
The Dose–Response Association between Nitrogen Dioxide Exposure and Serum Interleukin-6 Concentrations

 , , , , ,
, , , , ,  , add
Show full author list
, add
Show full author list
Abstract
:
1. Introduction
2. Results
2.1. Characteristics of TAHS Participants
2.2. Main Association between NO2 Exposure and Cytokines
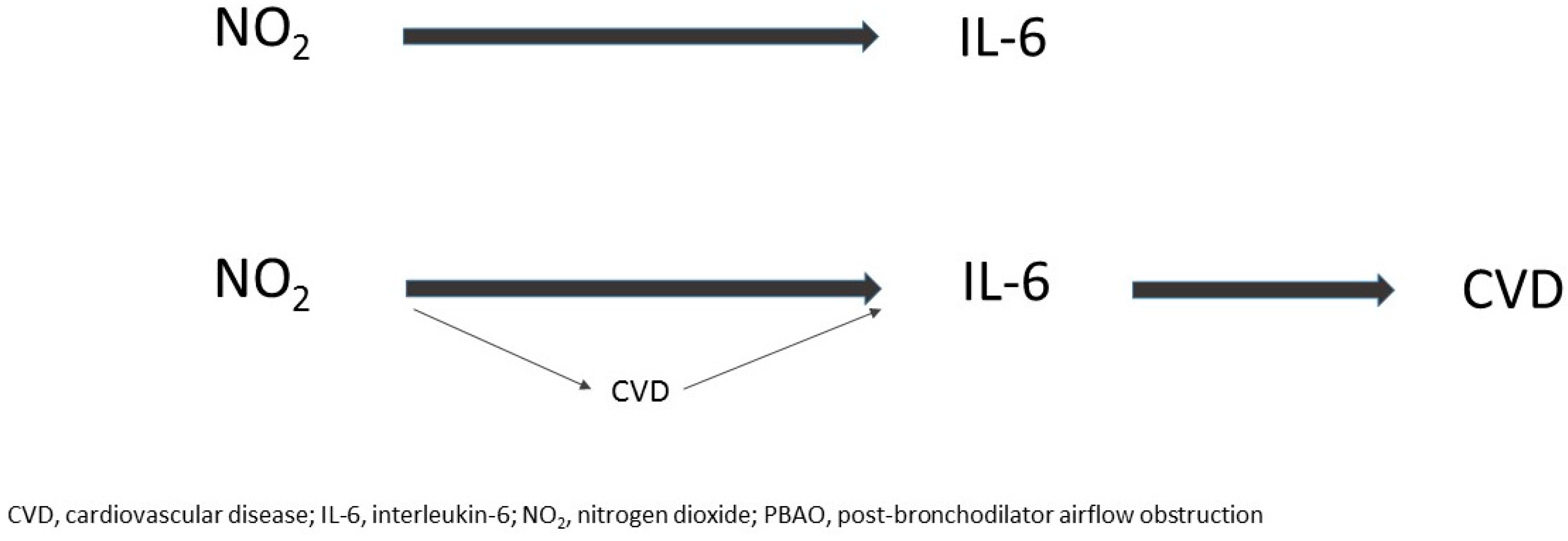
2.3. Mediation Analysis by Post-BD-AO
2.4. Mediation Analysis by Cardiovascular Disease (CVD)
3. Discussion
4. Materials and Methods
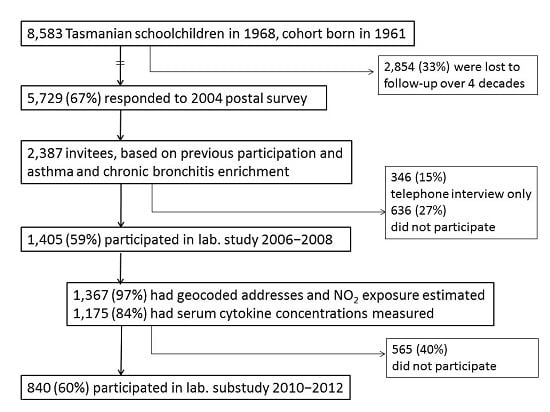
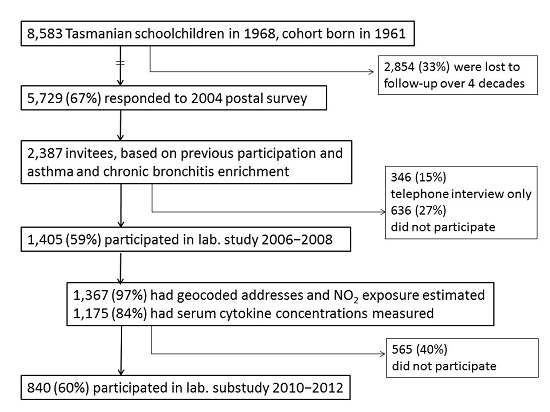
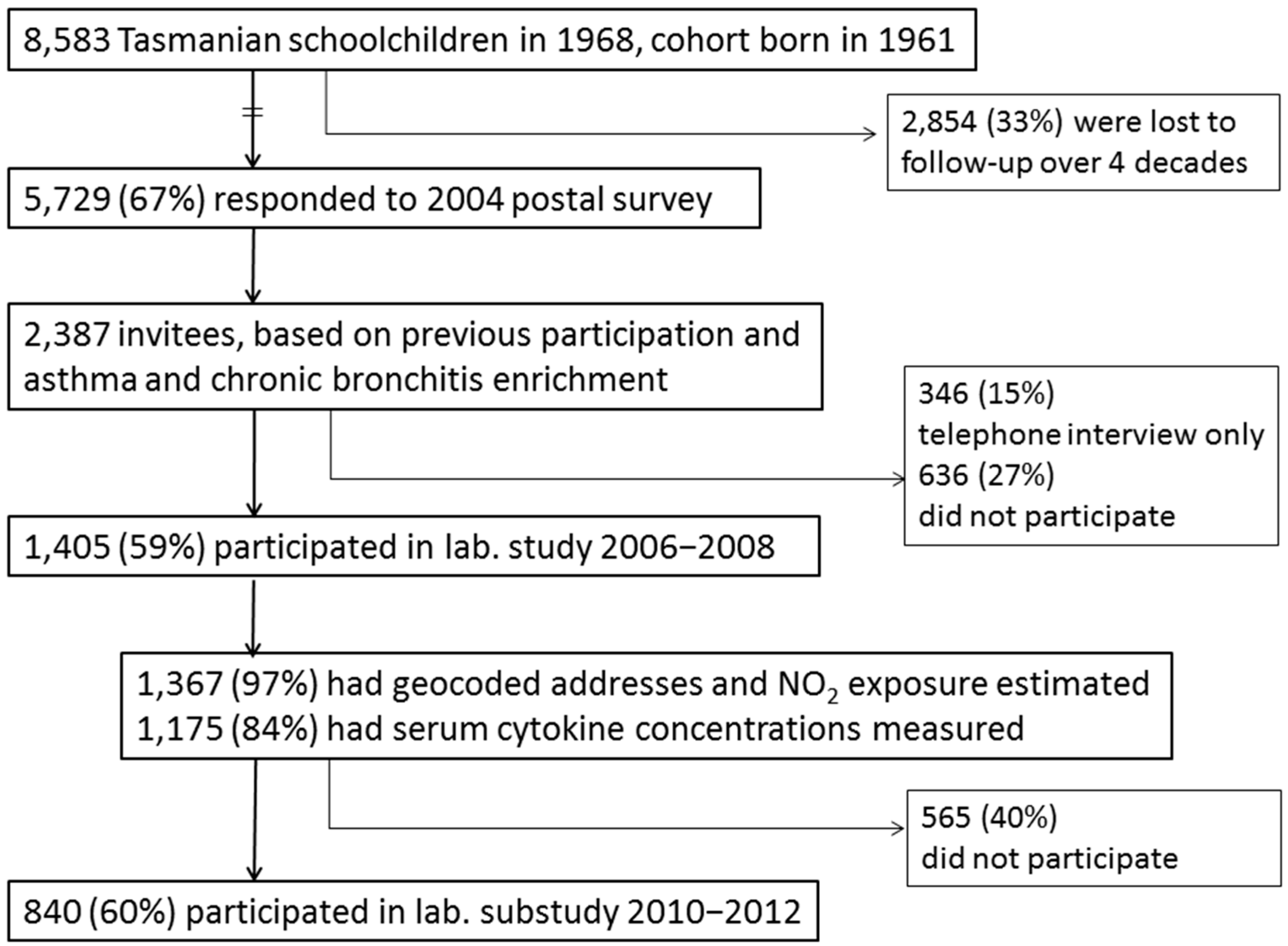
4.1. Study Design and Population
4.2. Data Collection Methods
4.3. Clinical Definitions
4.4. Cytokine Measurement
4.5. Statistical Analysis
4.6. Ethics
4.7. Data Sharing
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| AO | Airflow obstruction |
| BD | Bronchodilator |
| CI | Confidence interval |
| COPD | Chronic obstructive pulmonary disease |
| CRP | C-reactive protein |
| CVD | Cardiovascular disease |
| FEV1 | Forced expiratory volume in one second |
| FVC | Forced vital capacity |
| GM | Geometric mean |
| h | hours |
| IL | Interleukin |
| IQR | Interquartile range |
| ppb | Parts per billion |
| NO2 | Nitrogen dioxide |
| PM2.5 | Particulate matter less than 2.5 µm in diameter |
| RPM | Revolutions per minute |
| TAHS | Tasmanian longitudinal health study |
| TNF-α | Tumor necrosis factor-α |
| TRAP | Traffic-related air pollution |
| WHO | World Health Organization |
Appendix A

References
- Burki, T.K. Twice as bad: New estimates for mortality from air pollution. Lancet Respir. Med. 2014, 2, 355. [Google Scholar] [CrossRef]
- World Health Organization. Ambient (Outdoor) Air Quality and Health. Fact Sheet N°313. Updated September 2016. Available online: http://www.who.int/mediacentre/factsheets/fs313/en/ (accessed on 21 Apirl 2017).
- Faustini, A.; Rapp, R.; Forastiere, F. Nitrogen dioxide and mortality: Review and meta-analysis of long-term studies. Eur. Respir. J. 2014, 44, 744–753. [Google Scholar] [CrossRef] [PubMed]
- Brook, R.D. Cardiovascular effects of air pollution. Clin. Sci. 2008, 115, 175–187. [Google Scholar] [CrossRef] [PubMed]
- Brook, R.D.; Rajagopalan, S.; Pope, C.A., 3rd; Brook, J.R.; Bhatnagar, A.; Diez-Roux, A.V.; Holguin, F.; Hong, Y.; Luepker, R.V.; Mittleman, M.A.; et al. Particulate matter air pollution and cardiovascular disease: An update to the scientific statement from the American Heart Association. Circulation 2010, 121, 2331–2378. [Google Scholar] [CrossRef] [PubMed]
- Knibbs, L.D.; Hewson, M.G.; Bechle, M.J.; Marshall, J.D.; Barnett, A.G. A national satellite-based land-use regression model for air pollution exposure assessment in Australia. Environ. Res. 2014, 135, 204–211. [Google Scholar] [CrossRef] [PubMed]
- Richter, A.; Burrows, J.P.; Nuss, H.; Granier, C.; Niemeier, U. Increase in tropospheric nitrogen dioxide over China observed from space. Nature 2005, 437, 129–132. [Google Scholar] [CrossRef] [PubMed]
- Laden, F.; Schwartz, J.; Speizer, F.E.; Dockery, D.W. Reduction in fine particulate air pollution and mortality: Extended follow-up of the Harvard Six Cities study. Am. J. Respir. Crit. Care Med. 2006, 173, 667–672. [Google Scholar] [CrossRef] [PubMed]
- Dockery, D.W.; Pope, C.A., 3rd; Xu, X.; Spengler, J.D.; Ware, J.H.; Fay, M.E.; Ferris, B.G., Jr.; Speizer, F.E. An association between air pollution and mortality in six U.S. cities. N. Engl. J. Med. 1993, 329, 1753–1759. [Google Scholar] [CrossRef] [PubMed]
- Dadvand, P.; Nieuwenhuijsen, M.J.; Agusti, A.; de Batlle, J.; Benet, M.; Beelen, R.; Cirach, M.; Martinez, D.; Hoek, G.; Basagana, X.; et al. Air pollution and biomarkers of systemic inflammation and tissue repair in COPD patients. Eur. Respir. J. 2014, 44, 603–613. [Google Scholar] [CrossRef] [PubMed]
- Folsom, A.R.; Chambless, L.E.; Ballantyne, C.M.; Coresh, J.; Heiss, G.; Wu, K.K.; Boerwinkle, E.; Mosley, T.H., Jr.; Sorlie, P.; Diao, G.; et al. An assessment of incremental coronary risk prediction using C-reactive protein and other novel risk markers: The atherosclerosis risk in communities study. Arch. Intern. Med. 2006, 166, 1368–1373. [Google Scholar] [CrossRef] [PubMed]
- Pai, J.K.; Pischon, T.; Ma, J.; Manson, J.E.; Hankinson, S.E.; Joshipura, K.; Curhan, G.C.; Rifai, N.; Cannuscio, C.C.; Stampfer, M.J.; et al. Inflammatory markers and the risk of coronary heart disease in men and women. N. Engl. J. Med. 2004, 351, 2599–2610. [Google Scholar] [CrossRef] [PubMed]
- Rincon, M. Interleukin-6: From an inflammatory marker to a target for inflammatory diseases. Trends Immunol. 2012, 33, 571–577. [Google Scholar] [CrossRef] [PubMed]
- Ferrari, R.; Tanni, S.E.; Caram, L.M.; Correa, C.; Correa, C.R.; Godoy, I. Three-year follow-up of Interleukin 6 and C-reactive protein in chronic obstructive pulmonary disease. Respir. Res. 2013, 14, 24. [Google Scholar] [CrossRef] [PubMed]
- Agusti, A.; Edwards, L.D.; Rennard, S.I.; MacNee, W.; Tal-Singer, R.; Miller, B.E.; Vestbo, J.; Lomas, D.A.; Calverley, P.M.; Wouters, E.; et al. Persistent systemic inflammation is associated with poor clinical outcomes in COPD: A novel phenotype. PLoS ONE 2012, 7, e37483. [Google Scholar] [CrossRef] [PubMed]
- Agusti, A.; Faner, R. Systemic inflammation and comorbidities in chronic obstructive pulmonary disease. Proc. Am. Thorac. Soc. 2012, 9, 43–46. [Google Scholar] [CrossRef] [PubMed]
- Wei, J.; Xiong, X.F.; Lin, Y.H.; Zheng, B.X.; Cheng, D.Y. Association between serum interleukin-6 concentrations and chronic obstructive pulmonary disease: A systematic review and meta-analysis. PeerJ 2015, 3, e1199. [Google Scholar] [CrossRef] [PubMed]
- Maggio, M.; Guralnik, J.M.; Longo, D.L.; Ferrucci, L. Interleukin-6 in aging and chronic disease: A magnificent pathway. J. Gerontol. A Biol. Sci. Med. Sci. 2006, 61, 575–584. [Google Scholar] [CrossRef] [PubMed]
- Yanbaeva, D.G.; Dentener, M.A.; Creutzberg, E.C.; Wesseling, G.; Wouters, E.F. Systemic effects of smoking. Chest 2007, 131, 1557–1566. [Google Scholar] [CrossRef] [PubMed]
- Perret, J.L.; Dharmage, S.C.; Matheson, M.C.; Johns, D.P.; Gurrin, L.C.; Burgess, J.A.; Marrone, J.; Markos, J.; Morrison, S.; Feather, I.; et al. The Interplay between the Effects of Lifetime Asthma, Smoking, and Atopy on Fixed Airflow Obstruction in Middle Age. Am. J. Respir. Crit. Care Med. 2013, 187, 42–48. [Google Scholar] [CrossRef] [PubMed]
- VanderWeele, T.J. Explanation in Causal Inference: Methods for Mediation and Interaction; Oxford University Press: Oxford, UK, 2015. [Google Scholar]
- Diez Roux, A.V.; Auchincloss, A.H.; Astor, B.; Barr, R.G.; Cushman, M.; Dvonch, T.; Jacobs, D.R., Jr.; Kaufman, J.; Lin, X.; Samson, P. Recent exposure to particulate matter and C-reactive protein concentration in the multi-ethnic study of atherosclerosis. Am. J. Epidemiol. 2006, 164, 437–448. [Google Scholar] [CrossRef] [PubMed]
- Xie, W.; Li, G.; Zhao, D.; Xie, X.; Wei, Z.; Wang, W.; Wang, M.; Li, G.; Liu, W.; Sun, J.; et al. Relationship between fine particulate air pollution and ischaemic heart disease morbidity and mortality. Heart 2015, 101, 257–263. [Google Scholar] [CrossRef] [PubMed]
- Hoek, G.; Krishnan, R.M.; Beelen, R.; Peters, A.; Ostro, B.; Brunekreef, B.; Kaufman, J.D. Long-term air pollution exposure and cardio-respiratory mortality: A review. Environ. Health 2013, 12, 43. [Google Scholar] [CrossRef] [PubMed]
- Turner, M.C.; Jerrett, M.; Pope, C.A., 3rd; Krewski, D.; Gapstur, S.M.; Diver, W.R.; Beckerman, B.S.; Marshall, J.D.; Su, J.; Crouse, D.L.; et al. Long-Term Ozone Exposure and Mortality in a Large Prospective Study. Am. J. Respir. Crit. Care Med. 2016, 193, 1134–1142. [Google Scholar] [CrossRef] [PubMed]
- Donaldson, G.C.; Seemungal, T.A.; Patel, I.S.; Bhowmik, A.; Wilkinson, T.M.; Hurst, J.R.; Maccallum, P.K.; Wedzicha, J.A. Airway and systemic inflammation and decline in lung function in patients with COPD. Chest 2005, 128, 1995–2004. [Google Scholar] [CrossRef] [PubMed]
- Laumbach, R.J.; Kipen, H.M. Respiratory health effects of air pollution: Update on biomass smoke and traffic pollution. J. Allergy Clin. Immunol. 2012, 129, 3–11. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Lee, K.; Perez-Padilla, R.; Hudson, N.L.; Mannino, D.M. Outdoor and indoor air pollution and COPD-related diseases in high- and low-income countries. Int. J. Tuberc. Lung Dis. 2008, 12, 115–127. [Google Scholar] [PubMed]
- WHO. 7 Million Premature Deaths Annually Linked to Air Pollution. Available online: http://www.who.int/mediacentre/news/releases/2014/air-pollution/en/ (accessed on 13 December 2016).
- Geddes, J.A.; Martin, R.V.; Boys, B.L.; van Donkelaar, A. Long-Term Trends Worldwide in Ambient NO2 Concentrations Inferred from Satellite Observations. Environ. Health Perspect. 2016, 124, 281–289. [Google Scholar] [CrossRef] [PubMed]
- Dockery, D.W.; Brunekreef, B. Longitudinal studies of air pollution effects on lung function. Am. J. Respir. Crit. Care Med. 1996, 154, S250–S256. [Google Scholar] [CrossRef] [PubMed]
- Matheson, M.C.; Reece, J.C.; Kandane-Rathnayake, R.K.; Tang, M.L.; Simpson, J.A.; Feather, I.H.; Southey, M.C.; Tsimiklis, H.; Hopper, J.L.; Morrison, S.C.; et al. Mould-sensitized adults have lower Th2 cytokines and a higher prevalence of asthma than those sensitized to other aeroallergens. Allergy 2016, 71, 1701–1711. [Google Scholar] [CrossRef] [PubMed]
- De Jager, W.; Bourcier, K.; Rijkers, G.T.; Prakken, B.J.; Seyfert-Margolis, V. Prerequisites for cytokine measurements in clinical trials with multiplex immunoassays. BMC Immunol. 2009, 10, 52. [Google Scholar] [CrossRef] [PubMed]
- Duvigneau, J.C.; Hartl, R.T.; Teinfalt, M.; Gemeiner, M. Delay in processing porcine whole blood affects cytokine expression. J. Immunol. Methods 2003, 272, 11–21. [Google Scholar] [CrossRef]
- Greischel, A.; Zahn, G. Pharmacokinetics of recombinant human tumor necrosis factor α in rhesus monkeys after intravenous administration. J. Pharmacol. Exp. Ther. 1989, 251, 358–361. [Google Scholar] [PubMed]
- Gibson, H.B.; Silverstone, H.; Gandevia, B.; Hall, G.J. Respiratory disorders in seven-year-old children in Tasmania. Aims, methods and administration of the survey. Med. J. Aust. 1969, 2, 201–205. [Google Scholar] [PubMed]
- Giles, G.G.; Lickiss, N.; Gibson, H.B.; Shaw, K. Respiratory symptoms in Tasmanian adolescents: A follow up of the 1961 birth cohort. Aust. N. Z. J. Med. 1984, 14, 631–637. [Google Scholar] [CrossRef] [PubMed]
- Jenkins, M.A.; Hopper, J.L.; Bowes, G.; Carlin, J.B.; Flander, L.B.; Giles, G.G. Factors in childhood as predictors of asthma in adult life. BMJ 1994, 309, 90–93. [Google Scholar] [CrossRef] [PubMed]
- Matheson, M.C.; Abramson, M.J.; Allen, K.; Benke, G.; Burgess, J.A.; Dowty, J.G.; Erbas, B.; Feather, I.H.; Frith, P.A.; Giles, G.G.; et al. Cohort Profile: The Tasmanian Longitudinal Health Study (TAHS). Int. J. Epidemiol. 2016. [Google Scholar] [CrossRef] [PubMed]
- Bowatte, G.; Lodge, C.J.; Knibbs, L.D.; Lowe, A.J.; Erbas, B.; Dennekamp, M.; Marks, G.B.; Giles, G.; Morrison, S.; Thompson, B.; et al. Traffic-related air pollution exposure is associated with allergic sensitization, asthma, and poor lung function in middle age. J. Allergy Clin. Immunol. 2016, 139, 122–129. [Google Scholar] [CrossRef] [PubMed]
- Miller, M.R.; Hankinson, J.; Brusasco, V.; Burgos, F.; Casaburi, R.; Coates, A.; Crapo, R.; Enright, P.; van der Grinten, C.P.; Gustafsson, P.; et al. Standardisation of spirometry. Eur. Respir. J. 2005, 26, 319–338. [Google Scholar] [CrossRef] [PubMed]
- Quanjer, P.H.; Stanojevic, S.; Cole, T.J.; Baur, X.; Hall, G.L.; Culver, B.H.; Enright, P.L.; Hankinson, J.L.; Ip, M.S.; Zheng, J.; et al. Multi-ethnic reference values for spirometry for the 3–95-yr age range: The global lung function 2012 equations. Eur. Respir. J. 2012, 40, 1324–1343. [Google Scholar] [CrossRef] [PubMed]
- Public Sector Mapping Agencies Australia. Data Product Description: Geocoded National Address File. 2014. Available online: http://reference1.mapinfo.com/Data/Australia/G-NAF/2014–11/G-NAF_Product_Description.pdf (accessed on 3 October 2014).
- Quanjer, P.H.; Pretto, J.J.; Brazzale, D.J.; Boros, P.W. Grading the severity of airways obstruction: New wine in new bottles. Eur. Respir. J. 2014, 43, 505–512. [Google Scholar] [CrossRef] [PubMed]
- Kandane-Rathnayake, R.K.; Tang, M.L.; Simpson, J.A.; Burgess, J.A.; Meszaros, D.; Feather, I.; Southey, M.C.; Schroen, C.J.; Hopper, J.; Morrison, S.C.; et al. Adult serum cytokine concentrations and the persistence of asthma. Int. Arch. Allergy Immunol. 2013, 161, 342–350. [Google Scholar] [CrossRef] [PubMed]
- Rahman, S.A.; Castanon-Cervantes, O.; Scheer, F.A.; Shea, S.A.; Czeisler, C.A.; Davidson, A.J.; Lockley, S.W. Endogenous circadian regulation of pro-inflammatory cytokines and chemokines in the presence of bacterial lipopolysaccharide in humans. Brain Behav. Immun. 2015, 47, 4–13. [Google Scholar] [CrossRef] [PubMed]
- Hicks, R.; Tingley, D. Causal mediation analysis. Stata J. 2011, 11, 605–619. [Google Scholar]


{kind=link}
{kind=link}
{kind=link}
| Participant Characteristic | TAHS 5th Decade Laboratory Study | ||
|---|---|---|---|
| Post-BD Airflow Obstruction (n = 973) 1 | |||
| Yes (n = 97) | No (n = 876) | p-Value | |
| Demographic and other features | |||
| Age in years (mean (SD)) | 44.9 (0.8) | 44.9 (0.9) | 0.153 |
| Sex (n, % male) | 50 (52) | 438 (50) | 0.773 |
| BMI categories (kg/m2) | |||
| Underweight (<18.5) | 1 (1) | 3 (0.3) | 0.315 |
| Normal (18.5–24.9) | 30 (31) | 262 (30) | 0.835 |
| Overweight (25–29.9) | 36 (37) | 359 (41) | 0.462 |
| Obese (≥30) | 30 (31) | 252 (29) | 0.498 |
| Cigarette smoking | |||
| None | 21 (22) | 389 (45) | <0.001 |
| Past, <20 pack years | 16 (17) | 211 (24) | 0.107 |
| Past, ≥20 pack years | 4 (4) | 47 (5) | 0.624 |
| Current, <20 pack years | 17 (18) | 115 (13) | 0.206 |
| Current, ≥20 pack years | 37 (39) | 109 (13) | <0.001 |
| Annual mean NO2 exposure | |||
| Predicted ppb (median, IQR) | 4.1 (3.5, 5.9) | 4.2 (3.5, 5.6) | 0.733 |
| Per quartile (median, IQR) | |||
| 1 (lowest) | 3.1 (2.8, 3.5) | 3.1 (2.8, 3.4) | 0.727 |
| 2 | 3.8 (3.7, 4.1) | 3.9 (3.7, 4.1) | 0.668 |
| 3 | 4.7 (4.6, 5.1) | 4.9 (4.6, 5.4) | 0.350 |
| 4 (highest) | 6.7 (6.1, 7.3) | 7.1 (6.3, 9.4) | 0.066 |
| Post-BD spirometry 2 | |||
| FEV1 (L) | 2.84 (0.7) | 3.47 (0.7) | <0.001 |
| z-score | −1.44 (1.1) | 0.015 (1.0) | <0.001 |
| FVC (L) | 4.45 (1.1) | 4.34 (0.9) | 0.542 |
| z-score | +0.152 (1.1) | 0.008 (0.9) | 0.258 |
| FEV1/FVC (ratio) | 63.8 (7) | 80.2 (4.7) | <0.001 |
| z-score | −2.37 (0.7) | −0.041 (0.8) | <0.001 |
| Serum cytokine concentrations [GM 95% CI] pg/mL † | |||
| Interleukin-6 | 13.9 (12.2, 15.7) | 12.1 (8.2, 17.9) | 0.795 |
| Interleukin-8 | 275 (244, 308) | 353 (245, 509) | 0.861 |
| Tumor necrosis factor-α | 6.8 (6.5, 7.2) | 6.1 (5.2, 7.3) | 0.082 |
| Annual Mean NO2 Exposure | Serum Interleukin-6 (95% CI) | p-Value | |||
|---|---|---|---|---|---|
| Quartile | Range (ppb) | n | GM pg/mL | Ratio of GM 1 | |
| 1 | 2.41–3.54 | 340 | 11.4 (9.0 to 14.5) | 1 | |
| 2 | 3.54–4.30 | 340 | 12.9 (10.1 to 16.4) | 1.16 (0.1 to 1.6) | 0.400 |
| 3 | 4.31–5.81 | 338 | 16.0 (12.7 to 20.0) | 1.46 (1.2 to 2.0) | 0.024 |
| 4 | 5.81–23.8 | 337 | 17.5 (13.4 to 23.0) | 1.71 (2.0 to 2.4) | 0.003 |
| Mediation Analysis Effect | Post-BD Airflow Obstruction as the Mediator between NO2 and Serum Pro-Inflammatory Cytokines (Mean (95% CI)) | ||
|---|---|---|---|
| IL-6 | IL-8 | TNF-α | |
| Indirect Effect | 0.0003 (−0.007, 0.008) | 0.003 (−0.02, 0.01) | −0.001 (−0.004, 0.01) |
| Direct Effect | 0.175 (0.04, 0.30) | 0.090 (−0.03, 0.20) | 0.056 (0.003, 0.11) |
| Total Effect | 0.175 (0.04, 0.30) | 0.092 (−0.04, 0.20) | 0.055 (0.001, 0.11) |
| % of Total Effect mediated | 0.17% | 2.92% | 1.56% |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Perret, J.L.; Bowatte, G.; Lodge, C.J.; Knibbs, L.D.; Gurrin, L.C.; Kandane-Rathnayake, R.; Johns, D.P.; Lowe, A.J.; Burgess, J.A.; Thompson, B.R.; et al. The Dose–Response Association between Nitrogen Dioxide Exposure and Serum Interleukin-6 Concentrations. Int. J. Mol. Sci. 2017, 18, 1015. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms18051015
Perret JL, Bowatte G, Lodge CJ, Knibbs LD, Gurrin LC, Kandane-Rathnayake R, Johns DP, Lowe AJ, Burgess JA, Thompson BR, et al. The Dose–Response Association between Nitrogen Dioxide Exposure and Serum Interleukin-6 Concentrations. International Journal of Molecular Sciences. 2017; 18(5):1015. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms18051015
Chicago/Turabian StylePerret, Jennifer L., Gayan Bowatte, Caroline J. Lodge, Luke D. Knibbs, Lyle C. Gurrin, Rangi Kandane-Rathnayake, David P. Johns, Adrian J. Lowe, John A. Burgess, Bruce R. Thompson, and et al. 2017. "The Dose–Response Association between Nitrogen Dioxide Exposure and Serum Interleukin-6 Concentrations" International Journal of Molecular Sciences 18, no. 5: 1015. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms18051015