
Potential Modes of Intercellular α-Synuclein Transmission

Abstract
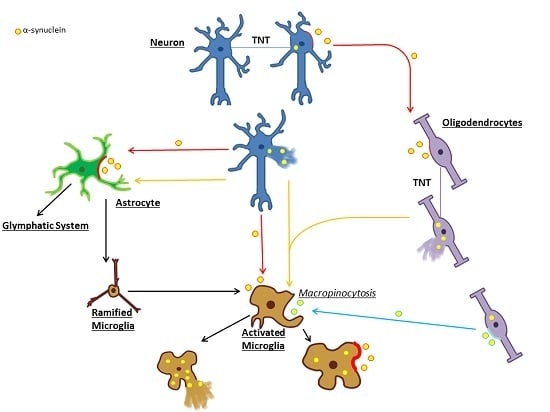
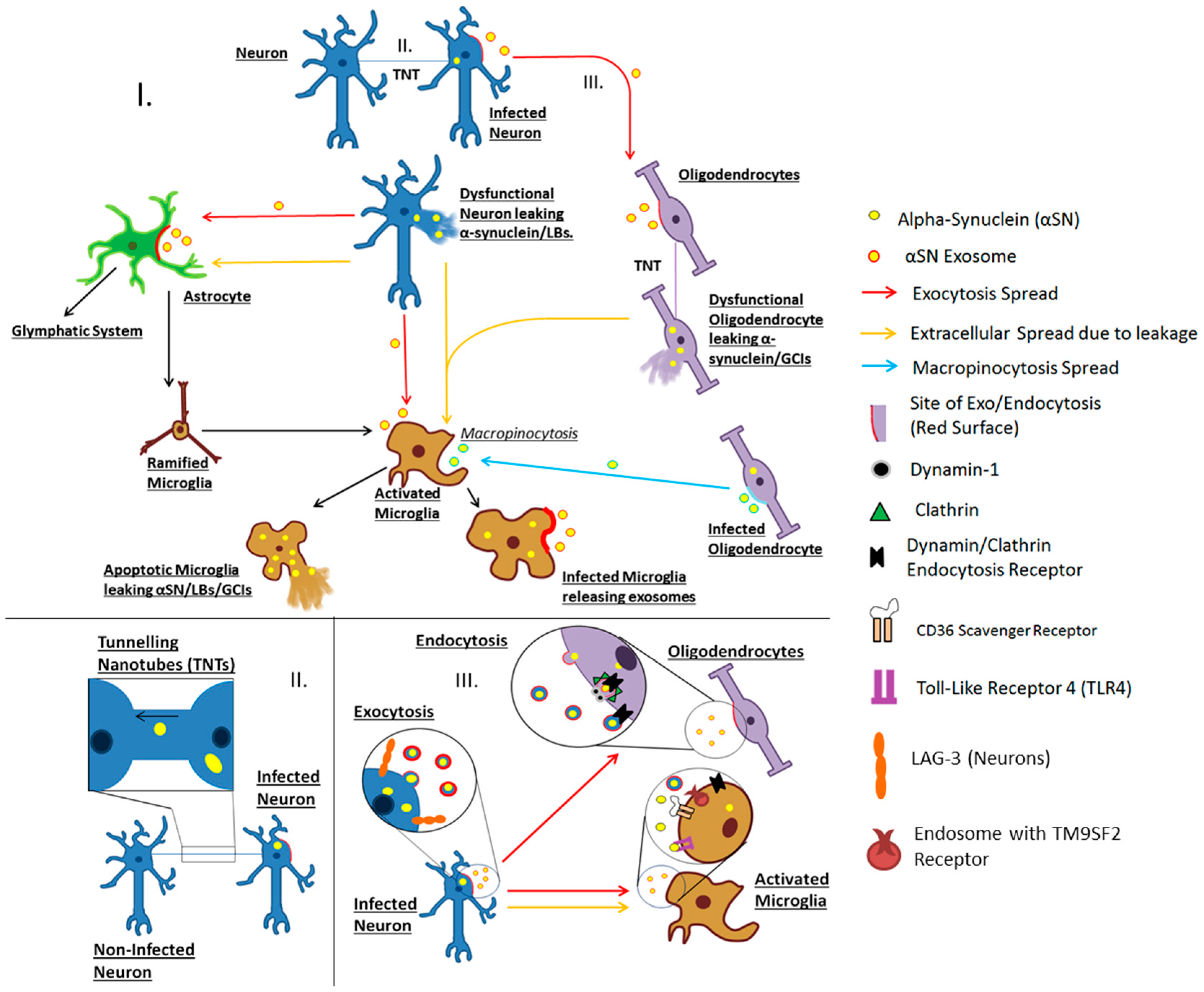
:
{kind=link}
{kind=link}
1. α-Synuclein
2. α-Synuclein in Disease
3. α-Synuclein and Disease Spread
4. Extracellular α-Syn, Secretion and Uptake
5. Tunneling Nanotubes
6. Microglia
7. Glymphatic Clearance
8. Summary and Concluding Remarks
Acknowledgments
Conflicts of Interest
Abbreviations
| α-syn | α-Synuclein |
| CNS | Central Nervous System |
| CSF | Cerebrospinal Fluid |
| DLB | Dementia with Lewy Bodies |
| GCI | Glial Cytoplasmic Inclusions |
| MSA | Multiple System Atrophy |
| PD | Parkinson’s disease |
| TNTs | Tunneling Nanotubes |
| TLR | Toll-Like Receptor |
| LAG-3 | Lymphocyte Activating Gene 3 |
| LN | Lewy Neurite |
| NAC | Non-Aβ Component of Alzheimer’s Disease Amyloid |
References
- Barbour, R.; Kling, K.; Anderson, J.; Banducci, K.; Cole, T.; Diep, L.; Fox, M.; Goldstein, J.; Soriano, F.; Seubert, P.; et al. Red Blood Cells Are the Major Source of α-Synuclein in Blood. Neurodegener. Dis. 2008, 5, 55–59. [Google Scholar] [CrossRef] [PubMed]
- Theillet, F.; Binolfi, A.; Bekei, B.; Martorana, A.; Rose, H.; Stuiver, M.; Verzini, S.; Lorenz, D.; van Rossum, M.; Goldfarb, D.; et al. Structural disorder of monomeric α-synuclein persists in mammalian cells. Nature 2016, 530, 45–50. [Google Scholar] [CrossRef] [PubMed]
- Weinreb, P.; Zhen, W.; Poon, A.; Conway, K.; Lansbury, P. NACP, A Protein Implicated in Alzheimer’s Disease and Learning, Is Natively Unfolded. Biochemistry 1996, 35, 13709–13715. [Google Scholar] [CrossRef] [PubMed]
- Cheng, F.; Vivacqua, G.; Yu, S. The role of α-synuclein in neurotransmission and synaptic plasticity. J. Chem. Neuroanat. 2011, 42, 242–248. [Google Scholar] [CrossRef] [PubMed]
- Burre, J.; Sharma, M.; Tsetsenis, T.; Buchman, V.; Etherton, M.; Sudhof, T. α-Synuclein Promotes SNARE-Complex Assembly in Vivo and in Vitro. Science 2010, 329, 1663–1667. [Google Scholar] [CrossRef] [PubMed]
- Perez, R.; Waymire, J.; Lin, E.; Liu, J.; Guo, F.; Zigmond, M. A role for α-synuclein in the regulation of dopamine biosynthesis. J. Neurosci. 2002, 22, 3090–3099. [Google Scholar] [PubMed]
- Larsen, K.; Schmitz, Y.; Troyer, M.; Mosharov, E.; Dietrich, P.; Quazi, A.; Savalle, M.; Nemani, V.; Chaudhry, F.; Edwards, R.; et al. α-Synuclein Overexpression in PC12 and Chromaffin Cells Impairs Catecholamine Release by Interfering with a Late Step in Exocytosis. J. Neurosci. 2006, 26, 11915–11922. [Google Scholar] [CrossRef] [PubMed]
- Yavich, L.; Jäkälä, P.; Tanila, H. Abnormal compartmentalization of norepinephrine in mouse dentate gyrus in α-synuclein knockout and A30P transgenic mice. J. Neurochem. 2006, 99, 724–732. [Google Scholar] [CrossRef] [PubMed]
- Yavich, L.; Tanilla, H.; Vepsalainen, S.; Jakala, P. Role of α-Synuclein in Presynaptic Dopamine Recruitment. J. Neurosci. 2004, 24, 11165–11170. [Google Scholar] [CrossRef] [PubMed]
- Bartels, T.; Choi, J.; Selkoe, D. α-Synuclein occurs physiologically as a helically folded tetramer that resists aggregation. Nature 2011, 477, 107–110. [Google Scholar] [CrossRef] [PubMed]
- Jakes, R.; Spillantini, M.; Goedert, M. Identification of two distinct synucleins from human brain. FEBS Lett. 1994, 345, 27–32. [Google Scholar] [CrossRef]
- Luth, E.; Bartels, T.; Dettmer, U.; Kim, N.; Selkoe, D. Purification of α-Synuclein from Human Brain Reveals an Instability of Endogenous Multimers as the Protein Approaches Purity. Biochemistry 2015, 54, 279–292. [Google Scholar] [CrossRef] [PubMed]
- Burré, J.; Vivona, S.; Diao, J.; Sharma, M.; Brunger, A.; Südhof, T. Properties of native brain α-synuclein. Nature 2013, 498, E4–E6. [Google Scholar] [CrossRef] [PubMed]
- McCann, H.; Stevens, C.; Cartwright, H.; Halliday, G. α-Synucleinopathy phenotypes. Park. Relat. Disord. 2014, 20, S62–S67. [Google Scholar] [CrossRef]
- Sveinbjornsdottir, S. The clinical symptoms of Parkinson’s disease. J. Neurochem. 2016, 139, 318–324. [Google Scholar] [CrossRef] [PubMed]
- Menza, M.; Robertson-Hoffman, D.; Bonapace, A. Parkinson’s disease and anxiety: Comorbidity with depression. Biol. Psychiatry 1993, 34, 465–470. [Google Scholar] [CrossRef]
- McKeith, I.; Mintzer, J.; Aarsland, D.; Burn, D.; Chiu, H.; Cohen-Mansfield, J.; Dickson, D.; Dubois, B.; Duda, J.; Feldman, H.; et al. Dementia with Lewy bodies. Lancet Neurol. 2004, 3, 19–28. [Google Scholar] [CrossRef]
- Longo, D.; Fanciulli, A.; Wenning, G. Multiple-System Atrophy. N. Engl. J. Med. 2015, 372, 249–263. [Google Scholar] [CrossRef] [PubMed]
- Ubhi, K.; Low, P.; Masliah, E. Multiple system atrophy: A clinical and neuropathological perspective. Trends Neurosci. 2011, 34, 581–590. [Google Scholar] [CrossRef] [PubMed]
- Braak, H.; Tredici, K.; Rüb, U.; de Vos, R.; Jansen Steur, E.; Braak, E. Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol. Aging 2003, 24, 197–211. [Google Scholar] [CrossRef]
- Marttila, R.; Rhine, U. Progression and survival in Parkinson’s disease. Acta Neurol. 1991, 136, 24–28. [Google Scholar] [CrossRef]
- Wenning, G.; Stefanova, N. Recent developments in multiple system atrophy. J. Neurol. 2009, 256, 1791–1808. [Google Scholar] [CrossRef] [PubMed]
- Williams, M.; Xiong, C.; Morris, J.; Galvin, J. Survival and mortality differences between dementia with Lewy bodies vs. Alzheimer disease. Neurology 2006, 67, 1935–1941. [Google Scholar] [CrossRef] [PubMed]
- Spillantini, M.; Schmidt, M.; Lee, V.; Trojanowski, J.; Jakes, R.; Goedert, M. α-Synuclein in Lewy bodies. Nature 1997, 388, 839–840. [Google Scholar] [CrossRef] [PubMed]
- Engelender, S. Ubiquitination of α-synuclein and autophagy in Parkinson’s disease. Autophagy 2008, 4, 372–374. [Google Scholar] [CrossRef] [PubMed]
- Braak, H.; Sandmann-Keil, D.; Gai, W.; Braak, E. Extensive axonal Lewy neurites in Parkinson’s disease: A novel pathological feature revealed by α-synuclein immunocytochemistry. Neurosci. Lett. 1999, 265, 67–69. [Google Scholar] [CrossRef]
- Galloway, P.; Mulvihill, P.; Perry, G. Filaments of Lewy bodies contain insoluble cytoskeletal elements. Am. J. Pathol. 1992, 140, 809–822. [Google Scholar] [PubMed]
- Kanazawa, T.; Uchihara, T.; Takahashi, A.; Nakamura, A.; Orimo, S.; Mizusawa, H. Three-Layered Structure Shared Between Lewy Bodies and Lewy Neurites—Three-Dimensional Reconstruction of Triple-Labeled Sections. Brain Pathol. 2008, 18, 415–422. [Google Scholar] [CrossRef] [PubMed]
- Miki, Y.; Mori, F.; Tanji, K.; Wakabayashi, K. Athology of neuro-glial α-synucleinopathy (Lewy body disease and multiple system atrophy). Hirosaki Med. J. 2010, 61, 80–88. [Google Scholar]
- Tu, P.; Galvin, J.; Baba, M.; Giasson, B.; Tomita, T.; Leight, S.; Nakajo, S.; Iwatsubo, T.; Trojanowski, J.; Lee, V. Glial cytoplasmic inclusions in white matter oligodendrocytes of multiple system atrophy brains contain insoluble α-synuclein. Ann. Neurol. 1998, 44, 415–422. [Google Scholar] [CrossRef] [PubMed]
- Grazia Spillantini, M.; Anthony Crowther, R.; Jakes, R.; Cairns, N.; Lantos, P.; Goedert, M. Filamentous α-synuclein inclusions link multiple system atrophy with Parkinson’s disease and dementia with Lewy bodies. Neurosci. Lett. 1998, 251, 205–208. [Google Scholar] [CrossRef]
- Ahmed, Z.; Asi, Y.; Sailer, A.; Lees, A.; Houlden, H.; Revesz, T.; Holton, J. The neuropathology, pathophysiology and genetics of multiple system atrophy. Neuropathol. Appl. Neurobiol. 2012, 38, 4–24. [Google Scholar] [CrossRef] [PubMed]
- Asi, Y.; Simpson, J.; Heath, P.; Wharton, S.; Lees, A.; Revesz, T.; Houlden, H.; Holton, J. α-Synuclein mRNA expression in oligodendrocytes in MSA. Glia 2014, 62, 964–970. [Google Scholar] [CrossRef] [PubMed]
- Djelloul, M.; Holmqvist, S.; Boza-Serrano, A.; Azevedo, C.; Yeung, M.; Goldwurm, S.; Frisén, J.; Deierborg, T.; Roybon, L. α-Synuclein Expression in the Oligodendrocyte Lineage: An In Vitro and In Vivo Study Using Rodent and Human Models. Stem Cell Rep. 2015, 5, 174–184. [Google Scholar] [CrossRef] [PubMed]
- Lesage, S.; Anheim, M.; Letournel, F.; Bousset, L.; Honoré, A.; Rozas, N.; Pieri, L.; Madiona, K.; Dürr, A.; Melki, R.; et al. G51D α-synuclein mutation causes a novel Parkinsonian-pyramidal syndrome. Ann. Neurol. 2013, 73, 459–471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Polymeropoulos, M. Mutation in the α-Synuclein Gene Identified in Families with Parkinson’s Disease. Science 1997, 276, 2045–2047. [Google Scholar] [CrossRef] [PubMed]
- Chartier-Harlin, M.; Kachergus, J.; Roumier, C.; Mouroux, V.; Douay, X.; Lincoln, S.; Levecque, C.; Larvor, L.; Andrieux, J.; Hulihan, M.; et al. α-Synuclein locus duplication as a cause of familial Parkinson’s disease. Lancet 2004, 364, 1167–1169. [Google Scholar] [CrossRef]
- Simón-Sánchez, J.; Schulte, C.; Bras, J.; Sharma, M.; Gibbs, J.; Berg, D.; Paisan-Ruiz, C.; Lichtner, P.; Scholz, S.; Hernandez, D.; et al. Genome-wide association study reveals genetic risk underlying Parkinson’s disease. Nat. Genet. 2009, 41, 1308–1312. [Google Scholar] [CrossRef] [PubMed]
- Nalls, M.; Pankratz, N.; Lill, C.; Do, C.; Hernandez, D.; Saad, M.; DeStefano, A.; Kara, E.; Bras, J.; Sharma, M.; et al. Large-scale meta-analysis of genome-wide association data identifies six new risk loci for Parkinson’s disease. Nat. Genet. 2014, 46, 989–993. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Satake, W.; Nakabayashi, Y.; Mizuta, I.; Hirota, Y.; Ito, C.; Kubo, M.; Kawaguchi, T.; Tsunoda, T.; Watanabe, M.; Takeda, A.; et al. Genome-wide association study identifies common variants at four loci as genetic risk factors for Parkinson’s disease. Nat. Genet. 2009, 41, 1303–1307. [Google Scholar] [CrossRef] [PubMed]
- Rcom-H’cheo-Gauthier, A.; Osborne, S.; Meedeniya, A.; Pountney, D. Calcium: α-Synuclein Interactions in α-Synucleinopathies. Front. Neurosci. 2016, 10, 570. [Google Scholar] [CrossRef] [PubMed]
- Fujiwara, H.; Hasegawa, M.; Dohmae, N.; Kawashima, A.; Masliah, E.; Goldberg, M.; Shen, J.; Takio, K.; Iwatsubo, T. α-Synuclein is phosphorylated in synucleinopathy lesions. Nat. Cell Biol. 2002, 4, 160–164. [Google Scholar] [PubMed]
- Reynolds, A.; Glanzer, J.; Kadiu, I.; Ricardo-Dukelow, M.; Chaudhuri, A.; Ciborowski, P.; Cerny, R.; Gelman, B.; Thomas, M.; Mosley, R.; et al. Nitrated α-synuclein-activated microglial profiling for Parkinson’s disease. J. Neurochem. 2007, 104, 1504–1525. [Google Scholar] [CrossRef] [PubMed]
- Cookson, M. α-Synuclein and neuronal cell death. Mol. Neurodegener. 2009, 4, 9. [Google Scholar] [CrossRef] [PubMed]
- Anderson, J.; Walker, D.; Goldstein, J.; de Laat, R.; Banducci, K.; Caccavello, R.; Barbour, R.; Huang, J.; Kling, K.; Lee, M.; et al. Phosphorylation of Ser-129 Is the Dominant Pathological Modification of α-Synuclein in Familial and Sporadic Lewy Body Disease. J. Biol. Chem. 2006, 281, 29739–29752. [Google Scholar] [CrossRef] [PubMed]
- Ueda, K.; Fukushima, H.; Masliah, E.; Xia, Y.; Iwai, A.; Yoshimoto, M.; Otero, D.; Kondo, J.; Ihara, Y.; Saitoh, T. Molecular cloning of cDNA encoding an unrecognized component of amyloid in Alzheimer disease. Proc. Natl. Acad. Sci. USA 1993, 90, 11282–11286. [Google Scholar] [CrossRef] [PubMed]
- Bousset, L.; Pieri, L.; Ruiz-Arlandis, G.; Gath, J.; Jensen, P.; Habenstein, B.; Madiona, K.; Olieric, V.; Böckmann, A.; Meier, B.; et al. Structural and functional characterization of two α-synuclein strains. Nat. Commun. 2013, 4, 2575. [Google Scholar] [CrossRef] [PubMed]
- Gai, W.; Pountney, D.; Power, J.; Li, Q.; Culvenor, J.; McLean, C.; Jensen, P.; Blumbergs, P. α-Synuclein fibrils constitute the central core of oligodendroglial inclusion filaments in multiple system atrophy. Exp. Neurol. 2003, 181, 68–78. [Google Scholar] [CrossRef]
- Pountney, D.; Lowe, R.; Quilty, M.; Vickers, J.; Voelcker, N.; Gai, W. Annular α-synuclein species from purified multiple system atrophy inclusions. J. Neurochem. 2004, 90, 502–512. [Google Scholar] [CrossRef] [PubMed]
- Paviour, D.; Price, S.; Lees, A.; Fox, N. MRI derived brain atrophy in PSP and MSA-P. J. Neurol. 2007, 254, 478–481. [Google Scholar] [CrossRef] [PubMed]
- Brettschneider, J.; Irwin, D.; Boluda, S.; Byrne, M.; Fang, L.; Lee, E.; Robinson, J.; Suh, E.; van Deerlin, V.; Toledo, J.; et al. Progression of α-synuclein pathology in multiple system atrophy of the cerebellar type. Neuropathol. Appl. Neurobiol. 2016. [Google Scholar] [CrossRef] [PubMed]
- McKeith, I.; Dickson, D.; Lowe, J.; Emre, M.; O’Brien, J.; Feldman, H.; Cummings, J.; Duda, J.; Lippa, C.; Perry, E.; et al. Diagnosis and management of dementia with Lewy bodies: Third report of the DLB consortium. Neurology 2005, 65, 1863–1872. [Google Scholar] [CrossRef] [PubMed]
- Peelaerts, W.; Bousset, L.; van der Perren, A.; Moskalyuk, A.; Pulizzi, R.; Giugliano, M.; van den Haute, C.; Melki, R.; Baekelandt, V. α-Synuclein strains cause distinct synucleinopathies after local and systemic administration. Nature 2015, 522, 340–344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luk, K.; Kehm, V.; Carroll, J.; Zhang, B.; O’Brien, P.; Trojanowski, J.; Lee, V. Pathological α-Synuclein Transmission Initiates Parkinson-like Neurodegeneration in Nontransgenic Mice. Science 2012, 338, 949–953. [Google Scholar] [CrossRef] [PubMed]
- Osterberg, V.; Spinelli, K.; Weston, L.; Luk, K.; Woltjer, R.; Unni, V. Progressive Aggregation of α-Synuclein and Selective Degeneration of Lewy Inclusion-Bearing Neurons in a Mouse Model of Parkinsonism. Cell Rep. 2015, 10, 1252–1260. [Google Scholar] [CrossRef] [PubMed]
- Paumier, K.; Luk, K.; Manfredsson, F.; Kanaan, N.; Lipton, J.; Collier, T.; Steece-Collier, K.; Kemp, C.; Celano, S.; Schulz, E.; et al. Intrastriatal injection of pre-formed mouse α-synuclein fibrils into rats triggers α-synuclein pathology and bilateral nigrostriatal degeneration. Neurobiol. Dis. 2015, 82, 185–199. [Google Scholar] [CrossRef] [PubMed]
- Sacino, A.; Brooks, M.; McGarvey, N.; McKinney, A.; Thomas, M.; Levites, Y.; Ran, Y.; Golde, T.; Giasson, B. Induction of CNS α-synuclein pathology by fibrillar and non-amyloidogenic recombinant α-synuclein. Acta Neuropathol. Commun. 2013, 1, 38. [Google Scholar] [CrossRef] [PubMed]
- Rey, N.L.; Steiner, J.A.; Maroof, N.; Luk, K.C.; Madaj, Z.; Trojanowski, J.Q.; Lee, V.M.; Brundin, P. Widespread transneuronal propagation of α-synucleinopathy triggered in olfactory bulb mimics prodromal Parkinson’s disease. J. Exp. Med. 2016, 213, 1759–1778. [Google Scholar] [CrossRef] [PubMed]
- Kovacs, G.; Budka, H. Prion Diseases: From Protein to Cell Pathology. Am. J. Pathol. 2008, 172, 555–565. [Google Scholar] [CrossRef] [PubMed]
- Goedert, M. Alzheimer’s and Parkinson’s diseases: The prion concept in relation to assembled Aβ, tau, and α-synuclein. Science 2015, 349. [Google Scholar] [CrossRef] [PubMed]
- Iwai, A. Properties of NACP/α-synuclein and its role in Alzheimer’s disease. Biochim. Biophys. Acta Mol. Basis Dis. 2000, 1502, 95–109. [Google Scholar] [CrossRef]
- Rodriguez, J.; Ivanova, M.; Sawaya, M.; Cascio, D.; Reyes, F.; Shi, D.; Sangwan, S.; Guenther, E.; Johnson, L.; Zhang, M.; et al. Structure of the toxic core of α-synuclein from invisible crystals. Nature 2015, 525, 486–490. [Google Scholar] [CrossRef] [PubMed]
- Radford, R.; Rcom-H’cheo-Gauthier, A.; Wong, M.B.; Eaton, E.D.; Quilty, M.; Blizzard, C.; Norazit, A.; Meedeniya, A.; Vickers, J.C.; Gai, W.P.; et al. The degree of astrocyte activation in multiple system atrophy is inversely proportional to the distance to α-synuclein inclusions. Mol. Cell Neurosci. 2015, 65, 68–81. [Google Scholar] [CrossRef] [PubMed]
- Recasens, A.; Dehay, B.; Bové, J.; Carballo-Carbajal, I.; Dovero, S.; Pérez-Villalba, A.; Fernagut, P.; Blesa, J.; Parent, A.; Perier, C.; et al. Lewy body extracts from Parkinson disease brains trigger α-synuclein pathology and neurodegeneration in mice and monkeys. Ann. Neurol. 2014, 75, 351–362. [Google Scholar] [CrossRef] [PubMed]
- Prusiner, S.; Woerman, A.; Mordes, D.; Watts, J.; Rampersaud, R.; Berry, D.; Patel, S.; Oehler, A.; Lowe, J.; Kravitz, S.; et al. Evidence for α-synuclein prions causing multiple system atrophy in humans with parkinsonism. Proc. Natl. Acad. Sci. USA 2015, 112, E5308–E5317. [Google Scholar] [CrossRef] [PubMed]
- Cykowski, M.; Coon, E.; Powell, S.; Jenkins, S.; Benarroch, E.; Low, P.; Schmeichel, A.; Parisi, J. Expanding the spectrum of neuronal pathology in multiple system atrophy. Brain 2015, 138, 2293–2309. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Englund, E.; Holton, J.; Soulet, D.; Hagell, P.; Lees, A.; Lashley, T.; Quinn, N.; Rehncrona, S.; Björklund, A.; et al. Lewy bodies in grafted neurons in subjects with Parkinson’s disease suggest host-to-graft disease propagation. Nat. Med. 2008, 14, 501–503. [Google Scholar] [CrossRef] [PubMed]
- Kordower, J.; Chu, Y.; Hauser, R.; Freeman, T.; Olanow, C. Lewy body–like pathology in long-term embryonic nigral transplants in Parkinson’s disease. Nat. Med. 2008, 14, 504–506. [Google Scholar] [CrossRef] [PubMed]
- Ahn, T.; Langston, J.; Aachi, V.; Dickson, D. Relationship of neighboring tissue and gliosis to α-synuclein pathology in a fetal transplant for Parkinson’s disease. Am. J. Neurodegener. Dis. 2012, 1, 49–59. [Google Scholar] [PubMed]
- El-Agnaf, O.; Salem, S.; Paleologou, K.; Curran, M.; Gibson, M.; Court, J.; Schlossmacher, M.; Allsop, D. Detection of oligomeric forms of α-synuclein protein in human plasma as a potential biomarker for Parkinson’s disease. FASEB J. 2006, 20, 419–425. [Google Scholar] [CrossRef] [PubMed]
- Danzer, K.; Ruf, W.; Putcha, P.; Joyner, D.; Hashimoto, T.; Glabe, C.; Hyman, B.; McLean, P. Heat-shock protein 70 modulates toxic extracellular α-synuclein oligomers and rescues trans-synaptic toxicity. FASEB J. 2011, 25, 326–336. [Google Scholar] [CrossRef] [PubMed]
- Fontaine, S.; Zheng, D.; Sabbagh, J.; Martin, M.; Chaput, D.; Darling, A.; Trotter, J.; Stothert, A.; Nordhues, B.; Lussier, A.; et al. DnaJ/Hsc70 chaperone complexes control the extracellular release of neurodegenerative-associated proteins. EMBO J. 2016, 35, 1537–1549. [Google Scholar] [CrossRef] [PubMed]
- Valadi, H.; Ekström, K.; Bossios, A.; Sjöstrand, M.; Lee, J.; Lötvall, J. Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat. Cell Biol. 2007, 9, 654–659. [Google Scholar] [CrossRef] [PubMed]
- Gibbings, D.; Ciaudo, C.; Erhardt, M.; Voinnet, O. Multivesicular bodies associate with components of miRNA effector complexes and modulate miRNA activity. Nat. Cell Biol. 2009, 11, 1143–1149. [Google Scholar] [CrossRef] [PubMed]
- Lancaster, G.; Febbraio, M. Exosome-dependent Trafficking of HSP70: A novel secretory pathway for cellular stress proteins. J. Biol. Chem. 2005, 280, 23349–23355. [Google Scholar] [CrossRef] [PubMed]
- Beaudoin, A.; Grondin, G. Shedding of vesicular material from the cell surface of eukaryotic cells: Different cellular phenomena. Biochim. Biophys. Acta Rev. Biomembr. 1991, 1071, 203–219. [Google Scholar] [CrossRef]
- Denzer, K.; Kleijmeer, M.; Heijnen, H.; Stoorvogel, W.; Geuze, H. Exosome: From internal vesicle of the multivesicular body to intercellular signaling device. J. Cell Sci. 2000, 113, 3365–3374. [Google Scholar] [PubMed]
- Fitzner, D.; Schnaars, M.; van Rossum, D.; Krishnamoorthy, G.; Dibaj, P.; Bakhti, M.; Regen, T.; Hanisch, U.; Simons, M. Selective transfer of exosomes from oligodendrocytes to microglia by macropinocytosis. J. Cell Sci. 2011, 124, 447–458. [Google Scholar] [CrossRef] [PubMed]
- Frühbeis, C.; Fröhlich, D.; Kuo, W.; Krämer-Albers, E. Extracellular vesicles as mediators of neuron-glia communication. Front. Cell. Neurosci. 2013, 7, 182. [Google Scholar] [CrossRef] [PubMed]
- Frühbeis, C.; Fröhlich, D.; Kuo, W.; Amphornrat, J.; Thilemann, S.; Saab, A.; Kirchhoff, F.; Möbius, W.; Goebbels, S.; Nave, K.; et al. Neurotransmitter-Triggered Transfer of Exosomes Mediates Oligodendrocyte–Neuron Communication. PLoS Biol. 2013, 11, e1001604. [Google Scholar] [CrossRef] [PubMed]
- Davis, C.; Kim, K.; Bushong, E.; Mills, E.; Boassa, D.; Shih, T.; Kinebuchi, M.; Phan, S.; Zhou, Y.; Bihlmeyer, N.; et al. Transcellular degradation of axonal mitochondria. Proc. Natl. Acad. Sci. USA 2014, 111, 9633–9638. [Google Scholar] [CrossRef] [PubMed]
- Hayakawa, K.; Esposito, E.; Wang, X.; Terasaki, Y.; Liu, Y.; Xing, C.; Ji, X.; Lo, E. Transfer of mitochondria from astrocytes to neurons after stroke. Nature 2016, 535, 551–555. [Google Scholar] [CrossRef] [PubMed]
- Von Bartheld, C.; Altick, A. Multivesicular bodies in neurons: Distribution, protein content, and trafficking functions. Prog. Neurobiol. 2011, 93, 313–340. [Google Scholar] [CrossRef] [PubMed]
- Danzer, K.; Kranich, L.; Ruf, W.; Cagsal-Getkin, O.; Winslow, A.; Zhu, L.; Vanderburg, C.; McLean, P. Exosomal cell-to-cell transmission of α synuclein oligomers. Mol. Neurodegener. 2012, 7, 42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alvarez-Erviti, L.; Seow, Y.; Schapira, A.; Gardiner, C.; Sargent, I.; Wood, M.; Cooper, J. Lysosomal dysfunction increases exosome-mediated α-synuclein release and transmission. Neurobiol. Dis. 2011, 42, 360–367. [Google Scholar] [CrossRef] [PubMed]
- Emmanouilidou, E.; Melachroinou, K.; Roumeliotis, T.; Garbis, S.; Ntzouni, M.; Margaritis, L.; Stefanis, L.; Vekrellis, K. Cell-Produced-Synuclein Is Secreted in a Calcium-Dependent Manner by Exosomes and Impacts Neuronal Survival. J. Neurosci. 2010, 30, 6838–6851. [Google Scholar] [CrossRef] [PubMed]
- Frohlich, D.; Kuo, W.; Fruhbeis, C.; Sun, J.; Zehendner, C.; Luhmann, H.; Pinto, S.; Toedling, J.; Trotter, J.; Kramer-Albers, E. Multifaceted effects of oligodendroglial exosomes on neurons: Impact on neuronal firing rate, signal transduction and gene regulation. Philos. Trans. R. Soc. B Biol. Sci. 2014, 369. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, K.; Mori, F.; Kon, T.; Tanji, K.; Miki, Y.; Tomiyama, M.; Kurotaki, H.; Toyoshima, Y.; Kakita, A.; Takahashi, H.; et al. Accumulation of phosphorylated α-synuclein in subpial and periventricular astrocytes in multiple system atrophy of long duration. Neuropathology 2016, 36, 157–167. [Google Scholar] [CrossRef] [PubMed]
- Braak, H.; Sastre, M.; Del Tredici, K. Development of α-synuclein immunoreactive astrocytes in the forebrain parallels stages of intraneuronal pathology in sporadic Parkinson’s disease. Acta Neuropathol. 2007, 114, 231–241. [Google Scholar] [CrossRef] [PubMed]
- Piao, Y.; Mori, F.; Hayashi, S.; Tanji, K.; Yoshimoto, M.; Kakita, A.; Wakabayashi, K.; Takahashi, H. α-Synuclein pathology affecting Bergmann glia of the cerebellum in patients with α-synucleinopathies. Acta Neuropathol. 2003, 105, 403–409. [Google Scholar] [PubMed]
- Reyes, J.; Rey, N.; Bousset, L.; Melki, R.; Brundin, P.; Angot, E. α-Synuclein transfers from neurons to oligodendrocytes. Glia 2013, 62, 387–398. [Google Scholar] [CrossRef] [PubMed]
- Desplats, P.; Lee, H.; Bae, E.; Patrick, C.; Rockenstein, E.; Crews, L.; Spencer, B.; Masliah, E.; Lee, S. Inclusion formation and neuronal cell death through neuron-to-neuron transmission of α-synuclein. Proc. Natl. Acad. Sci. USA 2009, 106, 13010–13015. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Zhou, Y.; Wang, Y.; Fong, H.; Murray, T.; Zhang, J. Identification of Proteins Involved in Microglial Endocytosis of α-Synuclein. J. Proteome Res. 2007, 6, 3614–3627. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Suk, J.; Bae, E.; Lee, J.; Paik, S.; Lee, S. Assembly-dependent endocytosis and clearance of extracellular α-synuclein. Int. J. Biochem. Cell Biol. 2008, 40, 1835–1849. [Google Scholar] [CrossRef] [PubMed]
- Konno, M.; Hasegawa, T.; Baba, T.; Miura, E.; Sugeno, N.; Kikuchi, A.; Fiesel, F.; Sasaki, T.; Aoki, M.; Itoyama, Y.; et al. Suppression of dynamin GTPase decreases α-synuclein uptake by neuronal and oligodendroglial cells: A potent therapeutic target for synucleinopathy. Mol. Neurodegener. 2012, 7, 38. [Google Scholar] [CrossRef] [PubMed]
- Mao, X.; Ou, M.; Karuppagounder, S.; Kam, T.; Yin, X.; Xiong, Y.; Ge, P.; Umanah, G.; Brahmachari, S.; Shin, J.; et al. Pathological α-synuclein transmission initiated by binding lymphocyte-activation gene 3. Science 2016, 353. [Google Scholar] [CrossRef] [PubMed]
- Schimmöller, F.; Dı́az, E.; Mühlbauer, B.; Pfeffer, S. Characterization of a 76kDa endosomal, multispanning membrane protein that is highly conserved throughout evolution. Gene 1998, 216, 311–318. [Google Scholar] [CrossRef]
- Wadman, M. Rogue protein’s partners offer hope in Parkinson’s disease. Science 2016, 354, 956. [Google Scholar] [CrossRef] [PubMed]
- Usenovic, M.; Tresse, E.; Mazzulli, J.; Taylor, J.; Krainc, D. Deficiency of ATP13A2 Leads to Lysosomal Dysfunction, α-Synuclein Accumulation, and Neurotoxicity. J. Neurosci. 2012, 32, 4240–4246. [Google Scholar] [CrossRef] [PubMed]
- Siebert, M.; Sidransky, E.; Westbroek, W. Glucocerebrosidase is shaking up the synucleinopathies. Brain 2014, 137, 1304–1322. [Google Scholar] [CrossRef] [PubMed]
- Holmes, B.; DeVos, S.; Kfoury, N.; Li, M.; Jacks, R.; Yanamandra, K.; Ouidja, M.; Brodsky, F.; Marasa, J.; Bagchi, D.; et al. Heparan sulfate proteoglycans mediate internalization and propagation of specific proteopathic seeds. Proc. Natl. Acad. Sci. USA 2013, 110, E3138–E3147. [Google Scholar] [CrossRef] [PubMed]
- Abounit, S.; Bousset, L.; Loria, F.; Zhu, S.; de Chaumont, F.; Pieri, L.; Olivo-Marin, J.; Melki, R.; Zurzolo, C. Tunneling nanotubes spread fibrillar α-synuclein by intercellular trafficking of lysosomes. EMBO J. 2016, 35, 2120–2138. [Google Scholar] [CrossRef] [PubMed]
- Agnati, L.; Fuxe, K. Extracellular-vesicle type of volume transmission and tunnelling-nanotube type of wiring transmission add a new dimension to brain neuro-glial networks. Philos. Trans. R. Soc. B Biol. Sci. 2014, 369. [Google Scholar] [CrossRef] [PubMed]
- Gousset, K.; Schiff, E.; Langevin, C.; Marijanovic, Z.; Caputo, A.; Browman, D.; Chenouard, N.; de Chaumont, F.; Martino, A.; Enninga, J.; et al. Prions hijack tunnelling nanotubes for intercellular spread. Nat. Cell Biol. 2009, 11, 328–336. [Google Scholar] [CrossRef] [PubMed]
- Eugenin, E.; Gaskill, P.; Berman, J. Tunneling nanotubes (TNT) are induced by HIV-infection of macrophages: A potential mechanism for intercellular HIV trafficking. Cell. Immunol. 2009, 254, 142–148. [Google Scholar] [CrossRef] [PubMed]
- Onfelt, B.; Nedvetzki, S.; Yanagi, K.; Davis, D. Cutting Edge: Membrane Nanotubes Connect Immune Cells. J. Immunol. 2004, 173, 1511–1513. [Google Scholar] [CrossRef] [PubMed]
- Onfelt, B.; Nedvetzki, S.; Benninger, R.; Purbhoo, M.; Sowinski, S.; Hume, A.; Seabra, M.; Neil, M.; French, P.; Davis, D. Structurally Distinct Membrane Nanotubes between Human Macrophages Support Long-Distance Vesicular Traffic or Surfing of Bacteria. J. Immunol. 2006, 177, 8476–8483. [Google Scholar] [CrossRef] [PubMed]
- Zhu, D.; Tan, K.; Zhang, X.; Sun, A.; Sun, G.; Lee, J. Hydrogen peroxide alters membrane and cytoskeleton properties and increases intercellular connections in astrocytes. J. Cell Sci. 2005, 118, 3695–3703. [Google Scholar] [CrossRef] [PubMed]
- Rustom, A.; Saffrich, R.; Markovic, I.; Walther, P.; Gerdes, H. Nanotubular Highways for Intercellular Organelle Transport. Science 2004, 303, 1007–1010. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Wang, Y.; Zhang, J.; Tu, J.; Wang, X.; Su, X.; Wang, L.; Zhang, Y. Tunneling-nanotube direction determination in neurons and astrocytes. Cell Death Dis. 2012, 3, e438. [Google Scholar] [CrossRef] [PubMed]
- McCoy-Simandle, K.; Hanna, S.; Cox, D. Exosomes and nanotubes: Control of immune cell communication. Int. J. Biochem. Cell Biol. 2016, 71, 44–54. [Google Scholar] [CrossRef] [PubMed]
- Kettenmann, H.; Hanisch, U.; Noda, M.; Verkhratsky, A. Physiology of Microglia. Physiol. Rev. 2011, 91, 461–553. [Google Scholar] [CrossRef] [PubMed]
- Jonas, R.; Yuan, T.; Liang, Y.; Jonas, J.; Tay, D.; Ellis-Behnke, R. The Spider Effect: Morphological and Orienting Classification of Microglia in Response to Stimuli in Vivo. PLoS ONE 2012, 7, e30763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perry, V.; Cunningham, C.; Holmes, C. Systemic infections and inflammation affect chronic neurodegeneration. Nat. Rev. Immunol. 2007, 7, 161–167. [Google Scholar] [CrossRef] [PubMed]
- Neumann, H.; Kotter, M.; Franklin, R. Debris clearance by microglia: An essential link between degeneration and regeneration. Brain 2008, 132, 288–295. [Google Scholar] [CrossRef] [PubMed]
- Fellner, L.; Stefanova, N. The Role of Glia in α-Synucleinopathies. Mol. Neurobiol. 2012, 47, 575–586. [Google Scholar] [CrossRef] [PubMed]
- Politis, M.; Su, P.; Piccini, P. Imaging of microglia in patients with neurodegenerative disorders. Front. Pharmacol. 2012, 3. [Google Scholar] [CrossRef] [PubMed]
- Kaufman, E.; Hall, S.; Surova, Y.; Widner, H.; Hansson, O.; Lindqvist, D. Proinflammatory Cytokines Are Elevated in Serum of Patients with Multiple System Atrophy. PLoS ONE 2013, 8, e62354. [Google Scholar] [CrossRef] [PubMed]
- Koga, S.; Aoki, N.; Uitti, R.; van Gerpen, J.; Cheshire, W.; Josephs, K.; Wszolek, Z.; Langston, J.; Dickson, D. When DLB, PD, and PSP masquerade as MSA. Neurology 2015, 85, 404–412. [Google Scholar] [CrossRef] [PubMed]
- Nagatsu, T.; Sawada, M. Inflammatory Process in Parkinsons Disease: Role for Cytokines. Curr. Pharm. Des. 2005, 11, 999–1016. [Google Scholar] [CrossRef] [PubMed]
- Tufekci, K.; Meuwissen, R.; Genc, S.; Genc, K. Inflammation in Parkinson’s Disease. Adv. Protein Chem. Struct. Biol. 2012, 88, 69–132. [Google Scholar] [PubMed]
- Higuchi, M.; Tashiro, M.; Arai, H.; Okamura, N.; Hara, S.; Higuchi, S.; Itoh, M.; Shin, R.; Trojanowski, J.; Sasaki, H. Glucose Hypometabolism and Neuropathological Correlates in Brains of Dementia with Lewy Bodies. Exp. Neurol. 2000, 162, 247–256. [Google Scholar] [CrossRef] [PubMed]
- Chu, Y.; Dodiya, H.; Aebischer, P.; Olanow, C.; Kordower, J. Alterations in lysosomal and proteasomal markers in Parkinson’s disease: Relationship to α-synuclein inclusions. Neurobiol. Dis. 2009, 35, 385–398. [Google Scholar] [CrossRef] [PubMed]
- Masliah, E.; Rockenstein, E.; Adame, A.; Alford, M.; Crews, L.; Hashimoto, M.; Seubert, P.; Lee, M.; Goldstein, J.; Chilicote, T.; et al. Effects of α-synuclein immunization in a mouse model of Parkinson’s disease. Neuron 2005, 46, 857–868. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Kim, K.; Lee, S.; Ryu, J.; Chung, K.; Choo, Y.; Jou, I.; Kim, J.; Park, S. On the mechanism of internalization of α-synuclein into microglia: Roles of ganglioside GM1 and lipid raft. J. Neurochem. 2009, 110, 400–411. [Google Scholar] [CrossRef] [PubMed]
- Fortin, D.; Troyer, M.; Nakamura, K.; Kubo, S.; Anthony, M.; Edwards, R. Lipid Rafts Mediate the Synaptic Localization of α-Synuclein. J. Neurosci. 2004, 24, 6715–6723. [Google Scholar] [CrossRef] [PubMed]
- Bar-On, P.; Crews, L.; Koob, A.; Mizuno, H.; Adame, A.; Spencer, B.; Masliah, E. Statins reduce neuronal α-synuclein aggregation in in vitro models of Parkinson’s disease. J. Neurochem. 2008, 105, 1656–1667. [Google Scholar] [CrossRef] [PubMed]
- Kubo, S.; Nemani, V.; Chalkley, R.; Anthony, M.; Hattori, N.; Mizuno, Y.; Edwards, R.; Fortin, D. A Combinatorial Code for the Interaction of α-Synuclein with Membranes. J. Biol. Chem. 2005, 280, 31664–31672. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Chu, C.; Stewart, T.; Ginghina, C.; Wang, Y.; Nie, H.; Guo, M.; Wilson, B.; Hong, J.; Zhang, J. α-Synuclein, a chemoattractant, directs microglial migration via H2O2-dependent Lyn phosphorylation. Proc. Natl. Acad. Sci. USA 2015, 112, E1926–E1935. [Google Scholar] [CrossRef] [PubMed]
- Su, X.; Maguire-Zeiss, K.; Giuliano, R.; Prifti, L.; Venkatesh, K.; Federoff, H. Synuclein activates microglia in a model of Parkinson’s disease. Neurobiol. Aging 2008, 29, 1690–1701. [Google Scholar] [CrossRef] [PubMed]
- Stefanova, N.; Fellner, L.; Reindl, M.; Masliah, E.; Poewe, W.; Wenning, G. Toll-Like Receptor 4 Promotes α-Synuclein Clearance and Survival of Nigral Dopaminergic Neurons. Am. J. Pathol. 2011, 179, 954–963. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.; Ho, D.; Suk, J.; You, S.; Michael, S.; Kang, J.; Joong Lee, S.; Masliah, E.; Hwang, D.; Lee, H.; et al. Neuron-released oligomeric α-synuclein is an endogenous agonist of TLR2 for paracrine activation of microglia. Nat. Commun. 2013, 4, 1562. [Google Scholar] [CrossRef] [PubMed]
- Roodveldt, C.; Labrador-Garrido, A.; Gonzalez-Rey, E.; Lachaud, C.; Guilliams, T.; Fernandez-Montesinos, R.; Benitez-Rondan, A.; Robledo, G.; Hmadcha, A.; Delgado, M.; et al. Preconditioning of Microglia by α-Synuclein Strongly Affects the Response Induced by Toll-like Receptor (TLR) Stimulation. PLoS ONE 2013, 8, e79160. [Google Scholar] [CrossRef] [PubMed]
- Fellner, L.; Irschick, R.; Schanda, K.; Reindl, M.; Klimaschewski, L.; Poewe, W.; Wenning, G.; Stefanova, N. Toll-like receptor 4 is required for α-synuclein dependent activation of microglia and astroglia. Glia 2013, 61, 349–360. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Le, W. Differential Roles of M1 and M2 Microglia in Neurodegenerative Diseases. Mol. Neurobiol. 2015, 53, 1181–1194. [Google Scholar] [CrossRef] [PubMed]
- Asai, H.; Ikezu, S.; Tsunoda, S.; Medalla, M.; Luebke, J.; Haydar, T.; Wolozin, B.; Butovsky, O.; Kügler, S.; Ikezu, T. Depletion of microglia and inhibition of exosome synthesis halt tau propagation. Nat. Neurosci. 2015, 18, 1584–1593. [Google Scholar] [CrossRef] [PubMed]
- Vieira, B.D.; Radford, R.A.; Chung, R.S.; Guillemin, G.J.; Pountney, D.L. Neuroinflammation in Multiple System Atrophy: Response to and Cause of α-Synuclein Aggregation. Front. Cell Neurosci. 2015, 9, 437. [Google Scholar] [CrossRef] [PubMed]
- Jessen, N.; Munk, A.; Lundgaard, I.; Nedergaard, M. The Glymphatic System: A Beginner’s Guide. Neurochem. Res. 2015, 40, 2583–2599. [Google Scholar] [CrossRef] [PubMed]
- Iliff, J.; Wang, M.; Liao, Y.; Plogg, B.; Peng, W.; Gundersen, G.; Benveniste, H.; Vates, G.; Deane, R.; Goldman, S.; et al. A Paravascular Pathway Facilitates CSF Flow Through the Brain Parenchyma and the Clearance of Interstitial Solutes, Including Amyloid β. Sci. Transl. Med. 2012, 4. [Google Scholar] [CrossRef] [PubMed]
- Iliff, J.; Chen, M.; Plog, B.; Zeppenfeld, D.; Soltero, M.; Yang, L.; Singh, I.; Deane, R.; Nedergaard, M. Impairment of Glymphatic Pathway Function Promotes Tau Pathology after Traumatic Brain Injury. J. Neurosci. 2014, 34, 16180–16193. [Google Scholar] [CrossRef] [PubMed]
- Hoshi, A.; Tsunoda, A.; Tada, M.; Nishizawa, M.; Ugawa, Y.; Kakita, A. Expression of aquaporin 1 and aquaporin 4 in the temporal neocortex of patients with Parkinson’s disease. Brain Pathol. 2016. [Google Scholar] [CrossRef] [PubMed]
- Gao, L.; Tang, H.; Nie, K.; Wang, L.; Zhao, J.; Gan, R.; Huang, J.; Zhu, R.; Feng, S.; Duan, Z.; et al. Cerebrospinal fluid α-synuclein as a biomarker for Parkinson’s disease diagnosis: A systematic review and meta-analysis. Int. J. Neurosci. 2014, 125, 645–654. [Google Scholar] [CrossRef] [PubMed]
- Lundgaard, I.; Lu, M.; Yang, E.; Peng, W.; Mestre, H.; Hitomi, E.; Deane, R.; Nedergaard, M. Glymphatic clearance controls state-dependent changes in brain lactate concentration. J. Cereb. Blood Flow Metab. 2016. [Google Scholar] [CrossRef] [PubMed]

© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Valdinocci, D.; Radford, R.A.W.; Siow, S.M.; Chung, R.S.; Pountney, D.L. Potential Modes of Intercellular α-Synuclein Transmission. Int. J. Mol. Sci. 2017, 18, 469. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms18020469
Valdinocci D, Radford RAW, Siow SM, Chung RS, Pountney DL. Potential Modes of Intercellular α-Synuclein Transmission. International Journal of Molecular Sciences. 2017; 18(2):469. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms18020469
Chicago/Turabian StyleValdinocci, Dario, Rowan A. W. Radford, Sue Maye Siow, Roger S. Chung, and Dean L. Pountney. 2017. "Potential Modes of Intercellular α-Synuclein Transmission" International Journal of Molecular Sciences 18, no. 2: 469. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms18020469