
Unbalance between Excitation and Inhibition in Phenylketonuria, a Genetic Metabolic Disease Associated with Autism

Abstract
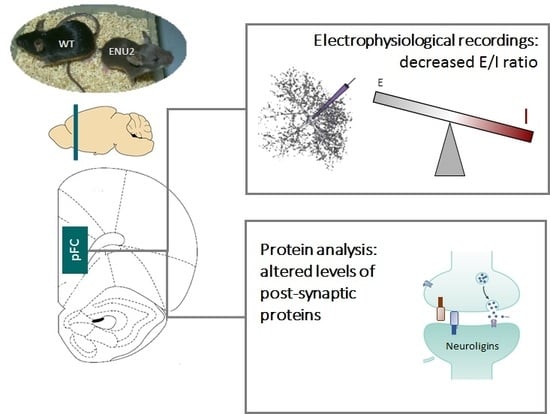
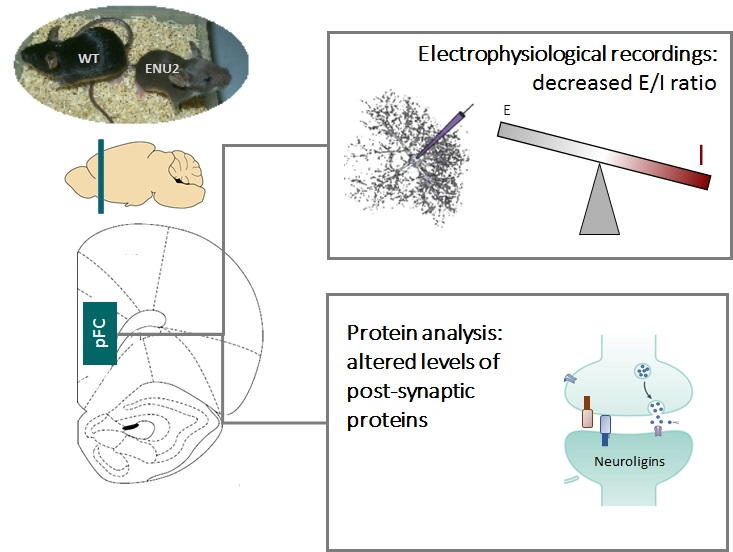
:
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
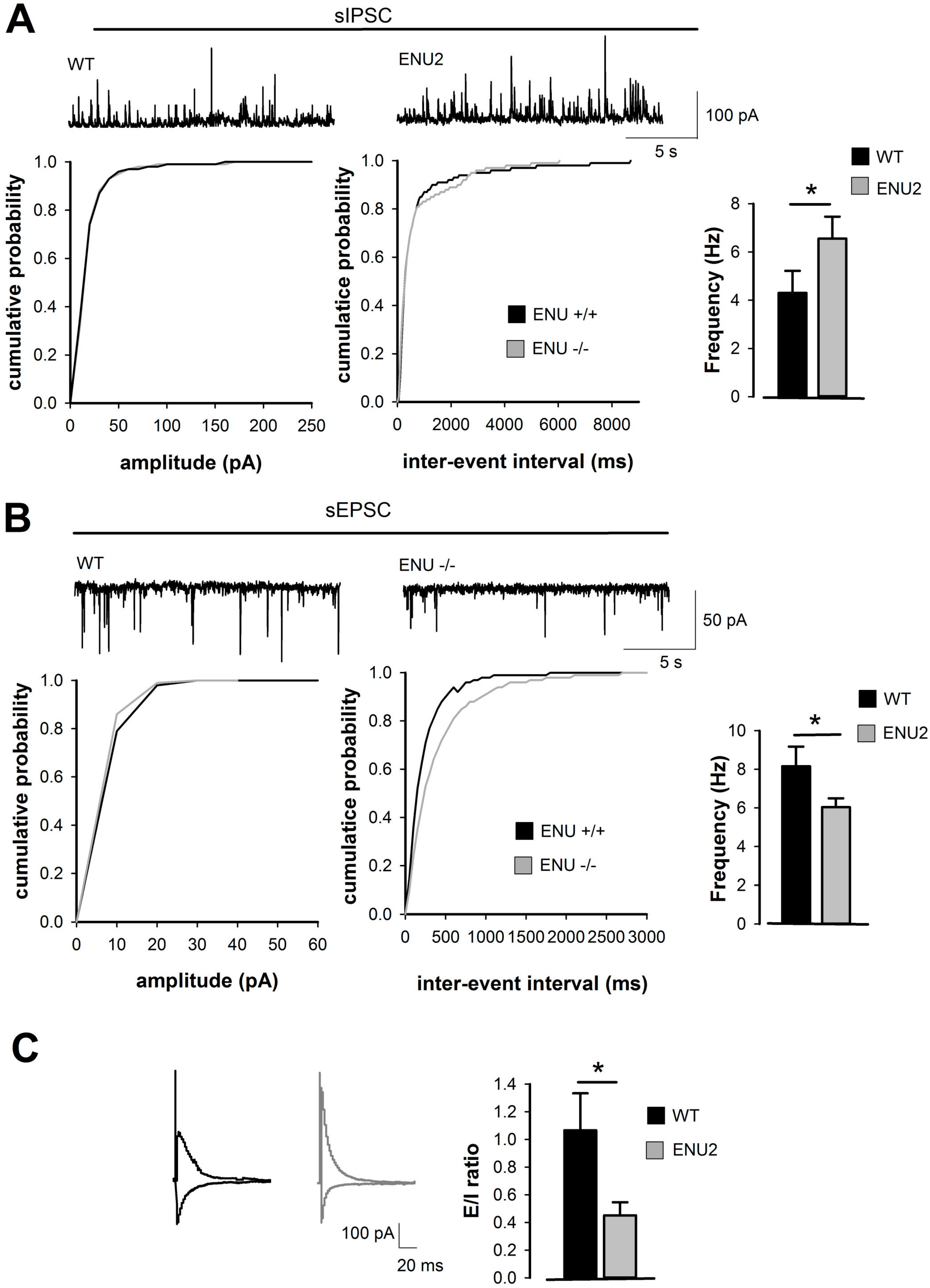
2.1. Analysis of Inhibitory and Excitatory Transmission in Layer II/III of ENU2 pFC
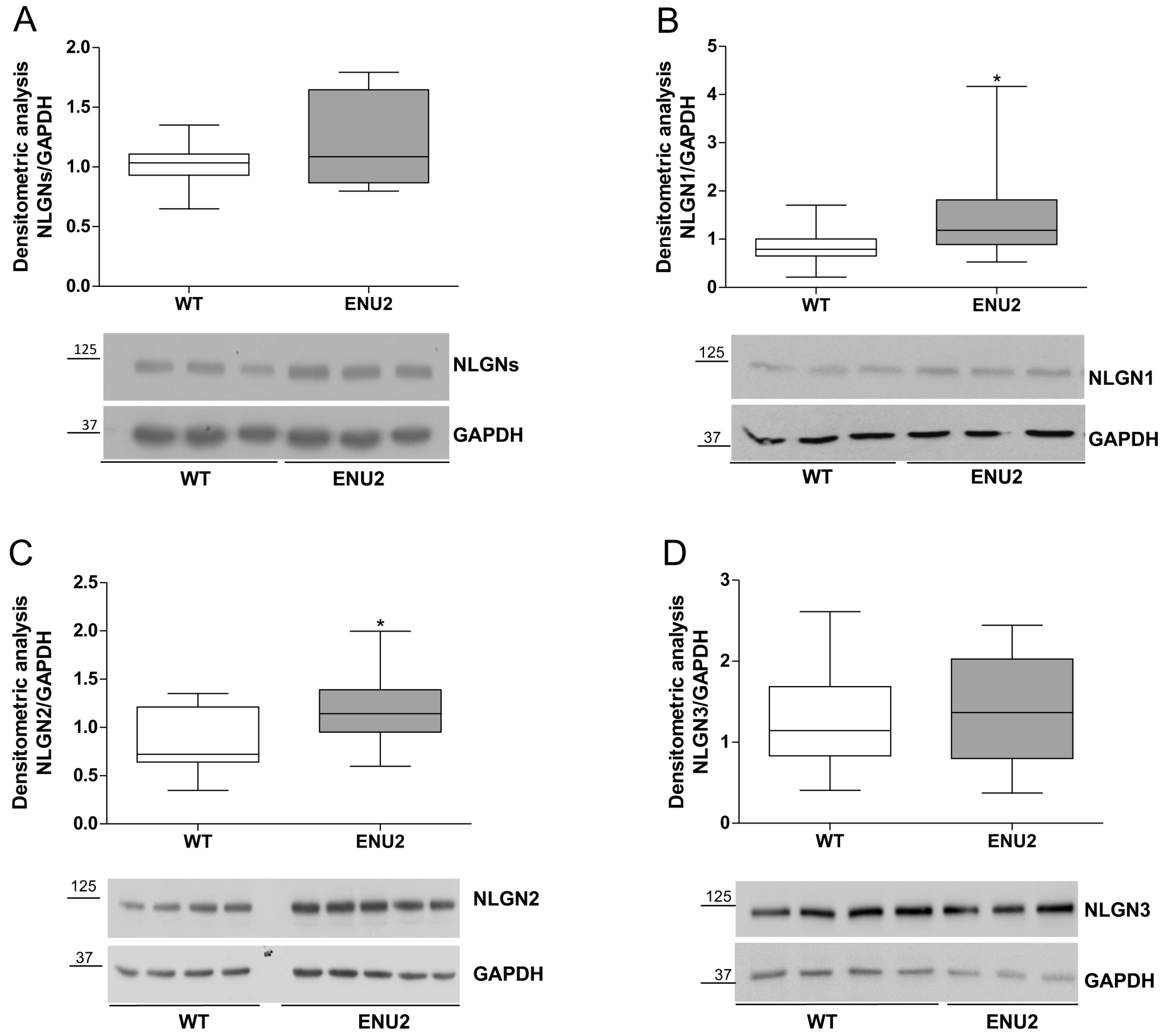
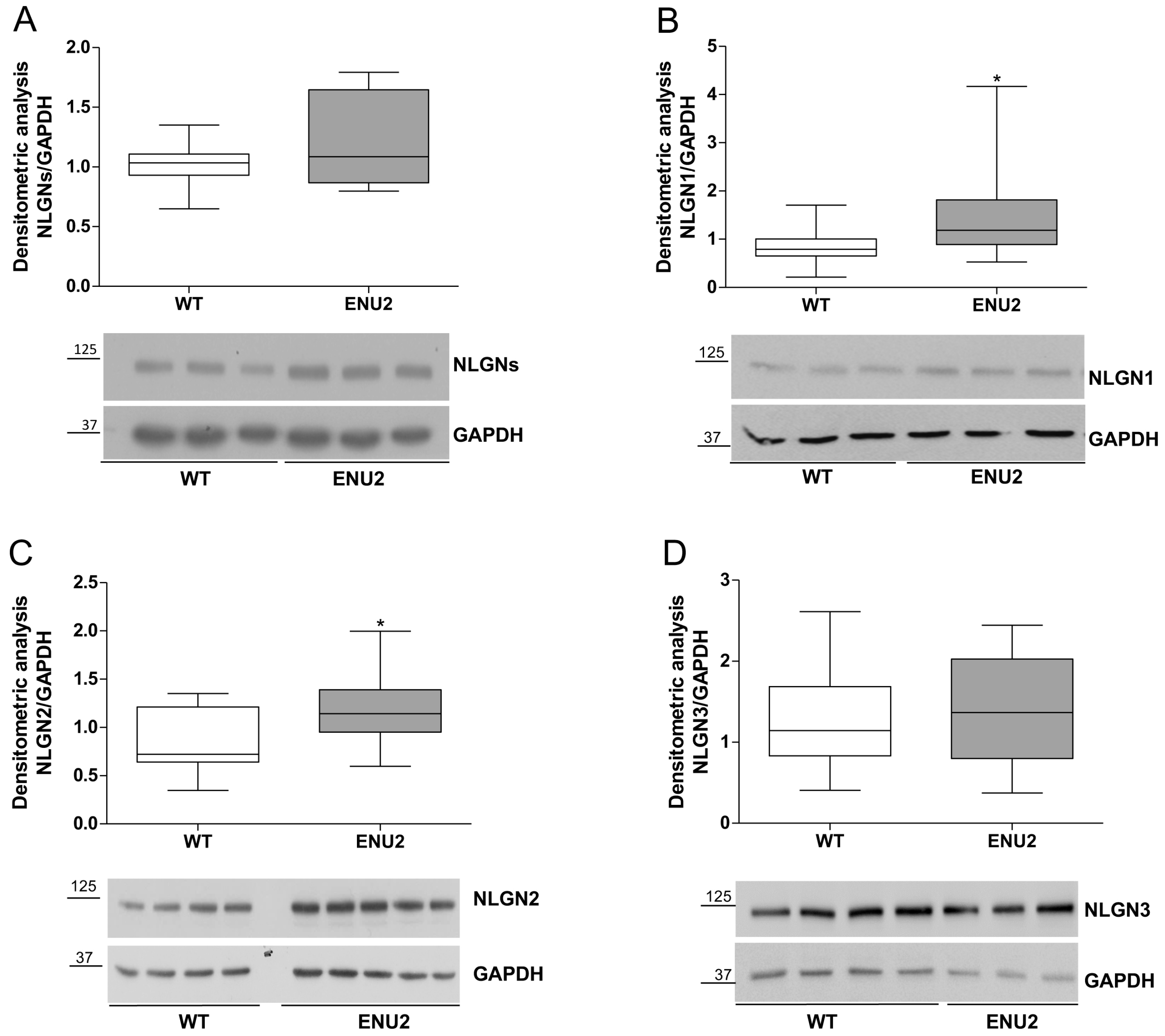
2.2. Protein Levels of Synaptic Cell Adhesion Molecules in the pFC of ENU2 Mice
3. Discussion
4. Materials and Methods
4.1. Animal Protocols and Housing
4.2. Slice Preparation for Electrophysiological Recordings
4.3. Whole-Cell Patch Clamp Recordings
4.4. SDS PAGE and Western Blot
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| sEPSC | spontaneous excitatory postsynaptic potential |
| sIPSC | spontaneous inhibitory postsynaptic potential |
| NLGNs | neuroligins |
| HRP | horseradish peroxidase |
| SDS | sodium dodecyl sulfate |
| PAGE | polyacrylamide gel electrophoresis |
| pFC | prefrontal cortex |
| LTP | long term potentiation |
| LTD | long term depression |
References
- Ghaziuddin, M.; Al-Owain, M. Autism spectrum disorders and inborn errors of metabolism: An update. Pediatr. Neurol. 2013, 49, 232–236. [Google Scholar] [CrossRef] [PubMed]
- DeRoche, K.; Welsh, M. Twenty-five years of research on neurocognitive outcomes in early-treated phenylketonuria: Intelligence and executive function. Dev. Neuropsychol. 2008, 33, 474–504. [Google Scholar] [CrossRef] [PubMed]
- Stemerdink, B.A.; Kalverboer, A.F.; van der Meere, J.J.; van der Molen, M.W.; Huisman, J.; de Jong, L.W.; Slijper, F.M.; Verkerk, P.H.; van Spronsen, F.J. Behaviour and school achievement in patients with early and continuously treated phenylketonuria. J. Inherit. Metab. Dis. 2000, 23, 548–562. [Google Scholar] [CrossRef] [PubMed]
- Diamond, A.; Prevor, M.B.; Callender, G.; Druin, D.P. Prefrontal cortex cognitive deficits in children treated early and continuously for PKU. Monogr. Soc. Res. Child Dev. 1997, 62. [Google Scholar] [CrossRef]
- Pascucci, T.; Andolina, D.; Mela, I.L.; Conversi, D.; Latagliata, C.; Ventura, R.; Puglisi-Allegra, S.; Cabib, S. 5-Hydroxytryptophan rescues serotonin response to stress in prefrontal cortex of hyperphenylalaninaemic mice. Int. J. Neuropsychopharmacol. 2009, 12, 1067–1079. [Google Scholar] [CrossRef] [PubMed]
- Andolina, D.; Conversi, D.; Cabib, S.; Trabalza, A.; Ventura, R.; Puglisi-Allegra, S.; Pascucci, T. 5-Hydroxytryptophan during critical postnatal period improves cognitive performances and promotes dendritic spine maturation in genetic mouse model of phenylketonuria. Int. J. Neuropsychopharmacol. 2011, 14, 479–489. [Google Scholar] [CrossRef] [PubMed]
- Nelson, S.B.; Valakh, V. Excitatory/Inhibitory Balance and Circuit Homeostasis in Autism Spectrum Disorders. Neuron 2015, 87, 684–698. [Google Scholar] [CrossRef] [PubMed]
- Bemben, M.A.; Shipman, S.L.; Nicoll, R.A.; Roche, K.W. The cellular and molecular landscape of neuroligins. Trends Neurosci. 2015, 38, 496–505. [Google Scholar] [CrossRef] [PubMed]
- Krueger, D.D.; Tuffy, L.P.; Papadopoulos, T.; Brose, N. The role of neurexins and neuroligins in the formation, maturation, and function of vertebrate synapses. Curr. Opin. Neurobiol. 2012, 22, 412–422. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.; Lee, J.; Kim, E. Excitation/Inhibition Imbalance in Animal Models of Autism Spectrum Disorders. Biol. Psychiatry 2017, 81, 838–847. [Google Scholar] [CrossRef] [PubMed]
- De la Torre-Ubieta, L.; Won, H.; Stein, J.L.; Geschwind, D.H. Advancing the understanding of autism disease mechanisms through genetics. Nat. Med. 2016, 22, 345–361. [Google Scholar] [CrossRef] [PubMed]
- Jamain, S.; Quach, H.; Betancur, C.; Råstam, M.; Colineaux, C.; Gillberg, I.C.; Soderstrom, H.; Giros, B.; Leboyer, M.; Gillberg, C.; et al. Paris Autism Research International Sibpair Study Mutations of the X-linked genes encoding neuroligins NLGN3 and NLGN4 are associated with autism. Nat. Genet. 2003, 34, 27–29. [Google Scholar] [CrossRef] [PubMed]
- Laumonnier, F.; Bonnet-Brilhault, F.; Gomot, M.; Blanc, R.; David, A.; Moizard, M.-P.; Raynaud, M.; Ronce, N.; Lemonnier, E.; Calvas, P.; et al. X-linked mental retardation and autism are associated with a mutation in the NLGN4 gene, a member of the neuroligin family. Am. J. Hum. Genet. 2004, 74, 552–557. [Google Scholar] [CrossRef] [PubMed]
- Chih, B.; Afridi, S.K.; Clark, L.; Scheiffele, P. Disorder-associated mutations lead to functional inactivation of neuroligins. Hum. Mol. Genet. 2004, 13, 1471–1477. [Google Scholar] [CrossRef] [PubMed]
- Comoletti, D.; de Jaco, A.; Jennings, L.L.; Flynn, R.E.; Gaietta, G.; Tsigelny, I.; Ellisman, M.H.; Taylor, P. The Arg451Cys-neuroligin-3 mutation associated with autism reveals a defect in protein processing. J. Neurosci. Off. J. Soc. Neurosci. 2004, 24, 4889–4893. [Google Scholar] [CrossRef] [PubMed]
- De Jaco, A.; Lin, M.Z.; Dubi, N.; Comoletti, D.; Miller, M.T.; Camp, S.; Ellisman, M.; Butko, M.T.; Tsien, R.Y.; Taylor, P. Neuroligin trafficking deficiencies arising from mutations in the α/β-hydrolase fold protein family. J. Biol. Chem. 2010, 285, 28674–28682. [Google Scholar] [CrossRef] [PubMed]
- Cao, X.; Tabuchi, K. Functions of synapse adhesion molecules neurexin/neuroligins and neurodevelopmental disorders. Neurosci. Res. 2017, 116, 3–9. [Google Scholar] [CrossRef] [PubMed]
- Siddiqui, T.J.; Craig, A.M. Synaptic organizing complexes. Curr. Opin. Neurobiol. 2011, 21, 132–143. [Google Scholar] [CrossRef] [PubMed]
- Südhof, T.C. Neuroligins and neurexins link synaptic function to cognitive disease. Nature 2008, 455, 903–911. [Google Scholar] [CrossRef] [PubMed]
- Maćkowiak, M.; Mordalska, P.; Wędzony, K. Neuroligins, synapse balance and neuropsychiatric disorders. Pharmacol. Rep. 2014, 66, 830–835. [Google Scholar] [CrossRef] [PubMed]
- Dani, V.S.; Chang, Q.; Maffei, A.; Turrigiano, G.G.; Jaenisch, R.; Nelson, S.B. Reduced cortical activity due to a shift in the balance between excitation and inhibition in a mouse model of Rett syndrome. Proc. Natl. Acad. Sci. USA 2005, 102, 12560–12565. [Google Scholar] [CrossRef] [PubMed]
- Tabuchi, K.; Blundell, J.; Etherton, M.R.; Hammer, R.E.; Liu, X.; Powell, C.M.; Südhof, T.C. A neuroligin-3 mutation implicated in autism increases inhibitory synaptic transmission in mice. Science 2007, 318, 71–76. [Google Scholar] [CrossRef] [PubMed]
- Belichenko, P.V.; Kleschevnikov, A.M.; Masliah, E.; Wu, C.; Takimoto-Kimura, R.; Salehi, A.; Mobley, W.C. Excitatory-inhibitory relationship in the fascia dentata in the Ts65Dn mouse model of Down syndrome. J. Comp. Neurol. 2009, 512, 453–466. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, F.; Garner, C.C. Over-inhibition: A model for developmental intellectual disability. Trends Neurosci. 2007, 30, 497–503. [Google Scholar] [CrossRef] [PubMed]
- Kleschevnikov, A.M.; Belichenko, P.V.; Villar, A.J.; Epstein, C.J.; Malenka, R.C.; Mobley, W.C. Hippocampal long-term potentiation suppressed by increased inhibition in the Ts65Dn mouse, a genetic model of Down syndrome. J. Neurosci. Off. J. Soc. Neurosci. 2004, 24, 8153–8160. [Google Scholar] [CrossRef] [PubMed]
- Yizhar, O.; Fenno, L.E.; Prigge, M.; Schneider, F.; Davidson, T.J.; O’Shea, D.J.; Sohal, V.S.; Goshen, I.; Finkelstein, J.; Paz, J.T.; et al. Neocortical excitation/inhibition balance in information processing and social dysfunction. Nature 2011, 477, 171–178. [Google Scholar] [CrossRef] [PubMed]
- De Groot, M.J.; Hoeksma, M.; Blau, N.; Reijngoud, D.J.; van Spronsen, F.J. Pathogenesis of cognitive dysfunction in phenylketonuria: Review of hypotheses. Mol. Genet. Metab. 2010, 99, S86–S89. [Google Scholar] [CrossRef] [PubMed]
- Levinson, J.N.; El-Husseini, A. Building excitatory and inhibitory synapses: Balancing neuroligin partnerships. Neuron 2005, 48, 171–174. [Google Scholar] [CrossRef] [PubMed]
- Hines, R.M.; Wu, L.; Hines, D.J.; Steenland, H.; Mansour, S.; Dahlhaus, R.; Singaraja, R.R.; Cao, X.; Sammler, E.; Hormuzdi, S.G.; et al. Synaptic imbalance, stereotypies, and impaired social interactions in mice with altered neuroligin 2 expression. J. Neurosci. 2008, 28, 6055–6067. [Google Scholar] [CrossRef] [PubMed]
- Liang, J.; Xu, W.; Hsu, Y.-T.; Yee, A.X.; Chen, L.; Südhof, T.C. Conditional neuroligin-2 knockout in adult medial prefrontal cortex links chronic changes in synaptic inhibition to cognitive impairments. Mol. Psychiatry 2015, 20, 850–859. [Google Scholar] [CrossRef] [PubMed]
- Varoqueaux, F.; Jamain, S.; Brose, N. Neuroligin 2 is exclusively localized to inhibitory synapses. Eur. J. Cell Biol. 2004, 83, 449–456. [Google Scholar] [CrossRef] [PubMed]
- Poulopoulos, A.; Aramuni, G.; Meyer, G.; Soykan, T.; Hoon, M.; Papadopoulos, T.; Zhang, M.; Paarmann, I.; Fuchs, C.; Harvey, K.; et al. Neuroligin 2 drives postsynaptic assembly at perisomatic inhibitory synapses through gephyrin and collybistin. Neuron 2009, 63, 628–642. [Google Scholar] [CrossRef] [PubMed]
- Soykan, T.; Schneeberger, D.; Tria, G.; Buechner, C.; Bader, N.; Svergun, D.; Tessmer, I.; Poulopoulos, A.; Papadopoulos, T.; Varoqueaux, F.; et al. A conformational switch in collybistin determines the differentiation of inhibitory postsynapses. EMBO J. 2014, 33, 2113–2133. [Google Scholar] [CrossRef] [PubMed]
- Chubykin, A.A.; Atasoy, D.; Etherton, M.R.; Brose, N.; Kavalali, E.T.; Gibson, J.R.; Südhof, T.C. Activity-dependent validation of excitatory versus inhibitory synapses by neuroligin-1 versus neuroligin-2. Neuron 2007, 54, 919–931. [Google Scholar] [CrossRef] [PubMed]
- Pascucci, T.; Ventura, R.; Puglisi-Allegra, S.; Cabib, S. Deficits in brain serotonin synthesis in a genetic mouse model of phenylketonuria. Neuroreport 2002, 13, 2561–2564. [Google Scholar] [CrossRef] [PubMed]
- Jones, K.A.; Srivastava, D.P.; Allen, J.A.; Strachan, R.T.; Roth, B.L.; Penzes, P. Rapid modulation of spine morphology by the 5-HT2A serotonin receptor through kalirin-7 signaling. Proc. Natl. Acad. Sci. USA 2009, 106, 19575–19580. [Google Scholar] [CrossRef] [PubMed]
- Prange, O.; Wong, T.P.; Gerrow, K.; Wang, Y.T.; El-Husseini, A. A balance between excitatory and inhibitory synapses is controlled by PSD-95 and neuroligin. Proc. Natl. Acad. Sci. USA 2004, 101, 13915–13920. [Google Scholar] [CrossRef] [PubMed]
- Ko, J.; Choii, G.; Um, J.W. The balancing act of GABAergic synapse organizers. Trends Mol. Med. 2015, 21, 256–268. [Google Scholar] [CrossRef] [PubMed]
- Graf, E.R.; Zhang, X.; Jin, S.-X.; Linhoff, M.W.; Craig, A.M. Neurexins induce differentiation of GABA and glutamate postsynaptic specializations via neuroligins. Cell 2004, 119, 1013–1026. [Google Scholar] [CrossRef] [PubMed]
- Pascucci, T.; Andolina, D.; Ventura, R.; Puglisi-Allegra, S.; Cabib, S. Reduced availability of brain amines during critical phases of postnatal development in a genetic mouse model of cognitive delay. Brain Res. 2008, 1217, 232–238. [Google Scholar] [CrossRef] [PubMed]
- Delattre, V.; La Mendola, D.; Meystre, J.; Markram, H.; Markram, K. NLGN4 knockout induces network hypo-excitability in juvenile mouse somatosensory cortex in vitro. Sci. Rep. 2013, 3, 2897. [Google Scholar] [CrossRef] [PubMed]
- Puglisi-Allegra, S.; Cabib, S.; Pascucci, T.; Ventura, R.; Cali, F.; Romano, V. Dramatic brain aminergic deficit in a genetic mouse model of phenylketonuria. Neuroreport 2000, 11, 1361–1364. [Google Scholar] [CrossRef] [PubMed]
- Ulbrich, L.; Favaloro, F.L.; Trobiani, L.; Marchetti, V.; Patel, V.; Pascucci, T.; Comoletti, D.; Marciniak, S.J.; De Jaco, A. Autism-associated R451C mutation in neuroligin3 leads to activation of the unfolded protein response in a PC12 Tet-On inducible system. Biochem. J. 2016, 473, 423–434. [Google Scholar] [CrossRef] [PubMed]

© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
De Jaco, A.; Mango, D.; De Angelis, F.; Favaloro, F.L.; Andolina, D.; Nisticò, R.; Fiori, E.; Colamartino, M.; Pascucci, T. Unbalance between Excitation and Inhibition in Phenylketonuria, a Genetic Metabolic Disease Associated with Autism. Int. J. Mol. Sci. 2017, 18, 941. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms18050941
De Jaco A, Mango D, De Angelis F, Favaloro FL, Andolina D, Nisticò R, Fiori E, Colamartino M, Pascucci T. Unbalance between Excitation and Inhibition in Phenylketonuria, a Genetic Metabolic Disease Associated with Autism. International Journal of Molecular Sciences. 2017; 18(5):941. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms18050941
Chicago/Turabian StyleDe Jaco, Antonella, Dalila Mango, Federica De Angelis, Flores Lietta Favaloro, Diego Andolina, Robert Nisticò, Elena Fiori, Marco Colamartino, and Tiziana Pascucci. 2017. "Unbalance between Excitation and Inhibition in Phenylketonuria, a Genetic Metabolic Disease Associated with Autism" International Journal of Molecular Sciences 18, no. 5: 941. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms18050941