
Transcriptome Sequencing of Dianthus spiculifolius and Analysis of the Genes Involved in Responses to Combined Cold and Drought Stress

 ,
,
Abstract
:1. Introduction
2. Results and Discussion
2.1. Transcriptome Sequencing of D. spiculifolius Subjected to Cold, Drought, and Combined Cold and Drought Stress
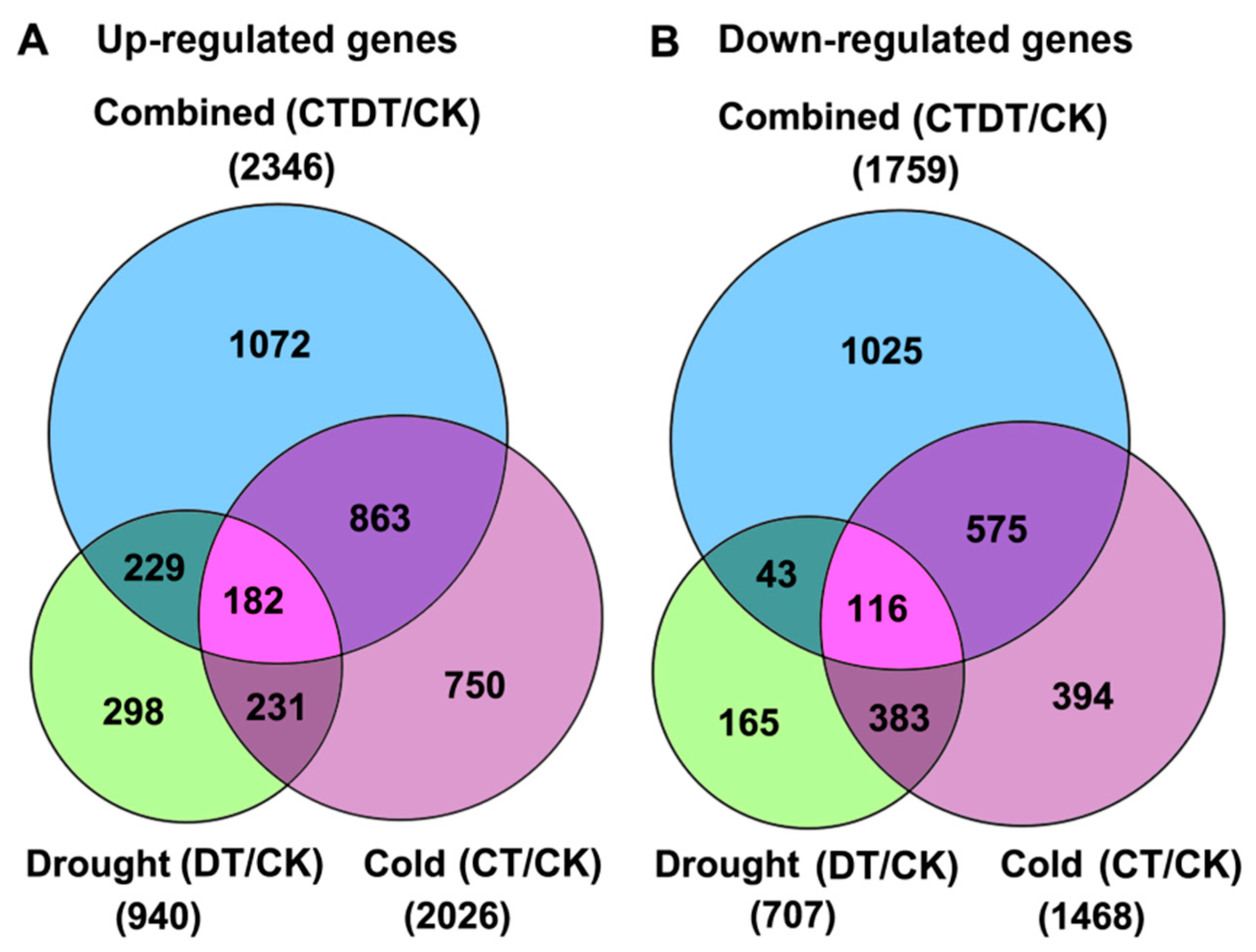
2.2. Identification of Differentially Expressed Genes (DEGs) Responding to Combined Cold and Drought Stress
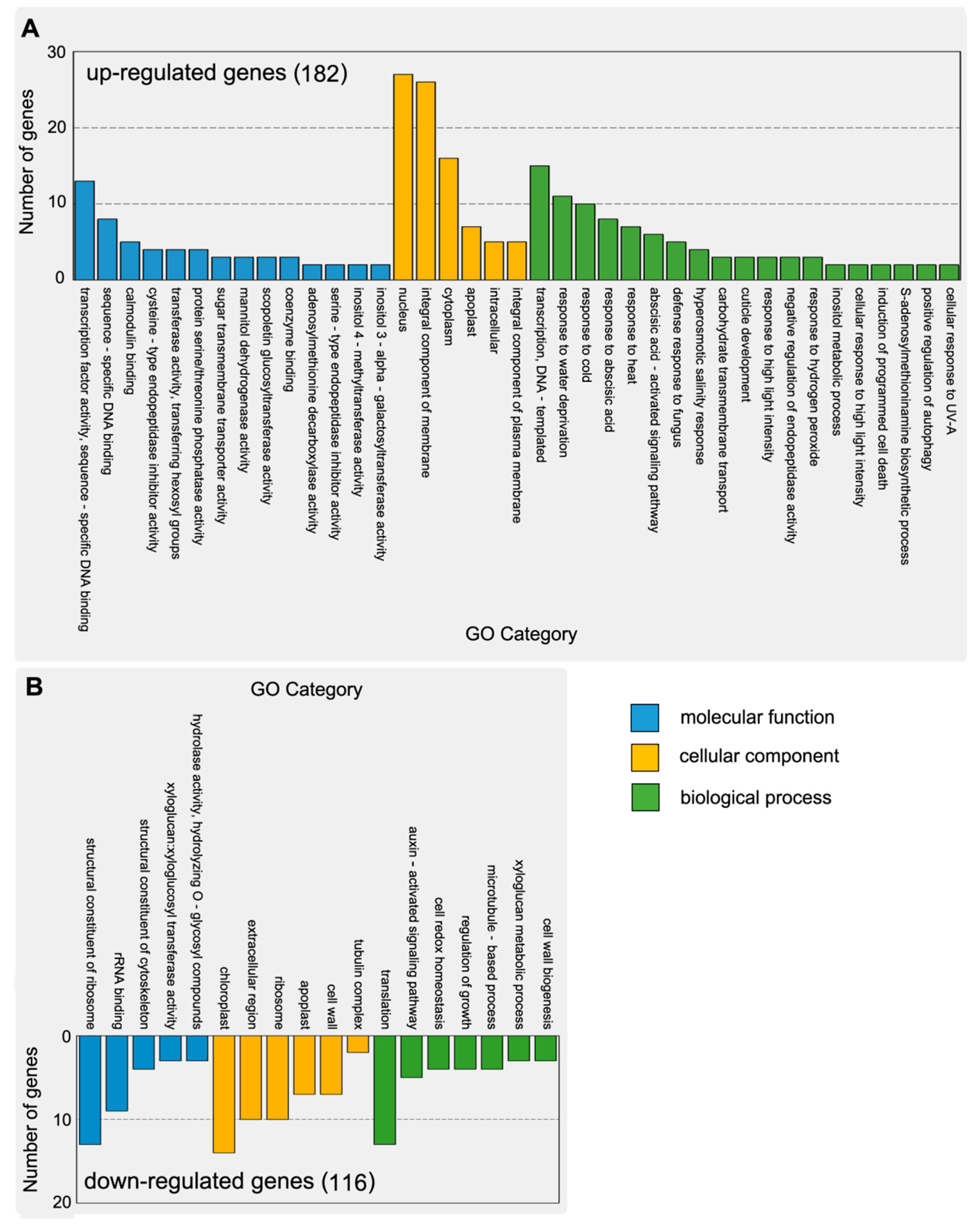
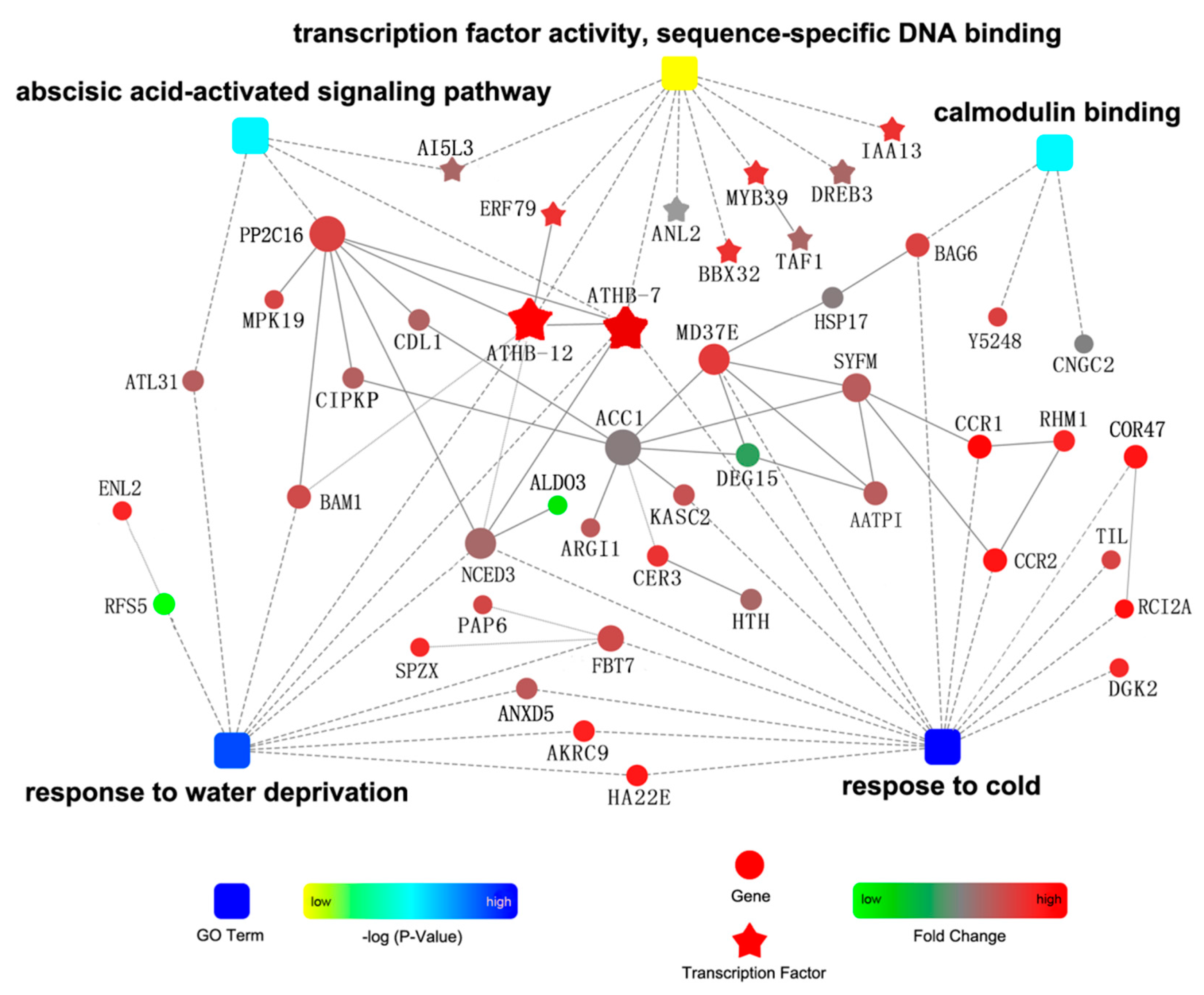
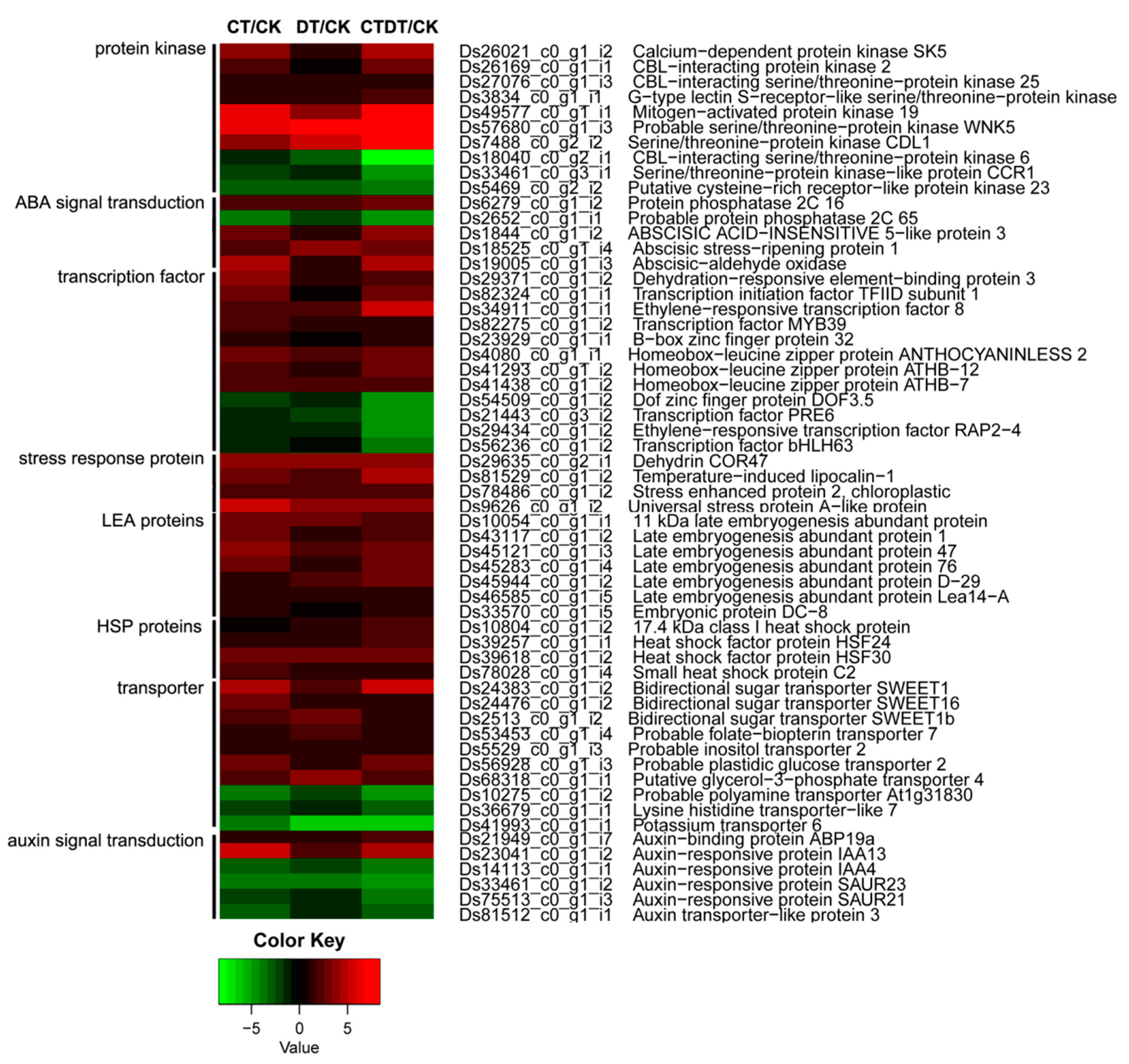
2.3. Functional Classification and Expression Analysis of DEGs
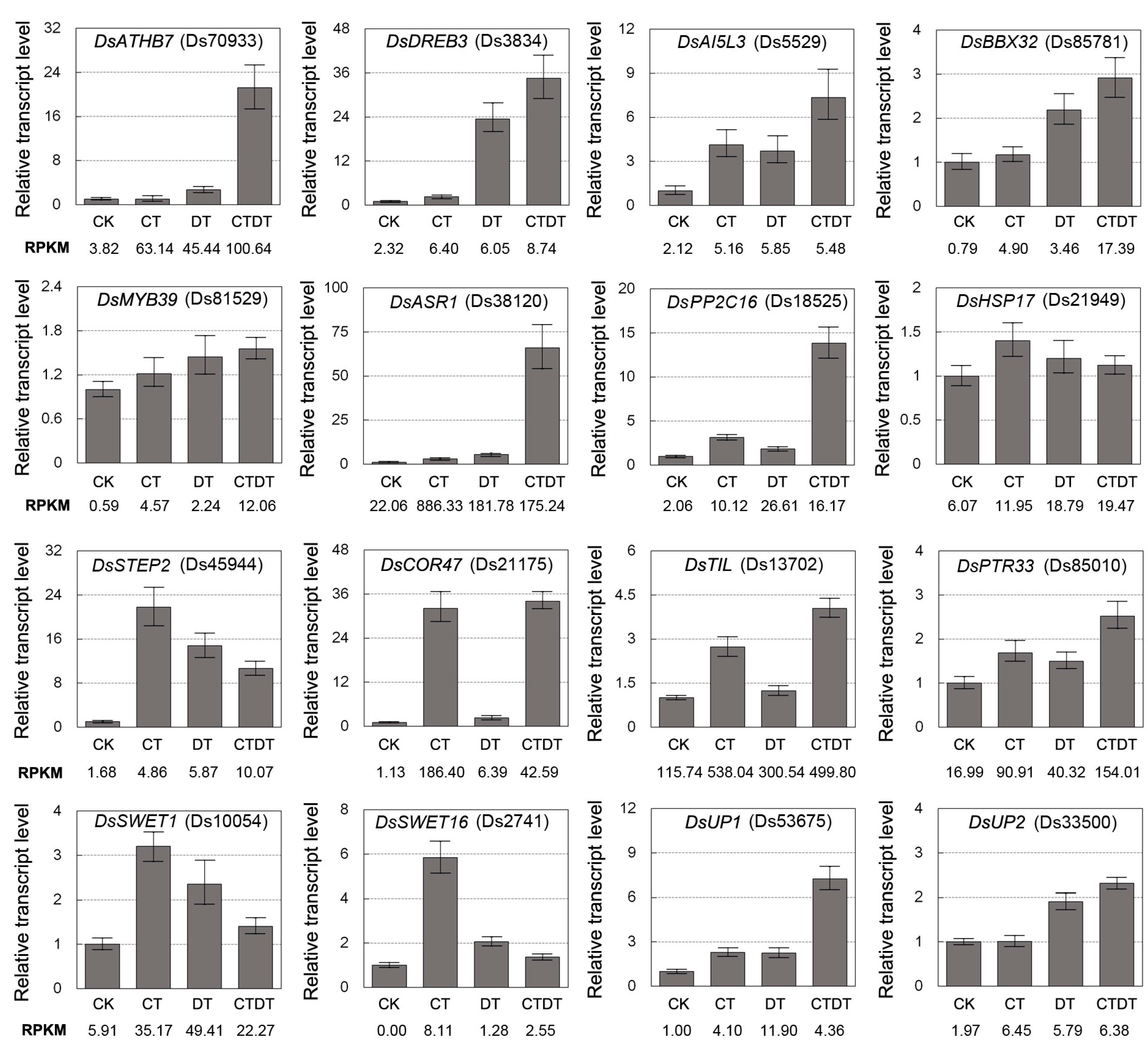
2.4. Validation of RNA-Seq Gene Expression
3. Materials and Methods
3.1. Plant Growth Conditions and Stress Treatments
3.2. RNA-Seq
3.3. Transcriptome Data Processing
3.4. Screening of Differentially Expressed Genes (DEGs)
3.5. Heatmap and Protein–Protein Interaction (PPI) Analysis of DEGs
3.6. Quantitative Real-Time PCR of DEGs
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Chinnusamy, V.; Zhu, J.; Zhu, J.K. Cold stress regulation of gene expression in plants. Trends Plant Sci. 2007, 12, 444–451. [Google Scholar] [CrossRef] [PubMed]
- Huang, G.T.; Ma, S.L.; Bai, L.P.; Zhang, L.; Ma, H.; Jia, P.; Liu, J.; Zhong, M.; Guo, Z.F. Signal transduction during cold, salt, and drought stresses in plants. Mol. Biol. Rep. 2012, 39, 969–987. [Google Scholar] [CrossRef] [PubMed]
- Su, L.; Dai, Z.; Li, S.; Xin, H. A novel system for evaluating drought-cold tolerance of grapevines using chlorophyll fluorescence. BMC Plant Biol. 2015, 15, 82. [Google Scholar] [CrossRef] [PubMed]
- Qu, Y.; Zhou, A.; Zhang, X.; Tang, H.; Liang, M.; Han, H.; Zuo, Y. De novo transcriptome sequencing of low temperature-treated Phlox subulata and analysis of the genes involved in cold stress. Int. J. Mol. Sci. 2015, 16, 9732–9748. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; Zhou, X.; Cao, Y.; Zhou, M.; McNeil, D.; Liang, S.; Yang, C. RNA-seq analysis of cold and drought responsive transcriptomes of Zea mays ssp. mexicana L. Front. Plant Sci. 2017, 8, 136. [Google Scholar] [CrossRef] [PubMed]
- Calzadilla, P.I.; Maiale, S.J.; Ruiz, O.A.; Escaray, F.J. Transcriptome response mediated by cold stress in Lotus japonicus. Front. Plant Sci. 2016, 7, 374. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Zhang, F.; Zhang, F.; Wang, W.; Zhou, Y.; Fu, B.; Li, Z. Comparative transcriptome sequencing of tolerant rice introgression line and its parents in response to drought stress. BMC Genom. 2014, 15, 1026. [Google Scholar] [CrossRef] [PubMed]
- Tian, D.Q.; Pan, X.Y.; Yu, Y.M.; Wang, W.Y.; Zhang, F.; Ge, Y.Y.; Shen, X.L.; Shen, F.Q.; Liu, X.J. De novo characterization of the Anthurium transcriptome and analysis of its digital gene expression under cold stress. BMC Genom. 2013, 14, 827. [Google Scholar] [CrossRef] [PubMed]
- Pang, T.; Ye, C.Y.; Xia, X.; Yin, W. De novo sequencing and transcriptome analysis of the desert shrub, Ammopiptanthus mongolicus, during cold acclimation using Illumina/Solexa. BMC Genom. 2013, 14, 488. [Google Scholar] [CrossRef] [PubMed]
- An, D.; Yang, J.; Zhang, P. Transcriptome profiling of low temperature-treated cassava apical shoots showed dynamic responses of tropical plant to cold stress. BMC Genom. 2012, 13, 64. [Google Scholar] [CrossRef] [PubMed]
- Kumar, D.; Hazra, S.; Datta, R.; Chattopadhyay, S. Transcriptome analysis of Arabidopsis mutants suggests a crosstalk between ABA, ethylene and GSH against combined cold and osmotic stress. Sci. Rep. 2016, 6, 36867. [Google Scholar] [CrossRef] [PubMed]
- Li, X.W.; Feng, Z.G.; Yang, H.M.; Zhu, X.P.; Liu, J.; Yuan, H.Y. A novel cold-regulated gene from Camellia sinensis, CsCOR1, enhances salt- and dehydration-tolerance in tobacco. Biochem. Biophys. Res. Commun. 2010, 394, 354–359. [Google Scholar] [CrossRef] [PubMed]
- Masand, S.; Yadav, S.K. Overexpression of MuHSP70 gene from Macrotyloma uniflorum confers multiple abiotic stress tolerance in transgenic Arabidopsis thaliana. Mol. Biol. Rep. 2016, 43, 53–64. [Google Scholar] [CrossRef] [PubMed]
- Zheng, C.; Wang, Y.; Ding, Z.; Zhao, L. Global transcriptional analysis reveals the complex relationship between tea quality, leaf senescence and the responses to cold-drought combined stress in Camellia sinensis. Front. Plant Sci. 2016, 7, 1858. [Google Scholar] [CrossRef] [PubMed]
- Holobiuc, M.; Blindu, R.; Mitoi, M.; Helepciuc, F.; Cristea, V. The establishment of an in vitro gene bank in Dianthus spiculifolius Schur. and D. glacialis ssp. gelidus (Schott Nym. et Kotschy) Tutin: I. The initiation of a tissue collection and the characterization of the cultures in minimal growth conditions. Ann. For. Res. 2009, 52, 117–128. [Google Scholar]
- Harris, M.A.; Clark, J.; Ireland, A.; Lomax, J.; Ashburner, M.; Foulger, R.; Eilbeck, K.; Lewis, S.; Marshall, B.; Mungall, C.; et al. The Gene Ontology (GO) database and informatics resource. Nucleic Acids Res. 2004, 32, D258–D261. [Google Scholar] [PubMed]
- Valdes, A.E.; Overnas, E.; Johansson, H.; Rada-Iglesias, A.; Engstrom, P. The homeodomain-leucine zipper (HD-Zip) class I transcription factors ATHB7 and ATHB12 modulate abscisic acid signalling by regulating protein phosphatase 2C and abscisic acid receptor gene activities. Plant Mol. Biol. 2012, 80, 405–418. [Google Scholar] [CrossRef] [PubMed]
- Danquah, A.; de Zelicourt, A.; Colcombet, J.; Hirt, H. The role of ABA and MAPK signaling pathways in plant abiotic stress responses. Biotechnol. Adv. 2014, 32, 40–52. [Google Scholar] [CrossRef] [PubMed]
- Eremina, M.; Rozhon, W.; Poppenberger, B. Hormonal control of cold stress responses in plants. Cell. Mol. Life Sci. CMLS 2016, 73, 797–810. [Google Scholar] [CrossRef] [PubMed]
- Luan, S. The CBL-CIPK network in plant calcium signaling. Trends Plant Sci. 2009, 14, 37–42. [Google Scholar] [CrossRef] [PubMed]
- Yu, Q.; An, L.; Li, W. The CBL-CIPK network mediates different signaling pathways in plants. Plant Cell Rep. 2014, 33, 203–214. [Google Scholar] [CrossRef] [PubMed]
- Yang, T.; Chaudhuri, S.; Yang, L.; Du, L.; Poovaiah, B.W. A calcium/calmodulin-regulated member of the receptor-like kinase family confers cold tolerance in plants. J. Biol. Chem. 2010, 285, 7119–7126. [Google Scholar] [CrossRef] [PubMed]
- Winfield, M.O.; Lu, C.; Wilson, I.D.; Coghill, J.A.; Edwards, K.J. Plant responses to cold: Transcriptome analysis of wheat. Plant Biotechnol. J. 2010, 8, 749–771. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.C.; Chung, W.S.; Yun, D.J.; Cho, M.J. Calcium and calmodulin-mediated regulation of gene expression in plants. Mol. Plant 2009, 2, 13–21. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Barrena, M.J.; Martinez-Ripoll, M.; Albert, A. Structural Biology of a Major Signaling Network that Regulates Plant Abiotic Stress: The CBL-CIPK Mediated Pathway. Int. J. Mol. Sci. 2013, 14, 5734–5749. [Google Scholar] [CrossRef] [PubMed]
- Lan, W.Z.; Lee, S.C.; Che, Y.F.; Jiang, Y.Q.; Luan, S. Mechanistic analysis of AKT1 regulation by the CBL-CIPK-PP2CA interactions. Mol. Plant 2011, 4, 527–536. [Google Scholar] [CrossRef] [PubMed]
- Soon, F.F.; Ng, L.M.; Zhou, X.E.; West, G.M.; Kovach, A.; Tan, M.H.; Suino-Powell, K.M.; He, Y.; Xu, Y.; Chalmers, M.J.; et al. Molecular mimicry regulates ABA signaling by SnRK2 kinases and PP2C phosphatases. Science 2012, 335, 85–88. [Google Scholar] [CrossRef] [PubMed]
- Novillo, F.; Medina, J.; Salinas, J. Arabidopsis CBF1 and CBF3 have a different function than CBF2 in cold acclimation and define different gene classes in the CBF regulon. Proc. Natl. Acad. Sci. USA 2007, 104, 21002–21007. [Google Scholar] [CrossRef] [PubMed]
- Shavrukov, Y.; Baho, M.; Lopato, S.; Langridge, P. The TaDREB3 transgene transferred by conventional crossings to different genetic backgrounds of bread wheat improves drought tolerance. Plant Biotechnol. J. 2016, 14, 313–322. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, M.; Hao, Y.; Kapoor, A.; Dong, C.H.; Fujii, H.; Zheng, X.; Zhu, J.K. A R2R3 type MYB transcription factor is involved in the cold regulation of CBF genes and in acquired freezing tolerance. J. Biol. Chem. 2006, 281, 37636–37645. [Google Scholar] [CrossRef] [PubMed]
- Licausi, F.; Ohme-Takagi, M.; Perata, P. APETALA2/ethylene responsive factor (AP2/ERF) transcription factors: Mediators of stress responses and developmental programs. New Phytol. 2013, 199, 639–649. [Google Scholar] [CrossRef] [PubMed]
- Gangappa, S.N.; Botto, J.F. The BBX family of plant transcription factors. Trends Plant Sci. 2014, 19, 460–470. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi-Shinozaki, K.; Shinozaki, K. Transcriptional regulatory networks in cellular responses and tolerance to dehydration and cold stresses. Ann. Rev. Plant Biol. 2006, 57, 781–803. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, T.; Mogami, J.; Yamaguchi-Shinozaki, K. ABA-dependent and ABA-independent signaling in response to osmotic stress in plants. Curr. Opin. Plant Biol. 2014, 21, 133–139. [Google Scholar] [CrossRef] [PubMed]
- Zhou, M.Q.; Shen, C.; Wu, L.H.; Tang, K.X.; Lin, J. CBF-dependent signaling pathway: A key responder to low temperature stress in plants. Crit. Rev. Biotechnol. 2011, 31, 186–192. [Google Scholar] [CrossRef] [PubMed]
- Qin, F.; Kakimoto, M.; Sakuma, Y.; Maruyama, K.; Osakabe, Y.; Tran, L.S.; Shinozaki, K.; Yamaguchi-Shinozaki, K. Regulation and functional analysis of ZmDREB2A in response to drought and heat stresses in Zea mays L. Plant J. 2007, 50, 54–69. [Google Scholar] [CrossRef] [PubMed]
- Tunnacliffe, A.; Wise, M.J. The continuing conundrum of the LEA proteins. Naturwissenschaften 2007, 94, 791–812. [Google Scholar] [CrossRef] [PubMed]
- Kosova, K.; Vitamvas, P.; Prasil, I.T. Wheat and barley dehydrins under cold, drought, and salinity—What can LEA-II proteins tell us about plant stress response? Front. Plant Sci. 2014, 5, 343. [Google Scholar] [CrossRef] [PubMed]
- Hu, T.Z.; Zhu, S.S.; Tan, L.L.; Qi, W.H.; He, S.; Wang, G.X. Overexpression of OsLEA4 enhances drought, high salt and heavy metal stress tolerance in transgenic rice (Oryza sativa L.). Environ. Exp. Bot. 2016, 123, 68–77. [Google Scholar] [CrossRef]
- Yu, J.; Lai, Y.; Wu, X.; Wu, G.; Guo, C. Overexpression of OsEm1 encoding a group I LEA protein confers enhanced drought tolerance in rice. Biochem. Biophys. Res. Commun. 2016, 478, 703–709. [Google Scholar] [CrossRef] [PubMed]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.; et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef] [PubMed]
- Ye, J.; Fang, L.; Zheng, H.; Zhang, Y.; Chen, J.; Zhang, Z.; Wang, J.; Li, S.; Li, R.; Bolund, L.; et al. WEGO: A web tool for plotting GO annotations. Nucleic Acids Res. 2006, 34, W293–W297. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Goto, S.; Sato, Y.; Furumichi, M.; Tanabe, M. KEGG for integration and interpretation of large-scale molecular data sets. Nucleic Acids Res. 2012, 40, D109–D114. [Google Scholar] [CrossRef] [PubMed]
- Mortazavi, A.; Williams, B.A.; McCue, K.; Schaeffer, L.; Wold, B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods 2008, 5, 621–628. [Google Scholar] [CrossRef] [PubMed]
- Saldanha, A.J. Java Treeview—Extensible visualization of microarray data. Bioinformatics 2004, 20, 3246–3248. [Google Scholar] [CrossRef] [PubMed]
- Franceschini, A.; Szklarczyk, D.; Frankild, S.; Kuhn, M.; Simonovic, M.; Roth, A.; Lin, J.; Minguez, P.; Bork, P.; von Mering, C.; et al. STRING v9.1: Protein-protein interaction networks, with increased coverage and integration. Nucleic Acids Res. 2013, 41, D808–D815. [Google Scholar] [CrossRef] [PubMed]
- Ren, D.; Madsen, J.S.; de la Cruz-Perera, C.I.; Bergmark, L.; Sorensen, S.J.; Burmolle, M. High-throughput screening of multispecies biofilm formation and quantitative PCR-based assessment of individual species proportions, useful for exploring interspecific bacterial interactions. Microb. Ecol. 2014, 68, 146–154. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Content | Control (CK) | Cold-Treated (CT) | Drought-Treated (DT) | Combined-Treated (CTDT) |
|---|---|---|---|---|
| Total reads | 30,100,666 | 29,709,677 | 30,182,572 | 29,577,457 |
| Total clean reads | 23,624,696 | 22,495,628 | 23,317,141 | 22,965,345 |
| Guanine and cytosine (GC) percentage | 46.45 | 46.32 | 46.62 | 46.10 |
| Total number of transcripts | 184,782 | |||
| Total number of unigenes | 112,015 | |||
| Mean length of unigenes (bp) | 819 |
| Annotated Database | Number of Unigenes | Percentage (%) |
|---|---|---|
| SwissProt | 36,142 | 32.27% |
| Protein family (PFAM) | 29,149 | 26.55% |
| Gene Ontology (GO) | 29,739 | 26.54% |
| euKaryotic Ortholog Groups (KOG) | 25,073 | 22.38% |
| KEGG Ortholog (KO) | 10,845 | 9.68% |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhou, A.; Ma, H.; Liu, E.; Jiang, T.; Feng, S.; Gong, S.; Wang, J. Transcriptome Sequencing of Dianthus spiculifolius and Analysis of the Genes Involved in Responses to Combined Cold and Drought Stress. Int. J. Mol. Sci. 2017, 18, 849. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms18040849
Zhou A, Ma H, Liu E, Jiang T, Feng S, Gong S, Wang J. Transcriptome Sequencing of Dianthus spiculifolius and Analysis of the Genes Involved in Responses to Combined Cold and Drought Stress. International Journal of Molecular Sciences. 2017; 18(4):849. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms18040849
Chicago/Turabian StyleZhou, Aimin, Hongping Ma, Enhui Liu, Tongtong Jiang, Shuang Feng, Shufang Gong, and Jingang Wang. 2017. "Transcriptome Sequencing of Dianthus spiculifolius and Analysis of the Genes Involved in Responses to Combined Cold and Drought Stress" International Journal of Molecular Sciences 18, no. 4: 849. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms18040849