
Plausible Roles for RAGE in Conditions Exacerbated by Direct and Indirect (Secondhand) Smoke Exposure



Abstract
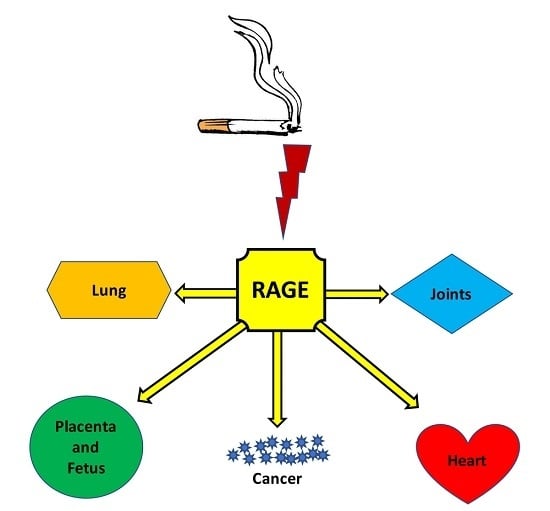
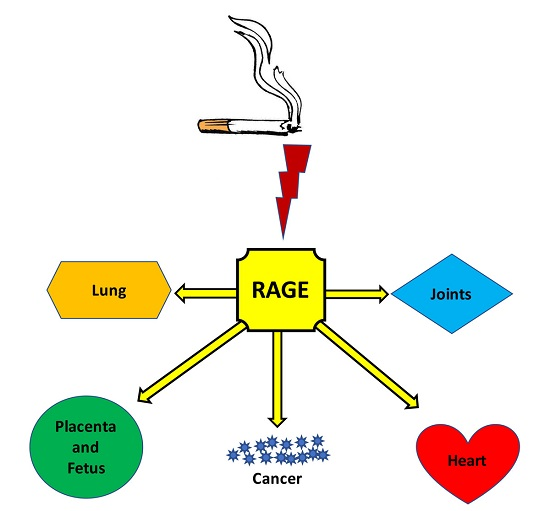
:
{kind=link}
1. Introduction
1.1. Global Burden
1.2. Tobacco Smoke
2. Health Outcomes and Comorbidities
2.1. Chronic Obstructive Pulmonary Disease
2.2. Cancer
2.3. Developmental Complications
2.4. Cardiometabolic Disorders
2.5. Joint and Movement Disorders
3. RAGE: A Plausible Unifying Mechanism
Acknowledgments
Conflicts of Interest
References
- Mackay, J.E.M.; Shafey, O. The Tobacco Atlas, 2nd ed.; American Cancer Society: Atlanta, GA, USA, 2006. [Google Scholar]
- Ekpu, V.U.; Brown, A.K. The economic impact of smoking and of reducing smoking prevalence: Review of evidence. Tob. Use Insights 2015, 8, 1–35. [Google Scholar] [CrossRef] [PubMed]
- Brett, J.; Schmidt, A.M.; Yan, S.D.; Zou, Y.S.; Weidman, E.; Pinsky, D.; Nowygrod, R.; Neeper, M.; Przysiecki, C.; Shaw, A.; et al. Survey of the distribution of a newly characterized receptor for advanced glycation end products in tissues. Am. J. Pathol. 1993, 143, 1699–1712. [Google Scholar] [PubMed]
- Buckley, S.T.; Ehrhardt, C. The receptor for advanced glycation end products (RAGE) and the lung. J. Biomed. Biotechnol. 2010, 2010, 917108. [Google Scholar] [CrossRef] [PubMed]
- Nelson, M.B.; Swensen, A.C.; Winden, D.R.; Bodine, J.S.; Bikman, B.T.; Reynolds, P.R. Cardiomyocyte mitochondrial respiration is reduced by receptor for advanced glycation end-product signaling in a ceramide-dependent manner. Am. J. Physiol. Heart Circ. Physiol. 2015, 309, H63–H69. [Google Scholar] [CrossRef] [PubMed]
- Moritsugu, K.P. The 2006 report of the surgeon general: The health consequences of involuntary exposure to tobacco smoke. Am. J. Prev. Med. 2007, 32, 542–543. [Google Scholar] [CrossRef] [PubMed]
- Fowles, J.; Dybing, E. Application of toxicological risk assessment principles to the chemical constituents of cigarette smoke. Tob. Control 2003, 12, 424–430. [Google Scholar] [CrossRef] [PubMed]
- Protano, C.; Andreoli, R.; Manini, P.; Guidotti, M.; Vitali, M. A tobacco-related carcinogen: Assessing the impact of smoking behaviours of cohabitants on benzene exposure in children. Tob. Control 2012, 21, 325–329. [Google Scholar] [CrossRef] [PubMed]
- Szumska, M.; Damasiewicz-Bodzek, A.; Tyrpien-Golder, K. Environmental tobacco smoke—Assessment of formaldehyde concentration in urine samples of exposed medicine students. Przegl. Lek. 2015, 72, 140–143. [Google Scholar] [PubMed]
- Abedin, Z.; Louis-Juste, M.; Stangl, M.; Field, J. The role of base excision repair genes OGG1, APN1 and APN2 in benzo[a]pyrene-7,8-dione induced p53 mutagenesis. Mutat. Res. 2013, 750, 121–128. [Google Scholar] [CrossRef] [PubMed]
- Leone, A. Toxics of tobacco smoke and cardiovascular system: From functional to cellular damage. Curr. Pharm. Des. 2015, 21, 4370–4379. [Google Scholar] [CrossRef] [PubMed]
- Noya, Y.; Seki, K.; Asano, H.; Mai, Y.; Horinouchi, T.; Higashi, T.; Terada, K.; Hatate, C.; Hoshi, A.; Nepal, P.; et al. Identification of stable cytotoxic factors in the gas phase extract of cigarette smoke and pharmacological characterization of their cytotoxicity. Toxicology 2013, 314, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Kilthau, G.F. Cancer risk in relation to radioactivity in tobacco. Radiol. Technol. 1996, 67, 217–222. [Google Scholar] [PubMed]
- Yuan, J.M.; Murphy, S.E.; Stepanov, I.; Wang, R.; Carmella, S.G.; Nelson, H.H.; Hatsukami, D.K.; Hecht, S.S. 2-Phenethyl isothiocyanate, glutathione S-transferase M1 and T1 polymorphisms, and detoxification of volatile organic carcinogens and toxicants in tobacco smoke. Cancer Prev. Res. 2016, 9, 598–606. [Google Scholar] [CrossRef] [PubMed]
- Nicholl, I.D.; Bucala, R. Advanced glycation endproducts and cigarette smoking. Cell. Mol. Biol. 1998, 44, 1025–1033. [Google Scholar] [PubMed]
- Cerami, C.; Founds, H.; Nicholl, I.; Mitsuhashi, T.; Giordano, D.; Vanpatten, S.; Lee, A.; Al-Abed, Y.; Vlassara, H.; Bucala, R.; et al. Tobacco smoke is a source of toxic reactive glycation products. Proc. Natl. Acad. Sci. USA 1997, 94, 13915–13920. [Google Scholar] [CrossRef] [PubMed]
- Robinson, A.B.; Stogsdill, J.A.; Lewis, J.B.; Wood, T.T.; Reynolds, P.R. Rage and tobacco smoke: Insights into modeling chronic obstructive pulmonary disease. Front. Physiol. 2012, 3, 301. [Google Scholar] [CrossRef] [PubMed]
- Prasad, K.; Dhar, I.; Caspar-Bell, G. Role of advanced glycation end products and its receptors in the pathogenesis of cigarette smoke-induced cardiovascular disease. Int. J. Angiol. 2015, 24, 75–80. [Google Scholar] [PubMed]
- Malik, P.; Chaudhry, N.; Mittal, R.; Mukherjee, T.K. Role of receptor for advanced glycation end products in the complication and progression of various types of cancers. Biochim. Biophys. Acta 2015, 1850, 1898–1904. [Google Scholar] [CrossRef] [PubMed]
- Nowotny, K.; Jung, T.; Hohn, A.; Weber, D.; Grune, T. Advanced glycation end products and oxidative stress in type 2 diabetes mellitus. Biomolecules 2015, 5, 194–222. [Google Scholar] [CrossRef] [PubMed]
- Espinet, C.; Gonzalo, H.; Fleitas, C.; Menal, M.J.; Egea, J. Oxidative stress and neurodegenerative diseases: A neurotrophic approach. Curr. Drug Targets 2015, 16, 20–30. [Google Scholar] [CrossRef] [PubMed]
- Guedes-Martins, L.; Matos, L.; Soares, A.; Silva, E.; Almeida, H. Ages, contributors to placental bed vascular changes leading to preeclampsia. Free Radic. Res. 2013, 47 (Suppl. S1), 70–80. [Google Scholar] [CrossRef] [PubMed]
- Alexander, K.L.; Mejia, C.A.; Jordan, C.; Nelson, M.B.; Howell, B.M.; Jones, C.M.; Reynolds, P.R.; Arroyo, J.A. Differential receptor for advanced glycation end products expression in preeclamptic, intrauterine growth restricted, and gestational diabetic placentas. Am. J. Reprod. Immunol. 2016, 75, 172–180. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.J.; Chan, D.C.; Chiang, C.K.; Wang, C.C.; Yang, T.H.; Lan, K.C.; Chao, S.C.; Tsai, K.S.; Yang, R.S.; Liu, S.H. Advanced glycation end-products induced VEGF production and inflammatory responses in human synoviocytes via RAGE-NF-κB pathway activation. J. Orthop. Res. 2015, 34, 791–800. [Google Scholar] [CrossRef] [PubMed]
- Van Puyvelde, K.; Mets, T.; Njemini, R.; Beyer, I.; Bautmans, I. Effect of advanced glycation end product intake on inflammation and aging: A systematic review. Nutr. Rev. 2014, 72, 638–650. [Google Scholar] [CrossRef] [PubMed]
- Ray, R.; Juranek, J.K.; Rai, V. RAGE axis in neuroinflammation, neurodegeneration and its emerging role in the pathogenesis of amyotrophic lateral sclerosis. Neurosci. Biobehav. Rev. 2016, 62, 48–55. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, Z.A.; Armour, C.L.; Phipps, S.; Sukkar, M.B. RAGE and TLRS: Relatives, friends or neighbours? Mol. Immunol. 2013, 56, 739–744. [Google Scholar] [CrossRef] [PubMed]
- Centers for Disease Control and Prevention (US). The Health Consequences of Involuntary Exposure to Tobacco Smoke: A Report of the Surgeon General; Centers for Disease Control and Prevention (US): Atlanta, GA, USA, 2006.
- Grimmer, G.; Naujack, K.W.; Dettbarn, G. Gaschromatographic determination of polycyclic aromatic hydrocarbons, aza-arenes, aromatic amines in the particle and vapor phase of mainstream and sidestream smoke of cigarettes. Toxicol. Lett. 1987, 35, 117–124. [Google Scholar] [CrossRef]
- Evans, W.H.; Thomas, N.C.; Boardman, M.C.; Nash, S.J. Relationships of polycyclic aromatic hydrocarbon yields with particulate matter (water and nicotine free) yields in mainstream and sidestream cigarette smoke. Sci. Total Environ. 1993, 136, 101–109. [Google Scholar] [CrossRef]
- Brunnemann, K.D.; Yu, L.; Hoffmann, D. Assessment of carcinogenic volatile N-nitrosamines in tobacco and in mainstream and sidestream smoke from cigarettes. Cancer Res. 1977, 37, 3218–3222. [Google Scholar] [PubMed]
- Ruhl, C.; Adams, J.D.; Hoffmann, D. Chemical studies on tobacco-specific N-nitrosamines in the smoke of selected cigarettes from the USA, west germany, and france. J. Anal. Toxicol. 1980, 4, 255–259. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, D.; Adams, J.D.; Brunnemann, K.D.; Hecht, S.S. Assessment of tobacco-specific N-nitrosamines in tobacco products. Cancer Res. 1979, 39 Pt 1, 2505–2509. [Google Scholar] [PubMed]
- Patrianakos, C.; Hoffmann, D. Chemical studies on tobacco smoke LXIV. On the analysis of aromatic amines in cigarette smoke. J. Anal. Toxicol. 1979, 3, 150–154. [Google Scholar] [CrossRef]
- Dong, M.; Schmeltz, I.; Jacobs, E.; Hoffmann, D. Aza-arenes in tobacco smoke. J. Anal. Toxicol. 1978, 2, 21–25. [Google Scholar] [CrossRef]
- Hoffmann, D.; Adams, J.D.; Wynder, E.L. Formation and analysis of carbon monoxide in cigarette mainstream and sidestream smoke. Prev. Med. 1979, 8, 344–350. [Google Scholar] [CrossRef]
- Krzych-Falta, E.; Modzelewska, D.; Samolinski, B. Levels of exhaled carbon monoxide in healthy active and passive smokers. Przegl. Lek. 2015, 72, 99–102. [Google Scholar] [PubMed]
- Daher, N.; Saleh, R.; Jaroudi, E.; Sheheitli, H.; Badr, T.; Sepetdjian, E.; Al Rashidi, M.; Saliba, N.; Shihadeh, A. Comparison of carcinogen, carbon monoxide, and ultrafine particle emissions from narghile waterpipe and cigarette smoking: Sidestream smoke measurements and assessment of second-hand smoke emission factors. Atmos. Environ. 2010, 44, 8–14. [Google Scholar] [CrossRef] [PubMed]
- Rickert, W.S.; Robinson, J.C.; Collishaw, N. Yields of tar, nicotine, and carbon monoxide in the sidestream smoke from 15 brands of Canadian cigarettes. Am. J. Public Health 1984, 74, 228–231. [Google Scholar] [CrossRef] [PubMed]
- Pakhale, S.; Maru, G. Distribution of major and minor alkaloids in tobacco, mainstream and sidestream smoke of popular Indian smoking products. Food Chem. Toxicol. 1998, 36, 1131–1138. [Google Scholar] [CrossRef]
- Brunnemann, K.D.; Hoffmann, D. Chemical studies on tobacco smoke XXXIV. Gas chromatographic determination of ammonia in cigarette and cigar smoke. J. Chromatogr. Sci. 1975, 13, 159–163. [Google Scholar] [CrossRef] [PubMed]
- Johnson, W.; Hale, R.; Clough, S.; Chen, P. Chemistry of the conversion of nitrate nitrogen to smoke products. Nature 1973, 243, 223–225. [Google Scholar] [CrossRef] [PubMed]
- Brunnemann, K.; Hoffmann, D. Chemical studies on tobacco smoke LIX. Analysis of volatile nitrosamines in tobacco smoke and polluted indoor environments. IARC Sci. Publ. 1977, 19, 343–356. [Google Scholar]
- Brunnemann, K.D.; Kagan, M.R.; Cox, J.E.; Hoffmann, D. Analysis of 1,3-butadiene and other selected gas-phase components in cigarette mainstream and sidestream smoke by gas chromatography-mass selective detection. Carcinogenesis 1990, 11, 1863–1868. [Google Scholar] [CrossRef] [PubMed]
- Ganjre, A.P.; Sarode, G.S. Third hand smoke—A hidden demon. Oral Oncol. 2016, 54, e3–e4. [Google Scholar] [CrossRef] [PubMed]
- Sleiman, M.; Gundel, L.A.; Pankow, J.F.; Jacob, P., 3rd; Singer, B.C.; Destaillats, H. Formation of carcinogens indoors by surface-mediated reactions of nicotine with nitrous acid, leading to potential thirdhand smoke hazards. Proc. Natl. Acad. Sci. USA 2010, 107, 6576–6581. [Google Scholar] [CrossRef] [PubMed]
- Ferrante, G.; Simoni, M.; Cibella, F.; Ferrara, F.; Liotta, G.; Malizia, V.; Corsello, G.; Viegi, G.; La Grutta, S. Third-hand smoke exposure and health hazards in children. Monaldi Arch. Chest. Dis. 2013, 79, 38–43. [Google Scholar] [CrossRef] [PubMed]
- Casey, G. Copd: Obstructed lungs. Nurs. N. Z. 2016, 22, 20–24. [Google Scholar] [PubMed]
- Hagstad, S.; Bjerg, A.; Ekerljung, L.; Backman, H.; Lindberg, A.; Ronmark, E.; Lundback, B. Passive smoking exposure is associated with increased risk of COPD in never smokers. Chest 2014, 145, 1298–1304. [Google Scholar] [CrossRef] [PubMed]
- Lomborg, B. Global Problems, Smart Solutions: Costs and Benefits; Cambridge University Press: Cambridge, UK, 2013. [Google Scholar]
- Rennard, S.I. Treatment of stable chronic obstructive pulmonary disease. Lancet 2004, 364, 791–802. [Google Scholar] [CrossRef]
- Sutherland, E.R.; Cherniack, R.M. Management of chronic obstructive pulmonary disease. N. Engl. J. Med. 2004, 350, 2689–2697. [Google Scholar] [CrossRef] [PubMed]
- Hogg, J.C.; Chu, F.; Utokaparch, S.; Woods, R.; Elliott, W.M.; Buzatu, L.; Cherniack, R.M.; Rogers, R.M.; Sciurba, F.C.; Coxson, H.O.; et al. The nature of small-airway obstruction in chronic obstructive pulmonary disease. N. Engl. J. Med. 2004, 350, 2645–2653. [Google Scholar] [CrossRef] [PubMed]
- Barnes, P.J. Small airways in COPD. N. Engl. J. Med. 2004, 350, 2635–2637. [Google Scholar] [CrossRef] [PubMed]
- Barnes, P.J.; Shapiro, S.D.; Pauwels, R.A. Chronic obstructive pulmonary disease: Molecular and cellular mechanisms. Eur. Respir. J. 2003, 22, 672–688. [Google Scholar] [CrossRef] [PubMed]
- Dawkins, P.A.; Stockley, R.A. Animal models of chronic obstructive pulmonary disease. Thorax 2001, 56, 972–977. [Google Scholar] [CrossRef] [PubMed]
- Shapiro, S.D. Animal models for COPD. Chest 2000, 117 (Suppl. S1), 223S–227S. [Google Scholar] [CrossRef] [PubMed]
- Mortaz, E.; Adcock, I.A. Limitation of COPD studies in animal modeling. Tanaffos 2012, 11, 7–8. [Google Scholar] [PubMed]
- Ravi, A.K.; Khurana, S.; Lemon, J.; Plumb, J.; Booth, G.; Healy, L.; Catley, M.; Vestbo, J.; Singh, D. Increased levels of soluble interleukin-6 receptor and CCL3 in COPD sputum. Respir. Res. 2014, 15, 103. [Google Scholar] [CrossRef] [PubMed]
- Menzies, D.; Nair, A.; Williamson, P.A.; Schembri, S.; Al-Khairalla, M.Z.; Barnes, M.; Fardon, T.C.; McFarlane, L.; Magee, G.J.; Lipworth, B.J. Respiratory symptoms, pulmonary function, and markers of inflammation among bar workers before and after a legislative ban on smoking in public places. JAMA 2006, 296, 1742–1748. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Yang, S.; Wu, X.; Zhao, J.; Zhao, J.; Ning, Q.; Xu, Y.; Xie, J. Interleukin-33/ST2 signaling promotes production of interleukin-6 and interleukin-8 in systemic inflammation in cigarette smoke-induced chronic obstructive pulmonary disease mice. Biochem. Biophys. Res. Commun. 2014, 450, 110–116. [Google Scholar] [CrossRef] [PubMed]
- Hubeau, C.; Kubera, J.E.; Masek-Hammerman, K.; Williams, C.M. Interleukin-6 neutralization alleviates pulmonary inflammation in mice exposed to cigarette smoke and poly(I:C). Clin. Sci. 2013, 125, 483–493. [Google Scholar] [CrossRef] [PubMed]
- Overbeek, S.A.; Braber, S.; Koelink, P.J.; Henricks, P.A.; Mortaz, E.; LoTam Loi, A.T.; Jackson, P.L.; Garssen, J.; Wagenaar, G.T.; Timens, W.; et al. Cigarette smoke-induced collagen destruction; key to chronic neutrophilic airway inflammation? PLoS ONE 2013, 8, e55612. [Google Scholar] [CrossRef]
- Kobayashi, S.D.; DeLeo, F.R. Role of neutrophils in innate immunity: A systems biology-level approach. Wiley Interdiscip. Rev. Syst. Biol. Med. 2009, 1, 309–333. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, S.D.; Voyich, J.M.; Burlak, C.; DeLeo, F.R. Neutrophils in the innate immune response. Arch. Immunol. Ther. Exp. (Warsz.) 2005, 53, 505–517. [Google Scholar]
- Keatings, V.M.; Collins, P.D.; Scott, D.M.; Barnes, P.J. Differences in interleukin-8 and tumor necrosis factor-alpha in induced sputum from patients with chronic obstructive pulmonary disease or asthma. Am. J. Respir. Crit. Care Med. 1996, 153, 530–534. [Google Scholar] [CrossRef] [PubMed]
- Tanino, M.; Betsuyaku, T.; Takeyabu, K.; Tanino, Y.; Yamaguchi, E.; Miyamoto, K.; Nishimura, M. Increased levels of interleukin-8 in BAL fluid from smokers susceptible to pulmonary emphysema. Thorax 2002, 57, 405–411. [Google Scholar] [CrossRef] [PubMed]
- Beeh, K.M.; Kornmann, O.; Buhl, R.; Culpitt, S.V.; Giembycz, M.A.; Barnes, P.J. Neutrophil chemotactic activity of sputum from patients with COPD: Role of interleukin 8 and leukotriene b4. Chest 2003, 123, 1240–1247. [Google Scholar] [CrossRef] [PubMed]
- Hodge, S.; Hodge, G.; Holmes, M.; Reynolds, P.N. Increased airway epithelial and t-cell apoptosis in COPD remains despite smoking cessation. Eur. Respir. J. 2005, 25, 447–454. [Google Scholar] [CrossRef] [PubMed]
- Thacker, E.L. Lung inflammatory responses. Vet. Res. 2006, 37, 469–486. [Google Scholar] [CrossRef] [PubMed]
- Churg, A.; Zhou, S.; Wang, X.; Wang, R.; Wright, J.L. The role of interleukin-1β in murine cigarette smoke-induced emphysema and small airway remodeling. Am. J. Respir. Cell Mol. Biol. 2009, 40, 482–490. [Google Scholar] [CrossRef] [PubMed]
- Barnes, P.J.; Karin, M. Nuclear factor-κB: A pivotal transcription factor in chronic inflammatory diseases. N. Engl. J. Med. 1997, 336, 1066–1071. [Google Scholar] [CrossRef]
- Merghani, T.H.; Saeed, A.; Alawad, A. Changes in plasma IL4, TNFA and CRP in response to regular passive smoking at home among healthy school children in khartoum, Sudan. Afr. Health Sci. 2012, 12, 41–47. [Google Scholar] [PubMed]
- Flouris, A.D.; Metsios, G.S.; Carrillo, A.E.; Jamurtas, A.Z.; Stivaktakis, P.D.; Tzatzarakis, M.N.; Tsatsakis, A.M.; Koutedakis, Y. Respiratory and immune response to maximal physical exertion following exposure to secondhand smoke in healthy adults. PLoS ONE 2012, 7, e31880. [Google Scholar] [CrossRef]
- Aldonyte, R.; Jansson, L.; Piitulainen, E.; Janciauskiene, S. Circulating monocytes from healthy individuals and COPD patients. Respir. Res. 2003, 4, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Finkelstein, R.; Fraser, R.S.; Ghezzo, H.; Cosio, M.G. Alveolar inflammation and its relation to emphysema in smokers. Am. J. Respir. Crit. Care Med. 1995, 152 Pt 1, 1666–1672. [Google Scholar] [CrossRef] [PubMed]
- Shapiro, S.D. The macrophage in chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 1999, 160, S29–S32. [Google Scholar] [CrossRef] [PubMed]
- Tsuji, H.; Fujimoto, H.; Lee, K.M.; Renne, R.; Iwanaga, A.; Okubo, C.; Onami, S.; Nomura, A.K.; Nishino, T.; Yoshimura, H. Characterization of biochemical, functional and structural changes in mice respiratory organs chronically exposed to cigarette smoke. Inhal. Toxicol. 2015, 27, 342–353. [Google Scholar] [CrossRef] [PubMed]
- Di Stefano, A.; Capelli, A.; Lusuardi, M.; Balbo, P.; Vecchio, C.; Maestrelli, P.; Mapp, C.E.; Fabbri, L.M.; Donner, C.F.; Saetta, M. Severity of airflow limitation is associated with severity of airway inflammation in smokers. Am. J. Respir. Crit. Care Med. 1998, 158, 1277–1285. [Google Scholar] [CrossRef] [PubMed]
- Majo, J.; Ghezzo, H.; Cosio, M.G. Lymphocyte population and apoptosis in the lungs of smokers and their relation to emphysema. Eur. Respir. J. 2001, 17, 946–953. [Google Scholar] [CrossRef] [PubMed]
- Podolin, P.L.; Foley, J.P.; Carpenter, D.C.; Bolognese, B.J.; Logan, G.A.; Long, E., 3rd; Harrison, O.J.; Walsh, P.T. T-cell depletion protects against alveolar destruction due to chronic cigarette smoke exposure in mice. Am. J. Physiol. Lung Cell. Mol. Physiol. 2013, 304, L312–L323. [Google Scholar] [CrossRef] [PubMed]
- Pfeifer, G.P.; Denissenko, M.F.; Olivier, M.; Tretyakova, N.; Hecht, S.S.; Hainaut, P. Tobacco smoke carcinogens, DNA damage and p53 mutations in smoking-associated cancers. Oncogene 2002, 21, 7435–7451. [Google Scholar] [CrossRef] [PubMed]
- Danaei, G.; Vander Hoorn, S.; Lopez, A.D.; Murray, C.J.; Ezzati, M. Causes of cancer in the world: Comparative risk assessment of nine behavioural and environmental risk factors. Lancet 2005, 366, 1784–1793. [Google Scholar] [CrossRef]
- Siegel, R.; Ma, J.; Zou, Z.; Jemal, A. Cancer statistics, 2014. CA Cancer J. Clin. 2014, 64, 9–29. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.S.; Vos, T.; Flaxman, A.D.; Danaei, G.; Shibuya, K.; Adair-Rohani, H.; Amann, M.; Anderson, H.R.; Andrews, K.G.; Aryee, M.; et al. A comparative risk assessment of burden of disease and injury attributable to 67 risk factors and risk factor clusters in 21 regions, 1990–2010: A systematic analysis for the global burden of disease study 2010. Lancet 2012, 380, 2224–2260. [Google Scholar] [CrossRef]
- Agudo, A.; Bonet, C.; Travier, N.; Gonzalez, C.A.; Vineis, P.; Bueno-de-Mesquita, H.B.; Trichopoulos, D.; Boffetta, P.; Clavel-Chapelon, F.; Boutron-Ruault, M.C.; et al. Impact of cigarette smoking on cancer risk in the European prospective investigation into cancer and nutrition study. J. Clin. Oncol. 2012, 30, 4550–4557. [Google Scholar] [CrossRef] [PubMed]
- Hori, M.; Tanaka, H.; Wakai, K.; Sasazuki, S.; Katanoda, K. Secondhand smoke exposure and risk of lung cancer in Japan: A systematic review and meta-analysis of epidemiologic studies. Jpn. J. Clin. Oncol. 2016, 46, 942–951. [Google Scholar] [CrossRef] [PubMed]
- Malik, A.; Jeyaraj, P.A.; Shankar, A.; Rath, G.K.; Mukhopadhyay, S.; Kamal, V.K. Passive smoking and breast cancer—A suspicious link. Asian Pac. J. Cancer Prev. 2015, 16, 5715–5719. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.H.; Hong, J.Y.; Lee, J.U.; Lee, D.R. Association between exposure to environmental tobacco smoke at the workplace and risk for developing a colorectal adenoma: A cross-sectional study. Ann. Coloproctol. 2016, 32, 51–57. [Google Scholar] [CrossRef] [PubMed]
- Shekari, M.; Kordi-Tamandani, D.M.; MalekZadeh, K.; Sobti, R.C.; Karimi, S.; Suri, V. Effect of anti-inflammatory (IL-4, IL-10) cytokine genes in relation to risk of cervical carcinoma. Am. J. Clin. Oncol. 2012, 35, 514–519. [Google Scholar] [CrossRef] [PubMed]
- Vrieling, A.; Bueno-de-Mesquita, H.B.; Boshuizen, H.C.; Michaud, D.S.; Severinsen, M.T.; Overvad, K.; Olsen, A.; Tjonneland, A.; Clavel-Chapelon, F.; Boutron-Ruault, M.C.; et al. Cigarette smoking, environmental tobacco smoke exposure and pancreatic cancer risk in the european prospective investigation into cancer and nutrition. Int. J. Cancer 2010, 126, 2394–2403. [Google Scholar] [CrossRef] [PubMed]
- Sleiman, M.; Maddalena, R.L.; Gundel, L.A.; Destaillats, H. Rapid and sensitive gas chromatography-ion-trap tandem mass spectrometry method for the determination of tobacco-specific N-nitrosamines in secondhand smoke. J. Chromatogr. A 2009, 1216, 7899–7905. [Google Scholar] [CrossRef] [PubMed]
- Church, T.R.; Anderson, K.E.; Caporaso, N.E.; Geisser, M.S.; Le, C.T.; Zhang, Y.; Benoit, A.R.; Carmella, S.G.; Hecht, S.S. A prospectively measured serum biomarker for a tobacco-specific carcinogen and lung cancer in smokers. Cancer Epidemiol. Biomark. Prev. 2009, 18, 260–266. [Google Scholar] [CrossRef] [PubMed]
- Balbo, S.; James-Yi, S.; Johnson, C.S.; O’Sullivan, M.G.; Stepanov, I.; Wang, M.; Bandyopadhyay, D.; Kassie, F.; Carmella, S.; Upadhyaya, P.; et al. (S)-N′-nitrosonornicotine, a constituent of smokeless tobacco, is a powerful oral cavity carcinogen in rats. Carcinogenesis 2013, 34, 2178–2183. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Jasso, E.; Lopez, T.; Lucas, D.; Berthou, F.; Manno, M.; Ortega, A.; Albores, A. CYP2E1 regulation by benzene and other small organic chemicals in rat liver and peripheral lymphocytes. Toxicol. Lett. 2003, 144, 55–67. [Google Scholar] [CrossRef]
- Joshi, M.; Tyndale, R.F. Induction and recovery time course of rat brain cyp2e1 after nicotine treatment. Drug Metab. Dispos. 2006, 34, 647–652. [Google Scholar] [CrossRef] [PubMed]
- Guengerich, F.P.; Shimada, T. Oxidation of toxic and carcinogenic chemicals by human cytochrome p-450 enzymes. Chem. Res. Toxicol. 1991, 4, 391–407. [Google Scholar] [CrossRef] [PubMed]
- Lieber, C.S. Cytochrome P-4502E1: Its physiological and pathological role. Physiol. Rev. 1997, 77, 517–544. [Google Scholar] [PubMed]
- Kase, S.; Sugio, K.; Yamazaki, K.; Okamoto, T.; Yano, T.; Sugimachi, K. Expression of E-cadherin and β-catenin in human non-small cell lung cancer and the clinical significance. Clin. Cancer Res. 2000, 6, 4789–4796. [Google Scholar] [CrossRef]
- Hecht, S.S. Tobacco smoke carcinogens and lung cancer. J. Natl. Cancer Inst. 1999, 91, 1194–1210. [Google Scholar] [CrossRef] [PubMed]
- Hecht, S.S. Tobacco carcinogens, their biomarkers and tobacco-induced cancer. Nat. Rev. Cancer 2003, 3, 733–744. [Google Scholar] [CrossRef] [PubMed]
- Hecht, S.S. Lung carcinogenesis by tobacco smoke. Int. J. Cancer 2012, 131, 2724–2732. [Google Scholar] [CrossRef] [PubMed]
- Thompson, C.A.; Burcham, P.C. Genome-wide transcriptional responses to acrolein. Chem. Res. Toxicol. 2008, 21, 2245–2256. [Google Scholar] [CrossRef] [PubMed]
- Cho, W.C.; Kwan, C.K.; Yau, S.; So, P.P.; Poon, P.C.; Au, J.S. The role of inflammation in the pathogenesis of lung cancer. Expert Opin. Ther. Targets 2011, 15, 1127–1137. [Google Scholar] [CrossRef] [PubMed]
- Erreni, M.; Mantovani, A.; Allavena, P. Tumor-associated macrophages (TAM) and inflammation in colorectal cancer. Cancer Microenviron. 2011, 4, 141–154. [Google Scholar] [CrossRef] [PubMed]
- Jang, J.Y.; Lee, J.K.; Jeon, Y.K.; Kim, C.W. Exosome derived from epigallocatechin gallate treated breast cancer cells suppresses tumor growth by inhibiting tumor-associated macrophage infiltration and M2 polarization. BMC Cancer 2013, 13, 421. [Google Scholar] [CrossRef] [PubMed]
- Mano, Y.; Aishima, S.; Fujita, N.; Tanaka, Y.; Kubo, Y.; Motomura, T.; Taketomi, A.; Shirabe, K.; Maehara, Y.; Oda, Y. Tumor-associated macrophage promotes tumor progression via STAT3 signaling in hepatocellular carcinoma. Pathobiology 2013, 80, 146–154. [Google Scholar] [CrossRef] [PubMed]
- Ho, C.C.; Liao, W.Y.; Wang, C.Y.; Lu, Y.H.; Huang, H.Y.; Chen, H.Y.; Chan, W.K.; Chen, H.W.; Yang, P.C. TREM-1 expression in tumor-associated macrophages and clinical outcome in lung cancer. Am. J. Respir. Crit. Care Med. 2008, 177, 763–770. [Google Scholar] [CrossRef] [PubMed]
- Ala-aho, R.; Kahari, V.M. Collagenases in cancer. Biochimie 2005, 87, 273–286. [Google Scholar] [CrossRef] [PubMed]
- Stamenkovic, I. Matrix metalloproteinases in tumor invasion and metastasis. Semin. Cancer Biol. 2000, 10, 415–433. [Google Scholar] [CrossRef] [PubMed]
- Stamenkovic, I. Extracellular matrix remodelling: The role of matrix metalloproteinases. J. Pathol. 2003, 200, 448–464. [Google Scholar] [CrossRef] [PubMed]
- Ozturk, F.; Sheldon, E.; Sharma, J.; Canturk, K.M.; Otu, H.H.; Nawshad, A. Nicotine exposure during pregnancy results in persistent midline epithelial seam with improper palatal fusion. Nicotine Tob. Res. 2016, 18, 604–612. [Google Scholar] [CrossRef] [PubMed]
- Arffin, F.; Al-Bayaty, F.H.; Hassan, J. Environmental tobacco smoke and stress as risk factors for miscarriage and preterm births. Arch. Gynecol. Obstet. 2012, 286, 1187–1191. [Google Scholar] [CrossRef] [PubMed]
- National Center for Chronic Disease Prevention and Health Promotion (US) Office on Smoking and Health. The Health Consequences of Smoking—50 Years of Progress: A Report of the Surgeon General; US Department of Health and Human Services, Centers for Disease Control and Prevention, National Center for Chronic Disease Prevention and Health Promotion, Office on Smoking and Health: Atlanta, GA, USA, 2014; Volume 17.
- Caspers, K.M.; Romitti, P.A.; Lin, S.; Olney, R.S.; Holmes, L.B.; Werler, M.M. Maternal periconceptional exposure to cigarette smoking and congenital limb deficiencies. Paediatr. Perinat. Epidemiol. 2013, 27, 509–520. [Google Scholar] [CrossRef] [PubMed]
- Gianicolo, E.A.; Cresci, M.; Ait-Ali, L.; Foffa, I.; Andreassi, M.G. Smoking and congenital heart disease: The epidemiological and biological link. Curr. Pharm. Des. 2010, 16, 2572–2577. [Google Scholar] [CrossRef] [PubMed]
- Salmasi, G.; Grady, R.; Jones, J.; McDonald, S.D. Environmental tobacco smoke exposure and perinatal outcomes: A systematic review and meta-analyses. Acta Obstet. Gynecol. Scand. 2010, 89, 423–441. [Google Scholar] [CrossRef] [PubMed]
- Dietz, P.M.; England, L.J.; Shapiro-Mendoza, C.K.; Tong, V.T.; Farr, S.L.; Callaghan, W.M. Infant morbidity and mortality attributable to prenatal smoking in the U.S. Am. J. Prev. Med. 2010, 39, 45–52. [Google Scholar] [CrossRef] [PubMed]
- Higgins, S.T.; Washio, Y.; Heil, S.H.; Solomon, L.J.; Gaalema, D.E.; Higgins, T.M.; Bernstein, I.M. Financial incentives for smoking cessation among pregnant and newly postpartum women. Prev. Med. 2012, 55, S33–S40. [Google Scholar] [CrossRef] [PubMed]
- Bickerstaff, M.; Beckmann, M.; Gibbons, K.; Flenady, V. Recent cessation of smoking and its effect on pregnancy outcomes. Aust. N. Z. J. Obstet. Gynaecol. 2012, 52, 54–58. [Google Scholar] [CrossRef] [PubMed]
- Akkar, O.B.; Yildiz, C.; Karakus, S.; Akkar, I.; Cetin, A.; Yanik, A.; Yenicesu, A.G.; Boztosun, A. Antenatal counseling against passive smoking may improve birth weight for gestational age. Clin. Exp. Obstet. Gynecol. 2015, 42, 805–809. [Google Scholar] [PubMed]
- Strulovici-Barel, Y.; Omberg, L.; O’Mahony, M.; Gordon, C.; Hollmann, C.; Tilley, A.E.; Salit, J.; Mezey, J.; Harvey, B.G.; Crystal, R.G. Threshold of biologic responses of the small airway epithelium to low levels of tobacco smoke. Am. J. Respir. Crit. Care Med. 2010, 182, 1524–1532. [Google Scholar] [CrossRef] [PubMed]
- Hellstrom-Lindahl, E.; Gorbounova, O.; Seiger, A.; Mousavi, M.; Nordberg, A. Regional distribution of nicotinic receptors during prenatal development of human brain and spinal cord. Brain Res. Dev. Brain Res. 1998, 108, 147–160. [Google Scholar] [CrossRef]
- Dwyer, J.B.; McQuown, S.C.; Leslie, F.M. The dynamic effects of nicotine on the developing brain. Pharmacol. Ther. 2009, 122, 125–139. [Google Scholar] [CrossRef] [PubMed]
- Luck, W.; Nau, H.; Hansen, R.; Steldinger, R. Extent of nicotine and cotinine transfer to the human fetus, placenta and amniotic fluid of smoking mothers. Dev. Pharmacol. Ther. 1985, 8, 384–395. [Google Scholar] [PubMed]
- Slotkin, T.A. Fetal nicotine or cocaine exposure: Which one is worse? J. Pharmacol. Exp. Ther. 1998, 285, 931–945. [Google Scholar] [PubMed]
- Slotkin, T.A.; Cho, H.; Whitmore, W.L. Effects of prenatal nicotine exposure on neuronal development: Selective actions on central and peripheral catecholaminergic pathways. Brain Res. Bull. 1987, 18, 601–611. [Google Scholar] [CrossRef]
- Dwyer, J.B.; Broide, R.S.; Leslie, F.M. Nicotine and brain development. Birth Defects Res. C Embryo Today 2008, 84, 30–44. [Google Scholar] [CrossRef] [PubMed]
- Palmer, R.H.; Bidwell, L.C.; Heath, A.C.; Brick, L.A.; Madden, P.A.; Knopik, V.S. Effects of maternal smoking during pregnancy on offspring externalizing problems: Contextual effects in a sample of female twins. Behav. Genet. 2016, 46, 403–415. [Google Scholar] [CrossRef] [PubMed]
- Niclasen, J.; Obel, C.; Homoe, P.; Korvel-Hanquist, A.; Dammeyer, J. Associations between otitis media and child behavioural and learning difficulties: Results from a Danish cohort. Int. J. Pediatr. Otorhinolaryngol. 2016, 84, 12–20. [Google Scholar] [CrossRef] [PubMed]
- Knopik, V.S.; Marceau, K.; Bidwell, L.C.; Palmer, R.H.; Smith, T.F.; Todorov, A.; Evans, A.S.; Heath, A.C. Smoking during pregnancy and ADHD risk: A genetically informed, multiple-rater approach. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2016, 171, 971–981. [Google Scholar] [CrossRef] [PubMed]
- De Alwis, D.; Tandon, M.; Tillman, R.; Luby, J. Nonverbal reasoning in preschool children: Investigating the putative risk of secondhand smoke exposure and attention-deficit/hyperactivity disorder as a mediator. Scand. J. Child. Adolesc. Psychiatr. Psychol. 2015, 3, 115–125. [Google Scholar] [CrossRef] [PubMed]
- Hanrahan, J.P.; Tager, I.B.; Segal, M.R.; Tosteson, T.D.; Castile, R.G.; Van Vunakis, H.; Weiss, S.T.; Speizer, F.E. The effect of maternal smoking during pregnancy on early infant lung function. Am. Rev. Respir. Dis. 1992, 145, 1129–1135. [Google Scholar] [CrossRef] [PubMed]
- Stillerman, K.P.; Mattison, D.R.; Giudice, L.C.; Woodruff, T.J. Environmental exposures and adverse pregnancy outcomes: A review of the science. Reprod. Sci. 2008, 15, 631–650. [Google Scholar] [CrossRef] [PubMed]
- Votavova, H.; Dostalova Merkerova, M.; Krejcik, Z.; Fejglova, K.; Vasikova, A.; Pastorkova, A.; Tabashidze, N.; Topinka, J.; Balascak, I.; Sram, R.J.; et al. Deregulation of gene expression induced by environmental tobacco smoke exposure in pregnancy. Nicotine Tob. Res. 2012, 14, 1073–1082. [Google Scholar] [CrossRef] [PubMed]
- Lodrup Carlsen, K.C.; Jaakkola, J.J.; Nafstad, P.; Carlsen, K.H. In utero exposure to cigarette smoking influences lung function at birth. Eur. Respir. J. 1997, 10, 1774–1779. [Google Scholar] [CrossRef] [PubMed]
- Stocks, J.; Dezateux, C. The effect of parental smoking on lung function and development during infancy. Respirology 2003, 8, 266–285. [Google Scholar] [CrossRef] [PubMed]
- Sekhon, H.S.; Jia, Y.; Raab, R.; Kuryatov, A.; Pankow, J.F.; Whitsett, J.A.; Lindstrom, J.; Spindel, E.R. Prenatal nicotine increases pulmonary α7 nicotinic receptor expression and alters fetal lung development in monkeys. J. Clin. Investig. 1999, 103, 637–647. [Google Scholar] [CrossRef] [PubMed]
- Sekhon, H.S.; Keller, J.A.; Proskocil, B.J.; Martin, E.L.; Spindel, E.R. Maternal nicotine exposure upregulates collagen gene expression in fetal monkey lung. Association with alpha7 nicotinic acetylcholine receptors. Am. J. Respir. Cell Mol. Biol. 2002, 26, 31–41. [Google Scholar] [CrossRef] [PubMed]
- Rehan, V.K.; Liu, J.; Naeem, E.; Tian, J.; Sakurai, R.; Kwong, K.; Akbari, O.; Torday, J.S. Perinatal nicotine exposure induces asthma in second generation offspring. BMC Med. 2012, 10, 129. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.F.; Langholz, B.; Salam, M.T.; Gilliland, F.D. Maternal and grandmaternal smoking patterns are associated with early childhood asthma. Chest 2005, 127, 1232–1241. [Google Scholar] [CrossRef]
- Leslie, F.M. Multigenerational epigenetic effects of nicotine on lung function. BMC Med. 2013, 11, 27. [Google Scholar] [CrossRef] [PubMed]
- Duskova, M.; Hruskovicova, H.; Simunkova, K.; Starka, L.; Parizek, A. The effects of smoking on steroid metabolism and fetal programming. J. Steroid Biochem. Mol. Biol. 2014, 139, 138–143. [Google Scholar] [CrossRef] [PubMed]
- Steptoe, A.; Ussher, M. Smoking, cortisol and nicotine. Int. J. Psychophysiol. 2006, 59, 228–235. [Google Scholar] [CrossRef] [PubMed]
- Stangenberg, S.; Chen, H.; Wong, M.G.; Pollock, C.A.; Saad, S. Fetal programming of chronic kidney disease: The role of maternal smoking, mitochondrial dysfunction, and epigenetic modfification. Am. J. Physiol. Renal Physiol. 2015, 308, F1189–F1196. [Google Scholar] [CrossRef] [PubMed]
- Kable, J.A.; Coles, C.D.; Lynch, M.E.; Carroll, J. The impact of maternal smoking on fast auditory brainstem responses. Neurotoxicol. Teratol. 2009, 31, 216–224. [Google Scholar] [CrossRef] [PubMed]
- Weitzman, M.; Govil, N.; Liu, Y.H.; Lalwani, A.K. Maternal prenatal smoking and hearing loss among adolescents. JAMA Otolaryngol. Head Neck Surg. 2013, 139, 669–677. [Google Scholar] [PubMed]
- Katbamna, B.; Klutz, N.; Pudrith, C.; Lavery, J.P.; Ide, C.F. Prenatal smoke exposure: Effects on infant auditory system and placental gene expression. Neurotoxicol. Teratol. 2013, 38, 61–71. [Google Scholar] [CrossRef] [PubMed]
- Wagijo, M.A.; Sheikh, A.; Duijts, L.; Been, J.V. Reducing tobacco smoking and smoke exposure to prevent preterm birth and its complications. Paediatr. Respir. Rev. 2015. [Google Scholar] [CrossRef] [PubMed]
- Suter, M.A.; Anders, A.M.; Aagaard, K.M. Maternal smoking as a model for environmental epigenetic changes affecting birthweight and fetal programming. Mol. Hum. Reprod. 2013, 19, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Horta, B.L.; Victora, C.G.; Menezes, A.M.; Halpern, R.; Barros, F.C. Low birthweight, preterm births and intrauterine growth retardation in relation to maternal smoking. Paediatr. Perinatal Epidemiol. 1997, 11, 140–151. [Google Scholar] [CrossRef]
- Bolehovska, P.; Sehnal, B.; Driak, D.; Halaska, M.; Magner, M.; Novotny, J.; Svandova, I. Changes in placental angiogenesis and their correlation with foetal intrauterine restriction. Ceska Gynekol. 2015, 80, 144–150. [Google Scholar] [PubMed]
- Goldenberg, R.L.; Rouse, D.J. Prevention of premature birth. N. Engl. J. Med. 1998, 339, 313–320. [Google Scholar] [CrossRef] [PubMed]
- Brar, H.S.; Rutherford, S.E. Classification of intrauterine growth retardation. Semin. Perinatol. 1988, 12, 2–10. [Google Scholar] [PubMed]
- Gray, P.H.; O’Callaghan, M.J.; Harvey, J.M.; Burke, C.J.; Payton, D.J. Placental pathology and neurodevelopment of the infant with intrauterine growth restriction. Dev. Med. Child Neurol. 1999, 41, 16–20. [Google Scholar] [CrossRef] [PubMed]
- Roger, V.L.; Go, A.S.; Lloyd-Jones, D.M.; Benjamin, E.J.; Berry, J.D.; Borden, W.B.; Bravata, D.M.; Dai, S.; Ford, E.S.; Fox, C.S.; et al. Heart disease and stroke statistics—2012 update: A report from the american heart association. Circulation 2012, 125, e2–e220. [Google Scholar] [PubMed]
- Menke, A.; Casagrande, S.; Geiss, L.; Cowie, C.C. Prevalence of and trends in diabetes among adults in the United States, 1988–2012. JAMA 2015, 314, 1021–1029. [Google Scholar] [CrossRef] [PubMed]
- Reaven, G.M. Do high carbohydrate diets prevent the development or attenuate the manifestations (or both) of syndrome X? A viewpoint strongly against. Curr. Opin. Lipidol. 1997, 8, 23–27. [Google Scholar] [CrossRef] [PubMed]
- Facchini, F.S.; Hollenbeck, C.B.; Jeppesen, J.; Chen, Y.D.; Reaven, G.M. Insulin resistance and cigarette smoking. Lancet 1992, 339, 1128–1130. [Google Scholar] [CrossRef]
- Ronnemaa, T.; Ronnemaa, E.M.; Puukka, P.; Pyorala, K.; Laakso, M. Smoking is independently associated with high plasma insulin levels in nondiabetic men. Diabetes Care 1996, 19, 1229–1232. [Google Scholar] [CrossRef] [PubMed]
- Attvall, S.; Fowelin, J.; Lager, I.; Von Schenck, H.; Smith, U. Smoking induces insulin resistance—A potential link with the insulin resistance syndrome. J. Intern. Med. 1993, 233, 327–332. [Google Scholar] [CrossRef] [PubMed]
- Thatcher, M.O.; Tippetts, T.S.; Nelson, M.B.; Swensen, A.C.; Winden, D.R.; Hansen, M.E.; Anderson, M.C.; Johnson, I.E.; Porter, J.P.; Reynolds, P.R.; et al. Ceramides mediate cigarette smoke-induced metabolic disruption in mice. Am. J. Physiol. Endocrinol. Metabol. 2014, 307, E919–E927. [Google Scholar] [CrossRef] [PubMed]
- DeFronzo, R.A.; Ferrannini, E. Insulin resistance. A multifaceted syndrome responsible for NIDDM, obesity, hypertension, dyslipidemia, and atherosclerotic cardiovascular disease. Diabetes Care 1991, 14, 173–194. [Google Scholar] [CrossRef] [PubMed]
- Ferrannini, E.; Buzzigoli, G.; Bonadonna, R.; Giorico, M.A.; Oleggini, M.; Graziadei, L.; Pedrinelli, R.; Brandi, L.; Bevilacqua, S. Insulin resistance in essential hypertension. N. Engl. J. Med. 1987, 317, 350–357. [Google Scholar] [CrossRef] [PubMed]
- Reaven, G.M. Insulin resistance and compensatory hyperinsulinemia: Role in hypertension, dyslipidemia, and coronary heart disease. Am. Heart J. 1991, 121 Pt 2, 1283–1288. [Google Scholar] [CrossRef]
- Witteles, R.M.; Tang, W.H.; Jamali, A.H.; Chu, J.W.; Reaven, G.M.; Fowler, M.B. Insulin resistance in idiopathic dilated cardiomyopathy: A possible etiologic link. J. Am. Coll. Cardiol. 2004, 44, 78–81. [Google Scholar] [CrossRef] [PubMed]
- Olefsky, J.M.; Glass, C.K. Macrophages, inflammation, and insulin resistance. Annu. Rev. Physiol. 2010, 72, 219–246. [Google Scholar] [CrossRef] [PubMed]
- Urakawa, H.; Katsuki, A.; Sumida, Y.; Gabazza, E.C.; Murashima, S.; Morioka, K.; Maruyama, N.; Kitagawa, N.; Tanaka, T.; Hori, Y.; et al. Oxidative stress is associated with adiposity and insulin resistance in men. J. Clin. Endocrinol. Metab. 2003, 88, 4673–4676. [Google Scholar] [CrossRef] [PubMed]
- Reaven, G.; Tsao, P.S. Insulin resistance and compensatory hyperinsulinemia: The key player between cigarette smoking and cardiovascular disease? J. Am. Coll. Cardiol. 2003, 41, 1044–1047. [Google Scholar] [CrossRef]
- Jeppesen, J.; Hein, H.O.; Suadicani, P.; Gyntelberg, F. Low triglycerides-high high-density lipoprotein cholesterol and risk of ischemic heart disease. Arch. Intern. Med. 2001, 161, 361–366. [Google Scholar] [CrossRef] [PubMed]
- Sijbrands, E.J.; Westendorp, R.G.; Hoffer, M.J.; Havekes, L.M.; Frants, R.R.; Meinders, A.E.; Frolich, M.; Smelt, A.H. Effect of insulin resistance, apoE2 allele, and smoking on combined hyperlipidemia. Arterioscleros. Thrombos. 1994, 14, 1576–1580. [Google Scholar] [CrossRef]
- Tahtinen, T.M.; Vanhala, M.J.; Oikarinen, J.A.; Keinanen-Kiukaanniemi, S.M. Effect of smoking on the prevalence of insulin resistance-associated cardiovascular risk factors among Finnish men in military service. J. Cardiovasc. Risk 1998, 5, 319–323. [Google Scholar] [CrossRef] [PubMed]
- Heitzer, T.; Yla-Herttuala, S.; Luoma, J.; Kurz, S.; Munzel, T.; Just, H.; Olschewski, M.; Drexler, H. Cigarette smoking potentiates endothelial dysfunction of forearm resistance vessels in patients with hypercholesterolemia. Role of oxidized LDL. Circulation 1996, 93, 1346–1353. [Google Scholar] [CrossRef] [PubMed]
- Adams, M.R.; Jessup, W.; Celermajer, D.S. Cigarette smoking is associated with increased human monocyte adhesion to endothelial cells: Reversibility with oral L-arginine but not vitamin c. J. Am. Coll. Cardiol. 1997, 29, 491–497. [Google Scholar] [CrossRef]
- Otsuka, R.; Watanabe, H.; Hirata, K.; Tokai, K.; Muro, T.; Yoshiyama, M.; Takeuchi, K.; Yoshikawa, J. Acute effects of passive smoking on the coronary circulation in healthy young adults. JAMA 2001, 286, 436–441. [Google Scholar] [CrossRef] [PubMed]
- Chen, N.G.; Holmes, M.; Reaven, G.M. Relationship between insulin resistance, soluble adhesion molecules, and mononuclear cell binding in healthy volunteers. J. Clin. Endocrinol. Metab. 1999, 84, 3485–3489. [Google Scholar] [CrossRef] [PubMed]
- Stuhlinger, M.C.; Abbasi, F.; Chu, J.W.; Lamendola, C.; McLaughlin, T.L.; Cooke, J.P.; Reaven, G.M.; Tsao, P.S. Relationship between insulin resistance and an endogenous nitric oxide synthase inhibitor. JAMA 2002, 287, 1420–1426. [Google Scholar] [CrossRef] [PubMed]
- Tippetts, T.S.; Winden, D.R.; Swensen, A.C.; Nelson, M.B.; Thatcher, M.O.; Saito, R.R.; Condie, T.B.; Simmons, K.J.; Judd, A.M.; Reynolds, P.R.; et al. Cigarette smoke increases cardiomyocyte ceramide accumulation and inhibits mitochondrial respiration. BMC Cardiovasc. Disord. 2014, 14, 165. [Google Scholar] [CrossRef] [PubMed]
- Barnoya, J.; Glantz, S.A. Cardiovascular effects of secondhand smoke: Nearly as large as smoking. Circulation 2005, 111, 2684–2698. [Google Scholar] [CrossRef] [PubMed]
- Faught, B.E.; Flouris, A.D.; Cairney, J. Epidemiological evidence associating secondhand smoke exposure with cardiovascular disease. Inflamm. Allergy Drug Targets 2009, 8, 321–327. [Google Scholar] [CrossRef] [PubMed]
- Institute of Medicine (US) Committee on Secondhand Smoke Exposure and Acute Coronary Events. Secondhand Smoke Exposure and Cardiovascular Effects: Making Sense of the Evidence; National Academy of Sciences: Washington, DC, USA, 2010. [Google Scholar]
- Law, M.R.; Morris, J.K.; Wald, N.J. Environmental tobacco smoke exposure and ischaemic heart disease: An evaluation of the evidence. BMJ 1997, 315, 973–980. [Google Scholar] [CrossRef] [PubMed]
- Rubenstein, D.A.; Morton, B.E.; Yin, W. The combined effects of sidestream smoke extracts and glycated serum albumin on endothelial cells and platelets. Cardiovasc. Diabetol. 2010, 9, 28. [Google Scholar] [CrossRef] [PubMed]
- Iso, H.; Shimamoto, T.; Sato, S.; Koike, K.; Iida, M.; Komachi, Y. Passive smoking and plasma fibrinogen concentrations. Am. J. Epidemiol. 1996, 144, 1151–1154. [Google Scholar] [CrossRef] [PubMed]
- Schmid, P.; Karanikas, G.; Kritz, H.; Pirich, C.; Stamatopoulos, Y.; Peskar, B.A.; Sinzinger, H. Passive smoking and platelet thromboxane. Thrombos. Res. 1996, 81, 451–460. [Google Scholar] [CrossRef]
- Barua, R.S.; Ambrose, J.A.; Eales-Reynolds, L.J.; deVoe, M.C.; Zervas, J.G.; Saha, D.C. Dysfunctional endothelial nitric oxide biosynthesis in healthy smokers with impaired endothelium-dependent vasodilatation. Circulation 2001, 104, 1905–1910. [Google Scholar] [CrossRef] [PubMed]
- Burke, A.; Fitzgerald, G.A. Oxidative stress and smoking-induced vascular injury. Prog. Cardiovasc. Dis. 2003, 46, 79–90. [Google Scholar] [CrossRef]
- Tribble, D.L.; Giuliano, L.J.; Fortmann, S.P. Reduced plasma ascorbic acid concentrations in nonsmokers regularly exposed to environmental tobacco smoke. Am. J. Clin. Nutr. 1993, 58, 886–890. [Google Scholar] [PubMed]
- Gotto, A.M., Jr.; Brinton, E.A. Assessing low levels of high-density lipoprotein cholesterol as a risk factor in coronary heart disease: A working group report and update. J. Am. Coll. Cardiol. 2004, 43, 717–724. [Google Scholar] [CrossRef] [PubMed]
- DeFaria Yeh, D.; Freeman, M.W.; Meigs, J.B.; Grant, R.W. Risk factors for coronary artery disease in patients with elevated high-density lipoprotein cholesterol. Am. J. Cardiol. 2007, 99, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Mack, W.J.; Islam, T.; Lee, Z.; Selzer, R.H.; Hodis, H.N. Environmental tobacco smoke and carotid arterial stiffness. Prev. Med. 2003, 37, 148–154. [Google Scholar] [CrossRef]
- El-Hakim, I.E.; Elyamani, A.O. Preliminary evaluation of histological changes found in a mechanical arthropatic temporomandibular joint (TMJ) exposed to an intra-articular hyaluronic acid (HA) injection, in a rat model. J. Cranio Maxill. Surg. 2011, 39, 610–614. [Google Scholar] [CrossRef] [PubMed]
- Abramson, S.; Krasnokutsky, S. Biomarkers in osteoarthritis. Bull. NYU Hosp. Jt. Dis. 2006, 64, 77–81. [Google Scholar] [PubMed]
- Xu, L.; Golshirazian, I.; Asbury, B.J.; Li, Y. Induction of high temperature requirement A1, a serine protease, by TGF-β1 in articular chondrocytes of mouse models of oa. Histol. Histopathol. 2014, 29, 609–618. [Google Scholar] [PubMed]
- Larkin, D.J.; Kartchner, J.Z.; Doxey, A.S.; Hollis, W.R.; Rees, J.L.; Wilhelm, S.K.; Draper, C.S.; Peterson, D.M.; Jackson, G.G.; Ingersoll, C.; et al. Inflammatory markers associated with osteoarthritis after destabilization surgery in young mice with and without receptor for advanced glycation end-products (RAGE). Front. Physiol. 2013, 4, 121. [Google Scholar] [CrossRef] [PubMed]
- Holt, D.W.; Henderson, M.L.; Stockdale, C.E.; Farrell, J.T.; Kooyman, D.L.; Bridgewater, L.C.; Seegmiller, R.E. Osteoarthritis-like changes in the heterozygous sedc mouse associated with the HtrA1-Ddr2-Mmp-13 degradative pathway: A new model of osteoarthritis. Osteoarthr. Cartil. 2012, 20, 430–439. [Google Scholar] [CrossRef] [PubMed]
- Matias, E.M.; Mecham, D.K.; Black, C.S.; Graf, J.W.; Steel, S.D.; Wilhelm, S.K.; Andersen, K.M.; Mitchell, J.A.; Macdonald, J.R.; Hollis, W.R.; et al. Malocclusion model of temporomandibular joint osteoarthritis in mice with and without receptor for advanced glycation end products. Arch. Oral Biol. 2016, 69, 47–62. [Google Scholar] [CrossRef] [PubMed]
- Mankin, H.J.; Dorfman, H.; Lippiello, L.; Zarins, A. Biochemical and metabolic abnormalities in articular cartilage from osteo-arthritic human hips. II. Correlation of morphology with biochemical and metabolic data. J. Bone Jt. Surg. Am. 1971, 53, 523–537. [Google Scholar] [CrossRef]
- Mankin, H.J. The reaction of articular cartilage to injury and osteoarthritis (first of two parts). N. Engl. J. Med. 1974, 291, 1285–1292. [Google Scholar] [CrossRef] [PubMed]
- Glasson, S.S.; Chambers, M.G.; Van Den Berg, W.B.; Little, C.B. The oarsi histopathology initiative—Recommendations for histological assessments of osteoarthritis in the mouse. Osteoarthr. Cartil. 2010, 18 (Suppl. S3), S17–S23. [Google Scholar] [CrossRef] [PubMed]
- Polur, I.; Lee, P.L.; Servais, J.M.; Xu, L.; Li, Y. Role of HTRA1, a serine protease, in the progression of articular cartilage degeneration. Histol. Histopathol. 2010, 25, 599–608. [Google Scholar] [PubMed]
- Felson, D.T.; Anderson, J.J.; Naimark, A.; Hannan, M.T.; Kannel, W.B.; Meenan, R.F. Does smoking protect against Osteo-arthritis. Arthritis Rheum. 1989, 32, 166–172. [Google Scholar] [CrossRef] [PubMed]
- Dube, C.E.; Liu, S.H.; Driban, J.B.; McAlindon, T.E.; Eaton, C.B.; Lapane, K.L. The relationship between smoking and knee osteoarthritis in the osteoarthritis initiative. Osteoarthr. Cartil. 2016, 24, 465–472. [Google Scholar] [CrossRef] [PubMed]
- Mnatzaganian, G.; Ryan, P.; Norman, P.; Davidson, D.; Hiller, J. Smoking, body weight, physical exercise and risk of lower limb total joint replacement in a population-based cohort of men. Arthritis Rheum. 2011, 63, 2523–2530. [Google Scholar] [CrossRef] [PubMed]
- Harrison, B.J.; Silman, A.J.; Wiles, N.J.; Scott, D.G.; Symmons, D.P. The association of cigarette smoking with disease outcome in patients with early inflammatory polyarthritis. Arthritis Rheum. 2001, 44, 323–330. [Google Scholar] [CrossRef]
- Kang, K.; Shin, J.S.; Lee, J.; Lee, Y.J.; Kim, M.R.; Park, K.B.; Ha, I.H. Association between direct and indirect smoking and osteoarthritis prevalence in Koreans: A cross-sectional study. BMJ Open 2016, 6, e010062. [Google Scholar] [CrossRef] [PubMed]
- Ding, C.; Cicuttini, F.; Blizzard, L.; Jones, G. Smoking interacts with family history with regard to change in knee cartilage volume and cartilage defect development. Arthritis Rheum. 2007, 56, 1521–1528. [Google Scholar] [CrossRef] [PubMed]
- Villaverde-Garcia, V.; Cobo-Ibanez, T.; Candelas-Rodriguez, G.; Seoane-Mato, D.; Campo-Fontecha, P.D.; Guerra, M.; Munoz-Fernandez, S.; Canete, J.D. The effect of smoking on clinical and structural damage in patients with axial spondyloarthritis: A systematic literature review. Semin. Arthritis Rheum. 2016. [Google Scholar] [CrossRef] [PubMed]
- Winden, D.R.; Barton, D.B.; Betteridge, B.C.; Bodine, J.S.; Jones, C.M.; Rogers, G.D.; Chavarria, M.; Wright, A.J.; Jergensen, Z.R.; Jimenez, F.R.; et al. Antenatal exposure of maternal secondhand smoke (SHS) increases fetal lung expression of rage and induces rage-mediated pulmonary inflammation. Respir. Res. 2014, 15, 129. [Google Scholar] [CrossRef] [PubMed]
- Wood, T.T.; Winden, D.R.; Marlor, D.R.; Wright, A.J.; Jones, C.M.; Chavarria, M.; Rogers, G.D.; Reynolds, P.R. Acute secondhand smoke-induced pulmonary inflammation is diminished in rage knockout mice. Am. J. Physiol. Lung Cell. Mol. Physiol. 2014, 307, L758–L764. [Google Scholar] [CrossRef] [PubMed]
- Robinson, A.B.; Johnson, K.D.; Bennion, B.G.; Reynolds, P.R. Rage signaling by alveolar macrophages influences tobacco smoke-induced inflammation. Am. J. Physiol. Lung Cell. Mol. Physiol. 2012, 302, L1192–L1199. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, P.R.; Kasteler, S.D.; Schmitt, R.E.; Hoidal, J.R. Receptor for advanced glycation end-products signals through Ras during tobacco smoke-induced pulmonary inflammation. Am. J. Respir. Cell Mol. Biol. 2011, 45, 411–418. [Google Scholar] [CrossRef] [PubMed]
- Bodine, B.G.; Bennion, B.G.; Leatham, E.; Jimenez, F.R.; Wright, A.J.; Jergensen, Z.R.; Erickson, C.J.; Jones, C.M.; Johnson, J.P.; Knapp, S.M.; et al. Conditionally induced RAGE expression by proximal airway epithelial cells in transgenic mice causes lung inflammation. Respir. Res. 2014, 15, 133. [Google Scholar] [CrossRef] [PubMed]
- Winden, D.R.; Ferguson, N.T.; Bukey, B.R.; Geyer, A.J.; Wright, A.J.; Jergensen, Z.R.; Robinson, A.B.; Stogsdill, J.A.; Reynolds, P.R. Conditional over-expression of rage by embryonic alveolar epithelium compromises the respiratory membrane and impairs endothelial cell differentiation. Respir. Res. 2013, 14, 108. [Google Scholar] [CrossRef] [PubMed]
- Stogsdill, M.P.; Stogsdill, J.A.; Bodine, B.G.; Fredrickson, A.C.; Sefcik, T.L.; Wood, T.T.; Kasteler, S.D.; Reynolds, P.R. Conditional overexpression of receptors for advanced glycation end-products in the adult murine lung causes airspace enlargement and induces inflammation. Am. J. Respir. Cell Mol. Biol. 2013, 49, 128–134. [Google Scholar] [CrossRef] [PubMed]
- Stogsdill, J.A.; Stogsdill, M.P.; Porter, J.L.; Hancock, J.M.; Robinson, A.B.; Reynolds, P.R. Embryonic overexpression of receptors for advanced glycation end-products by alveolar epithelium induces an imbalance between proliferation and apoptosis. Am. J. Respir. Cell Mol. Biol. 2012, 47, 60–66. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, P.R.; Stogsdill, J.A.; Stogsdill, M.P.; Heimann, N.B. Up-regulation of receptors for advanced glycation end-products by alveolar epithelium influences cytodifferentiation and causes severe lung hypoplasia. Am. J. Respir. Cell Mol. Biol. 2011, 45, 1195–1202. [Google Scholar] [CrossRef] [PubMed]
- Reddy, M.A.; Li, S.L.; Sahar, S.; Kim, Y.S.; Xu, Z.G.; Lanting, L.; Natarajan, R. Key role of src kinase in S100B-induced activation of the receptor for advanced glycation end products in vascular smooth muscle cells. J. Biol. Chem. 2006, 281, 13685–13693. [Google Scholar] [CrossRef] [PubMed]
- Cai, C.; Dai, X.; Zhu, Y.; Lian, M.; Xiao, F.; Dong, F.; Zhang, Q.; Huang, Y.; Zheng, Q. A specific rage-binding peptide biopanning from phage display random peptide library that ameliorates symptoms in amyloid β peptide-mediated neuronal disorder. Appl. Microbiol. Biotechnol. 2016, 100, 825–835. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.; Zhao, H.; Dong, H.; Wu, Y.; Yao, L.; Zou, F.; Cai, S. High-mobility group box 1 impairs airway epithelial barrier function through the activation of the RAGE/ERK pathway. Int. J. Mol. Med. 2016, 37, 1189–1198. [Google Scholar] [PubMed]
- Khodeer, D.M.; Zaitone, S.A.; Farag, N.E.; Moustafa, Y.M. Cardioprotective effect of pioglitazone in diabetic and non-diabetic rats subjected to acute myocardial infarction involves suppression of AGE-RAGE axis and inhibition of apoptosis. Can. J. Physiol. Pharmacol. 2016, 94, 463–476. [Google Scholar] [CrossRef] [PubMed]
- Kim, V.; Rogers, T.J.; Criner, G.J. New concepts in the pathobiology of chronic obstructive pulmonary disease. Proc. Am. Thoracic. Soc. 2008, 5, 478–485. [Google Scholar] [CrossRef] [PubMed]
- Toure, F.; Zahm, J.M.; Garnotel, R.; Lambert, E.; Bonnet, N.; Schmidt, A.M.; Vitry, F.; Chanard, J.; Gillery, P.; Rieu, P. Receptor for advanced glycation end-products (RAGE) modulates neutrophil adhesion and migration on glycoxidated extracellular matrix. Biochem. J. 2008, 416, 255–261. [Google Scholar] [CrossRef] [PubMed]
- Hudson, B.I.; Kalea, A.Z.; Del Mar Arriero, M.; Harja, E.; Boulanger, E.; D’Agati, V.; Schmidt, A.M. Interaction of the rage cytoplasmic domain with diaphanous-1 is required for ligand-stimulated cellular migration through activation of Rac1 and Cdc42. J. Biol. Chem. 2008, 283, 34457–34468. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, A.M.; Yan, S.D.; Yan, S.F.; Stern, D.M. The multiligand receptor rage as a progression factor amplifying immune and inflammatory responses. J. Clin. Investig. 2001, 108, 949–955. [Google Scholar] [CrossRef] [PubMed]
- Bianchi, R.; Giambanco, I.; Donato, R. S100B/RAGE-dependent activation of microglia via NF-κB and AP-1 Co-regulation of COX-2 expression by S100B, IL-1β and TNF-α. Neurobiol. Aging 2010, 31, 665–677. [Google Scholar] [CrossRef] [PubMed]
- Lin, L.; Park, S.; Lakatta, E.G. Rage signaling in inflammation and arterial aging. Front. Biosci. 2009, 14, 1403–1413. [Google Scholar] [CrossRef]
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lewis, J.B.; Hirschi, K.M.; Arroyo, J.A.; Bikman, B.T.; Kooyman, D.L.; Reynolds, P.R. Plausible Roles for RAGE in Conditions Exacerbated by Direct and Indirect (Secondhand) Smoke Exposure. Int. J. Mol. Sci. 2017, 18, 652. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms18030652
Lewis JB, Hirschi KM, Arroyo JA, Bikman BT, Kooyman DL, Reynolds PR. Plausible Roles for RAGE in Conditions Exacerbated by Direct and Indirect (Secondhand) Smoke Exposure. International Journal of Molecular Sciences. 2017; 18(3):652. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms18030652
Chicago/Turabian StyleLewis, Joshua B., Kelsey M. Hirschi, Juan A. Arroyo, Benjamin T. Bikman, David L. Kooyman, and Paul R. Reynolds. 2017. "Plausible Roles for RAGE in Conditions Exacerbated by Direct and Indirect (Secondhand) Smoke Exposure" International Journal of Molecular Sciences 18, no. 3: 652. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms18030652