
Ginsenoside Rb2 Alleviates Hepatic Lipid Accumulation by Restoring Autophagy via Induction of Sirt1 and Activation of AMPK

Abstract
:1. Introduction
2. Results
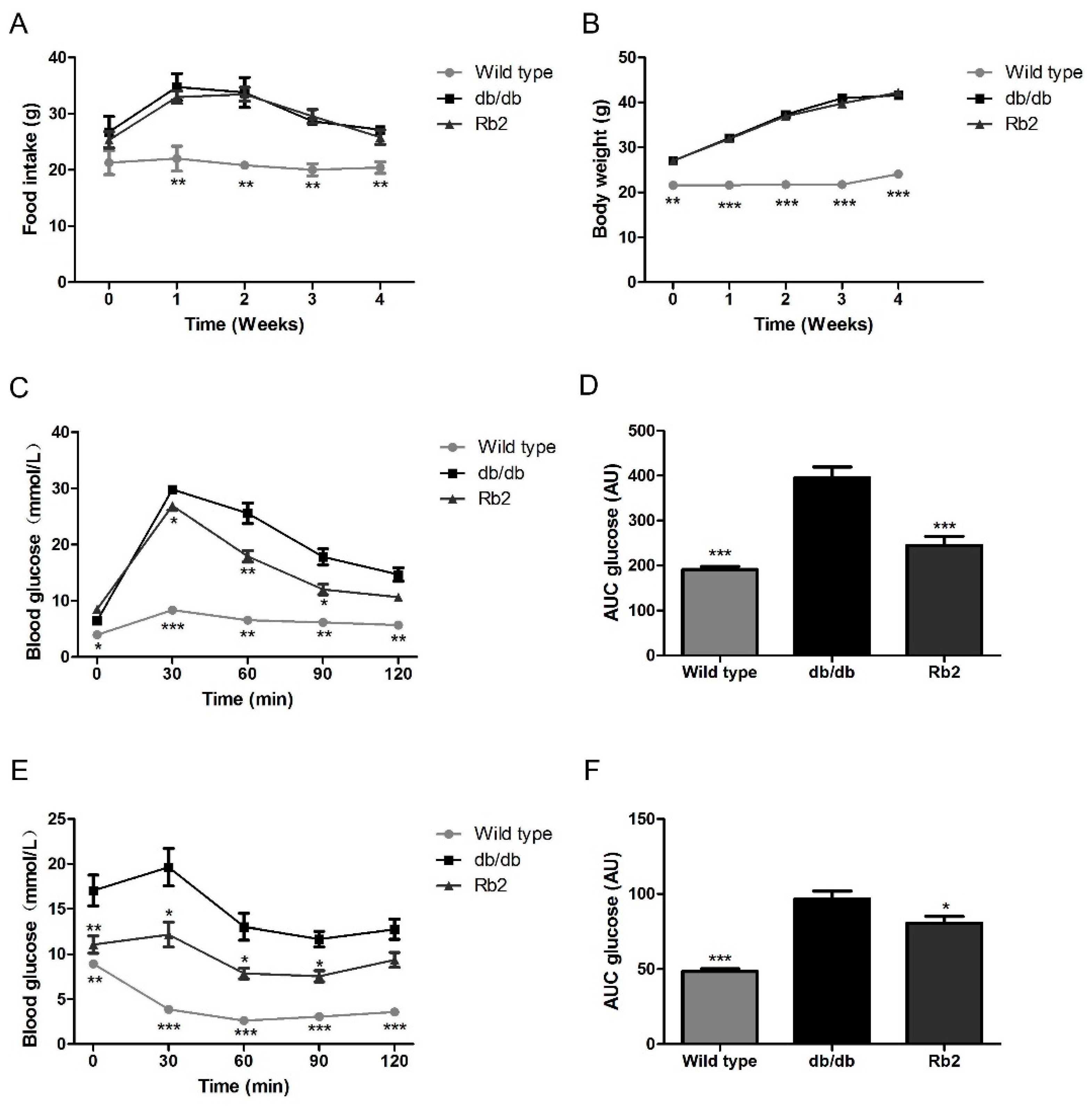
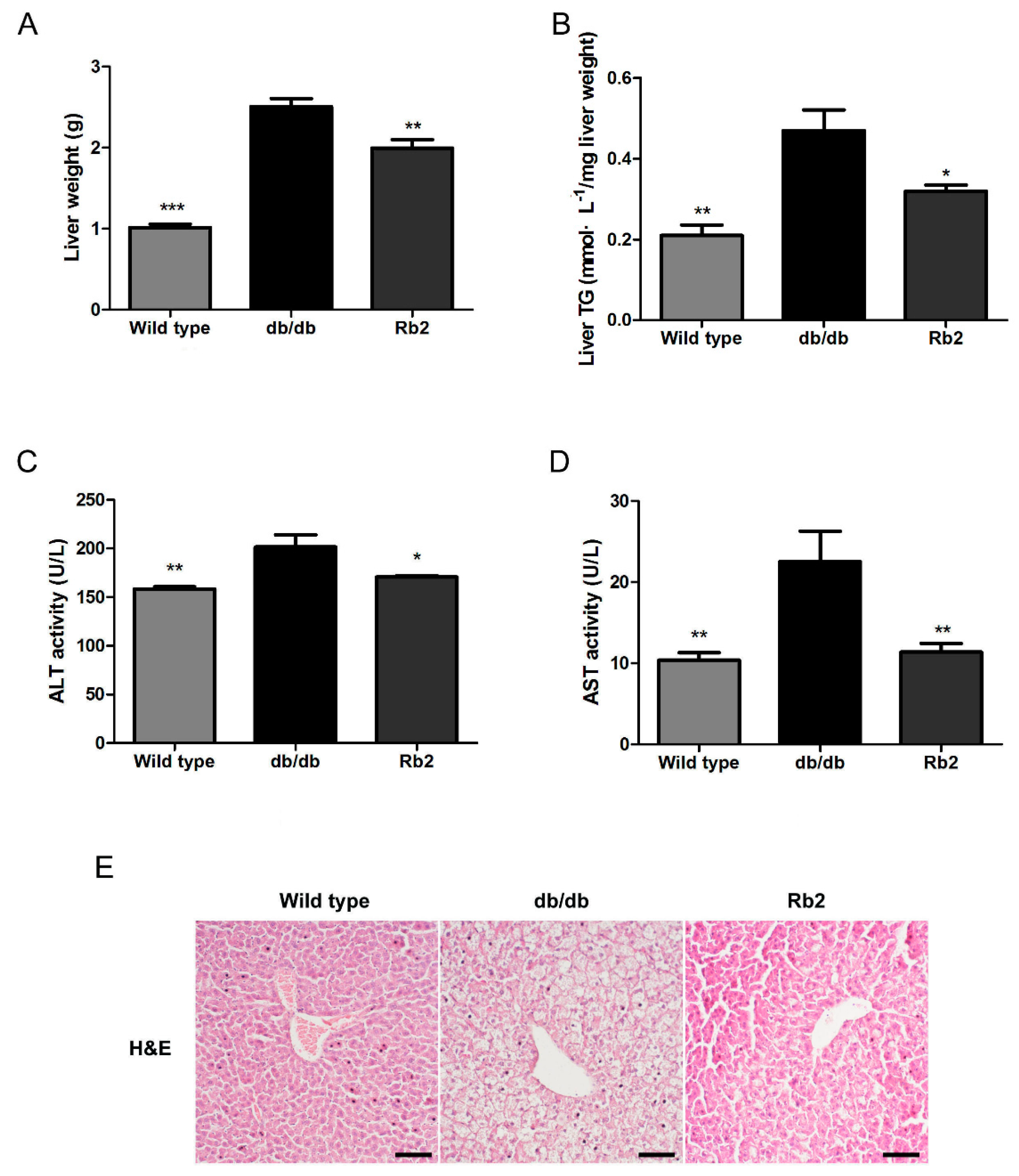
2.1. Rb2 Alleviated Hepatic Steatosis and Improved Glucose Tolerance in db/db Mice
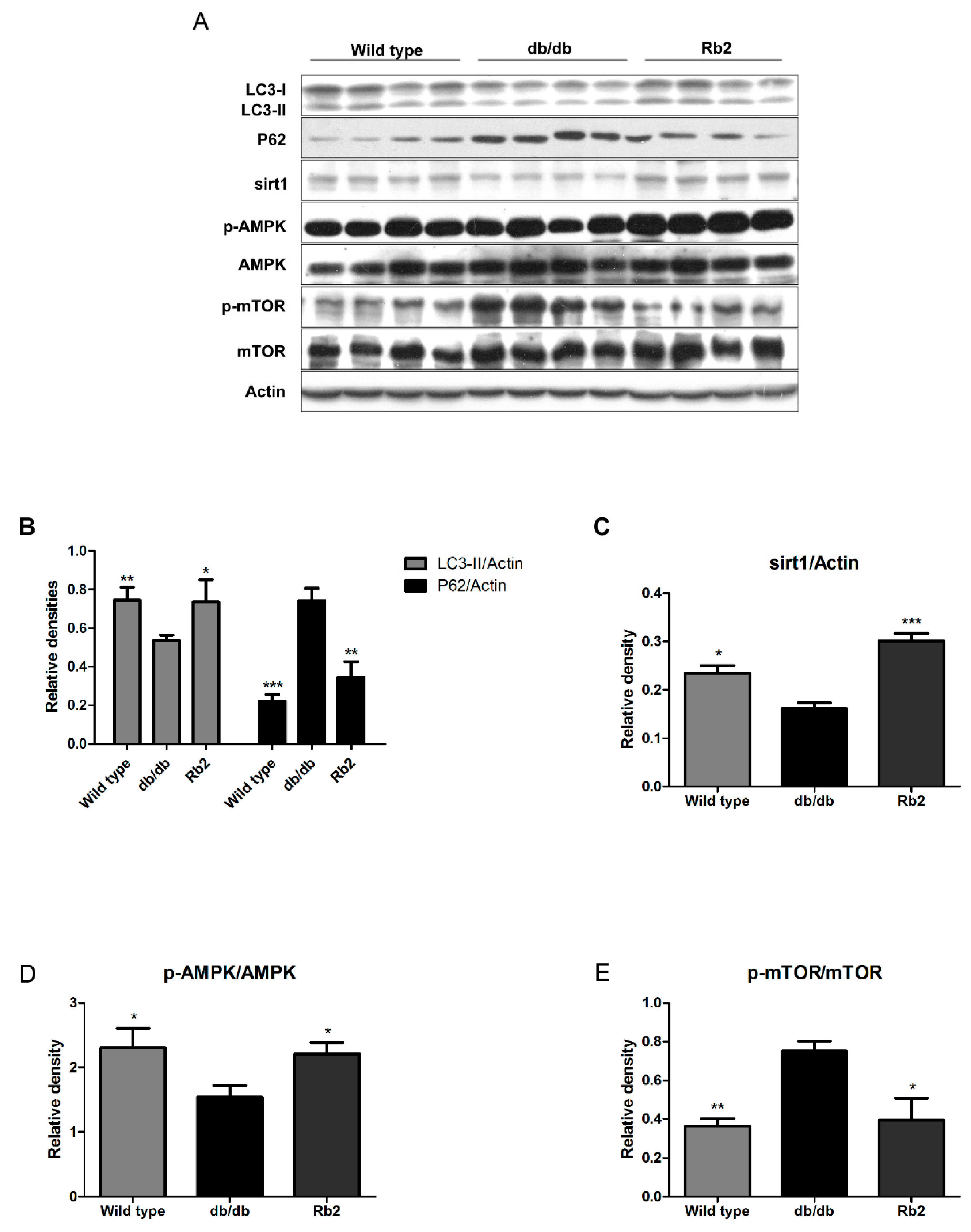
2.2. Rb2 Increased Expression of Sirt1 and Phosphorylation of AMP-Activated Protein Kinases (AMPK) and Restored the Impaired Hepatic Autophagy in db/db Mice
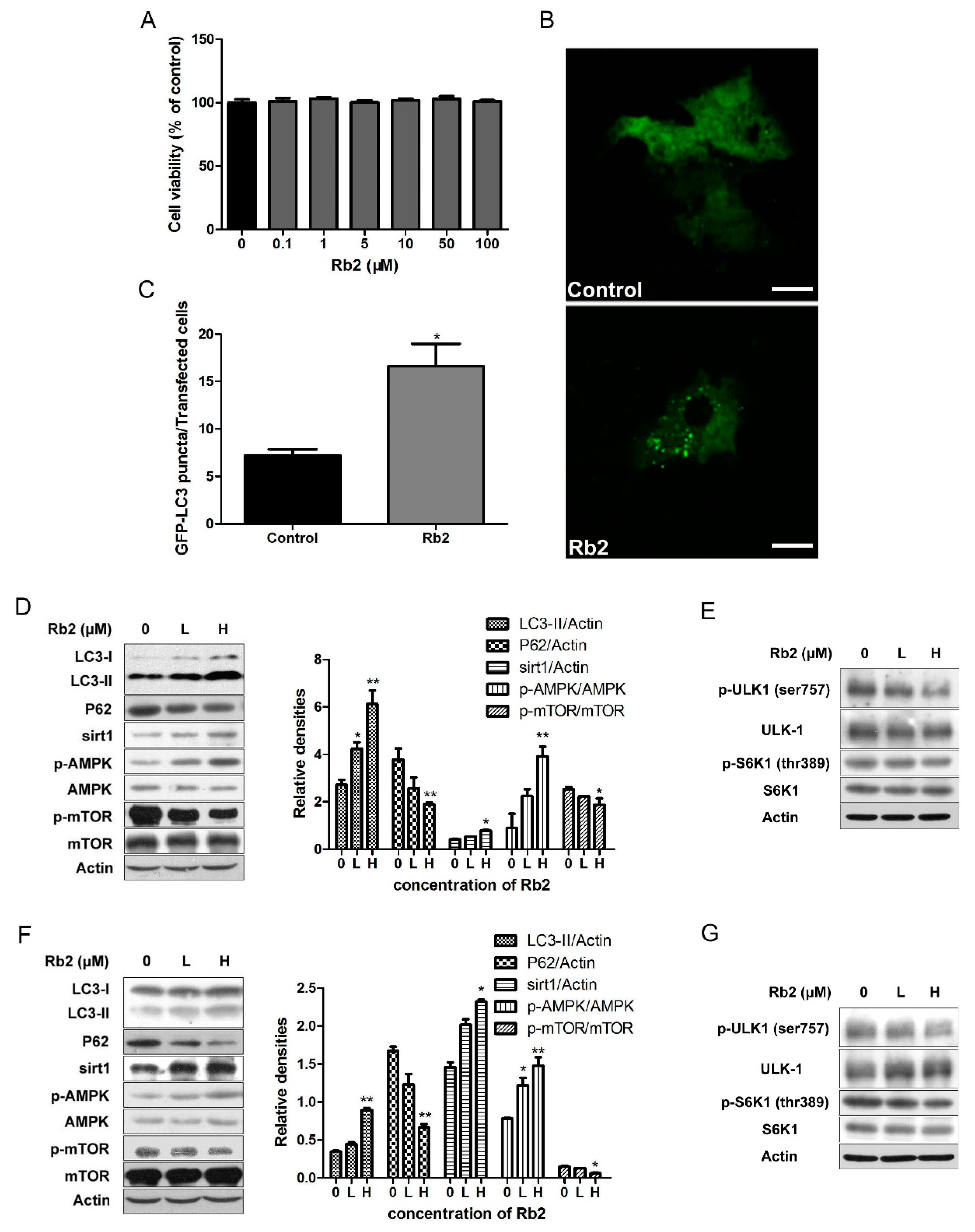
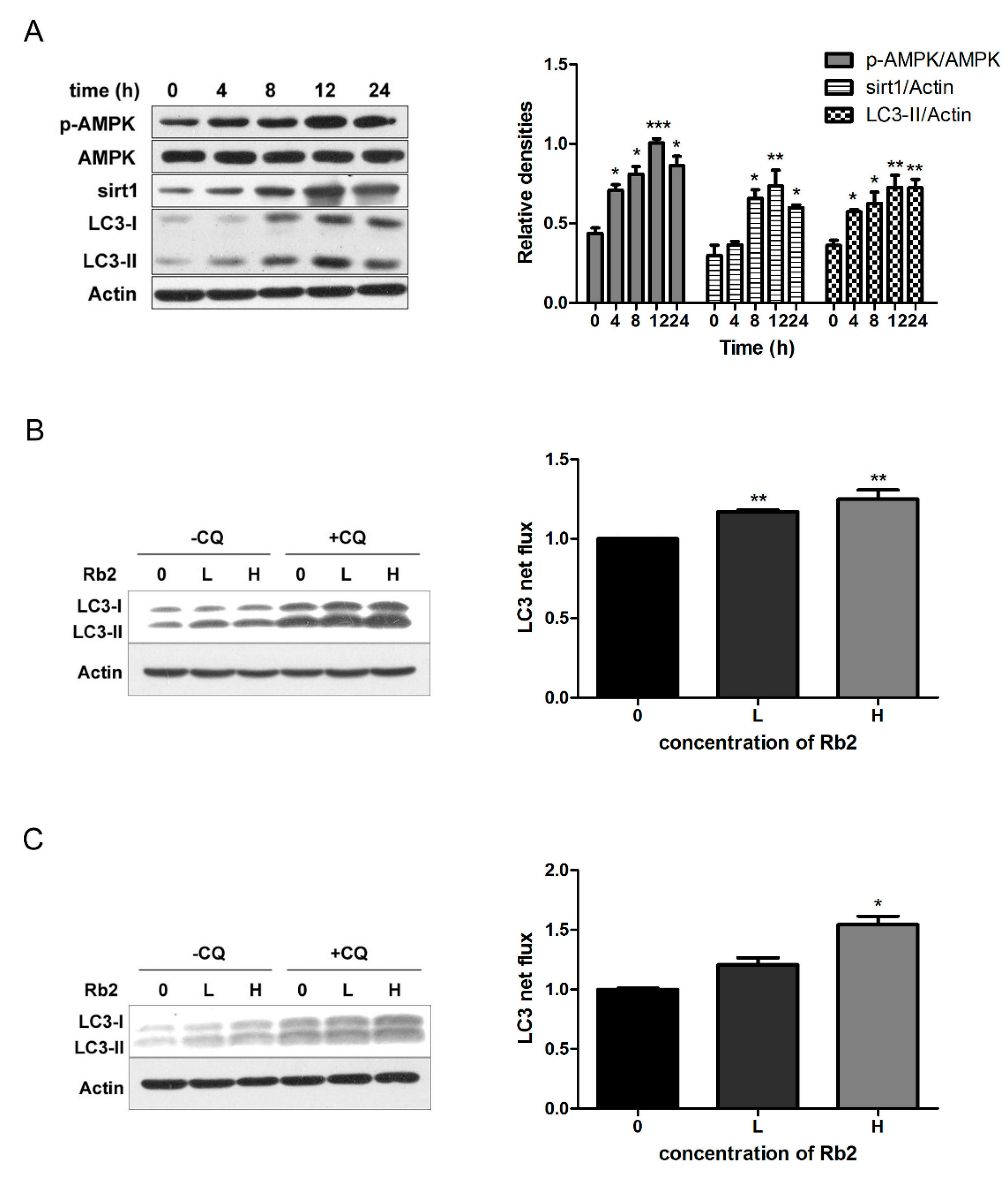
2.3. Rb2 Dose- and Time-Dependently Promoted Autophagic Flux in Hepatic Cells
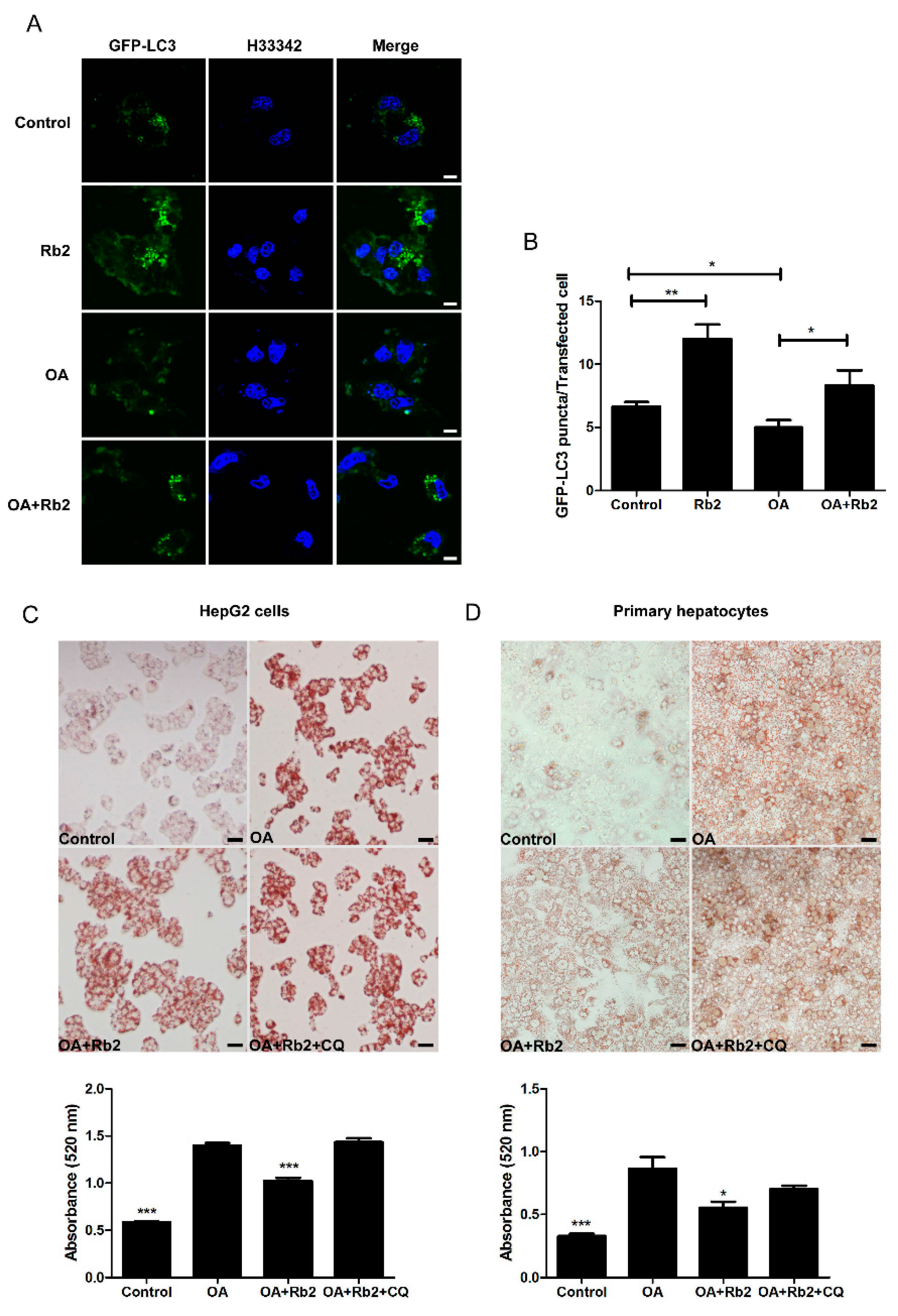
2.4. The Inhibitory Effect of Rb2 on Lipid Accumulation Depends on a Coordinate Increase in Hepatic Autophagy
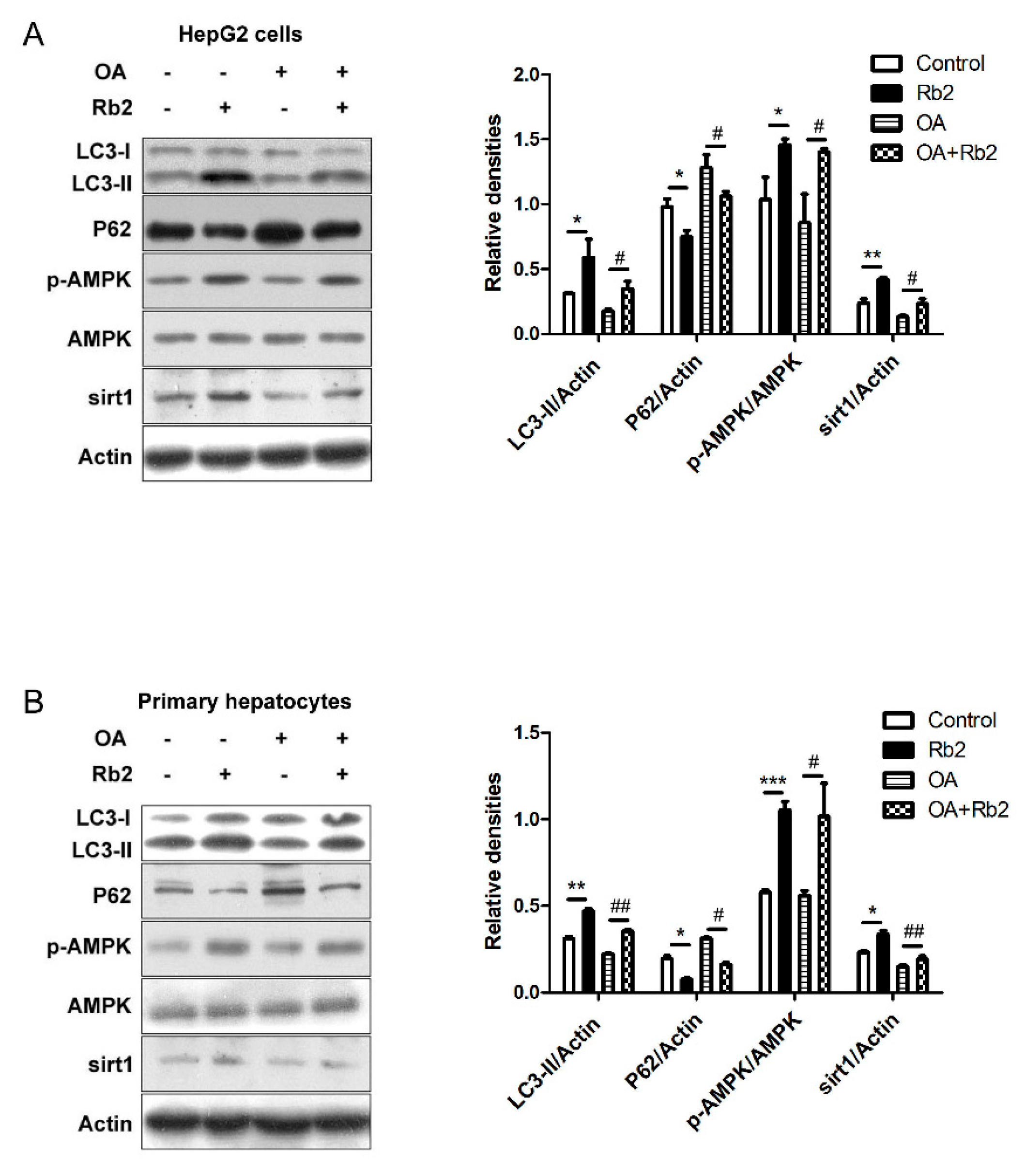
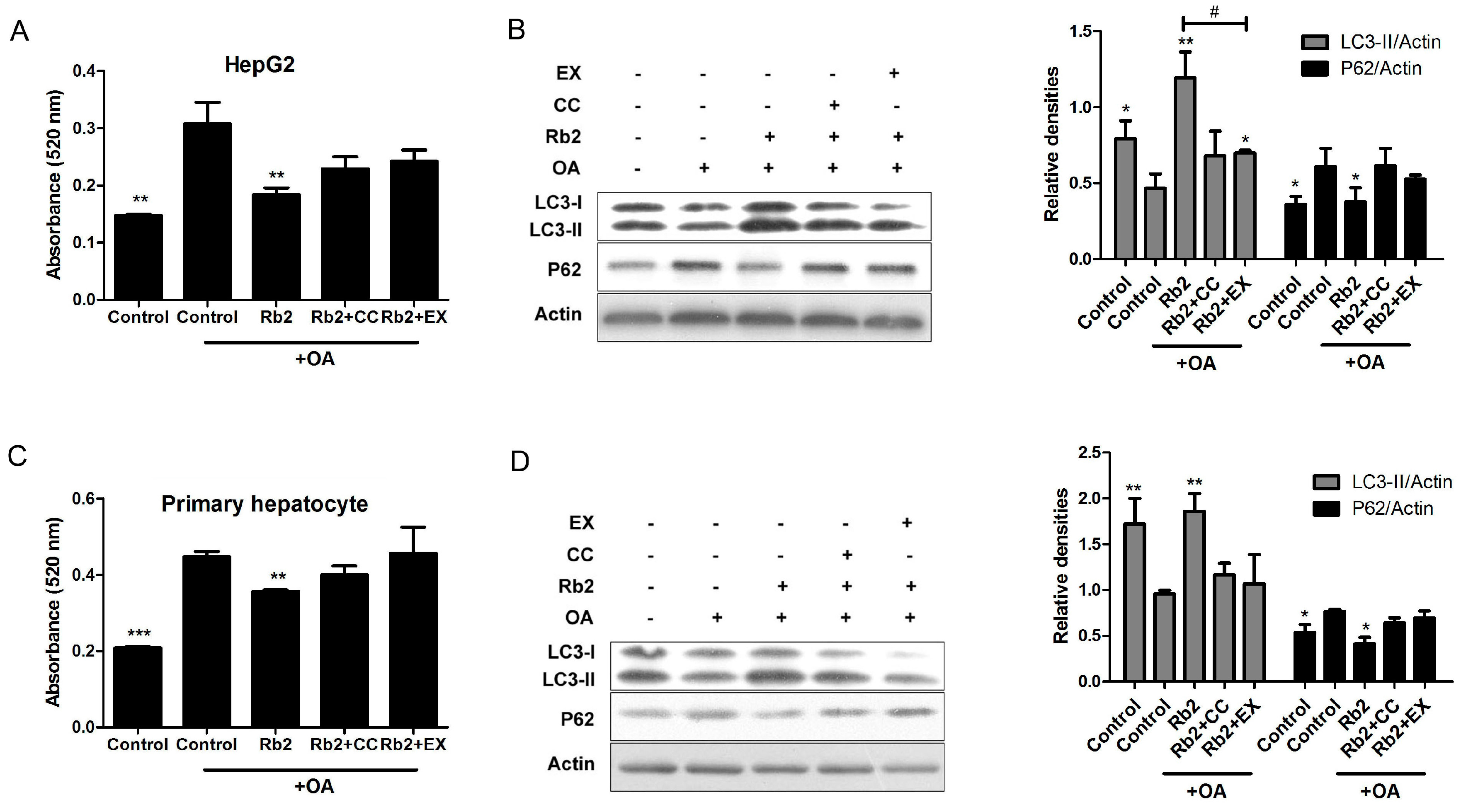
2.5. Rb2 Restored Impaired Hepatic Autophagy through Increasing Expression of Sirt1 and Phosphorylation of AMPK in Cultured Steatotic Hepatocytes
3. Discussion
4. Materials and Methods
4.1. Reagents
4.2. Animals
4.3. Metabolic and Biochemical Analysis
4.4. Cell Culture
4.5. 5-Diphenyltetrazoliumbromide (MTT) Assay
4.6. Western Blot Analysis
4.7. Detection of Autophagic Flux
4.8. Oil Red O (ORO) Staining and Quantification of Hepatic TG Contents
4.9. Liver Histology Examination
4.10. Statistical Analysis
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Im, D.S.; Nah, S.Y. Yin and Yang of ginseng pharmacology: Ginsenosides vs. gintonin. Acta Pharmacol. Sin. 2013, 34, 1367–1373. [Google Scholar] [CrossRef] [PubMed]
- Choi, K.T. Botanical characteristics, pharmacological effects and medicinal components of Korean Panax ginseng C A Meyer. Acta Pharmacol. Sin. 2008, 29, 1109–1118. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.M.; Yao, Q.; Chen, C. Ginseng compounds: An update on their molecular mechanisms and medical applications. Curr. Vasc. Pharmacol. 2009, 7, 293–302. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Meng, Y.; Sun, Q.; Zhang, Z.; Guo, X.; Sheng, X.; Tai, G.; Cheng, H.; Zhou, Y. Ginsenoside compound K sensitizes human colon cancer cells to TRAIL-induced apoptosis via autophagy-dependent and -independent DR5 upregulation. Cell Death Dis. 2016, 7, e2334. [Google Scholar] [CrossRef] [PubMed]
- Attele, A.S.; Zhou, Y.P.; Xie, J.T.; Wu, J.A.; Zhang, L.; Dey, L.; Pugh, W.; Rue, P.A.; Polonsky, K.S.; Yuan, C.S. Antidiabetic effects of Panax ginseng berry extract and the identification of an effective component. Diabetes 2002, 51, 1851–1858. [Google Scholar] [CrossRef] [PubMed]
- Yokozawa, T.; Kobayashi, T.; Oura, H.; Kawashima, Y. Hyperlipemia-improving effects of ginsenoside-Rb2 in streptozotocin-diabetic rats. Chem. Pharm. Bull. 1985, 33, 3893–3898. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.T.; Jung, T.W.; Lee, H.J.; Kim, S.G.; Shin, Y.S.; Whang, W.K. The antidiabetic effect of ginsenoside Rb2 via activation of AMPK. Arch. Pharm. Res. 2011, 34, 1201–1208. [Google Scholar] [CrossRef] [PubMed]
- Han, J.Y.; Lee, S.; Yang, J.H.; Kim, S.; Sim, J.; Kim, M.G.; Jeong, T.C.; Ku, S.K.; Cho, I.J.; Ki, S.H. Korean Red Ginseng attenuates ethanol-induced steatosis and oxidative stress via AMPK/Sirt1 activation. J. Ginseng Res. 2015, 39, 105–115. [Google Scholar] [CrossRef] [PubMed]
- Hu, C.F.; Sun, L.P.; Yang, Q.H.; Lu, D.X.; Luo, S. Ginsenosides from stems and leaves of Ginseng prevent ethanol-induced lipid accumulation in human L02 hepatocytes. Chin. J. Integr. Med. 2016. [Google Scholar] [CrossRef] [PubMed]
- Anstee, Q.M.; Targher, G.; Day, C.P. Progression of NAFLD to diabetes mellitus, cardiovascular disease or cirrhosis. Nat. Rev. Gastroenterol. Hepatol. 2013, 10, 330–344. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.; Kaushik, S.; Wang, Y.; Xiang, Y.; Novak, I.; Komatsu, M.; Tanaka, K.; Cuervo, A.M.; Czaja, M.J. Autophagy regulates lipid metabolism. Nature 2009, 458, 1131–1135. [Google Scholar] [CrossRef] [PubMed]
- Sinha, R.A.; You, S.H.; Zhou, J.; Siddique, M.M.; Bay, B.H.; Zhu, X.; Privalsky, M.L.; Cheng, S.Y.; Stevens, R.D.; Summers, S.A.; et al. Thyroid hormone stimulates hepatic lipid catabolism via activation of autophagy. J. Clin. Investig. 2012, 122, 2428–2438. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Rodriguez, A.; Mayoral, R.; Agra, N.; Valdecantos, M.P.; Pardo, V.; Miquilena-Colina, M.E.; Vargas-Castrillon, J.; lo Iacono, O.; Corazzari, M.; Fimia, G.M.; et al. Impaired autophagic flux is associated with increased endoplasmic reticulum stress during the development of NAFLD. Cell Death Dis. 2014, 5, e1179. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Lopez, N.; Singh, R. Autophagy and lipid droplets in the liver. Annu. Rev. Nutr. 2015, 35, 215–237. [Google Scholar] [CrossRef] [PubMed]
- Salminen, A.; Kaarniranta, K. AMP-activated protein kinase (AMPK) controls the aging process via an integrated signaling network. Ageing Res. Rev. 2012, 11, 230–241. [Google Scholar] [CrossRef] [PubMed]
- Pallauf, K.; Rimbach, G. Autophagy, polyphenols and healthy ageing. Ageing Res. Rev. 2013, 12, 237–252. [Google Scholar] [CrossRef] [PubMed]
- Sinha, R.A.; Farah, B.L.; Singh, B.K.; Siddique, M.M.; Li, Y.; Wu, Y.; Ilkayeva, O.R.; Gooding, J.; Ching, J.; Zhou, J.; et al. Caffeine stimulates hepatic lipid metabolism by the autophagy-lysosomal pathway in mice. Hepatology 2014, 59, 1366–1380. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.Y.; Xiong, X.Q.; Ren, X.S.; Zhao, M.X.; Shi, C.X.; Wang, J.J.; Zhou, Y.B.; Zhang, F.; Han, Y.; Gao, X.Y.; et al. FNDC5 Alleviates Hepatosteatosis by Restoring AMPK/mTOR-Mediated Autophagy, Fatty Acid Oxidation and Lipogenesis in Mice. Diabetes 2016, 65, 3262–3275. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Farah, B.L.; Sinha, R.A.; Wu, Y.; Singh, B.K.; Bay, B.H.; Yang, C.S.; Yen, P.M. Epigallocatechin-3-gallate (EGCG), a green tea polyphenol, stimulates hepatic autophagy and lipid clearance. PLoS ONE 2014, 9, e87161. [Google Scholar] [CrossRef] [PubMed]
- Trak-Smayra, V.; Paradis, V.; Massart, J.; Nasser, S.; Jebara, V.; Fromenty, B. Pathology of the liver in obese and diabetic ob/ob and db/db mice fed a standard or high-calorie diet. Int. J. Exp. Pathol. 2011, 92, 413–421. [Google Scholar] [CrossRef] [PubMed]
- Lancha, A.; Fruhbeck, G.; Gomez-Ambrosi, J. Peripheral signalling involved in energy homeostasis control. Nutr. Res. Rev. 2012, 25, 223–248. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.M.; Lee, Y.H.; Kim, J.W.; Ham, D.S.; Kang, E.S.; Cha, B.S.; Lee, H.C.; Lee, B.W. Metformin alleviates hepatosteatosis by restoring SIRT1-mediated autophagy induction via an AMP-activated protein kinase-independent pathway. Autophagy 2015, 11, 46–59. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.C.; Guan, K.L. mTOR: A pharmacologic target for autophagy regulation. J. Clin. Investig. 2015, 125, 25–32. [Google Scholar] [CrossRef] [PubMed]
- Cheon, J.M.; Kim, D.I.; Kim, K.S. Insulin sensitivity improvement of fermented Korean Red Ginseng (Panax ginseng) mediated by insulin resistance hallmarks in old-aged ob/ob mice. J. Ginseng Res. 2015, 39, 331–337. [Google Scholar] [CrossRef] [PubMed]
- Shishtar, E.; Sievenpiper, J.L.; Djedovic, V.; Cozma, A.I.; Ha, V.; Jayalath, V.H.; Jenkins, D.J.; Meija, S.B.; de Souza, R.J.; Jovanovski, E.; et al. The effect of ginseng (the genus panax) on glycemic control: A systematic review and meta-analysis of randomized controlled clinical trials. PLoS ONE 2014, 9, e107391. [Google Scholar] [CrossRef] [PubMed]
- Yao, H.; Qiao, Y.J.; Zhao, Y.L.; Tao, X.F.; Xu, L.N.; Yin, L.H.; Qi, Y.; Peng, J.Y. Herbal medicines and nonalcoholic fatty liver disease. World J. Gastroenterol. 2016, 22, 6890–6905. [Google Scholar] [CrossRef] [PubMed]
- Leamy, A.K.; Egnatchik, R.A.; Young, J.D. Molecular mechanisms and the role of saturated fatty acids in the progression of non-alcoholic fatty liver disease. Prog. Lipid Res. 2013, 52, 165–174. [Google Scholar] [CrossRef] [PubMed]
- Malaguarnera, M.; di Rosa, M.; Nicoletti, F.; Malaguarnera, L. Molecular mechanisms involved in NAFLD progression. J. Mol. Med. 2009, 87, 679–695. [Google Scholar] [CrossRef] [PubMed]
- Musso, G.; Gambino, R.; Cassader, M. Recent insights into hepatic lipid metabolism in non-alcoholic fatty liver disease (NAFLD). Prog. Lipid Res. 2009, 48, 1–26. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Li, P.; Fu, S.; Calay, E.S.; Hotamisligil, G.S. Defective hepatic autophagy in obesity promotes ER stress and causes insulin resistance. Cell Metab. 2010, 11, 467–478. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.; Cuervo, A.M. Autophagy in the cellular energetic balance. Cell Metab. 2011, 13, 495–504. [Google Scholar] [CrossRef] [PubMed]
- Greenberg, A.S.; Coleman, R.A.; Kraemer, F.B.; McManaman, J.L.; Obin, M.S.; Puri, V.; Yan, Q.W.; Miyoshi, H.; Mashek, D.G. The role of lipid droplets in metabolic disease in rodents and humans. J. Clin. Investig. 2011, 121, 2102–2110. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.; Cuervo, A.M. Lipophagy: Connecting autophagy and lipid metabolism. Int. J. Cell Biol. 2012, 2012, 282041. [Google Scholar] [CrossRef] [PubMed]
- Cuervo, A.M.; Bergamini, E.; Brunk, U.T.; Droge, W.; Ffrench, M.; Terman, A. Autophagy and aging: The importance of maintaining “clean” cells. Autophagy 2005, 1, 131–140. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Zhao, T.; Liu, H.; Zhang, L. Ginsenoside Rh2 inhibits hepatocellular carcinoma through β-catenin and autophagy. Sci. Rep. 2016, 6, 19383. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.; Wang, N.; Rocchi, A.; Zhang, W.; Vassar, R.; Zhou, Y.; He, C. Identification of natural products with neuronal and metabolic benefits through autophagy induction. Autophagy 2017, 13, 41–56. [Google Scholar] [CrossRef] [PubMed]
- Mao, N.; Tan, R.Z.; Wang, S.Q.; Wei, C.; Shi, X.L.; Fan, J.M.; Wang, L. Ginsenoside Rg1 inhibits angiotensin II-induced podocyte autophagy via AMPK/mTOR/PI3K pathway. Cell Biol. Int. 2016, 40, 917–925. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Mao, N.; Tan, R.Z.; Wang, H.L.; Wen, J.; Liu, Y.H.; Furhad, M.; Fan, J.M. Ginsenoside Rg1 reduces aldosterone-induced autophagy via the AMPK/mTOR pathway in NRK-52E cells. Int. J. Mol. Med. 2015, 36, 518–526. [Google Scholar] [CrossRef] [PubMed]
- Luo, T.; Liu, G.; Ma, H.; Lu, B.; Xu, H.; Wang, Y.; Wu, J.; Ge, P.; Liang, J. Inhibition of autophagy via activation of PI3K/Akt pathway contributes to the protection of ginsenoside Rb1 against neuronal death caused by ischemic insults. Int. J. Mol. Sci. 2014, 15, 15426–15442. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Lu, T.; Yue, X.; Wei, N.; Jiang, Y.; Chen, M.; Ni, G.; Liu, X.; Xu, G. Neuroprotective effect of ginsenoside Rb1 on glutamate-induced neurotoxicity: With emphasis on autophagy. Neurosci. Lett. 2010, 482, 264–268. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Sun, P.; Chen, L.; Huang, Q.; Chen, K.; Jia, Q.; Li, Y.; Wang, H. Cinnamtannin D-1 protects pancreatic β-cells from palmitic acid-induced apoptosis by attenuating oxidative stress. J. Agric. Food Chem. 2014, 62, 5038–5045. [Google Scholar] [CrossRef] [PubMed]
- Seglen, P.O. Hepatocyte suspensions and cultures as tools in experimental carcinogenesis. J. Toxicol. Environ. Health 1979, 5, 551–560. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.J.; Chen, S.; Huang, K.X.; Le, W.D. Why should autophagic flux be assessed? Acta Pharmacol. Sin. 2013, 34, 595–599. [Google Scholar] [CrossRef] [PubMed]
- Salmon, D.M.; Flatt, J.P. Effect of dietary fat content on the incidence of obesity among ad libitum fed mice. Int. J. Obes. 1985, 9, 443–449. [Google Scholar] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | Wild Type | db/db | Rb2 |
|---|---|---|---|
| Epididymal fat weight (g) | 0.18 ± 0.05 *** | 2.43 ± 0.31 | 2.50 ± 0.27 |
| Perirenal fat weight (g) | 0.1 ± 0.05 ** | 1.32 ± 0.21 | 1.40 ± 0.14 |
| Serum NEFA (mM) | 0.23 ± 0.06 * | 0.68 ± 0.14 | 0.57 ± 0.08 |
| Serum triacylglycerol (mM) | 0.98 ± 0.29 ** | 1.50 ± 0.33 | 1.16 ± 0.12 * |
| Serum cholesterol (mM) | 1.97 ± 0.53 ** | 2.77 ± 0.14 | 2.24 ± 0.17 * |
| Serum glucose (mM) | 6.08 ± 0.90 ** | 19.27 ± 2.56 | 15.15 ± 4.46 * |
| Serum insulin (ng/ml) | 2.6 ± 0.51 *** | 20.58 ± 2.51 | 17.54 ± 1.83 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huang, Q.; Wang, T.; Yang, L.; Wang, H.-Y. Ginsenoside Rb2 Alleviates Hepatic Lipid Accumulation by Restoring Autophagy via Induction of Sirt1 and Activation of AMPK. Int. J. Mol. Sci. 2017, 18, 1063. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms18051063
Huang Q, Wang T, Yang L, Wang H-Y. Ginsenoside Rb2 Alleviates Hepatic Lipid Accumulation by Restoring Autophagy via Induction of Sirt1 and Activation of AMPK. International Journal of Molecular Sciences. 2017; 18(5):1063. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms18051063
Chicago/Turabian StyleHuang, Qi, Ting Wang, Liu Yang, and He-Yao Wang. 2017. "Ginsenoside Rb2 Alleviates Hepatic Lipid Accumulation by Restoring Autophagy via Induction of Sirt1 and Activation of AMPK" International Journal of Molecular Sciences 18, no. 5: 1063. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms18051063