 1993 | First report of the use of SELDI for analysis of marcomolecules | [45] |
| Proteome and proteomics | 1995 | First use of the terms “proteome” and “proteomics” to denote the full complement of an organism’s proteins and their study, respectively | [18] |
| [46] | |||
| [47] | |||
| Isotope coded affinity tags (ICAT) | 1999 | First report of use of ICAT in a proteomic study | [48] |
| Reverse phase protein array | 2001 | First described in 2001 | [49] |
| Stable isotopic labeling by amino acids in cell culture (SILAC) | 2002/3 | First reports of use of SILAC in proteomic studies | [50] |
| [51] | |||
| Pharmacoproteomics | 2002 | First use of the term “pharmacoproteomics” in peer-reviewed literature to indicate the use of proteomics in the study of drug effects | [19] |
| [20] | |||
| Isobaric tags for relative and absolute quantification (iTRAQ) | 2004 | First report of the use of iTRAQ in the quantification of Saccharomyces cerevisiae | [52] |
| PubMed Publication | Drug | Phase | Condition | Proteomic Objectives | Proteomic Analytics | Findings |
|---|---|---|---|---|---|---|
| Coleman et al. 2016 [54] | PepCan | 1 | HPV | Safety and efficacy of HPV vaccination, Proteomic analysis was used as exploratory endpoint to determine feasibility of biomarker identification | LC-MS/MS, LTQ-FT-Orbitrap | Differences in protein expression between baseline and post vaccination were detected. Feasibility of using the PBMC samples for proteomic analysis was established. Requirement to be consistent with the sample processing after blood draws was realized |
| Cheraghchi-Bashi et al. 2015 [55] | GSK2141795 | 1 | Ovarian Cancer | Validating ovarian cancer proteomic signatures identified in preclinical xenograft and cell line studies | ELISA, Protein arrays | Proteomic signature was established as a predictive biomarker and could be used in patient stratification in larger studies. Importance of noninvasive methods to obtain samples for biomarker assessment was emphasized |
| Corcoran et al. 2015 [56] | Dabrafenib + Tramatenib | 1 | mCRC | Proteomic biomarker assessment for treatment of mCRC | Protein arrays | No correlation established between protein markers and mCRC treatment effects |
| Buscail et al. 2015 [57] | CYL-02 | 1 | Pancreatic cancer | Safety, PK, and efficacy in pancreatic cancer. High throughput proteomic study conducted as exploratory endpoint | LC-MS/MS | Proteomic signature identified as predictive biomarker and correlated with good and poor treatment responders |
| Hare et al. 2015 [58] | Telaprevir in combination with peg-interferon and ribavirin | 2 | HCV | High throughput proteomic analysis conducted on samples from 3 phase-2 treatment studies for HCV | LC-MS/MS | Proteomic signature established as potential predictive biomarker. Proteomic analysis enhances understanding of biological mechanisms leading to response |
| Lee et al. 2014 [59] | Olaparib and carboplatin | 1/2 | Breast/ovarian cancer | Exploratory proteomic analysis for breast/ovarian cancer treatment efficacy | Protein arrays | pS209-eIF4E and FOXO3a may be predictive of response. Prospective studies are required for validation |
| Cardin et al. 2014 [60] | Erlotinib with sorafenib | 2 | Pancreatic adenocarcinoma | VeriStrat® testing of pre-treatment samples to predict outcomes in treatment of pancreatic adenocarcinoma | MALSI-MS (VeriStrat) | Proteomic classification demonstrated correlation with clinical outcomes and could be useful in designing future therapeutic pancreatic cancer studies |
| Maitland et al. 2014 [61] | Cetuximab and Pemetrexed | 2 | NSCLC | Development of proteomic biomarkers for EGFR inhibitor efficacy in NSCLC | MALSI-MS (VeriStrat) | Serum proteomic markers may be predictive of NSCLC outcomes |
| Templeton et al. 2013 [62] | Everolimus | 2 | mCRPC | Proteomic analysis used to explore serum biomarkers in treatment of mCRPC | Immuno-histo-chemistry; hybrid LTQ-FT-MS | Proteomic biomarkers could be predictive of treatment outcomes but need further validation |
| Azad et al. 2013 [63] | Sorafenib and Bevacizumab | 1 | Solid tumors | Identifying proteomic biomarkers of response to treatment of solid tumors | Protein array; immune-histo-chemistry | Proteomic biomarkers that are potentially predictive of treatment effects were identified and will be used for stratification in larger studies |
| Stinchcombe et al. 2013 [64] | Gemcitabine and Erlotinib | 2 | NSCLC | Exploratory examination of the potential of proteomic test VeriStrat® to predict treatment outcomes of NSCLC | MALSI-MS (VeriStrat) | VeriStrat® can predict treatment outcomes in NSCLC patients |
| Akerley et al. 2013 [65] | Bevacizumab and Erlotinib | 2 | NSCLC | Prospective evaluation of proteomic serum biomarkers in the prediction of response to NSCLC treatment | MALSI-MS (VeriStrat) | Proteomic biomarkers (VeriStrat®) can be used for patient selection and are predictive of NSCLC treatment outcomes |
| Chinnaiyan et al. 2011 [66] | Vorinostat & Bevacizumab and CPT-11 | 1 | Glioblastoma | Use of proteomic profiling to identify serum biomarkers of glioblastoma treatment outcomes. Serum proteomic profiling was an exploratory endpoint | Protein array | Proteomic analysis provided preliminary information on predictive and prognostic biomarkers (PFS and recurrence) |
| Jensen et al. 2011 [67] | Cetuximab and IMRT plus C12 heavy ion boost | 2 | ACC | Predict treatment efficacy in ACC. Well known markers for angiogenesis and tumorigenesis will be assessed from collected samples | ELISA | Not Applicable—description of an ongoing study protocol |
| Dalenc et al. 2010 [68] | Tipifarnib | 2 | Metastatic breast cancer | Identifying markers of therapeutic response in breast cancer patients treated with FTIs | SELDI-TOF, LTQ-FT-Orbitrap | Proteomic analysis identified a peptide of fibrinogen α that correlated with disease progression |
| Tabernero et al. 2010 [69] | Cetuximab | 1 | mCRC | Identifying biomarkers of cetuximab-responsive disease in plasma and tissue samples | Immuno-assays for 97 proteins | Candidate predictive biomarkers of response to cetuximab treatment were identified including inhibition of signaling proteins |
| Debucquoy et al. 2009 [70] | Cetuximab with chemo-radiotherapy | 2 | Rectal cancer | Identifying biomarkers predictive of response in plasma and tissue samples | Immune-assay | Proteomic profile of patients is predictive of disease-free survival in cetuximab-treated rectal cancer patients |
| Schilder et al. 2009 [71] | Cetuximab | 2 | Ovarian or peritoneal carcinoma | Prediction of response using proteomic serum biomarkers | ELISA and bead-based immune-assays | Serologic biomarkers were identified and patients with elevated levels are more likely to have earlier disease progression versus stable disease or partial remission |
| O'Byrne et al. 2007 [72] | Gefitinib and Rofecoxib | 2 | NSCLC | Identifying proteomic markers of response to EGFR TKIs | MALDI | Proteomic biomarkers were identified which could identify patients most likely to benefit from treatment from those with stable illness or progressive disease |
| Posadas et al. 2007 [73] | Imatinib | 2 | Ovarian cancer | Identifying proteomic biomarkers of response to treatment in tumor biopsies | Protein array | Though the study did not meet the primary endpoints of response to imatinib treatment, biomarker correlation with treatment was consistent with in vitro molecular signaling findings |
| Dragovich et al. 2006 [74] | Erlotinib | 2 | Gastric adenocarcinoma | Identifying plasma proteomic markers of response to erlotinib treatment | ELISA, SELDI | No biomarker correlations with treatment response were identified |
| Challenges in the Application of Pharmacoproteomics Approaches in Early-Phase Development | |
|---|---|
| Methodological [13] |
|
| Operational [80,81,82] |
|
| Regulatory [82] |
|
| Financial and legal [82] |
|
| Ethical challenges [83] |
|
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nandal, S.; Burt, T. Integrating Pharmacoproteomics into Early-Phase Clinical Development: State-of-the-Art, Challenges, and Recommendations. Int. J. Mol. Sci. 2017, 18, 448. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms18020448
Nandal S, Burt T. Integrating Pharmacoproteomics into Early-Phase Clinical Development: State-of-the-Art, Challenges, and Recommendations. International Journal of Molecular Sciences. 2017; 18(2):448. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms18020448
Chicago/Turabian StyleNandal, Savita, and Tal Burt. 2017. "Integrating Pharmacoproteomics into Early-Phase Clinical Development: State-of-the-Art, Challenges, and Recommendations" International Journal of Molecular Sciences 18, no. 2: 448. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms18020448