
High-Mobility Group Box 1 Disrupts Metabolic Function with Cigarette Smoke Exposure in a Ceramide-Dependent Manner


 and
and 
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
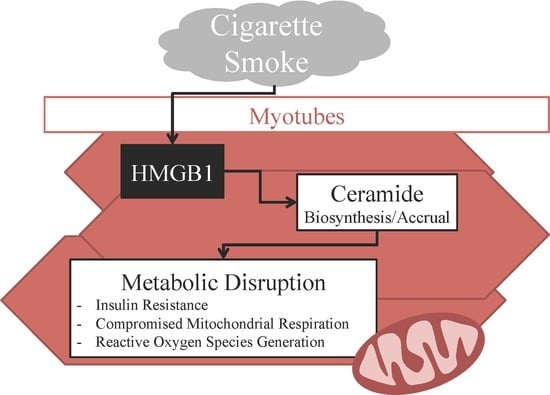
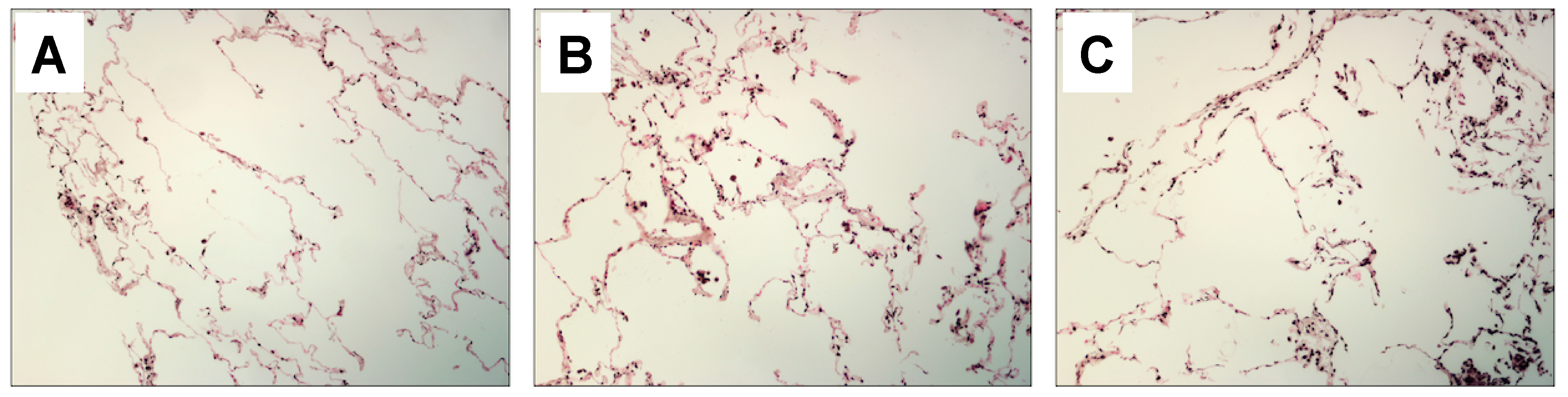
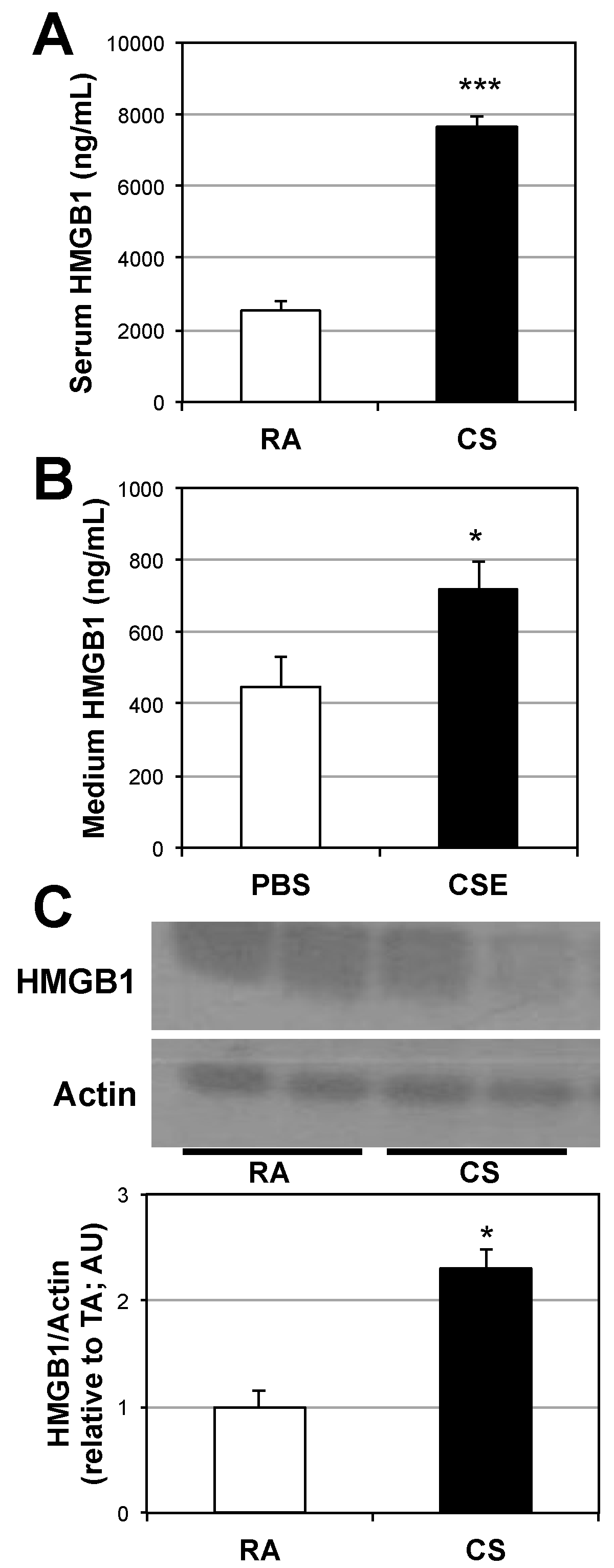
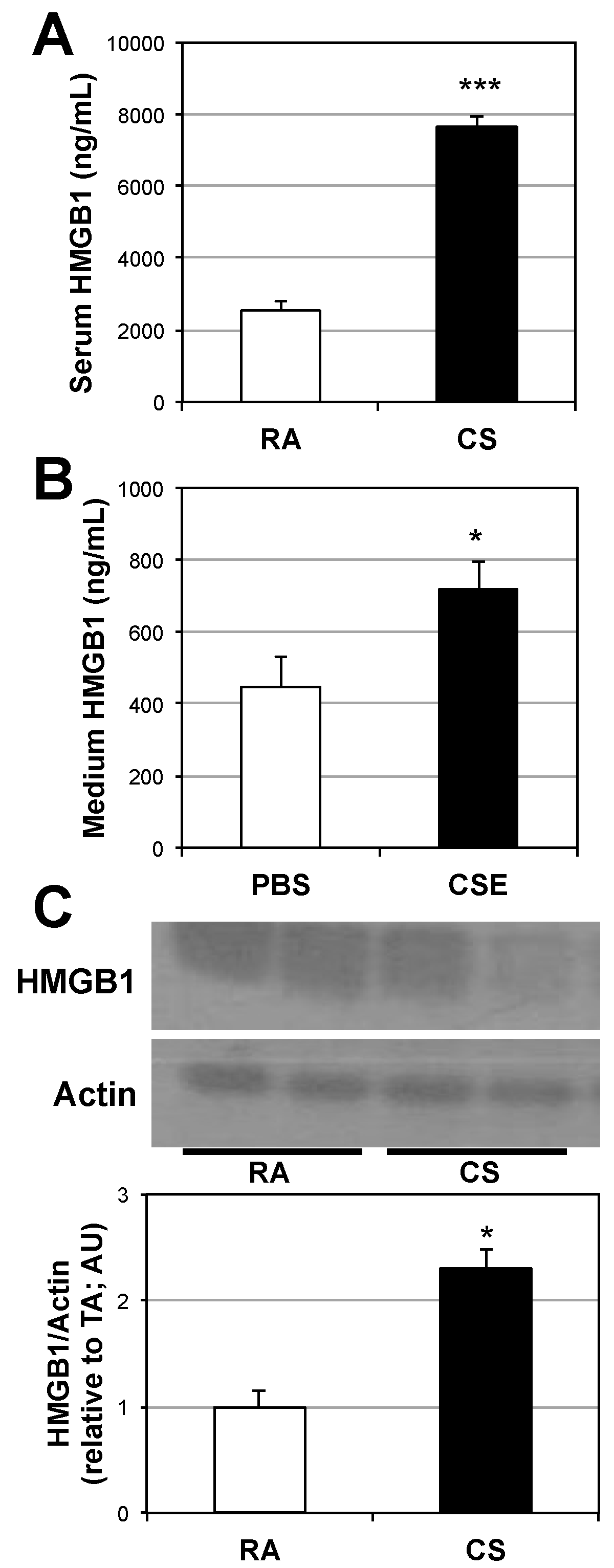
2.1. Lung and Serum HMGB1 Is Elevated Following Cigarette Smoke Exposure
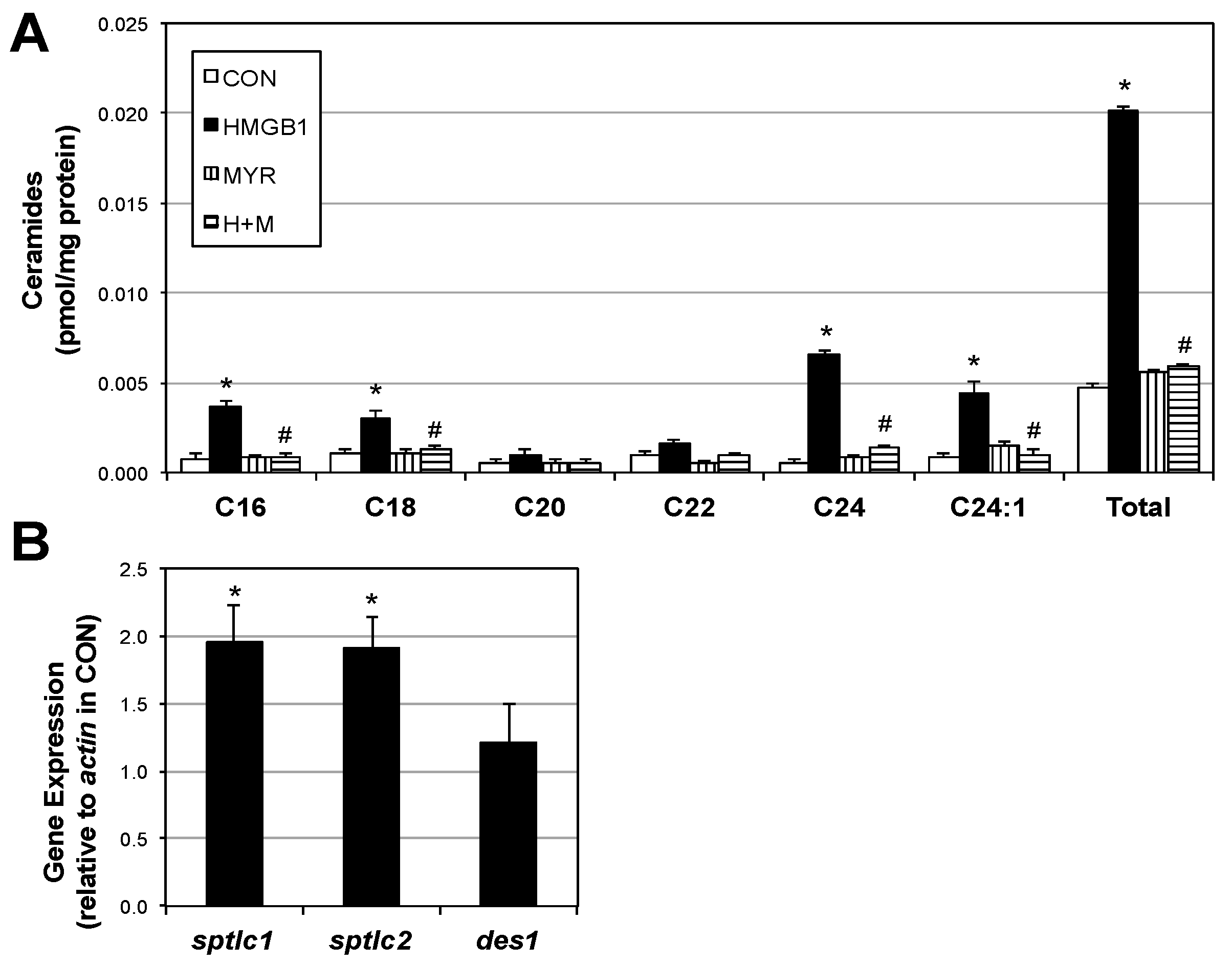
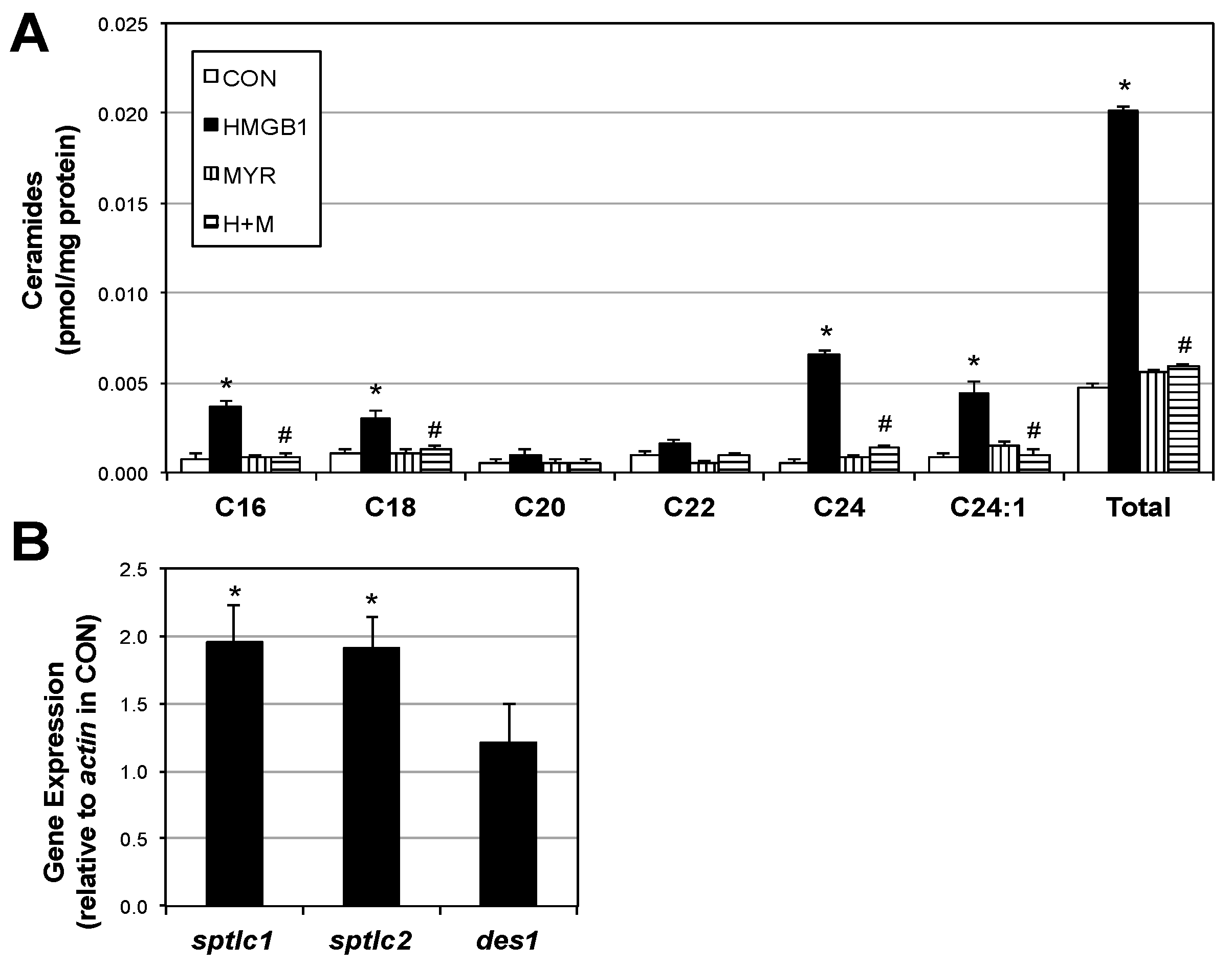
2.2. HMGB1 Increases Myotube Ceramide Biosynthesis
2.3. HMGB1 Disrupts Myotube Mitochondrial Function and Insulin Signaling via Ceramides
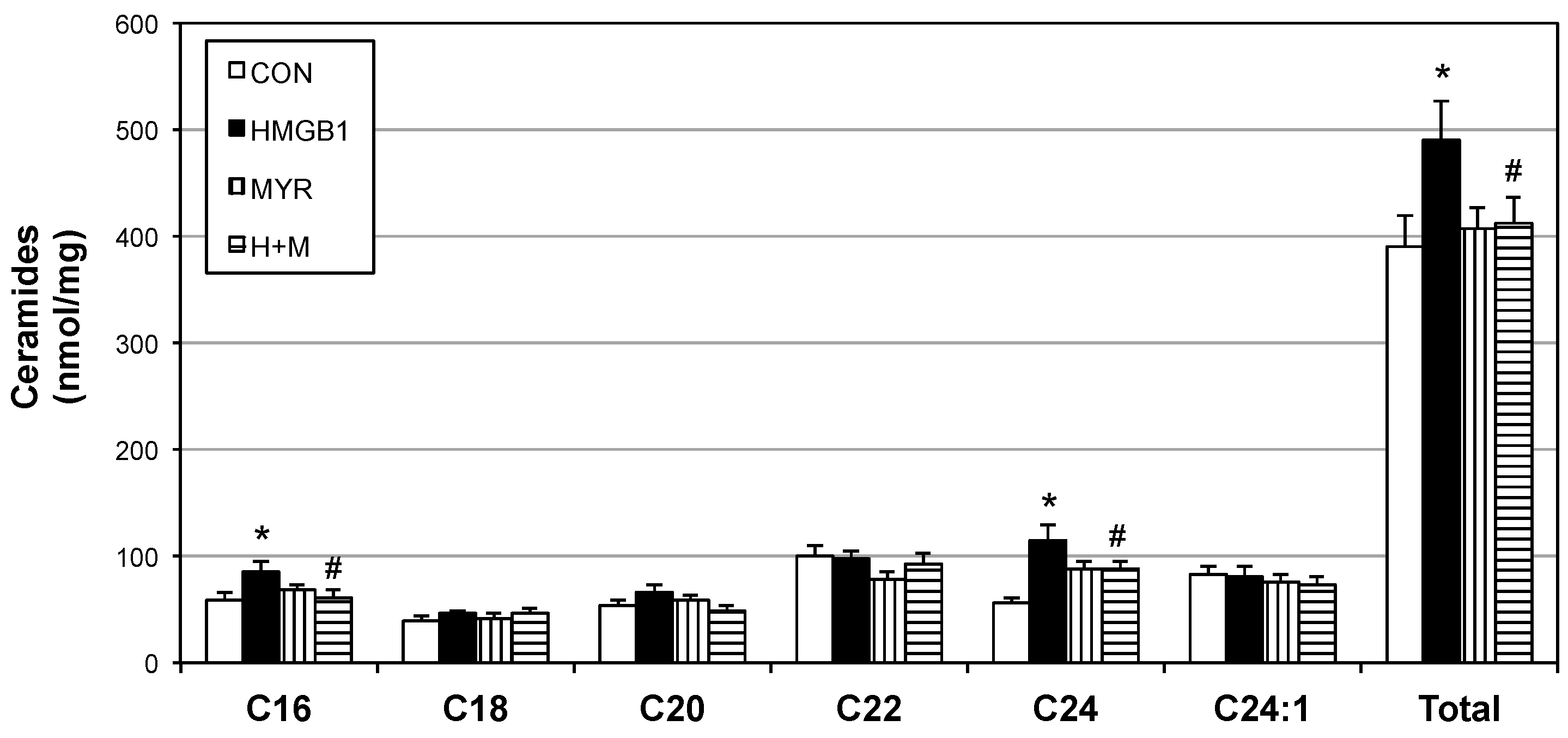
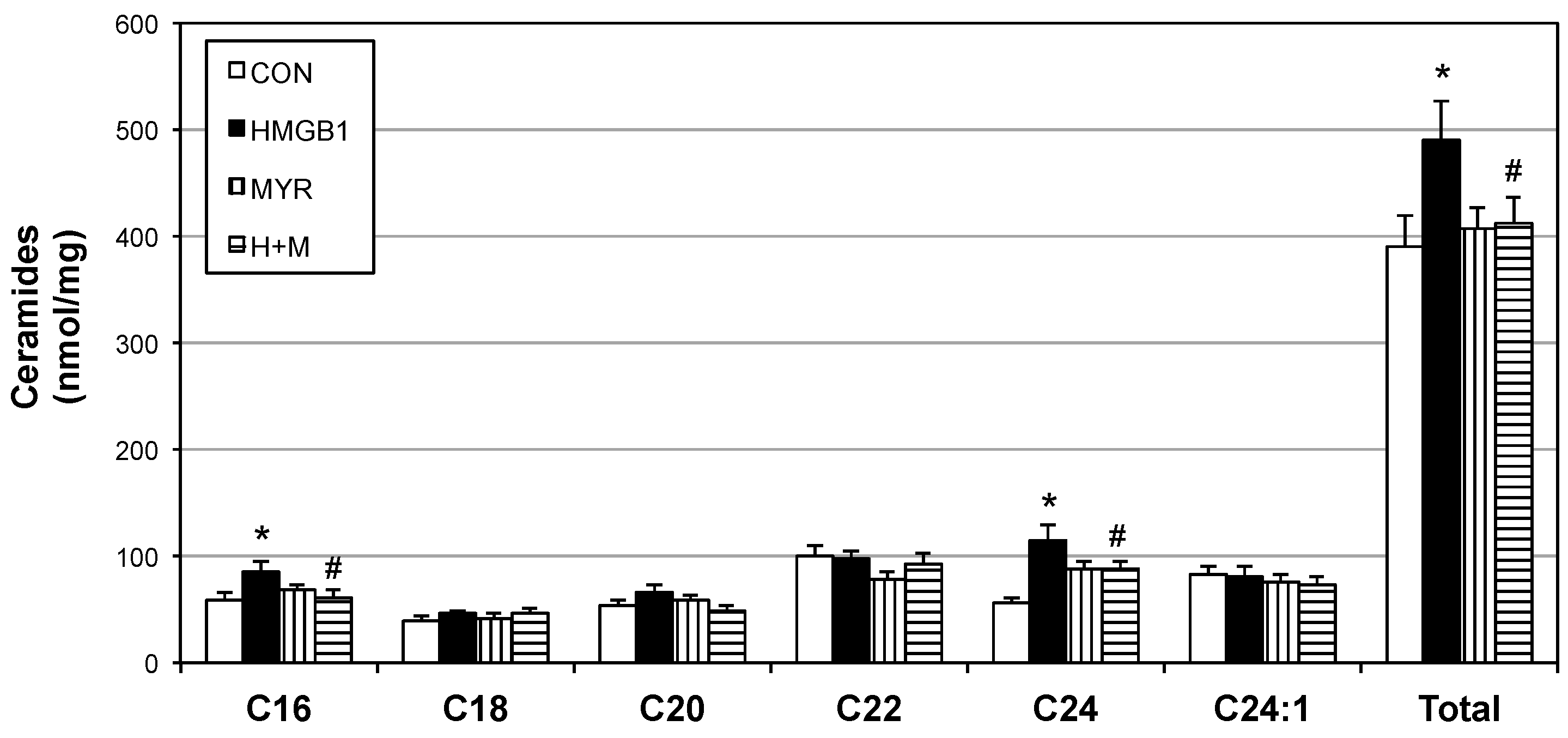
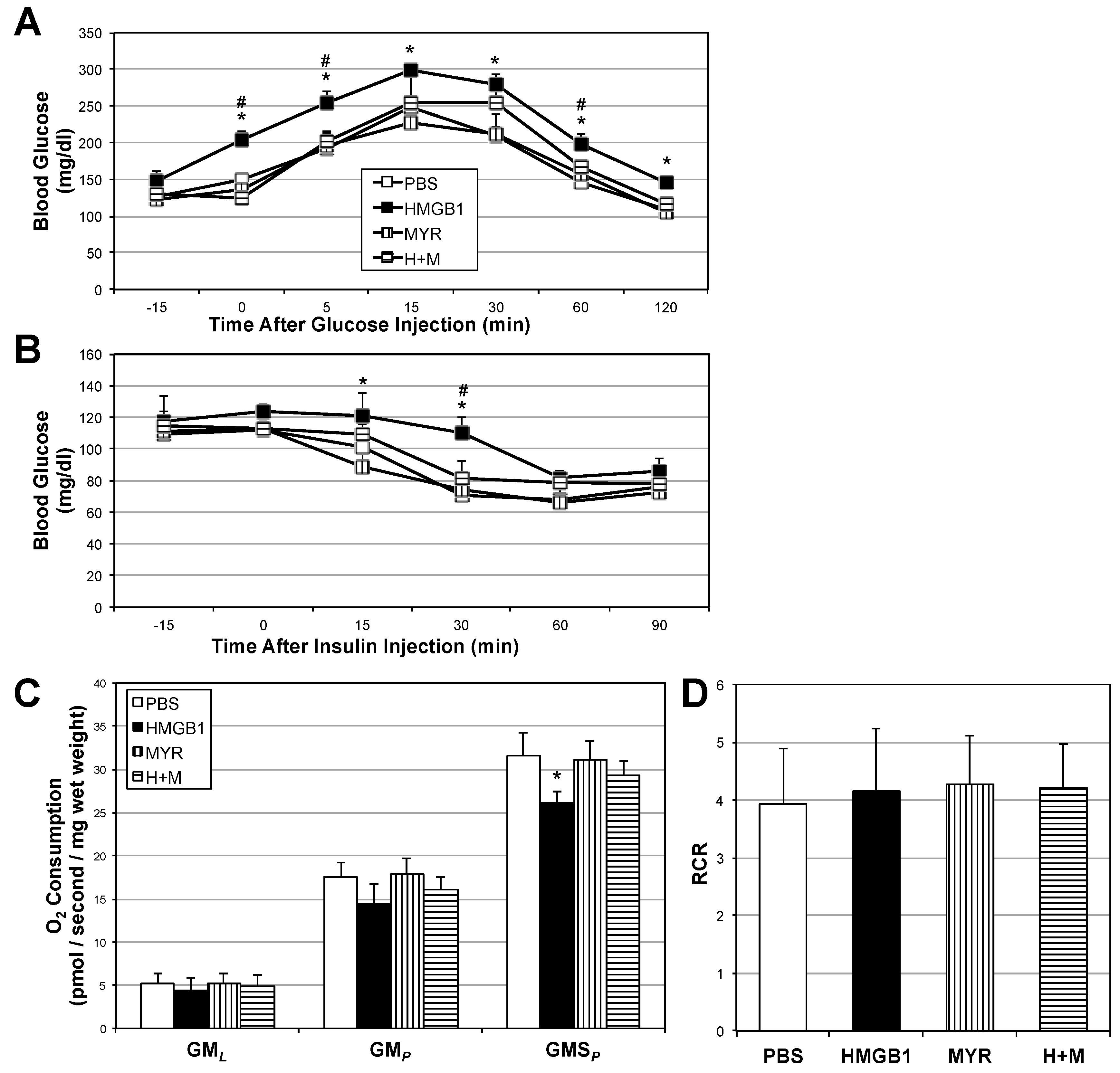
2.4. HMGB1 Injections Increase Muscle Ceramides and Disrupt Metabolic Function
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Animals
4.3. Serum and Culture Medium HMGB1 Analysis
4.4. Lipid Analysis
4.5. Cell and Muscle Fiber Bundle Permeabilization
4.6. Mitochondrial Respiration Protocol
4.7. Oxidative Stress
4.8. Human Samples
4.9. Immunohistochemistry
4.10. qRT-PCR
4.11. Statistics
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Doll, R.; Peto, R.; Boreham, J.; Sutherland, I. Mortality in relation to smoking: 50 years’ observations on male british doctors. BMJ 2004, 328, 1519. [Google Scholar] [CrossRef] [PubMed]
- Oberg, M.; Jaakkola, M.S.; Woodward, A.; Peruga, A.; Pruss-Ustun, A. Worldwide burden of disease from exposure to second-hand smoke: A retrospective analysis of data from 192 countries. Lancet 2011, 377, 139–146. [Google Scholar] [CrossRef]
- Thatcher, M.O.; Tippetts, T.S.; Nelson, M.B.; Swensen, A.C.; Winden, D.R.; Hansen, M.E.; Anderson, M.C.; Johnson, I.E.; Porter, J.P.; Reynolds, P.R.; et al. Ceramides mediate cigarette smoke-induced metabolic disruption in mice. Am. J. Physiol. Endocrinol. Metab. 2014, 307, E919–E927. [Google Scholar] [CrossRef] [PubMed]
- Bikman, B.T. A role for sphingolipids in the pathophysiology of obesity-induced inflammation. Cell. Mol. Life Sci. 2012, 69, 2135–2146. [Google Scholar] [CrossRef] [PubMed]
- Nelson, M.B.; Swensen, A.C.; Winden, D.R.; Bodine, J.S.; Bikman, B.T.; Reynolds, P.R. Cardiomyocyte mitochondrial respiration is reduced by receptor for advanced glycation end-product signaling in a ceramide-dependent manner. Am. J. Physiol. Heart Circ. Physiol. 2015, 309, H63–H69. [Google Scholar] [CrossRef] [PubMed]
- Tippetts, T.S.; Winden, D.R.; Swensen, A.C.; Nelson, M.B.; Thatcher, M.O.; Saito, R.R.; Condie, T.B.; Simmons, K.J.; Judd, A.M.; Reynolds, P.R.; et al. Cigarette smoke increases cardiomyocyte ceramide accumulation and inhibits mitochondrial respiration. BMC Cardiovasc. Disord. 2014, 14, 165. [Google Scholar] [CrossRef] [PubMed]
- Bikman, B.T.; Guan, Y.; Shui, G.; Siddique, M.M.; Holland, W.L.; Kim, J.Y.; Fabrias, G.; Wenk, M.R.; Summers, S.A. Fenretinide prevents lipid-induced insulin resistance by blocking ceramide biosynthesis. J. Biol. Chem. 2012, 287, 17426–17437. [Google Scholar] [CrossRef] [PubMed]
- Baeder, A.C.; Napa, K.; Richardson, S.T.; Taylor, O.J.; Andersen, S.G.; Wilcox, S.H.; Winden, D.R.; Reynolds, P.R.; Bikman, B.T. Oral gingival cell cigarette smoke exposure induces muscle cell metabolic disruption. Int. J. Dent. 2016, 2016, 2763160. [Google Scholar] [CrossRef] [PubMed]
- Summers, S.A. Ceramides in insulin resistance and lipotoxicity. Prog. Lipid Res. 2006, 45, 42–72. [Google Scholar] [CrossRef] [PubMed]
- Smith, M.E.; Tippetts, T.S.; Brassfield, E.S.; Tucker, B.J.; Ockey, A.; Swensen, A.C.; Anthonymuthu, T.S.; Washburn, T.D.; Kane, D.A.; Prince, J.T.; et al. Mitochondrial fission mediates ceramide-induced metabolic disruption in skeletal muscle. Biochem. J. 2013, 456, 427–439. [Google Scholar] [CrossRef] [PubMed]
- Robinson, A.B.; Johnson, K.D.; Bennion, B.G.; Reynolds, P.R. Rage signaling by alveolar macrophages influences tobacco smoke-induced inflammation. Am. J. Physiol. Lung Cell. Mol. Physiol. 2012, 302, L1192–L1199. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, P.R.; Kasteler, S.D.; Cosio, M.G.; Sturrock, A.; Huecksteadt, T.; Hoidal, J.R. Rage: Developmental expression and positive feedback regulation by EGR-1 during cigarette smoke exposure in pulmonary epithelial cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 2008, 294, L1094–L1101. [Google Scholar] [CrossRef] [PubMed]
- Musumeci, D.; Roviello, G.N.; Montesarchio, D. An overview on HMGB1 inhibitors as potential therapeutic agents in HMGB1-related pathologies. Pharmacol. Ther. 2014, 141, 347–357. [Google Scholar] [CrossRef] [PubMed]
- Carlson, G.L. Insulin resistance in sepsis. Br. J. Surg. 2003, 90, 259–260. [Google Scholar] [CrossRef] [PubMed]
- Holland, W.L.; Bikman, B.T.; Wang, L.P.; Yuguang, G.; Sargent, K.M.; Bulchand, S.; Knotts, T.A.; Shui, G.; Clegg, D.J.; Wenk, M.R.; et al. Lipid-induced insulin resistance mediated by the proinflammatory receptor tlr4 requires saturated fatty acid-induced ceramide biosynthesis in mice. J. Clin. Investig. 2011, 121, 1858–1870. [Google Scholar] [CrossRef] [PubMed]
- Rauvala, H.; Rouhiainen, A. Rage as a receptor of HMGB1 (amphoterin): Roles in health and disease. Curr. Mol. Med. 2007, 7, 725–734. [Google Scholar] [CrossRef] [PubMed]
- Volz, H.C.; Kaya, Z.; Katus, H.A.; Andrassy, M. The role of HMGB1/RAGE in inflammatory cardiomyopathy. Semin. Thromb. Hemost. 2010, 36, 185–194. [Google Scholar] [CrossRef] [PubMed]
- Lin, L.; Zhong, K.; Sun, Z.; Wu, G.; Ding, G. Receptor for advanced glycation end products (RAGE) partially mediates HMGB1-ERKs activation in clear cell renal cell carcinoma. J. Cancer Res. Clin. Oncol. 2012, 138, 11–22. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.C.; Yi, P.P.; Zhou, R.R.; Xiao, M.F.; Huang, Z.B.; Tang, D.L.; Huang, Y.; Fan, X.G. The role of HMGB1-RAGE axis in migration and invasion of hepatocellular carcinoma cell lines. Mol. Cell. Biochem. 2014, 390, 271–280. [Google Scholar] [CrossRef] [PubMed]
- Kang, R.; Tang, D.; Schapiro, N.E.; Loux, T.; Livesey, K.M.; Billiar, T.R.; Wang, H.; Van Houten, B.; Lotze, M.T.; Zeh, H.J. The HMGB1/RAGE inflammatory pathway promotes pancreatic tumor growth by regulating mitochondrial bioenergetics. Oncogene 2014, 33, 567–577. [Google Scholar] [CrossRef] [PubMed]
- Tang, D.; Kang, R.; Zeh, H.J., 3rd.; Lotze, M.T. High-mobility group box 1, oxidative stress, and disease. Antioxid. Redox Signal. 2011, 14, 1315–1335. [Google Scholar] [CrossRef] [PubMed]
- Lo Coco, D.; Veglianese, P.; Allievi, E.; Bendotti, C. Distribution and cellular localization of high mobility group box protein 1 (HMGB1) in the spinal cord of a transgenic mouse model of als. Neurosci. Lett. 2007, 412, 73–77. [Google Scholar] [CrossRef] [PubMed]
- Tang, D.; Kang, R.; Cheh, C.W.; Livesey, K.M.; Liang, X.; Schapiro, N.E.; Benschop, R.; Sparvero, L.J.; Amoscato, A.A.; Tracey, K.J.; et al. HMGB1 release and redox regulates autophagy and apoptosis in cancer cells. Oncogene 2010, 29, 5299–5310. [Google Scholar] [CrossRef] [PubMed]
- Poyton, R.O.; McEwen, J.E. Crosstalk between nuclear and mitochondrial genomes. Annu. Rev. Biochem. 1996, 65, 563–607. [Google Scholar] [CrossRef] [PubMed]
- Tang, D.; Kang, R.; Livesey, K.M.; Kroemer, G.; Billiar, T.R.; van Houten, B.; Zeh, H.J., 3rd.; Lotze, M.T. High-mobility group box 1 is essential for mitochondrial quality control. Cell Metab. 2011, 13, 701–711. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Wang, T.; Guo, L.; Shen, Y.; Yang, T.; Wan, C.; Liao, Z.; Xu, D.; Wen, F. Overexpression of rage contributes to cigarette smoke-induced nitric oxide generation in copd. Lung 2014, 192, 267–275. [Google Scholar] [CrossRef] [PubMed]
- Rao, N.V.; Argyle, B.; Xu, X.; Reynolds, P.R.; Walenga, J.M.; Prechel, M.; Prestwich, G.D.; MacArthur, R.B.; Walters, B.B.; Hoidal, J.R.; et al. Low anticoagulant heparin targets multiple sites of inflammation, suppresses heparin-induced thrombocytopenia, and inhibits interaction of rage with its ligands. Am. J. Phys. Cell Phys. 2010, 299, C97–C110. [Google Scholar] [CrossRef] [PubMed]
- Peltz, E.D.; Moore, E.E.; Eckels, P.C.; Damle, S.S.; Tsuruta, Y.; Johnson, J.L.; Sauaia, A.; Silliman, C.C.; Banerjee, A.; Abraham, E. HMGB1 is markedly elevated within 6 hours of mechanical trauma in humans. Shock 2009, 32, 17–22. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Yang, H.; Tracey, K.J. Extracellular role of HMGB1 in inflammation and sepsis. J. Intern. Med. 2004, 255, 320–331. [Google Scholar] [CrossRef] [PubMed]
- Bikman, B.T.; Summers, S.A. Ceramides as modulators of cellular and whole-body metabolism. J. Clin. Investig. 2011, 121, 4222–4230. [Google Scholar] [CrossRef] [PubMed]
- Hansen, M.E.; Tippetts, T.S.; Anderson, M.C.; Holub, Z.E.; Moulton, E.R.; Swensen, A.C.; Prince, J.T.; Bikman, B.T. Insulin increases ceramide synthesis in skeletal muscle. J. Diabetes Res. 2014, 2014, 765784. [Google Scholar] [CrossRef] [PubMed]
- Xia, J.Y.; Holland, W.L.; Kusminski, C.M.; Sun, K.; Sharma, A.X.; Pearson, M.J.; Sifuentes, A.J.; McDonald, J.G.; Gordillo, R.; Scherer, P.E. Targeted induction of ceramide degradation leads to improved systemic metabolism and reduced hepatic steatosis. Cell Metab. 2015, 22, 266–278. [Google Scholar] [CrossRef] [PubMed]
- Hansen, M.E.; Simmons, K.J.; Tippetts, T.S.; Thatcher, M.O.; Saito, R.R.; Hubbard, S.T.; Trumbull, A.M.; Parker, B.A.; Taylor, O.J.; Bikman, B.T. Lipopolysaccharide disrupts mitochondrial physiology in skeletal muscle via disparate effects on sphingolipid metabolism. Shock 2015, 44, 585–592. [Google Scholar] [CrossRef] [PubMed]
- Pfaffl, M.W. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001, 29, e45. [Google Scholar] [CrossRef] [PubMed]


© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Taylor, O.J.; Thatcher, M.O.; Carr, S.T.; Gibbs, J.L.; Trumbull, A.M.; Harrison, M.E.; Winden, D.R.; Pearson, M.J.; Tippetts, T.S.; Holland, W.L.; et al. High-Mobility Group Box 1 Disrupts Metabolic Function with Cigarette Smoke Exposure in a Ceramide-Dependent Manner. Int. J. Mol. Sci. 2017, 18, 1099. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms18051099
Taylor OJ, Thatcher MO, Carr ST, Gibbs JL, Trumbull AM, Harrison ME, Winden DR, Pearson MJ, Tippetts TS, Holland WL, et al. High-Mobility Group Box 1 Disrupts Metabolic Function with Cigarette Smoke Exposure in a Ceramide-Dependent Manner. International Journal of Molecular Sciences. 2017; 18(5):1099. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms18051099
Chicago/Turabian StyleTaylor, Oliver J., Mikayla O. Thatcher, Sheryl T. Carr, Jonathan L. Gibbs, Annie M. Trumbull, Mitchell E. Harrison, Duane R. Winden, Mackenzie J. Pearson, Trevor S. Tippetts, William L. Holland, and et al. 2017. "High-Mobility Group Box 1 Disrupts Metabolic Function with Cigarette Smoke Exposure in a Ceramide-Dependent Manner" International Journal of Molecular Sciences 18, no. 5: 1099. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms18051099