
Proline Residues as Switches in Conformational Changes Leading to Amyloid Fibril Formation

Abstract
:1. Introduction
2. Results
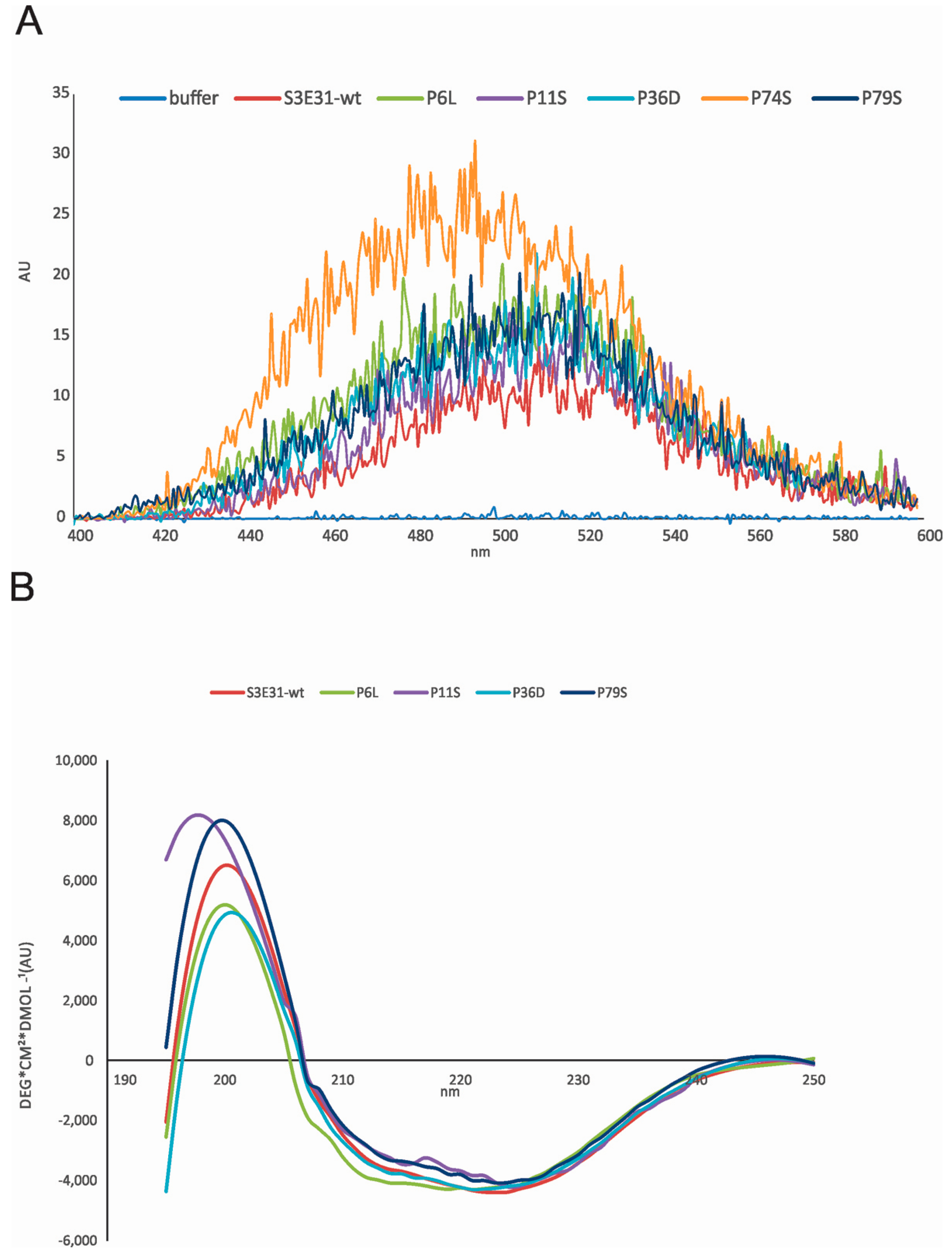
2.1. Influence of Prolines on Folding and Stability of Stefin B
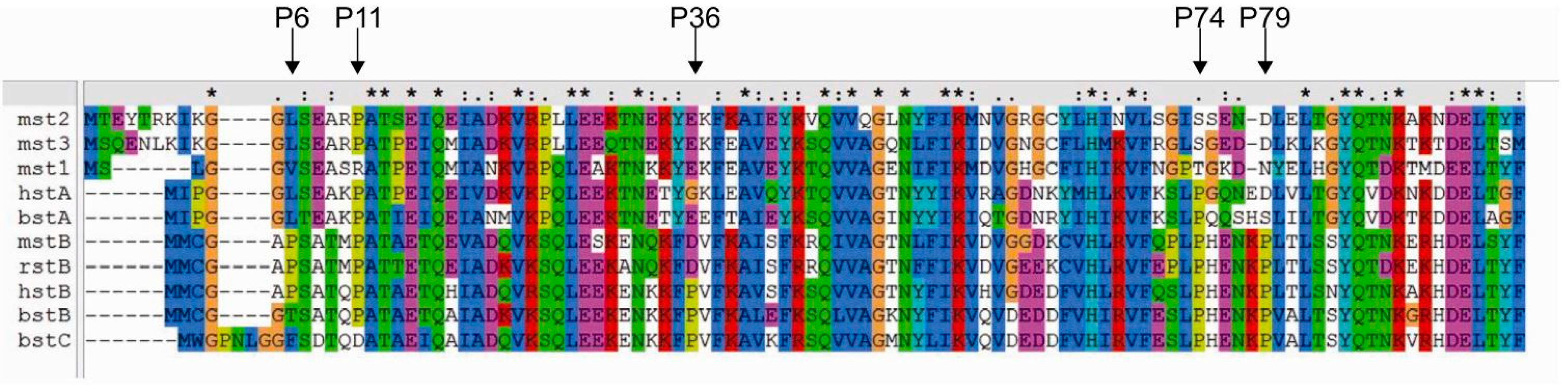
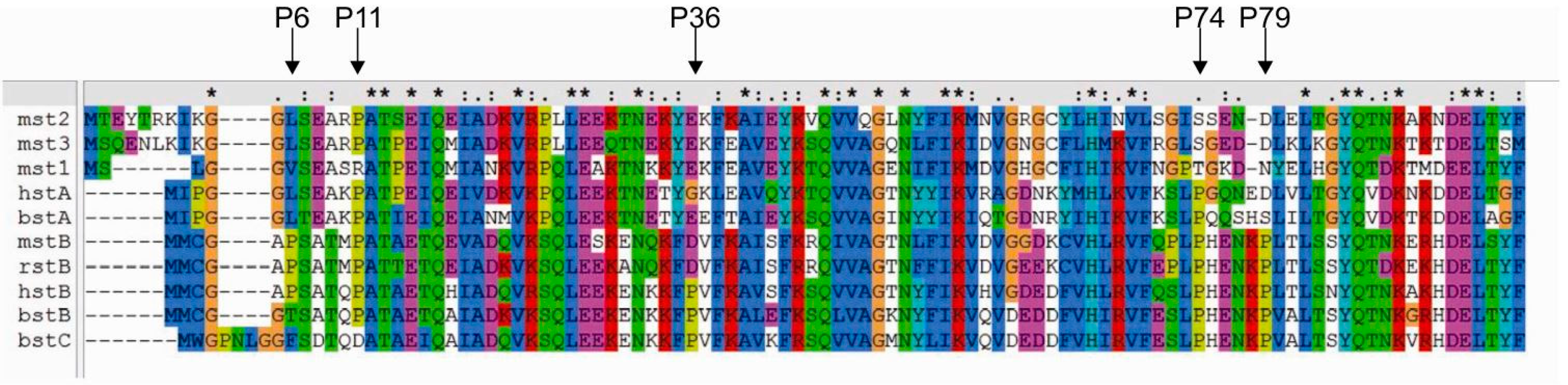
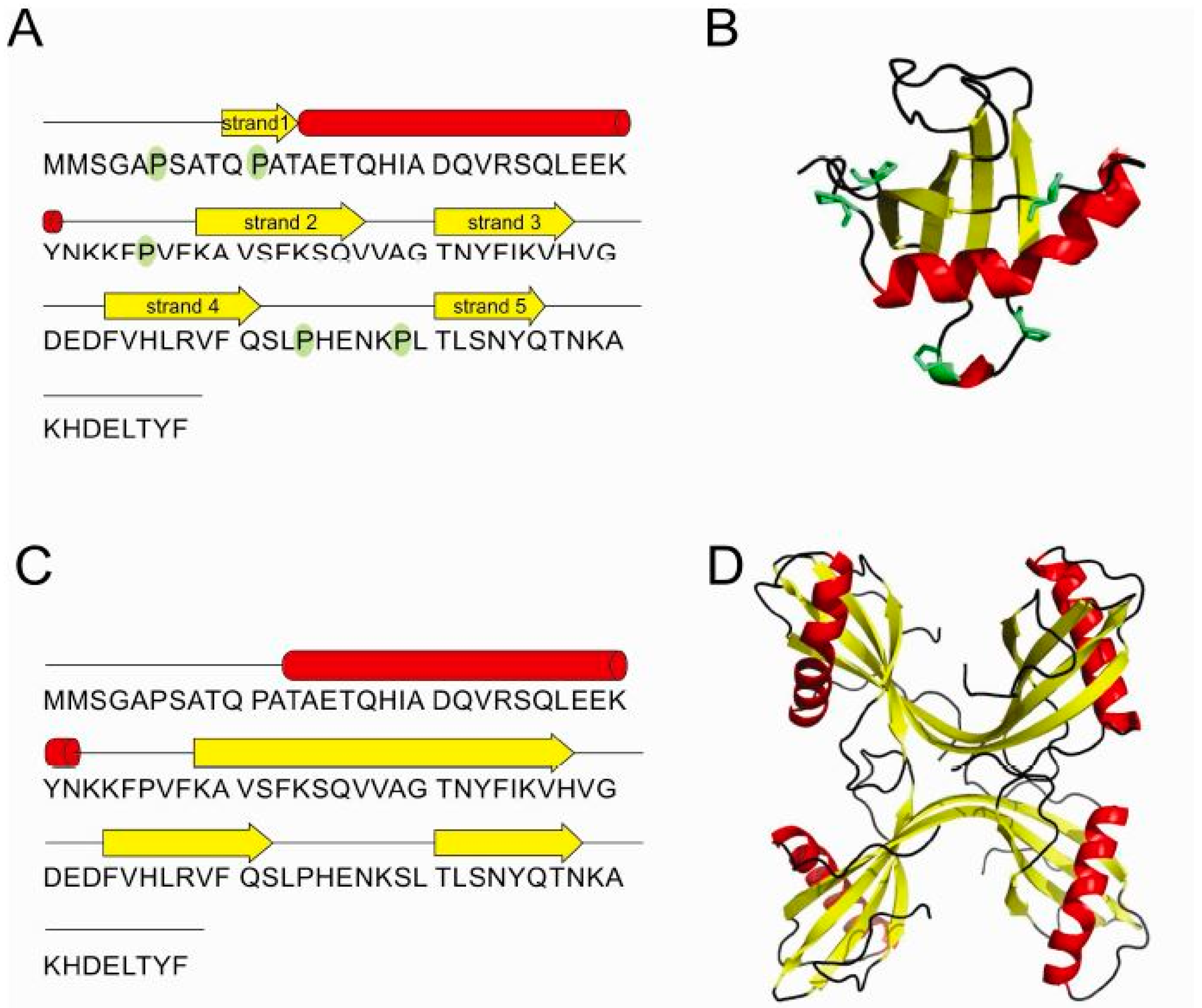
2.2. Influence of Prolines on Conformation and Oligomerization of Stefin B
2.3. Prediction of the Effects of Proline Mutations on Human Stefin B Stability
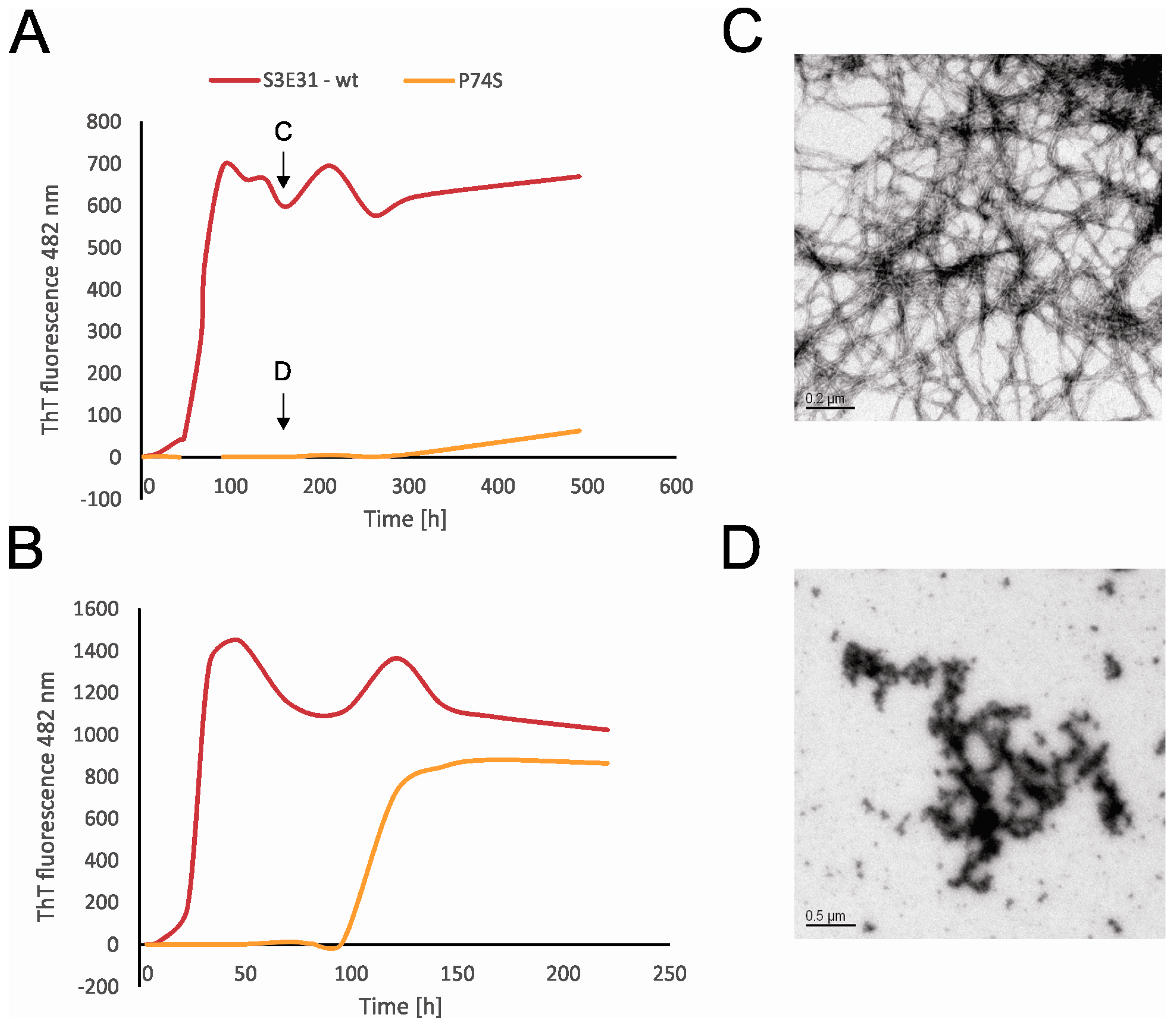
2.4. Influence of Prolines on Amyloid Fibril Formation of Human Stefin B
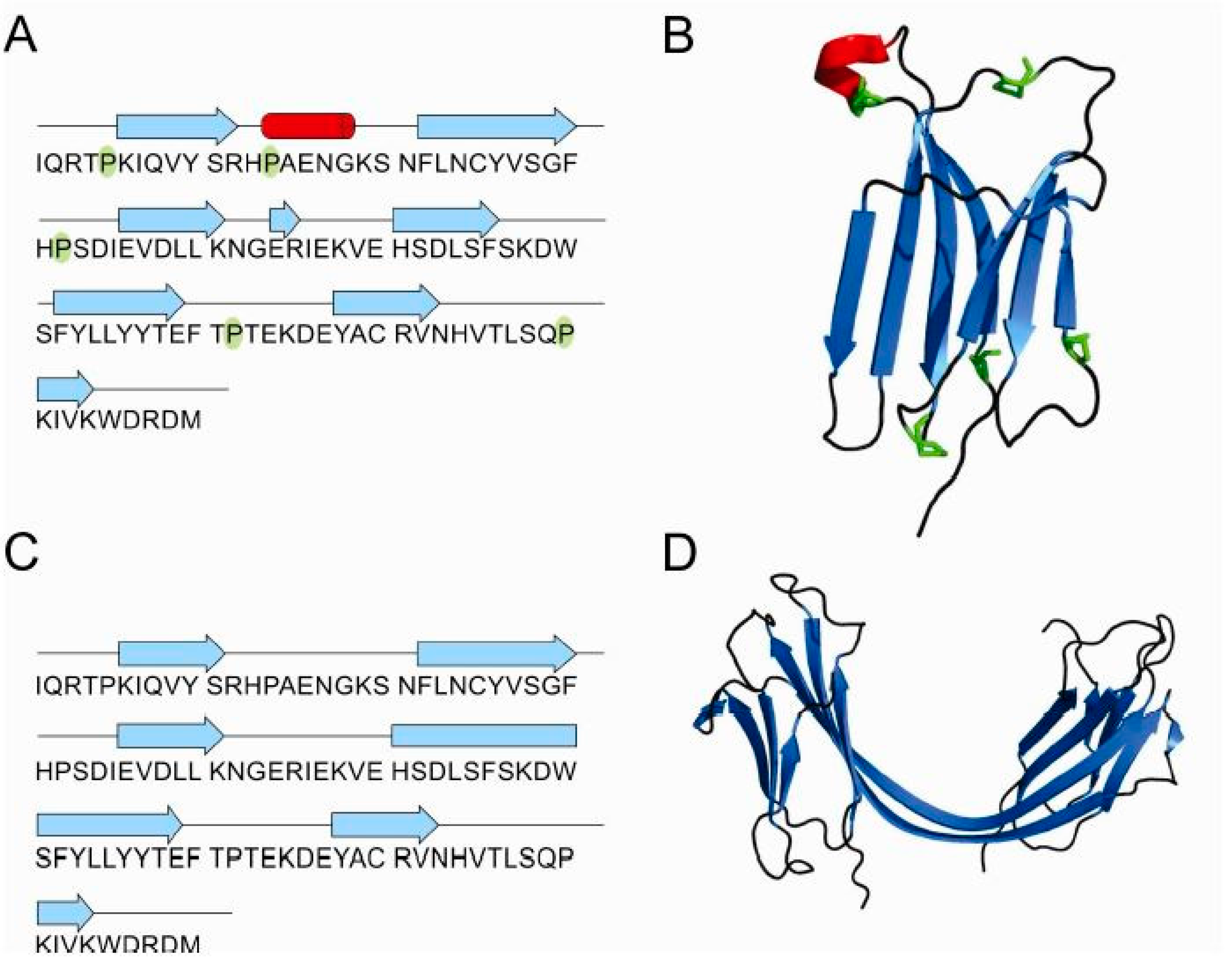
2.5. Structure of Monomer and Tetramer Composed of Domain-Swapped Dimers of Stefin B
3. Discussion
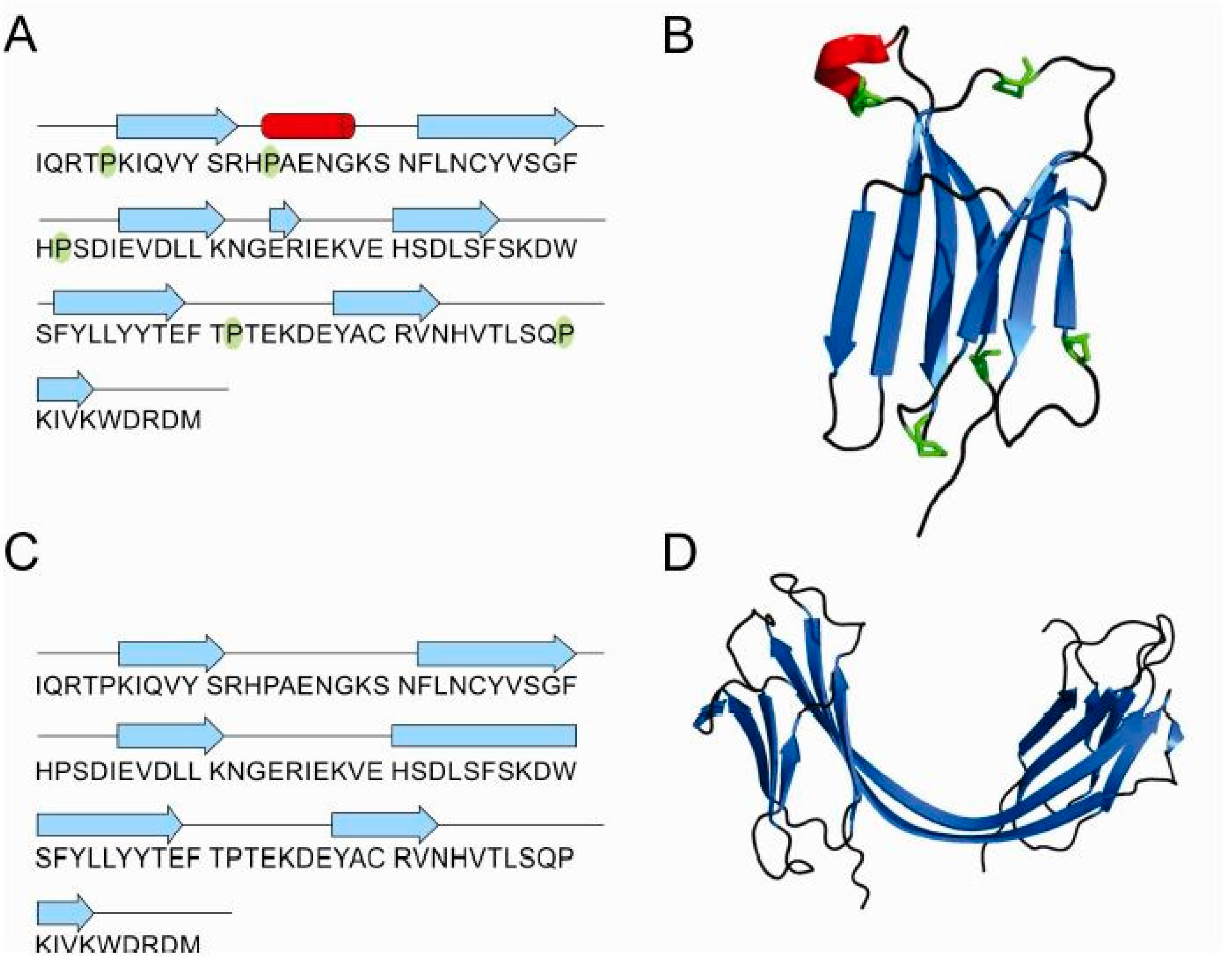
3.1. Influence of Prolines in β2-Microglobulin: Folding and Oligomerization
3.2. Aggregation and Amyloid-Fibril Formation of β2-Microglobulin
3.3. Prediction of Stability of β2-Microglobulin and Its Proline Mutants
3.4. Structures of β2-Microglobulin Monomer and Domain-Swapped Dimer
4. Materials and Methods
4.1. Protein Isolation
4.2. Fluorescence Spectra
4.3. Circular Dicroism Specta
4.4. Size-Exclusion Chromatography (SEC)
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Englander, S.W.; Mayne, L. The nature of protein folding pathways. Proc. Natl. Acad. Sci. USA 2014, 111, 15873–15880. [Google Scholar] [CrossRef]
- Wedemeyer, W.J.; Welker, E.; Scheraga, H.A. Proline cis-trans isomerization and protein folding. Biochemistry 2002, 41, 14637–14644. [Google Scholar] [CrossRef]
- Borgia, A.; Kemplen, K.R.; Borgia, M.B.; Soranno, A.; Shammas, S.; Wunderlich, B.; Nettels, D.; Best, R.B.; Clarke, J.; Schuler, B. Transient misfolding dominates multidomain protein folding. Nat. Commun. 2015, 6, 8861. [Google Scholar] [CrossRef]
- Rousseau, F.; Schymkowitz, J.; Itzhaki, L.S. Implications of 3D domain swapping for protein folding, misfolding and function. Adv. Exp. Med. Biol. 2012, 747, 137–152. [Google Scholar]
- Rousseau, F.; Schymkowitz, J.W.; Itzhaki, L.S. The unfolding story of three-dimensional domain swapping. Structure 2003, 11, 243–251. [Google Scholar] [CrossRef]
- Rousseau, F.; Schymkowitz, J.W.; Wilkinson, H.R.; Itzhaki, L.S. Three-dimensional domain swapping in p13suc1 occurs in the unfolded state and is controlled by conserved proline residues. Proc. Natl. Acad. Sci. USA 2001, 98, 5596–5601. [Google Scholar] [CrossRef]
- Eakin, C.M.; Berman, A.J.; Miranker, A.D. A native to amyloidogenic transition regulated by a backbone trigger. Nat. Struct. Mol. Biol. 2006, 13, 202–208. [Google Scholar] [CrossRef]
- Jahn, T.R.; Parker, M.J.; Homans, S.W.; Radford, S.E. Amyloid formation under physiological conditions proceeds via a native-like folding intermediate. Nat. Struct. Mol. Biol. 2006, 13, 195–201. [Google Scholar] [CrossRef]
- Pedersen, J.S.; Christensen, G.; Otzen, D.E. Modulation of S6 fibrillation by unfolding rates and gatekeeper residues. J. Mol. Biol. 2004, 341, 575–588. [Google Scholar] [CrossRef]
- Lummis, S.C.; Beene, D.L.; Lee, L.W.; Lester, H.A.; Broadhurst, R.W.; Dougherty, D.A. Cis-trans isomerization at a proline opens the pore of a neurotransmitter-gated ion channel. Nature 2005, 438, 248–252. [Google Scholar] [CrossRef]
- Ryo, A.; Togo, T.; Nakai, T.; Hirai, A.; Nishi, M.; Yamaguchi, A.; Suzuki, K.; Hirayasu, Y.; Kobayashi, H.; Perrem, K.; et al. Prolyl-isomerase Pin1 accumulates in lewy bodies of parkinson disease and facilitates formation of alpha-synuclein inclusions. J. Biol. Chem. 2006, 281, 4117–4125. [Google Scholar] [CrossRef]
- Dobson, C.M. Protein Folding and its Links with Human Disease. Biochem. Soc. Symp. 2001, 68, 1–26. [Google Scholar]
- Guijarro, J.I.; Sunde, M.; Jones, J.A.; Campbell, I.D.; Dobson, C.M. Amyloid Fibril Formation by an SH3 Domain. Proc. Natl. Acad. Sci USA 1998, 95, 4224–4228. [Google Scholar] [CrossRef]
- Turk, B.; Bieth, J.G.; Bjork, I.; Dolenc, I.; Turk, D.; Cimerman, N.; Kos, J.; Colic, A.; Stoka, V.; Turk, V. Regulation of the Activity of Lysosomal Cysteine Proteinases by Ph-Induced Inactivation and/or Endogenous Protein Inhibitors, Cystatins. Biol. Chem. Hoppe Seyler 1995, 376, 225–230. [Google Scholar] [CrossRef] [PubMed]
- Turk, B.; Turk, D.; Salvesen, G.S. Regulating cysteine protease activity: Essential role of protease inhibitors as guardians and regulators. Curr. Pharm. Des. 2002, 8, 1623–1637. [Google Scholar] [CrossRef] [PubMed]
- Turk, V.; Stoka, V.; Turk, D. Cystatins: Biochemical and structural properties, and medical relevance. Front. Biosci. 2008, 13, 5406–5420. [Google Scholar] [CrossRef] [PubMed]
- Turk, B.; Colic, A.; Stoka, V.; Turk, V. Kinetics of Inhibition of Bovine Cathepsin-S by Bovine Stefin-B. FEBS Lett. 1994, 339, 155–159. [Google Scholar] [CrossRef]
- Turk, B.; Krizaj, I.; Kralj, B.; Dolenc, I.; Popovic, T.; Bieth, J.G.; Turk, V. Bovine Stefin-C, a New Member of the Stefin Family. J. Biol. Chem. 1993, 268, 7323–7329. [Google Scholar] [PubMed]
- Jenko Kokalj, S.; Guncar, G.; Stern, I.; Morgan, G.; Rabzelj, S.; Kenig, M.; Staniforth, R.A.; Waltho, J.P.; Zerovnik, E.; Turk, D. Essential role of proline isomerization in stefin B tetramer formation. J. Mol. Biol. 2007, 366, 1569–1579. [Google Scholar] [CrossRef] [PubMed]
- Smajlovic, A.; Berbic, S.; Schiene-Fischer, C.; Tusek-Znidaric, M.; Taler, A.; Jenko-Kokalj, S.; Turk, D.; Zerovnik, E. Essential role of Pro 74 in stefin B amyloid-fibril formation: Dual action of cyclophilin A on the process. FEBS Lett. 2009, 583, 1114–1120. [Google Scholar] [CrossRef] [PubMed]
- Taler-Vercic, A.; Kirsipuu, T.; Friedemann, M.; Noormagi, A.; Polajnar, M.; Smirnova, J.; Znidaric, M.T.; Zganec, M.; Skarabot, M.; Vilfan, A.; et al. The role of initial oligomers in amyloid fibril formation by human stefin B. Int. J. Mol. Sci. 2013, 14, 18362–18384. [Google Scholar] [CrossRef] [PubMed]
- Zerovnik, E.; Pompe-Novak, M.; Skarabot, M.; Ravnikar, M.; Musevic, I.; Turk, V. Human stefin B readily forms amyloid fibrils in vitro. Biochim. Biophys. Acta 2002, 1594, 1–5. [Google Scholar] [CrossRef]
- Zerovnik, E.; Virden, R.; Jerala, R.; Kroon- Zitko, L.; Turk, V.; Waltho, J.P. Differences in the effects of TFE on the folding pathways of human stefins A and B. Proteins Struct. Funct. Bioinf. 1999, 36, 205–216. [Google Scholar] [CrossRef]
- Kenig, M.; Jenko-Kokalj, S.; Tusek-Znidaric, M.; Pompe-Novak, M.; Guncar, G.; Turk, D.; Waltho, J.P.; Staniforth, R.A.; Avbelj, F.; Zerovnik, E. Folding and amyloid-fibril formation for a series of human stefins’ chimeras: Any correlation? Proteins 2006, 62, 918–927. [Google Scholar] [CrossRef] [PubMed]
- Staniforth, R.A.; Dean, J.L.; Zhong, Q.; Zerovnik, E.; Clarke, A.R.; Waltho, J.P. The major transition state in folding need not involve the immobilization of side chains. Proc. Natl. Acad. Sci. USA 2000, 97, 5790–5795. [Google Scholar] [CrossRef] [PubMed]
- Staniforth, R.A.; Giannini, S.; Higgins, L.D.; Conroy, M.J.; Hounslow, A.M.; Jerala, R.; Craven, C.J.; Waltho, J.P. Three-dimensional domain swapping in the folded and molten-globule states of cystatins, an amyloid-forming structural superfamily. EMBO J. 2001, 20, 4774–4781. [Google Scholar] [CrossRef] [PubMed]
- Ceru, S.; Konjar, S.; Maher, K.; Repnik, U.; Krizaj, I.; Bencina, M.; Renko, M.; Nepveu, A.; Zerovnik, E.; Turk, B.; et al. Stefin B interacts with histones and cathepsin L in the nucleus. J. Biol. Chem. 2010, 285, 10078–10086. [Google Scholar] [CrossRef] [PubMed]
- Joensuu, T.; Lehesjoki, A.E.; Kopra, O. Molecular background of EPM1-Unverricht-Lundborg disease. Epilepsia 2008, 49, 557–563. [Google Scholar] [CrossRef] [PubMed]
- Korja, M.; Kaasinen, V.; Lamusuo, S.; Parkkola, R.; Nagren, K.; Marttila, R.J. Substantial thalamostriatal dopaminergic defect in Unverricht-Lundborg disease. Epilepsia 2007, 48, 1768–1773. [Google Scholar] [CrossRef] [PubMed]
- Kopitar-Jerala, N.; Schweiger, A.; Myers, R.M.; Turk, V.; Turk, B. Sensitization of stefin B-deficient thymocytes towards staurosporin-induced apoptosis is independent of cysteine cathepsins. FEBS Lett. 2005, 579, 2149–2155. [Google Scholar] [CrossRef] [PubMed]
- Lehtinen, M.K.; Tegelberg, S.; Schipper, H.; Su, H.; Zukor, H.; Manninen, O.; Kopra, O.; Joensuu, T.; Hakala, P.; Bonni, A.; et al. Cystatin B deficiency sensitizes neurons to oxidative stress in progressive myoclonus epilepsy, EPM1. J. Neurosci. 2009, 29, 5910–5915. [Google Scholar] [CrossRef] [PubMed]
- Polajnar, M.; Zavasnik-Bergant, T.; Skerget, K.; Vizovisek, M.; Vidmar, R.; Fonovic, M.; Kopitar-Jerala, N.; Petrovic, U.; Navarro, S.; Ventura, S.; et al. Human Stefin B Role in Cell’s Response to Misfolded Proteins and Autophagy. PLoS ONE 2014, 9, e102500. [Google Scholar] [CrossRef] [PubMed]
- Zerovnik, E. Putative alternative functions of human stefin B (cystatin B): Binding to amyloid-β, membranes, and copper. J. Mol. Recognit. 2017, 30, e2562. [Google Scholar] [CrossRef] [PubMed]
- Bode, W.; Engh, R.; Musil, D.; Thiele, U.; Huber, R.; Karshikov, A.; Brzin, J.; Kos, J.; Turk, V. The 2.0 A X-ray crystal structure of chicken egg white cystatin and its possible mode of interaction with cysteine proteinases. EMBO J. 1988, 7, 2593–2599. [Google Scholar] [PubMed]
- Engh, R.A.; Dieckmann, T.; Bode, W.; Auerswald, E.A.; Turk, V.; Huber, R.; Oschkinat, H. Conformational variability of chicken cystatin. Comparison of structures determined by X-ray diffraction and NMR spectroscopy. J. Mol. Biol. 1993, 234, 1060–1069. [Google Scholar] [CrossRef] [PubMed]
- Stubbs, M.T.; Laber, B.; Bode, W.; Huber, R.; Jerala, R.; Lenarcic, B.; Turk, V. The refined 2.4 A X-ray crystal structure of recombinant human stefin B in complex with the cysteine proteinase papain: A novel type of proteinase inhibitor interaction. EMBO J. 1990, 9, 1939–1947. [Google Scholar] [PubMed]
- Zerovnik, E.; Jerala, R.; Virden, R.; Kroon Zitko, L.; Turk, V.; Waltho, J.P. On the mechanism of human stefin B folding: II. Folding from GuHCl unfolded, TFE denatured, acid denatured, and acid intermediate states. Proteins 1998, 32, 304–313. [Google Scholar] [CrossRef]
- Zerovnik, E.; Virden, R.; Jerala, R.; Turk, V.; Waltho, J.P. On the mechanism of human stefin B folding: I. Comparison to homologous stefin A. Influence of pH and trifluoroethanol on the fast and slow folding phases. Proteins 1998, 32, 296–303. [Google Scholar] [CrossRef]
- Ceru, S.; Zerovnik, E. Similar toxicity of the oligomeric molten globule state and the prefibrillar oligomers. FEBS Lett. 2008, 582, 203–209. [Google Scholar] [CrossRef] [PubMed]
- Kenig, M.; Berbic, S.; Krijestorac, A.; Kroon-Zitko, L.; Tusek, M.; Pompe-Novak, M.; Zerovnik, E. Differences in aggregation properties of three site-specific mutants of recombinant human stefin B. Protein Sci. 2004, 13, 63–70. [Google Scholar] [CrossRef] [PubMed]
- Jerala, R.; Trstenjak, M.; Lenarcic, B.; Turk, V. Cloning a synthetic gene for human stefin B and its expression in E. coli. FEBS Lett. 1988, 239, 41–44. [Google Scholar] [CrossRef]
- Sievers, F.; Wilm, A.; Dineen, D.; Gibson, T.J.; Karplus, K.; Li, W.Z.; Lopez, R.; McWilliam, H.; Remmert, M.; Soding, J.; et al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 2011, 7, 539. [Google Scholar] [CrossRef] [PubMed]
- Ritonja, A.; Machleidt, W.; Barrett, A.J. Amino-Acid Sequence of the Intracellular Cysteine Proteinase-Inhibitor Cystatin-B from Human-Liver. Biochem. Biophys. Res. Commun. 1985, 131, 1187–1192. [Google Scholar] [CrossRef]
- Renko, M.; Taler-Vercic, A.; Mihelic, M.; Zerovnik, E.; Turk, D. Partial rotational lattice order-disorder in stefin B crystals. Acta Crystallogr. D Biol Crystallogr. 2014, 70, 1015–1025. [Google Scholar] [CrossRef] [PubMed]
- Capriotti, E.; Fariselli, P.; Casadio, R. I-Mutant2.0: Predicting stability changes upon mutation from the protein sequence or structure. Nucleic Acids Res. 2005, 33, W306–W310. [Google Scholar] [CrossRef] [PubMed]
- Debelouchina, G.T.; Platt, G.W.; Bayro, M.J.; Radford, S.E.; Griffin, R.G. Intermolecular alignment in β2-microglobulin amyloid fibrils. J. Am. Chem. Soc. 2010, 132, 17077–17079. [Google Scholar] [CrossRef] [PubMed]
- Eichner, T.; Kalverda, A.P.; Thompson, G.S.; Homans, S.W.; Radford, S.E. Conformational conversion during amyloid formation at atomic resolution. Mol. Cell 2011, 41, 161–172. [Google Scholar] [CrossRef] [PubMed]
- Gronenborn, A.M. Protein acrobatics in Pairs—Dimerization via domain swapping. Curr. Opin. Struct. Biol. 2009, 19, 39–49. [Google Scholar] [CrossRef] [PubMed]
- Janowski, R.; Kozak, M.; Jankowska, E.; Grzonka, Z.; Grubb, A.; Abrahamson, M.; Jaskolski, M. Human cystatin C, an amyloidogenic protein, dimerizes through three-dimensional domain swapping. Nat. Struct. Biol. 2001, 8, 316–320. [Google Scholar] [CrossRef] [PubMed]
- Adams, E.J.; Luoma, A.M. The adaptable major histocompatibility complex (MHC) fold: Structure and function of nonclassical and MHC class I-like molecules. Annu. Rev. Immunol. 2013, 31, 529–561. [Google Scholar] [CrossRef] [PubMed]
- Floege, J.; Ketteler, M. B2-microglobulin-derived amyloidosis: An update. Kidney Int. Suppl. 2001, 78, S164–S171. [Google Scholar] [CrossRef] [PubMed]
- Verdone, G.; Corazza, A.; Viglino, P.; Pettirossi, F.; Giorgetti, S.; Mangione, P.; Andreola, A.; Stoppini, M.; Bellotti, V.; Esposito, G. The solution structure of human β2-microglobulin reveals the prodromes of its amyloid transition. Protein Sci. 2002, 11, 487–499. [Google Scholar] [CrossRef] [PubMed]
- Eichner, T.; Radford, S.E. Understanding the complex mechanisms of β2-microglobulin amyloid assembly. FEBS J. 2011, 278, 3868–3883. [Google Scholar] [CrossRef] [PubMed]
- Chiti, F.; Mangione, P.; Andreola, A.; Giorgetti, S.; Stefani, M.; Dobson, C.M.; Bellotti, V.; Taddei, N. Detection of two partially structured species in the folding process of the amyloidogenic protein β 2-microglobulin. J. Mol. Biol. 2001, 307, 379–391. [Google Scholar] [CrossRef] [PubMed]
- Kameda, A.; Hoshino, M.; Higurashi, T.; Takahashi, S.; Naiki, H.; Goto, Y. Nuclear magnetic resonance characterization of the refolding intermediate of β2-microglobulin trapped by non-native prolyl peptide bond. J. Mol. Biol. 2005, 348, 383–397. [Google Scholar] [CrossRef] [PubMed]
- Xue, W.F.; Homans, S.W.; Radford, S.E. Systematic analysis of nucleation-dependent polymerization reveals new insights into the mechanism of amyloid self-assembly. Proc. Natl. Acad. Sci. USA 2008, 105, 8926–8931. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kad, N.M.; Thomson, N.H.; Smith, D.P.; Smith, D.A.; Radford, S.E. B2-microglobulin and its deamidated variant, N17D form amyloid fibrils with a range of morphologies in vitro. J. Mol. Biol. 2001, 313, 559–571. [Google Scholar] [CrossRef] [PubMed]
- Chiba, T.; Hagihara, Y.; Higurashi, T.; Hasegawa, K.; Naiki, H.; Goto, Y. Amyloid fibril formation in the context of full-length protein: Effects of proline mutations on the amyloid fibril formation of β2-microglobulin. J. Biol. Chem. 2003, 278, 47016–47024. [Google Scholar] [CrossRef] [PubMed]
- Walsh, D.M.; Klyubin, I.; Fadeeva, J.V.; Cullen, W.K.; Anwyl, R.; Wolfe, M.S.; Rowan, M.J.; Selkoe, D.J. Naturally secreted oligomers of amyloid β protein potently inhibit hippocampal long-term potentiation in vivo. Nature 2002, 416, 535–539. [Google Scholar] [CrossRef] [PubMed]
- Hoshi, M.; Sato, M.; Matsumoto, S.; Noguchi, A.; Yasutake, K.; Yoshida, N.; Sato, K. Spherical aggregates of β-amyloid (amylospheroid) show high neurotoxicity and activate tau protein kinase I/glycogen synthase kinase-3β. Proc. Natl. Acad. Sci. USA 2003, 100, 6370–6375. [Google Scholar] [CrossRef] [PubMed]
- Kayed, R.; Head, E.; Thompson, J.L.; McIntire, T.M.; Milton, S.C.; Cotman, C.W.; Glabe, C.G. Common structure of soluble amyloid oligomers implies common mechanism of pathogenesis. Science 2003, 300, 486–489. [Google Scholar] [CrossRef] [PubMed]
- Bucciantini, M.; Giannoni, E.; Chiti, F.; Baroni, F.; Formigli, L.; Zurdo, J.; Taddei, N.; Ramponi, G.; Dobson, C.M.; Stefani, M. Inherent toxicity of aggregates implies a common mechanism for protein misfolding diseases. Nature 2002, 416, 507–511. [Google Scholar] [CrossRef] [PubMed]
- Hirakura, Y.; Kagan, B.L. Pore formation by β2-microglobulin: A mechanism for the pathogenesis of dialysis associated amyloidosis. Amyloid 2001, 8, 94–100. [Google Scholar] [CrossRef] [PubMed]
- Mustata, M.; Capone, R.; Jang, H.; Arce, F.T.; Ramachandran, S.; Lal, R.; Nussinov, R. K3 fragment of amyloidogenic β(2)-microglobulin forms ion channels: Implication for dialysis related amyloidosis. J. Am. Chem. Soc. 2009, 131, 14938–14945. [Google Scholar] [CrossRef] [PubMed]
- Luhrs, T.; Ritter, C.; Adrian, M.; Riek-Loher, D.; Bohrmann, B.; Dobeli, H.; Schubert, D.; Riek, R. 3D structure of Alzheimer's amyloid-β(1–42) fibrils. Proc. Natl. Acad. Sci. USA 2005, 102, 17342–17347. [Google Scholar] [CrossRef] [PubMed]
- Petkova, A.T.; Yau, W.M.; Tycko, R. Experimental constraints on quaternary structure in Alzheimer’s β-amyloid fibrils. Biochemistry 2006, 45, 498–512. [Google Scholar] [CrossRef] [PubMed]
- Ferguson, N.; Becker, J.; Tidow, H.; Tremmel, S.; Sharpe, T.D.; Krause, G.; Flinders, J.; Petrovich, M.; Berriman, J.; Oschkinat, H.; et al. General structural motifs of amyloid protofilaments. Proc. Natl. Acad. Sci. USA 2006, 103, 16248–16253. [Google Scholar] [CrossRef] [PubMed]
- Saper, M.A.; Bjorkman, P.J.; Wiley, D.C. Refined structure of the human histocompatibility antigen HLA-A2 at 2.6 A resolution. J. Mol. Biol. 1991, 219, 277–319. [Google Scholar] [CrossRef]
- Hasegawa, K.; Ohhashi, Y.; Yamaguchi, I.; Takahashi, N.; Tsutsumi, S.; Goto, Y.; Gejyo, F.; Naiki, H. Amyloidogenic synthetic peptides of β2-microglobulin—A role of the disulfide bond. Biochem. Biophys. Res. Commun. 2003, 304, 101–106. [Google Scholar] [CrossRef]
- Pawar, A.P.; Dubay, K.F.; Zurdo, J.; Chiti, F.; Vendruscolo, M.; Dobson, C.M. Prediction of “aggregation-prone” and “aggregation-susceptible” regions in proteins associated with neurodegenerative diseases. J. Mol. Biol. 2005, 350, 379–392. [Google Scholar] [CrossRef] [PubMed]
- Jones, S.; Manning, J.; Kad, N.M.; Radford, S.E. Amyloid-forming peptides from β2-microglobulin-Insights into the mechanism of fibril formation in vitro. J. Mol. Biol. 2003, 325, 249–257. [Google Scholar] [CrossRef]
- Eakin, C.M.; Miranker, A.D. From chance to frequent encounters: Origins of β2-microglobulin fibrillogenesis. Biochim. Biophys. Acta 2005, 1753, 92–99. [Google Scholar] [CrossRef] [PubMed]
- Ivanova, M.I.; Thompson, M.J.; Eisenberg, D. A systematic screen of β2-microglobulin and insulin for amyloid-like segments. Proc. Natl. Acad. Sci. USA 2006, 103, 4079–4082. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, S.; Hasegawa, K.; Yamaguchi, I.; Tsutsumi, S.; Kardos, J.; Goto, Y.; Gejyo, F.; Naiki, H. Low concentrations of sodium dodecyl sulfate induce the extension of β 2-microglobulin-related amyloid fibrils at a neutral pH. Biochemistry 2004, 43, 11075–11082. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, S.; Yamaguchi, I.; Hasegawa, K.; Tsutsumi, S.; Goto, Y.; Gejyo, F.; Naiki, H. Glycosaminoglycans enhance the trifluoroethanol-induced extension of β 2-microglobulin-related amyloid fibrils at a neutral pH. J. Am. Soc. Nephrol. 2004, 15, 126–133. [Google Scholar] [CrossRef] [PubMed]
- Ohhashi, Y.; Kihara, M.; Naiki, H.; Goto, Y. Ultrasonication-induced amyloid fibril formation of β2-microglobulin. J. Biol. Chem. 2005, 280, 32843–32848. [Google Scholar] [CrossRef] [PubMed]
- Sasahara, K.; Yagi, H.; Sakai, M.; Naiki, H.; Goto, Y. Amyloid nucleation triggered by agitation of β2-microglobulin under acidic and neutral pH conditions. Biochemistry 2008, 47, 2650–2660. [Google Scholar] [CrossRef] [PubMed]
- Rennella, E.; Corazza, A.; Giorgetti, S.; Fogolari, F.; Viglino, P.; Porcari, R.; Verga, L.; Stoppini, M.; Bellotti, V.; Esposito, G. Folding and fibrillogenesis: Clues from β2-microglobulin. J. Mol. Biol. 2010, 401, 286–297. [Google Scholar] [CrossRef] [PubMed]
- Platt, G.W.; Radford, S.E. Glimpses of the molecular mechanisms of β2-microglobulin fibril formation in vitro: Aggregation on a complex energy landscape. FEBS Lett. 2009, 583, 2623–2629. [Google Scholar] [CrossRef] [PubMed]
- Barbet-Massin, E.; Ricagno, S.; Lewandowski, J.R.; Giorgetti, S.; Bellotti, V.; Bolognesi, M.; Emsley, L.; Pintacuda, G. Fibrillar vs. crystalline full-length β-2-microglobulin studied by high-resolution solid-state NMR spectroscopy. J. Am. Chem. Soc. 2010, 132, 5556–5557. [Google Scholar] [CrossRef] [PubMed]
- Routledge, K.E.; Tartaglia, G.G.; Platt, G.W.; Vendruscolo, M.; Radford, S.E. Competition between intramolecular and intermolecular interactions in an amyloid-forming protein. J. Mol. Biol. 2009, 389, 776–786. [Google Scholar] [CrossRef] [PubMed]
- Ivanova, M.I.; Sawaya, M.R.; Gingery, M.; Attinger, A.; Eisenberg, D. An amyloid-forming segment of β2-microglobulin suggests a molecular model for the fibril. Proc. Natl. Acad. Sci. USA 2004, 101, 10584–10589. [Google Scholar] [CrossRef] [PubMed]
- Platt, G.W.; Routledge, K.E.; Homans, S.W.; Radford, S.E. Fibril growth kinetics reveal a region of β2-microglobulin important for nucleation and elongation of aggregation. J. Mol. Biol. 2008, 378, 251–263. [Google Scholar] [CrossRef] [PubMed]
- Estacio, S.G.; Krobath, H.; Vila-Vicosa, D.; Machuqueiro, M.; Shakhnovich, E.I.; Faisca, P.F. A simulated intermediate state for folding and aggregation provides insights into DeltaN6 β2-microglobulin amyloidogenic behavior. PLoS Comput. Biol. 2014, 10, e1003606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Torbeev, V.; Ebert, M.O.; Dolenc, J.; Hilvert, D. Substitution of proline32 by alpha-methylproline preorganizes β2-microglobulin for oligomerization but not for aggregation into amyloids. J. Am. Chem. Soc. 2015, 137, 2524–2535. [Google Scholar] [CrossRef] [PubMed]
- Jahn, T.R.; Tennent, G.A.; Radford, S.E. A common β-sheet architecture underlies in vitro and in vivo β2-microglobulin amyloid fibrils. J. Biol. Chem. 2008, 283, 17279–17286. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Sawaya, M.R.; Eisenberg, D. β2-microglobulin forms three-dimensional domain-swapped amyloid fibrils with disulfide linkages. Nat. Struct. Mol. Biol. 2011, 18, 49–55. [Google Scholar] [CrossRef] [PubMed]
- Trinh, C.H.; Smith, D.P.; Kalverda, A.P.; Phillips, S.E.V.; Radford, S.E. Crystal structure of monomeric human β2-microglobulin reveals clues to its amyloidogenic properties. Proc. Natl. Acad. Sci. USA 2002, 99, 9771–9776. [Google Scholar] [CrossRef] [PubMed]
- Calabrese, M.F.; Eakin, C.M.; Wang, J.M.; Miranker, A.D. A regulatable switch mediates self-association in an immunoglobulin fold. Nat. Struct. Mol. Biol. 2008, 15, 965–971. [Google Scholar] [CrossRef] [PubMed]
- Rabzelj, S.; Turk, V.; Zerovnik, E. In vitro study of stability and amyloid-fibril formation of two mutants of human stefin B (cystatin B) occurring in patients with EPM1. Protein Sci. 2005, 14, 2713–2722. [Google Scholar] [CrossRef] [PubMed]
- Skerget, K.; Vilfan, A.; Pompe-Novak, M.; Turk, V.; Waltho, J.P.; Turk, D.; Zerovnik, E. The mechanism of amyloid-fibril formation by stefin B: Temperature and protein concentration dependence of the rates. Proteins 2009, 74, 425–436. [Google Scholar] [CrossRef] [PubMed]
- Kraus, A. Proline and lysine residues provide modulatory switches in amyloid formation: Insights from prion protein. Prion 2016, 10, 57–62. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, K.; Greenwood, A.; Binder, L.; Bigio, E.H.; Denial, S.; Nicholson, L.; Zhou, X.Z.; Lu, K.P. Proline Isomer-Specific Antibodies Reveal the Early Pathogenic Tau Conformation in Alzheimer’s Disease. Cell 2012, 149, 232–244. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mutation | ΔΔGP04080|C3S (Kcal/mol) | ΔΔG1STF:I (Kcal/mol) | ΔΔG4N6V:0 (Kcal/mol) | ΔΔG2OCT:A (Kcal/mol) |
|---|---|---|---|---|
| P6L | 1.23 | P11L 1.47 | NA | 1.17 |
| P11S | −0.71 | P16S -0.20 | −0.30 | −0.42 |
| P36D | −0.89 | P43D -0.83 | −0.69 | −0.69 |
| P74S | −1.64 | P103S -0.82 | −1.44 | −0.48 |
| P79S | −1.88 | P107S -0.67 | −1.19 | - |
| Mutation | ΔΔGB2MG|21-119 (Kcal/mol) | ΔΔG1LDS:A (Kcal/mol) | ΔΔG3LOW:A (Kcal/mol) |
|---|---|---|---|
| P5S | −0.99 | −2.50 | −1.93 |
| P14S | −1.85 | −0.55 | −0.94 |
| P32G * | −2.62 | −2.15 | −1.74 |
| P32L * | −2.05 | 0.79 | 1.05 |
| P32S | −2.24 | −0.90 | −0.57 |
| P72S | −1.74 | −0.61 | −0.81 |
| P90S | −1.28 | −1.92 | −1.81 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Taler-Verčič, A.; Hasanbašić, S.; Berbić, S.; Stoka, V.; Turk, D.; Žerovnik, E. Proline Residues as Switches in Conformational Changes Leading to Amyloid Fibril Formation. Int. J. Mol. Sci. 2017, 18, 549. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms18030549
Taler-Verčič A, Hasanbašić S, Berbić S, Stoka V, Turk D, Žerovnik E. Proline Residues as Switches in Conformational Changes Leading to Amyloid Fibril Formation. International Journal of Molecular Sciences. 2017; 18(3):549. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms18030549
Chicago/Turabian StyleTaler-Verčič, Ajda, Samra Hasanbašić, Selma Berbić, Veronika Stoka, Dušan Turk, and Eva Žerovnik. 2017. "Proline Residues as Switches in Conformational Changes Leading to Amyloid Fibril Formation" International Journal of Molecular Sciences 18, no. 3: 549. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms18030549