
High-Throughput Screening Methodology to Identify Alpha-Synuclein Aggregation Inhibitors

 and
and 
Abstract
:1. Introduction
2. Results
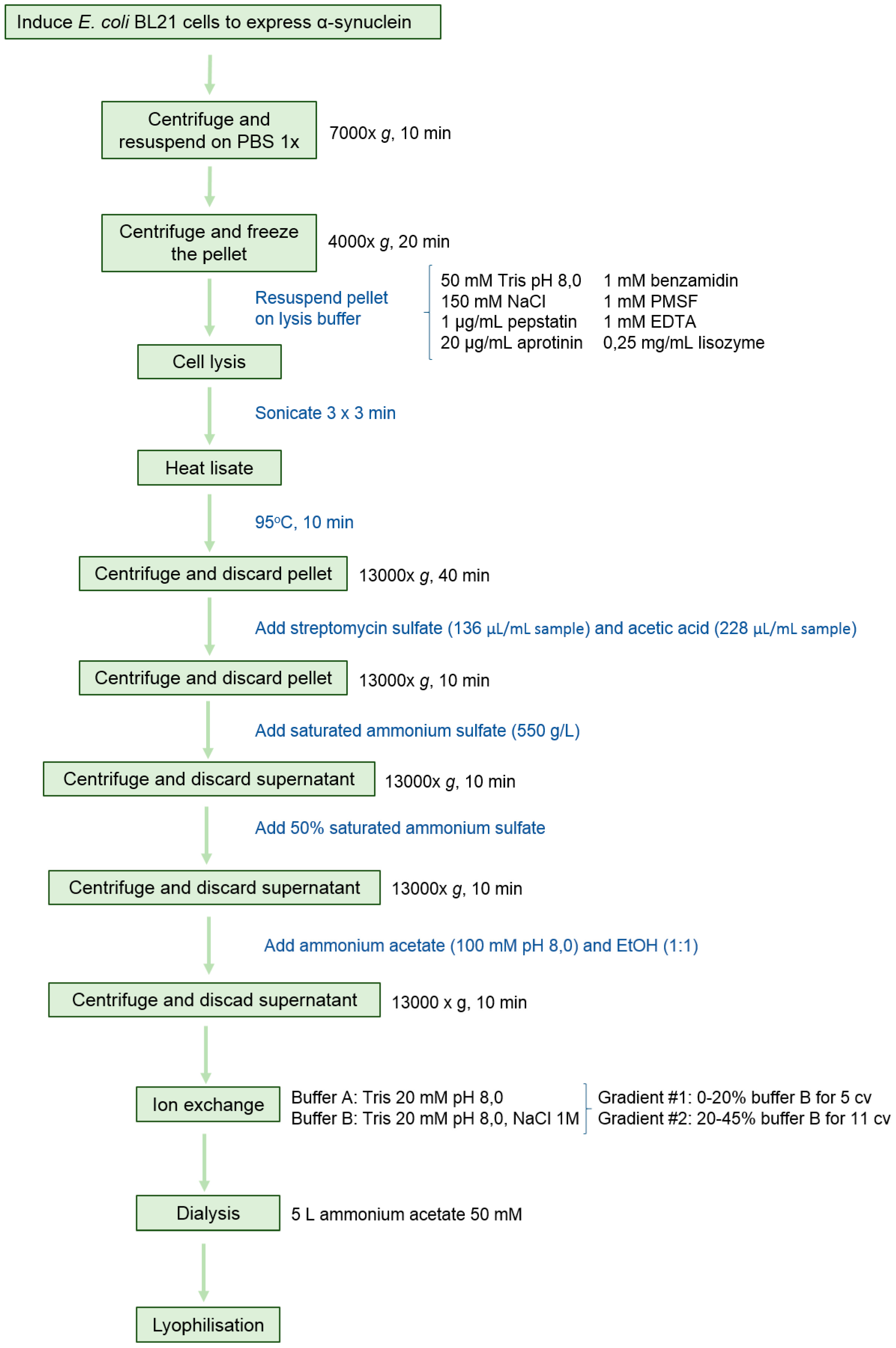
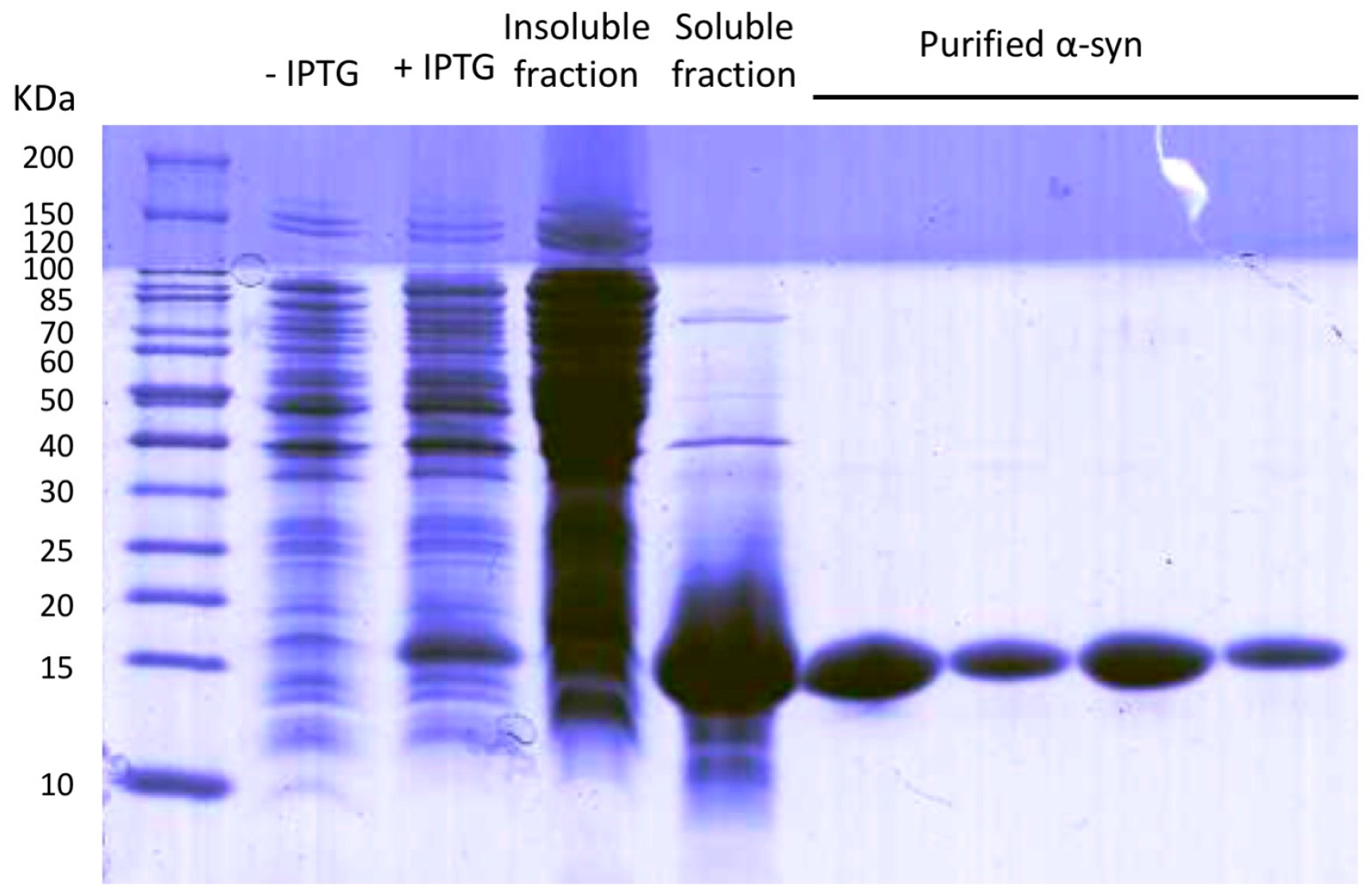
2.1. Protein Expression and Purification
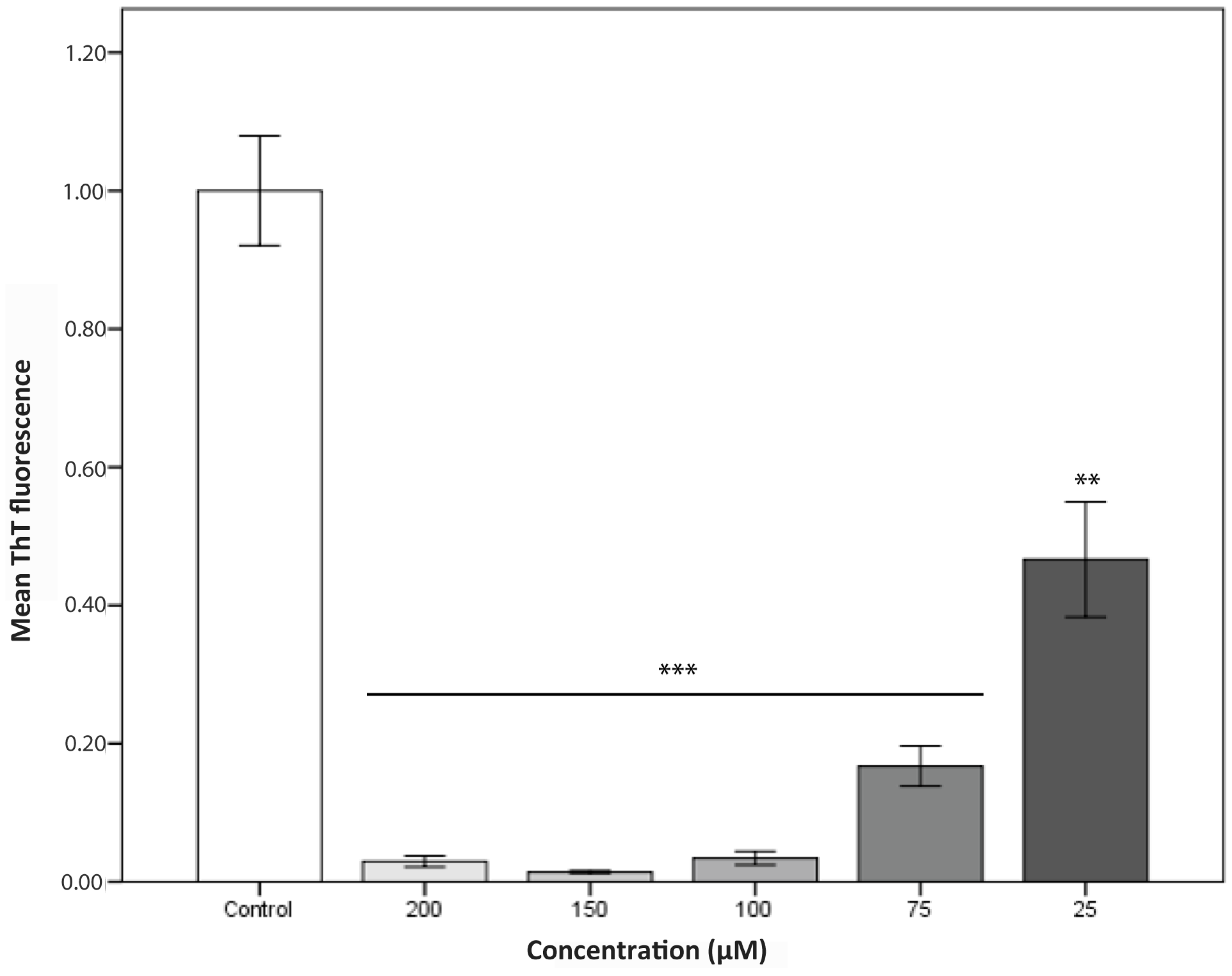
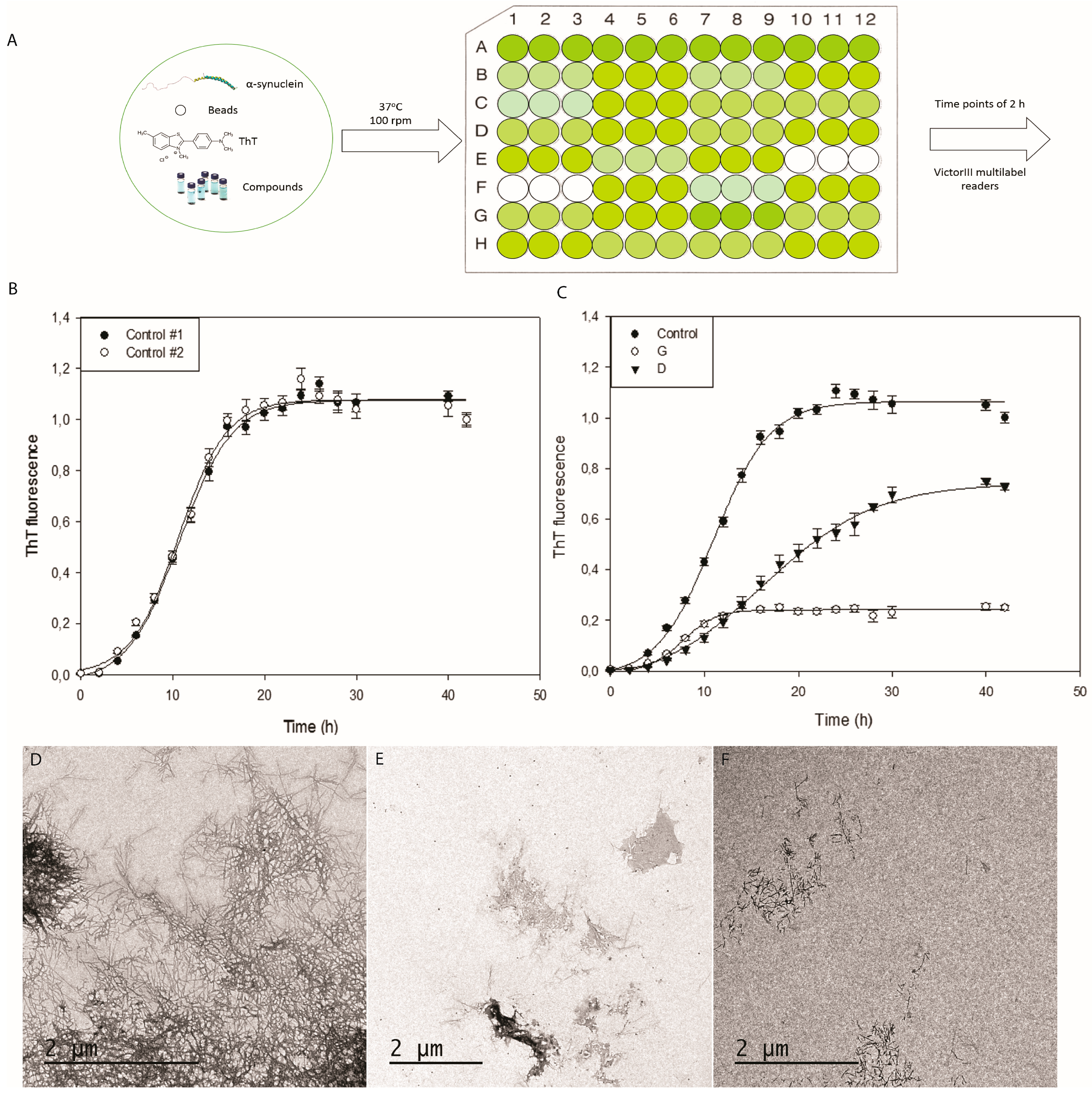
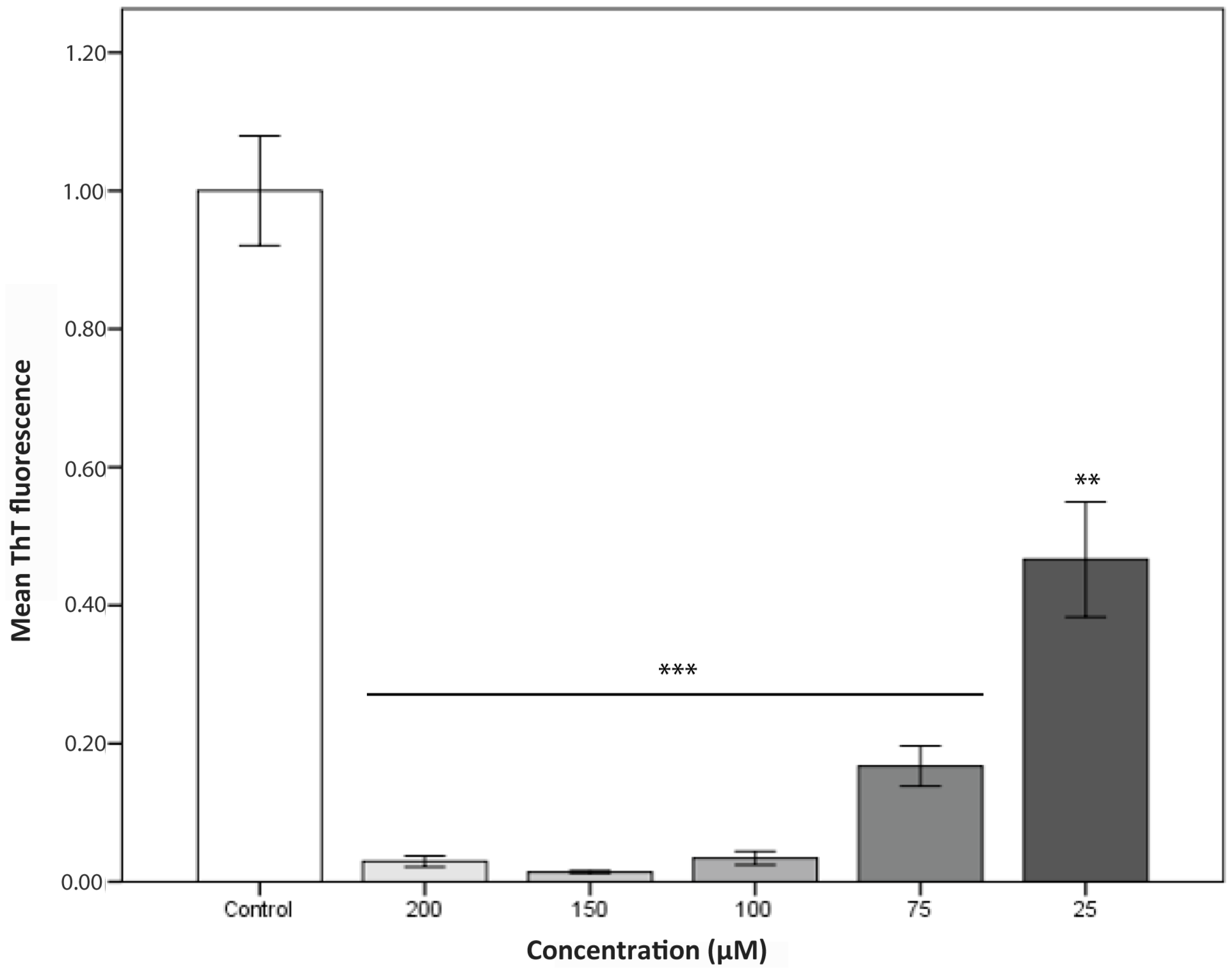
2.2. Aggregation Kinetics
3. Discussion
4. Materials and Methods
4.1. Expression and Purification of Human α-Synuclein
4.2. Quenching Analysis of Compounds
4.3. α-Synuclein Aggregation and Thioflavin-T Assays
4.4. Transmission Electron Microscopy (TEM)
4.5. Statistical Analysis
Acknowledgments
Author Contributions
Conflicts of Interest
Abreviations
| α-syn | α-synuclein |
| PD | Parkinson Disease |
| AD | Alzheimer’s disease |
| Th-T | Thioflavin T |
| TEM | Transmission Electron Microscopy |
| cv | column volumes |
References
- Invernizzi, G.; Papaleo, E.; Sabate, R.; Ventura, S. Protein aggregation: Mechanisms and functional consequences. Int. J. Biochem. Cell Biol. 2012, 44, 1541–1554. [Google Scholar] [CrossRef] [PubMed]
- Houlden, H.; Singleton, A.B. The genetics and neuropathology of parkinson’s disease. Acta Neuropathol. 2012, 124, 325–338. [Google Scholar] [CrossRef] [PubMed]
- Bendor, J.T.; Logan, T.P.; Edwards, R.H. The function of α-synuclein. Neuron 2013, 79, 1044–1066. [Google Scholar] [CrossRef] [PubMed]
- Spillantini, M.G.; Schmidt, M.L.; Lee, V.M.; Trojanowski, J.Q.; Jakes, R.; Goedert, M. α-Synuclein in lewy bodies. Nature 1997, 388, 839–840. [Google Scholar] [CrossRef] [PubMed]
- Maroteaux, L.; Campanelli, J.T.; Scheller, R.H. Synuclein: A neuron-specific protein localized to the nucleus and presynaptic nerve terminal. J. Neurosci. 1988, 8, 2804–2815. [Google Scholar] [PubMed]
- Goedert, M. α-Synuclein and neurodegenerative diseases. Nat. Rev. Neurosci. 2001, 2, 492–501. [Google Scholar] [CrossRef] [PubMed]
- Peelaerts, W.; Bousset, L.; van der Perren, A.; Moskalyuk, A.; Pulizzi, R.; Giugliano, M.; van den Haute, C.; Melki, R.; Baekelandt, V. α-Synuclein strains cause distinct synucleinopathies after local and systemic administration. Nature 2015, 522, 340–344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Serpell, L.C.; Berriman, J.; Jakes, R.; Goedert, M.; Crowther, R.A. Fiber diffraction of synthetic α-synuclein filaments shows amyloid-like cross-β conformation. Proc. Natl. Acad. Sci. USA 2000, 97, 4897–4902. [Google Scholar] [CrossRef] [PubMed]
- Conway, K.A.; Harper, J.D.; Lansbury, P.T., Jr. Fibrils formed in vitro from α-synuclein and two mutant forms linked to Parkinson’s disease are typical amyloid. Biochemistry 2000, 39, 2552–2563. [Google Scholar] [CrossRef] [PubMed]
- Conway, K.A.; Lee, S.J.; Rochet, J.C.; Ding, T.T.; Williamson, R.E.; Lansbury, P.T., Jr. Acceleration of oligomerization, not fibrillization, is a shared property of both α-synuclein mutations linked to early-onset Parkinson’s disease: Implications for pathogenesis and therapy. Proc. Natl. Acad. Sci. USA 2000, 97, 571–576. [Google Scholar] [CrossRef] [PubMed]
- Winner, B.; Jappelli, R.; Maji, S.K.; Desplats, P.A.; Boyer, L.; Aigner, S.; Hetzer, C.; Loher, T.; Vilar, M.; Campioni, S.; et al. In vivo demonstration that α-synuclein oligomers are toxic. Proc. Natl Acad. Sci. USA 2011, 108, 4194–4199. [Google Scholar] [CrossRef] [PubMed]
- Karpinar, D.P.; Balija, M.B.; Kugler, S.; Opazo, F.; Rezaei-Ghaleh, N.; Wender, N.; Kim, H.Y.; Taschenberger, G.; Falkenburger, B.H.; Heise, H.; et al. Pre-fibrillar α-synuclein variants with impaired β-structure increase neurotoxicity in Parkinson’s disease models. EMBO J. 2009, 28, 3256–3268. [Google Scholar] [CrossRef] [PubMed]
- Hansen, C.; Angot, E.; Bergstrom, A.L.; Steiner, J.A.; Pieri, L.; Paul, G.; Outeiro, T.F.; Melki, R.; Kallunki, P.; Fog, K.; et al. α-synuclein propagates from mouse brain to grafted dopaminergic neurons and seeds aggregation in cultured human cells. J. Clin. Investig. 2011, 121, 715–725. [Google Scholar] [CrossRef] [PubMed]
- Luk, K.C.; Kehm, V.; Carroll, J.; Zhang, B.; O’Brien, P.; Trojanowski, J.Q.; Lee, V.M. Pathological α-synuclein transmission initiates parkinson-like neurodegeneration in nontransgenic mice. Science 2012, 338, 949–953. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, T.; McLaurin, J. α-Synuclein aggregation, seeding and inhibition by scyllo-inositol. Biochem. Biophys. Res. Commun. 2016, 469, 529–534. [Google Scholar] [CrossRef] [PubMed]
- Deeg, A.A.; Reiner, A.M.; Schmidt, F.; Schueder, F.; Ryazanov, S.; Ruf, V.C.; Giller, K.; Becker, S.; Leonov, A.; Griesinger, C.; et al. Anle138b and related compounds are aggregation specific fluorescence markers and reveal high affinity binding to α-synuclein aggregates. Biochim. Biophys. Acta 2015, 1850, 1884–1890. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Zheng, L.L.; Wang, F.; Hu, Z.L.; Wu, W.N.; Gu, J.; Chen, J.G. Tanshinone iia attenuates neuronal damage and the impairment of long-term potentiation induced by hydrogen peroxide. J. Ethnopharmacol. 2011, 134, 147–155. [Google Scholar] [CrossRef] [PubMed]
- Jiang, M.; Porat-Shliom, Y.; Pei, Z.; Cheng, Y.; Xiang, L.; Sommers, K.; Li, Q.; Gillardon, F.; Hengerer, B.; Berlinicke, C.; et al. Baicalein reduces E46K α-synuclein aggregation in vitro and protects cells against E46K α-synuclein toxicity in cell models of familiar parkinsonism. J. Neurochem. 2010, 114, 419–429. [Google Scholar] [CrossRef] [PubMed]
- Ji, K.; Zhao, Y.; Yu, T.; Wang, Z.; Gong, H.; Yang, X.; Liu, Y.; Huang, K. Inhibition effects of tanshinone on the aggregation of α-synuclein. Food Funct. 2016, 7, 409–416. [Google Scholar] [CrossRef] [PubMed]
- Ahsan, N.; Mishra, S.; Jain, M.K.; Surolia, A.; Gupta, S. Curcumin pyrazole and its derivative (N-(3-nitrophenylpyrazole) curcumin inhibit aggregation, disrupt fibrils and modulate toxicity of wild type and mutant α-synuclein. Sci. Rep. 2015, 5, 9862. [Google Scholar] [CrossRef] [PubMed]
- Vazquez, J.A. Modeling of chemical inhibition from amyloid protein aggregation kinetics. BMC Pharmacol. Toxicol. 2014, 15, 9. [Google Scholar] [CrossRef] [PubMed]
- Lam, H.T.; Graber, M.C.; Gentry, K.A.; Bieschke, J. Stabilization of α-synuclein fibril clusters prevents fragmentation and reduces seeding activity and toxicity. Biochemistry 2016, 55, 675–685. [Google Scholar] [CrossRef] [PubMed]
- Celej, M.S.; Sarroukh, R.; Goormaghtigh, E.; Fidelio, G.D.; Ruysschaert, J.M.; Raussens, V. Toxic prefibrillar α-synuclein amyloid oligomers adopt a distinctive antiparallel β-sheet structure. Biochem. J. 2012, 443, 719–726. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Griggs, A.; Rochet, J.C.; Stanciu, L.A. In vitro study of α-synuclein protofibrils by cryo-EM suggests a Cu2+-dependent aggregation pathway. Biophys. J. 2013, 104, 2706–2713. [Google Scholar] [CrossRef] [PubMed]
- Planchard, M.S.; Exley, S.E.; Morgan, S.E.; Rangachari, V. Dopamine-induced α-synuclein oligomers show self- and cross-propagation properties. Protein Sci. 2014, 23, 1369–1379. [Google Scholar] [CrossRef]
- Ghosh, D.; Singh, P.K.; Sahay, S.; Jha, N.N.; Jacob, R.S.; Sen, S.; Kumar, A.; Riek, R.; Maji, S.K. Structure based aggregation studies reveal the presence of helix-rich intermediate during α-synuclein aggregation. Sci. Rep. 2015, 5, 9228. [Google Scholar] [CrossRef] [PubMed]
- Lorenzen, N.; Nielsen, S.B.; Buell, A.K.; Kaspersen, J.D.; Arosio, P.; Vad, B.S.; Paslawski, W.; Christiansen, G.; Valnickova-Hansen, Z.; Andreasen, M.; et al. The role of stable α-synuclein oligomers in the molecular events underlying amyloid formation. J. Am. Chem. Soc. 2014, 136, 3859–3868. [Google Scholar] [CrossRef] [PubMed]
- Bai, J.; Zhang, Z.; Liu, M.; Li, C. α-Synuclein-lanthanide metal ions interaction: Binding sites, conformation and fibrillation. BMC Biophys. 2015, 9, 1. [Google Scholar] [CrossRef] [PubMed]
- Kardani, J.; Roy, I. Understanding caffeine’s role in attenuating the toxicity of α-synuclein aggregates: Implications for risk of Parkinson’s disease. ACS Chem. Neurosci. 2015, 6, 1613–1625. [Google Scholar] [CrossRef] [PubMed]
- Shaykhalishahi, H.; Gauhar, A.; Wordehoff, M.M.; Gruning, C.S.; Klein, A.N.; Bannach, O.; Stoldt, M.; Willbold, D.; Hard, T.; Hoyer, W. Contact between the β1 and β2 segments of α-synuclein that inhibits amyloid formation. Angew. Chem. Int. Ed. Engl. 2015, 54, 8837–8840. [Google Scholar] [CrossRef] [PubMed]
- Shvadchak, V.V.; Claessens, M.M.; Subramaniam, V. Fibril breaking accelerates α-synuclein fibrillization. J. Phys. Chem. B 2015, 119, 1912–1918. [Google Scholar] [CrossRef] [PubMed]
- Volles, M.J.; Lansbury, P.T., Jr. Relationships between the sequence of α-synuclein and its membrane affinity, fibrillization propensity, and yeast toxicity. J. Mol. Biol. 2007, 366, 1510–1522. [Google Scholar] [CrossRef] [PubMed]
- Giehm, L.; Otzen, D.E. Strategies to increase the reproducibility of protein fibrillization in plate reader assays. Anal. Biochem. 2010, 400, 270–281. [Google Scholar] [CrossRef] [PubMed]
- Pronchik, J.; He, X.; Giurleo, J.T.; Talaga, D.S. In vitro formation of amyloid from α-synuclein is dominated by reactions at hydrophobic interfaces. J. Am. Chem. Soc. 2010, 132, 9797–9803. [Google Scholar] [CrossRef] [PubMed]
- Sluzky, V.; Tamada, J.A.; Klibanov, A.M.; Langer, R. Kinetics of insulin aggregation in aqueous solutions upon agitation in the presence of hydrophobic surfaces. Proc. Natl. Acad. Sci. USA 1991, 88, 9377–9381. [Google Scholar] [CrossRef] [PubMed]
- Ulrih, N.P.; Barry, C.H.; Fink, A.L. Impact of Tyr to Ala mutations on α-synuclein fibrillation and structural properties. Biochim. Biophys. Acta 2008, 1782, 581–585. [Google Scholar] [CrossRef] [PubMed]
- Janowska, M.K.; Wu, K.P.; Baum, J. Unveiling transient protein-protein interactions that modulate inhibition of α-synuclein aggregation by β-synuclein, a pre-synaptic protein that co-localizes with α-synuclein. Sci. Rep. 2015, 5, 15164. [Google Scholar] [CrossRef] [PubMed]
- Morris, A.M.; Watzky, M.A.; Agar, J.N.; Finke, R.G. Fitting neurological protein aggregation kinetic data via a 2-step, minimal/“Ockham’s Razor” model: The Finke-Watzky mechanism of nucleation followed by autocatalytic surface growth. Biochemistry 2008, 47, 2413–2427. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Chatani, E.; Goto, Y.; Paik, S.R. Seed-dependent accelerated fibrillation of α-synuclein induced by periodic ultrasonication treatment. J. Microbiol. Biotechnol. 2007, 17, 2027–2032. [Google Scholar] [PubMed]
- Malisauskas, R.; Botyriute, A.; Cannon, J.G.; Smirnovas, V. Flavone derivatives as inhibitors of insulin amyloid-like fibril formation. PLoS ONE 2015, 10, e0121231. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Code | Structure | % Inhibition a | t50 (h) b |
|---|---|---|---|---|
| N-(1-benzothiophen-2-yl)-N′-[3,5-bis(trifluoromethyl)phenyl]urea | A |  | 79.6 | 1 |
| 5-bromo-N-(3-chloro-4-fluorophenyl)-2-hydroxybenzamide | B |  | 74.4 | 5 |
| 1-[4-(4-chlorophenyl)-2,5-dihydro-1,3-thiazol-2-yliden]-2-(1-methylethylidene)hydrazine | C |  | 76.4 | −3 |
| 2-hydroxy-5-nitro-6-(3-nitrophenyl)-4-(trifluoromethyl)nicotinonitrile | D |  | 32.4 | 3 |
| 4-(4-methoxyphenyl)-5-(2-thienyl)-2,4-dihydro-3H-1,2,4-triazole-3-thione | E |  | 59.8 | 3 |
| methyl 6-(4-methoxyphenyl)-2H-thiopyran-3-carboxylate | F |  | 93.0 | 1 |
| 4-cyclohexyl-2-{[2-nitro-4-(trifluoromethyl)phenyl]thio}-6-oxo-1,6-dihydropyrimidine-5-carbonitrile | G |  | 72.7 | −2 |
| 3-(2-methyl-2,3-dihydro-1-benzofuran-5-yl)-5-(trifluoromethyl)-4,5-dihydro-1H-pyrazol-5-ol | H |  | 31.7 | 1 |
| 4-[4-(1H-pyrrol-1-yl)phenyl]-6-(trifluoromethyl)pyrimidin-2-amine | I |  | 0 | 11 |
| 5-chloro-2-(4-nitrophenyl)benzo[b]furan | J |  | 44.2 | 1 |
| benzyl N-{[3,5-bis(methylsulfanyl)-4-isothiazolyl]carbonyl}carbamate | K |  | 67.7 | 5 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pujols, J.; Peña-Díaz, S.; Conde-Giménez, M.; Pinheiro, F.; Navarro, S.; Sancho, J.; Ventura, S. High-Throughput Screening Methodology to Identify Alpha-Synuclein Aggregation Inhibitors. Int. J. Mol. Sci. 2017, 18, 478. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms18030478
Pujols J, Peña-Díaz S, Conde-Giménez M, Pinheiro F, Navarro S, Sancho J, Ventura S. High-Throughput Screening Methodology to Identify Alpha-Synuclein Aggregation Inhibitors. International Journal of Molecular Sciences. 2017; 18(3):478. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms18030478
Chicago/Turabian StylePujols, Jordi, Samuel Peña-Díaz, María Conde-Giménez, Francisca Pinheiro, Susanna Navarro, Javier Sancho, and Salvador Ventura. 2017. "High-Throughput Screening Methodology to Identify Alpha-Synuclein Aggregation Inhibitors" International Journal of Molecular Sciences 18, no. 3: 478. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms18030478