
Incomplete Segregation of MSH6 Frameshift Variants with Phenotype of Lynch Syndrome


Abstract
:1. Introduction
2. Results
3. Discussion
4. Patients and Methods
4.1. Patients
4.2. Isolation of Genomic DNA
4.3. DNA Amplification and Microsatellite Analysis
4.4. Mutation Analysis
4.5. In Silico Analysis
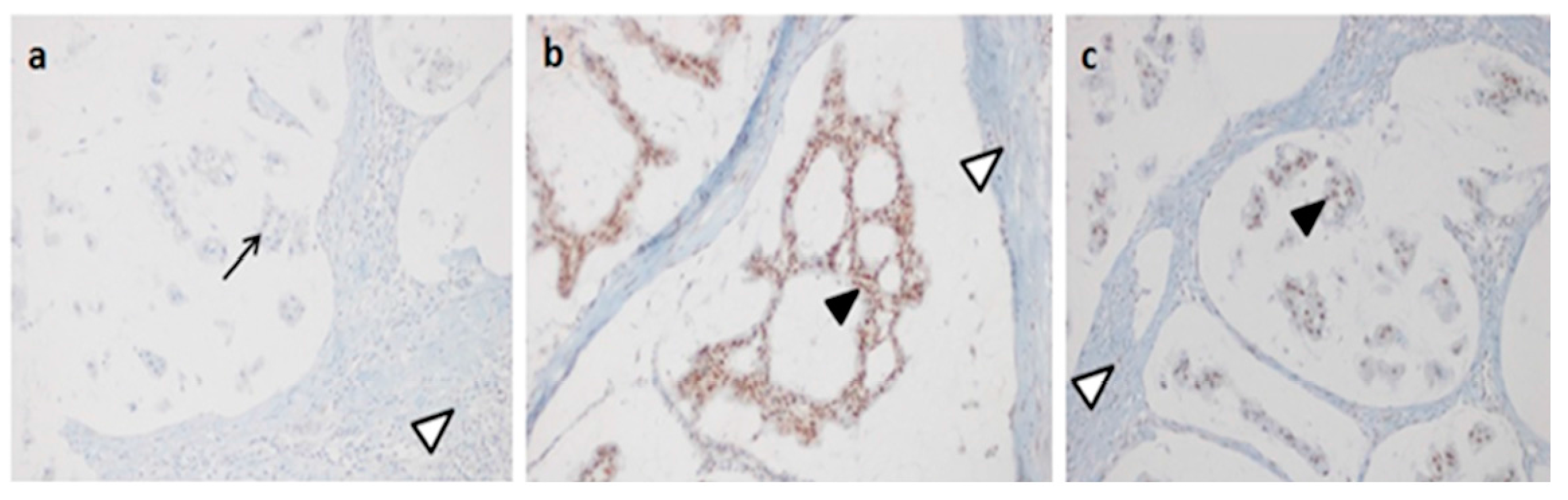
4.6. Immunohistochemistry
5. Conclusions
Acknowledgments
Author Contributions
Conflict of interest
References
- Lynch, H.T.; Drescher, K.; Knezetic, J.; Lanspa, S. Genetics, biomarkers, hereditary cancer syndrome diagnosis, heterogeneity and treatment: A review. Curr. Treat. Options Oncol. 2014, 15, 429–442. [Google Scholar] [CrossRef] [PubMed]
- De Rosa, M.; Pace, U.; Rega, D.; Costabile, V.; Duraturo, F.; Izzo, P.; Delrio, P. Genetics, diagnosis and management of colorectal cancer (Review). Oncol. Rep. 2015, 34, 1087–1096. [Google Scholar] [CrossRef] [PubMed]
- De Rosa, M.; Galatola, M.; Borriello, S.; Duraturo, F.; Masone, S.; Izzo, P. Implication of adenomatous polyposis coli and MUTYH mutations in familial colorectal polyposis. Dis. Colon. Rectum. 2009, 52, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Lynch, P.M. The hMSH2 and hMLH1 genes in hereditary nonpolyposis colorectal cancer. Surg. Oncol. Clin. N. Am. 2009, 18, 611–624. [Google Scholar] [CrossRef] [PubMed]
- Tiwari, A.K.; Roy, H.K.; Lynch, H.T. Lynch syndrome in the 21st century: Clinical perspectives. QJM 2016, 109, 151–158. [Google Scholar] [CrossRef] [PubMed]
- Duraturo, F.; Liccardo, R.; Izzo, P. Coexistence of MLH3 germline variants in colon cancer patients belonging to families with Lynch syndrome-associated brain tumors. J. Neurooncol. 2016, 129, 577–578. [Google Scholar] [CrossRef] [PubMed]
- Duraturo, F.; Liccardo, R.; Cavallo, A.; de Rosa, M.; Grosso, M.; Izzo, P. Association of low-risk MSH3 and MSH2 variant alleles with Lynch syndrome: Probability of synergistic effects. Int. J. Cancer 2011, 129, 1643–1650. [Google Scholar] [CrossRef] [PubMed]
- Sinicrope, F.A.; Sargent, D.J. Molecular pathways: Microsatellite instability in colorectal cancer: Prognostic, predictive, and therapeutic implications. Clin. Cancer Res. 2012, 18, 1506–1512. [Google Scholar] [CrossRef] [PubMed]
- Poulogiannis, G.; Frayling, I.M.; Arends, M.J. DNA mismatch repair deficiency in sporadic colorectal cancer and Lynch syndrome. Histopathology 2010, 56, 167–179. [Google Scholar] [CrossRef] [PubMed]
- Vasen, H.F.A.; Watson, P.; Mecklin, J.P.; Lynch, H.T. New clinical criteria for hereditary nonpolyposis colorectal cancer (HNPCC, Lynch syndrome) proposed by the International Collaborative group on HNPCC. Gastroenterology 1999, 116, 1453–1456. [Google Scholar] [CrossRef]
- Boland, C.R.; Thibodeau, S.N.; Hamilton, S.R.; Sidransky, D.; Eshleman, J.R.; Burt, R.W.; Meltzer, S.J.; Rodriguez-Bigas, M.A.; Fodde, R.; Ranzani, G.N.; et al. A national cancer institute workshop on microsatellite instability for cancer detection and familial predisposition: Development of international criteria for the determination of microsatellite instability in colorectal cancer. Cancer Res. 1998, 58, 5248–5257. [Google Scholar] [PubMed]
- Umar, A.; Boland, C.R.; Terdiman, J.P.; Syngal, S.; de la Chapelle, A.; Rüschoff, J.; Fishel, R.; Lindor, N.M.; Burgart, L.J.; Hamelin, R.; et al. Revised bethesda guidelines for hereditary non polyposis colorectal cancer (Lynch Syndrome) and microsatellite instability. J. Natl. Cancer Inst. 2004, 96, 261–268. [Google Scholar] [CrossRef] [PubMed]
- Rubenstein, J.H.; Enns, R.; Heidelbaugh, J.; Barkun, A. Clinical Guidelines Committee. American gastroenterological association institute guideline on the diagnosis and management of Lynch Syndrome. Gastroenterology 2015, 149, 777–782. [Google Scholar] [CrossRef] [PubMed]
- Palombo, F.; Gallinari, P.; Iaccarino, I.; Lettieri, T.; Hughes, M.; D’Arrigo, A.; Truong, O.; Hsuan, J.J.; Jiricny, J. GTBP, a 160-kilodalton protein essential for mismatch-binding activity in human cells. Science 1995, 268, 1912–1914. [Google Scholar] [CrossRef] [PubMed]
- Drummond, J.T.; Li, G.M.; Longley, M.J.; Modrich, P. Isolation of an hMSH2-p160 heterodimer that restores DNA mismatch repair to tumor cells. Science 1995, 268, 1909–1912. [Google Scholar] [CrossRef] [PubMed]
- Edelmann, W.; Yang, K.; Umar, A.; Heyer, J.; Lau, K.; Fan, K.; Liedtke, W.; Cohen, P.E.; Kane, M.F.; Lipford, J.R.; et al. Mutation in the mismatch repair gene MSH6 causes cancer susceptibility. Cell 1997, 91, 467–477. [Google Scholar] [CrossRef]
- Miyaki, M.; Konishi, M.; Tanaka, K.; Kikuchi-Yanoshita, R.; Muraoka, M.; Yasuno, M.; Igari, T; Koike, M.; Chiba, M.; Mori, T. Germline mutation of MSH6 as the cause of hereditary nonpolyposis colorectal cancer. Nat. Genet. 1997, 17, 271–272. [Google Scholar] [CrossRef] [PubMed]
- Lucci-Cordisco, E.; Rovella, V.; Carrara, S.; Percesepe, A.; Pedroni, M.; Bellacosa, A.; Caluseriu, O.; Forasarig, M.; Anti, M.; Neri, G.; et al. Mutations of the “minor” mismatch repair gene MSH6 in typical and atypical hereditary nonpolyposis colorectal cancer. Fam. Cancer 2001, 1, 93–99. [Google Scholar] [CrossRef] [PubMed]
- Long, Q.; Peng, Y.; Tang, Z.; Wu, C. Role of endometrial cancer abnormal MMR protein in screening Lynch-syndrome families. Int. J. Clin. Exp. Pathol. 2014, 7, 7297–7303. [Google Scholar] [PubMed]
- Chu, M.M.; Liu, S.S.; Tam, K.F.; Ip, P.P.; Cheung, A.N.; Ngan, H.Y. The significance of mismatch repair deficiency in young patients with endometrial cancer. Int. J. Gynecol. Pathol. 2015, 34, 403–410. [Google Scholar] [CrossRef] [PubMed]
- Ou, J.; Niessen, R.C.; Vonk, J.; Westers, H; Hofstra, R.M.W.; Sijmons, R.H. A database to support the interpretation of human mismatch repair gene variants. Hum. Mutat. 2008, 29, 1337–1341. [Google Scholar] [CrossRef] [PubMed]
- Bonk, T.; Humeny, A.; Gebert, J.; Sutter, C.; von Knebel Doeberitz, M.; Becker, C.M. Matrix-assisted laser desorption/ionization time-of-flight mass spectrometry-based detection of microsatellite instabilities in coding DNA sequences: A novel approach to identify DNA-mismatch repair-deficient cancer cells. Clin. Chem. 2003, 49, 552–561. [Google Scholar] [CrossRef] [PubMed]
- Duraturo, F.; Cavallo, A.; Liccardo, R.; Cudia, B.; de Rosa, M.; Diana, G.; Izzo, P. Contribution of large genomic rearrangements in Italian Lynch syndrome patients: Characterization of a novel alu-mediated deletion. Biomed. Res. Int. 2013, 2013, 219897. [Google Scholar] [CrossRef] [PubMed]
- Paparo, L.; Rossi, G.B.; Delrio, P.; Rega, D.; Duraturo, F.; Liccardo, R.; Debellis, M; Izzo, P.; de Rosa, M. Differential expression of PTEN gene correlates with phenotypic heterogeneity in three cases of patients showing clinical manifestations of PTEN hamartoma tumour syndrome. Hered. Cancer Clin. Pract. 2013, 11, 8. [Google Scholar] [CrossRef] [PubMed]
- Wong, A.; Kuick, C.H.; Wong, W.L.; Tham, J.M.; Mansor, S.; Loh, E.; Jain, S.; Vikas, N.N.; Tan, S.H; Chan, S.H.; et al. Mutation spectrum of POLE and POLD1 mutations in South East Asian women presenting with grade 3 endometrioid endometrial carcinomas. Gynecol. Oncol. 2016, 141, 113–120. [Google Scholar] [CrossRef] [PubMed]
- Colebatch, A.; Hitchins, M.; Williams, R.; Meagher, A; Hawkins, N.J.; Ward, R.L. The role of MYH and microsatellite instability in the development of sporadic colorectal cancer. Br. J. Cancer 2006, 95, 1239–1243. [Google Scholar] [CrossRef] [PubMed]
- Bilbao, C.; Rodríguez, G.; Ramírez, R.; Falcón, O.; León, L; Chirino, R; Rivero, J.F.; Falcón, O., Jr.; Díaz-Chico, B.N.; Díaz-Chico, J.C; et al. The relationship between microsatellite instability and PTEN gene mutations in endometrial cancer. Int. J. Cancer 2006, 119, 563–570. [Google Scholar] [CrossRef] [PubMed]
- Lawes, D.A.; SenGupta, S.B.; Boulos, P.B. Pathogenesis and clinical management of hereditary non-polyposis colorectal cancer. Br. J. Surg. 2002, 89, 1357–1369. [Google Scholar] [CrossRef] [PubMed]
- Jun, S.H.; Kim, T.G.; Ban, C. DNA mismatch repair system. Classical and fresh roles. FEBS J. 2006, 273, 1609–1619. [Google Scholar] [CrossRef] [PubMed]
- Wiesendanger, M.; Kneitz, B.; Edelmann, W.; Scharff, M.D. Somatic hypermutation in MutS homologue (MSH)3-, MSH6-, and MSH3/MSH6-deficient mice reveals a role for the MSH2-MSH6 heterodimer in modulating the base substitution pattern. J. Exp. Med. 2000, 7, 579–584. [Google Scholar] [CrossRef]
- Edelmann, W.; Umar, A.; Yang, K.; Heyer, J.; Kucherlapati, M.; Lia, M.; Kneitz, B.; Avdievich, E.; Fan, K.; Wong, E.; et al. The DNA mismatch repair genes Msh3 and Msh6 cooperate in intestinal tumor suppression. Cancer Res. 2000, 15, 803–807. [Google Scholar]
- Yang, G.; Scherer, S.J.; Shell, S.S.; Yang, K.; Kim, M.; Lipkin, M.; Kucherlapati, R.; Kolodner, R.D; Edelmann, W. Dominant effects of an MSH6 missense mutation on DNA repair and cancer susceptibility. Cancer Cell 2004, 6, 139–150. [Google Scholar] [CrossRef] [PubMed]
- Martinez, S.L.; Kolodner, R.D. Functional analysis of human mismatch repair gene mutations identifies weak alleles and polymorphisms capable of polygenic interactions. Proc. Natl. Acad. Sci. USA 2010, 107, 5070–5075. [Google Scholar] [CrossRef] [PubMed]
- Kumar, C.; Piacente, S.C.; Sibert, J; Bukata, A.R.; O’Connor, J.; Alani, E.; Surtees, J.A. Multiple factors insulate Msh2–Msh6 mismatch repair activity from defects in Msh2 domain I. J. Mol. Biol. 2011, 411, 765–780. [Google Scholar] [CrossRef] [PubMed]
- Talseth-Palmer, B.A.; Bauer, D.C.; Sjursen, W.; Evans, T.J.; McPhillips, M.; Proietto, A.; Otton, G.; Spigelman, A.D.; Scott, R.J. Targeted next-generation sequencing of 22 mismatch repair genes identifies Lynch syndrome families. Cancer Med. 2016, 5, 929–941. [Google Scholar] [CrossRef] [PubMed]
- Carethers, J.M.; Stoffel, E.M. Lynch syndrome and Lynch syndrome mimics: The growing complex landscape of hereditary colon cancer. World J. Gastroenterol. 2015, 21, 9253–9261. [Google Scholar] [CrossRef] [PubMed]
- Cudia, B.; Liccardo, R.; di Carlo, G.; Damiano, G; Lo Monte, A.I.; Izzo, P; Duraturo, F. Clinical and anamnestic evaluation role for the diagnosis and treatment of families affected by Lynch syndrome. Case report and review of the literature. Eur. J. Oncol. 2015, 19, 265–271. [Google Scholar]
- Duraturo, F.; Liccardo, R.; Cavallo, A.; de Rosa, M; Rossi, G.B.; Izzo, P. Multivariate analysis as a method for evaluating the pathogenicity of novel genetic MLH1 variants in patients with colorectal cancer and microsatellite instability. Int. J. Mol. Med. 2015, 36, 511–517. [Google Scholar] [CrossRef] [PubMed]
- Suraweera, N.; Duval, A.; Reperant, M.; Vaury, C.; Furlan, D.; Leroy, K.; Seruca, R.; Iacopetta, B.; Hamelin, R. Evaluation of tumor microsatellite instability using five quasimonomorphic nucleotide repeats and pentaplex PCR. Gastroenterology 2002, 123, 1804–1811. [Google Scholar] [CrossRef] [PubMed]
- Xicola, R.M.; Llor, X.; Pons, E; Castells, A.; Alenda, C.; Piñol, V.; Andreu, M.; Castellví-Bel, S.; Payá, A.; Jover, R.; et al. Performance of different microsatellite marker panels for detection of mismatch repair-deficient colorectal tumors gastrointestinal oncology group of the Spanish gastroenterological association. J. Natl. Cancer Inst. 2007, 99, 244–252. [Google Scholar] [CrossRef] [PubMed]
- Vilar, E.; Mork, M.E.; Cuddy, A.; Borras, E.; Bannon, S.A.; Taggart, M.W.; Ying, J.; Broaddus, R.R.; Luthra, R.; Rodriguez-Bigas, M.A.; et al. Role of microsatellite instability-low as a diagnostic biomarker of Lynch syndrome in colorectal cancer. Cancer Genet. 2014, 207, 495–502. [Google Scholar] [CrossRef] [PubMed]
- Kumar, P.; Henikoff, S.; Ng, P.C. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat. Protoc. 2009, 4, 1073–1081. [Google Scholar] [CrossRef] [PubMed]
- Adzhubei, I.; Jordan, D.M.; Sunyaev, S.R. Predicting functional effect of human missense mutations using PolyPhen-2. Curr. Protoc. Hum. Genet. 2013, 7, 7–20. [Google Scholar]
- Desmet, F.O.; Hamroun, D.; Lalande, M.; Collod-Beroud, G.; Claustres, M.; Beroud, C. Human splicing finder: An online bioinformatics tool to predict splicing signals. Nucleic Acids Res. 2009, 37, e67. [Google Scholar] [CrossRef] [PubMed]
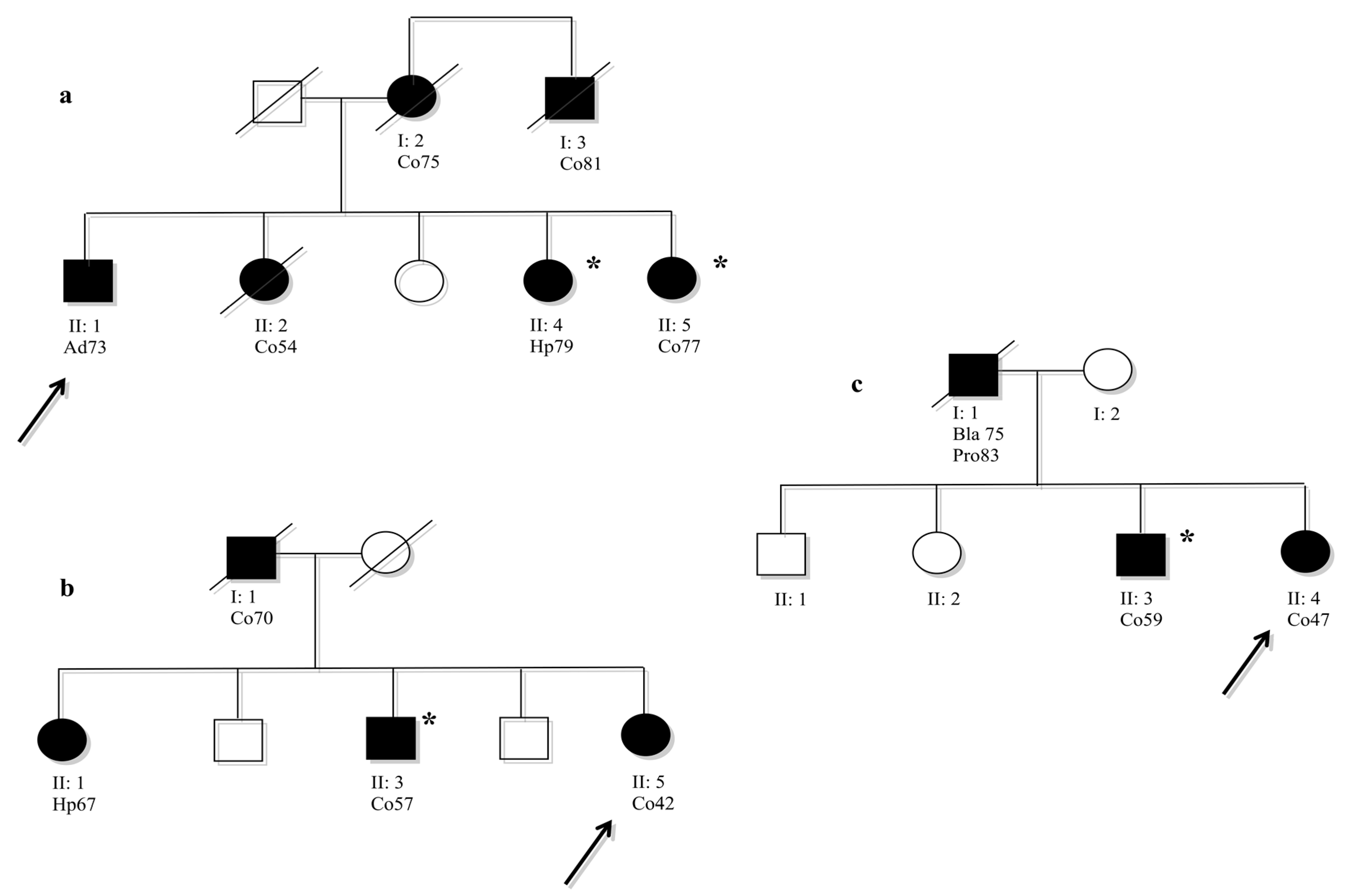
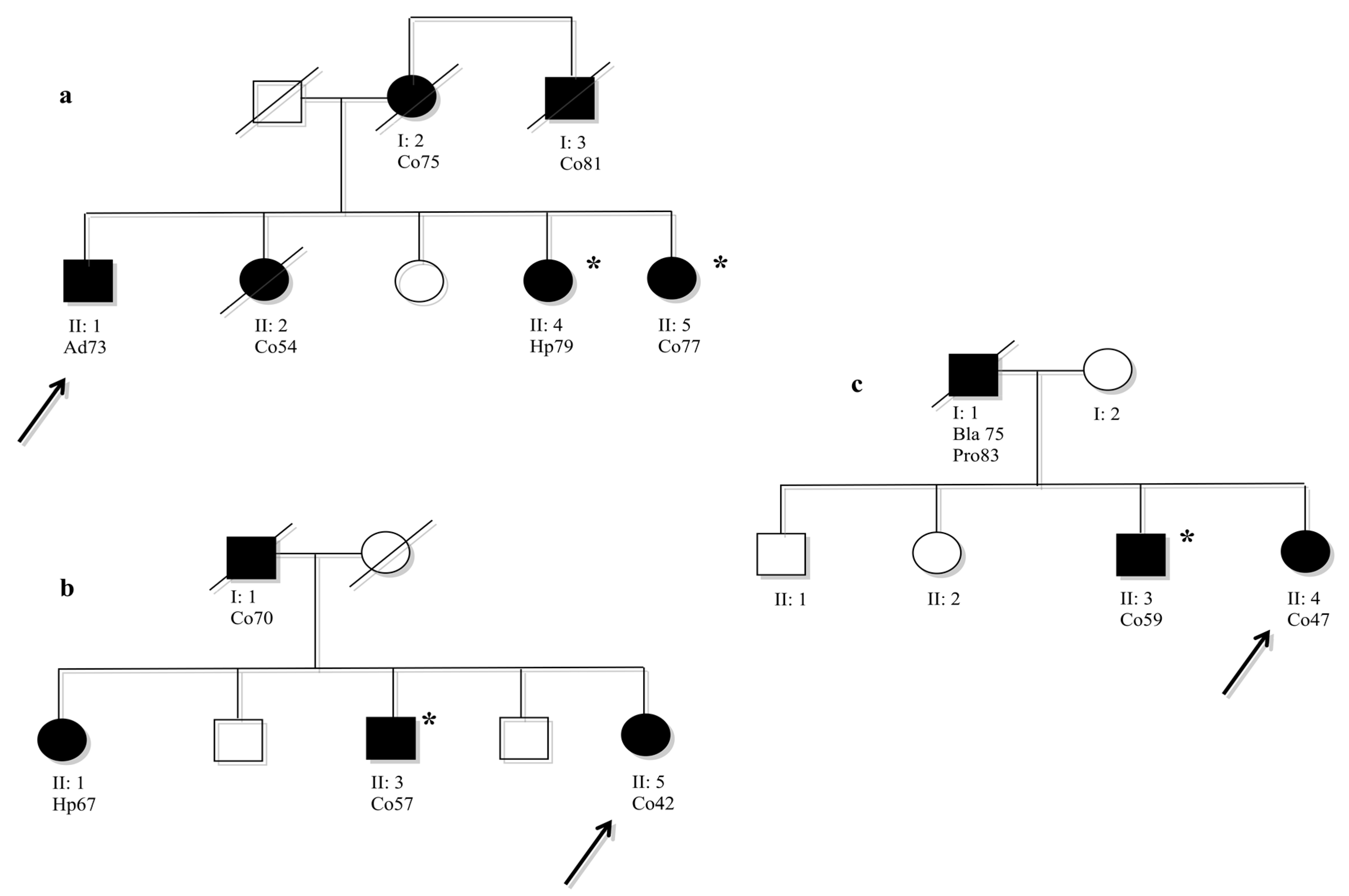
 Analysed members that were not carried of mutation.
Analysed members that were not carried of mutation.
Analysed members that were not carried of mutation.
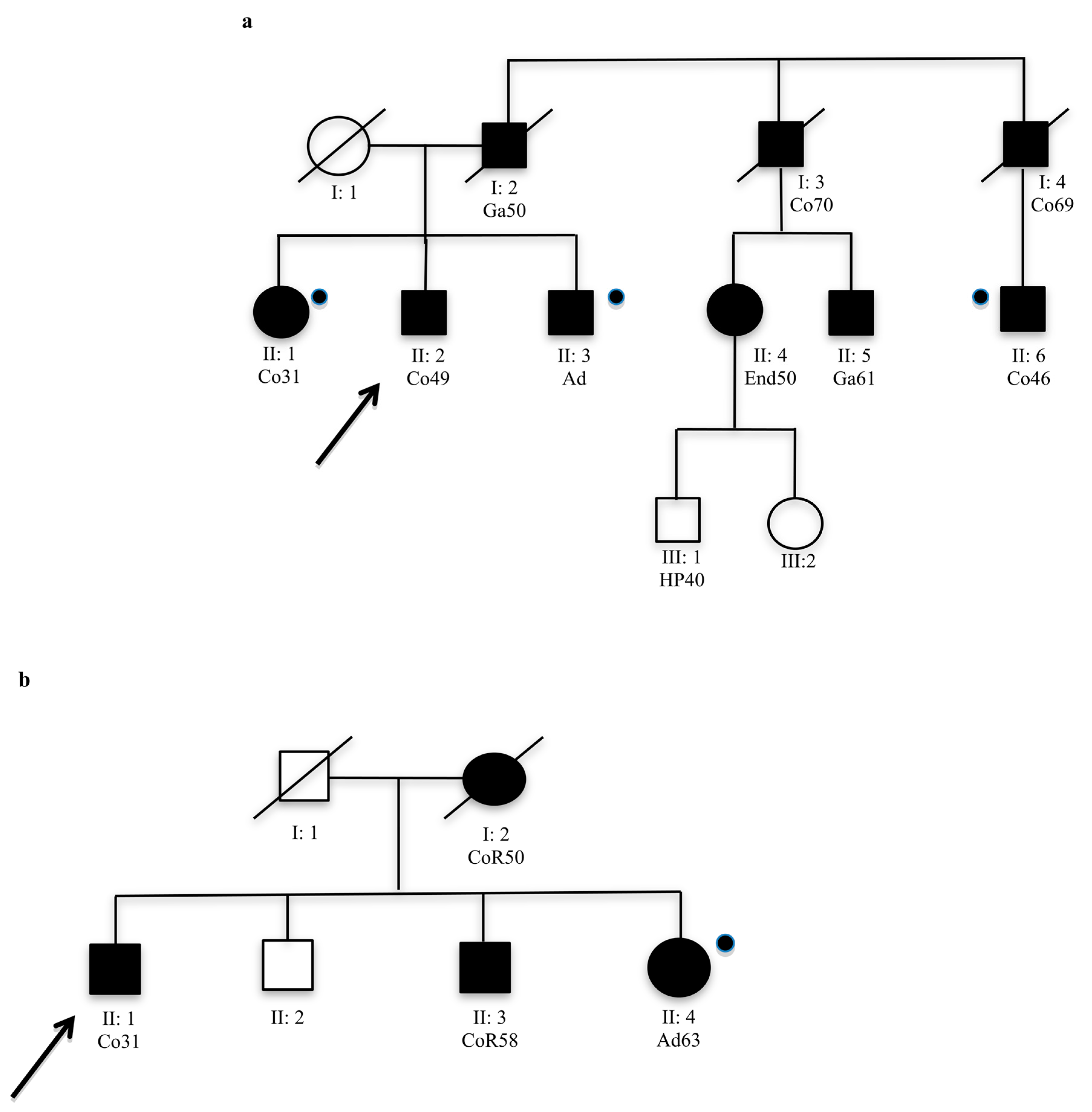
Analysed members that were not carried of mutation.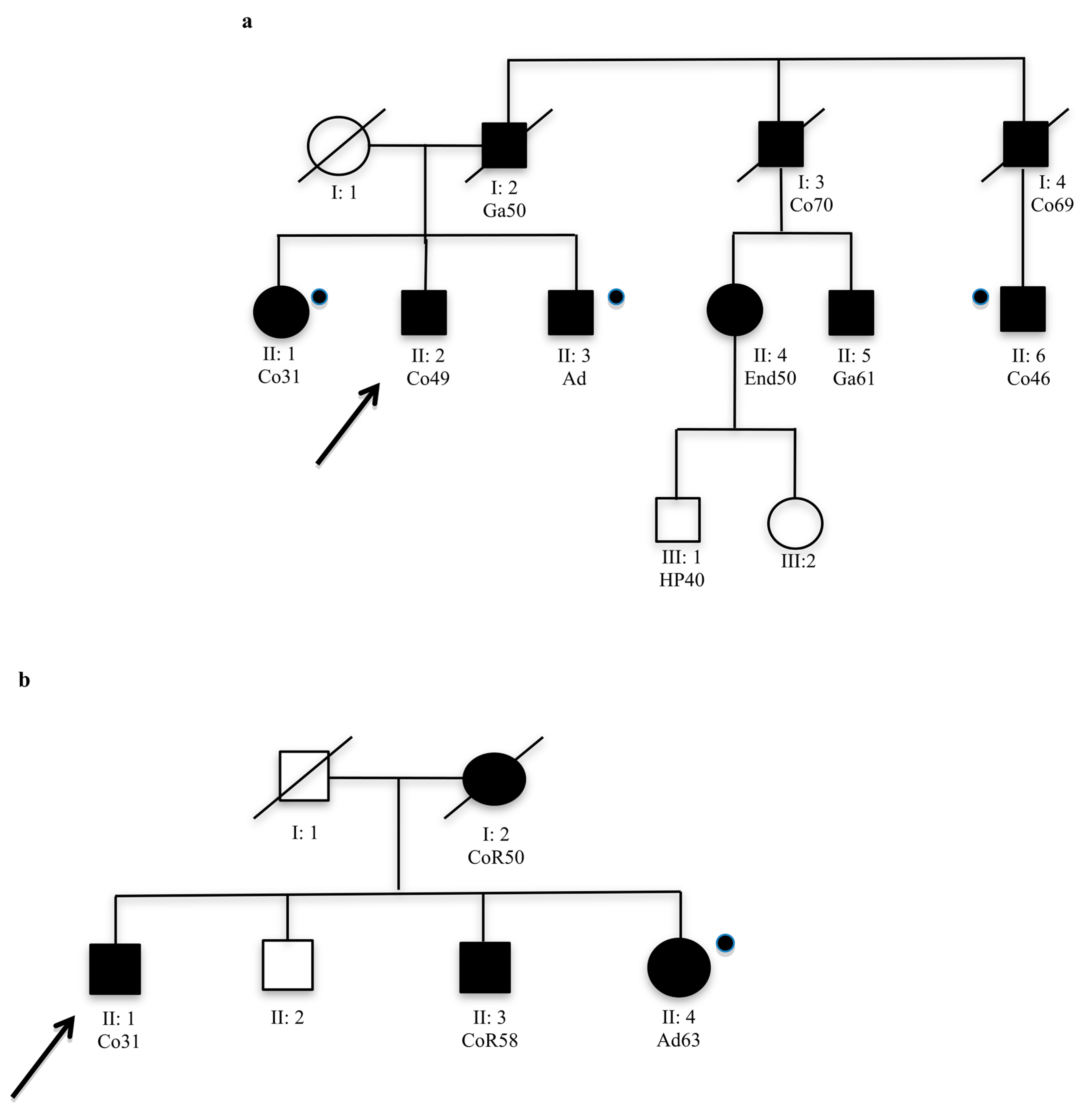


{kind=link}
{kind=link}
{kind=link}
| Exon | Nucleotide Change | Amino Acid Change | Frequency in Hereditary CRC | Reference (Reportage) | Other Studies | ||
|---|---|---|---|---|---|---|---|
| Segregation Analysis | MSI | IHC | |||||
| 1 | c.116 G>A | p.Gly39Glu | 42 families | dbSNP-rs1042821 | ND | ND | ND |
| 1 | c.186 A>C | p. = (Arg) | 40 families | Nicolaides et al. 1996 (29 times) | ND | ND | ND |
| 1 | c.260 +22 C>G | 29 families | Kolodner et al. 1999 (8 times) | ND | ND | ND | |
| 2 | c.261 −46 A>G | 1 family | this study | ND | ND | ND | |
| 2 | c.276 A>G | p. = (Pro) | 20 families | dbSNP-rs1800932 | ND | ND | ND |
| 2 | c.431 G>T | p.Ser144Ile | 1 family | Wu et al. 1999 (26 times) | ND | MSI-H | ND |
| 2 | c.457 +33_+34insGTGT | 1 family | Identified in this study | (+) | MSI-L | ND | |
| 2 | c.457 +50 T>A | 1 family | Identified in this study | ND | MSI-H | ND | |
| 2 | c.457 +52 T>A | 3 families | Plaschke et al. 2000 (25 times) | ND | ND; ND; MSI-H | ND | |
| 3 | c.540 T>C | p. = (Asp) | 11 families | dbSNP-rs1800935 | ND | ND | ND |
| 4 | c.642 C>T | p. = (Tyr) | 6 families | Wijnen et al. 1999 (26 times) | ND | ND | ND |
| 4 | c.663 A>C | p.Glu221Asp | 1 family | Devlin et al. 2008 (7 times) | ND | ND | ND |
| 4 | c.990 A>T | p. = (Ser) | 1 family | Identified in this study | ND | MSI-H | ND |
| 4 | c.1164 C>T | p. = (His) | 1 family | Kolodner et al. 1999 (4 times) | (−) | MSI-H | ND |
| 4 | c.1395 A>T | p. = (Ala) | 1 family | Identified in this study | (−) | MSI-H | ND |
| 4 | c.2049_2050insAGT | p.Ala683_Leu684insSer | 1 family | Identified in this study | (+) | MSI-H | MSH6-; MSH2+; MLH1+ |
| 4 | c.2398 G>C | p.Val800Leu | 1 family | Kolodner et al. 1999 (3 times) | (+) | MSI-H | ND |
| 4 | c.2633 T>C | p.Val878Ala | 2 families | dbSNP-rs2020912 | ND | MSI-H, ND | ND |
| 4 | c.2941 A>G | p.Ile981Val | 1 family | Identified in this study | (+) | MSI-H | ND |
| 5 | c.3226 C>T | p.Arg1076Cys | 1 family | Plaschke et al. 2000 (8 times) | ND | MSI-H | ND |
| 5 | c.3261dup | p.Phe1088Argfs*3 | 1 family | Bonk et al. (2 times) | (−) | MSI-L | MSH6-; MSH2+; MLH1+ |
| 5 | c.3296_3297delTT | p.Ile1099delinsAsnfs*8 | 1 family | Identified in this study | (−) | MSI-H | ND |
| 5 | c.3438 +14 A>T | 15 families | dbSNP-rs2020911 | ND | ND | ND | |
| 7 | c.3639 T>A | p.Asp1214Glu | 1 family | Identified in this study | ND | ND | ND |
| 7 | c.3646 +31_+34del | 16 families | dbSNP-rs1805181 | ND | ND | ND | |
| 8_9 | c.3802−42insT | 4 families | Plaschke et al. 2000 (2 times) | ND | ND; ND; ND; ND | ND | |
| 8_9 | c.3801 +54C>G | 8 families | Kolodner et al. 1999 (10 times) | ND | ND | ND | |
| Family | ID | Mutation | Protein Effect | In Silico Analysis | Frequency in Healthy Controls | Phenotype MSI | Segregation Analysis in Affected Subjects | ||
|---|---|---|---|---|---|---|---|---|---|
| PolyPhen (Score) | SIFT (Score) | HSF | |||||||
| 31 | 808 | Ex2 c.261 -46 A>G | ND | ND | +3’ss; +BP | 0/100 | AM−; ND | ND | |
| 33 | 409 | Ex2 c.457+33_+34insGTGT | ND | ND | +3’ss × 2; +ESE | 0/100 | AM−; MSI-L | 3/3 | |
| 10 | 9529 | Ex2 c.457 +50 T>A | ND | ND | +3’ss | 0/100 | AM+; MSI-H | ND | |
| 34 | 410 | Ex4 c.990 A>T | p. = (Ser) | ND | ND | −SRp55; −EIE | 0/100 | AM+; MSI-H | ND |
| 26 | 210 | Ex4 c.1395 A>T | p. = (Ala) | ND | ND | −SRp55; +ESS × 2; +ESR | 2/100 | AM+; MSI-H | 1/2 |
| 102 | 1454 | Ex4 c.2049_2050insAGT | p.Ala683_Leu684insSer | ND | ND | +3’ss × 2; +BP | 0/100 | AM−; MSI-H | 2/2 |
| 26 | 210 | Ex4 c.2941 A>G | p.Ile981Val | Benign (0.181) | Tolerated (1) | +3’ss; +ESE × 2; −EIE × 2; +ESS; +9G8; +ESR | 0/100 | AM+; MSI-H | 2/2 |
| 21 | 105 | Ex5 c.3296_97delTT | p.Ile1099delinsAsnfs*8 | ND | ND | ND | 0/100 | AM+; MSI-H | 1/4 |
| 18 | 013 | Ex7 c.3639 T>A | p.Asp1214Glu | Probably damaging (1) | Damaging (0) | +3’ss × 2; +ESE × 5; +EIE; +Tra2β; −IIE × 3; +ESR | 0/100 | AM+; ND | ND |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liccardo, R.; De Rosa, M.; Rossi, G.B.; Carlomagno, N.; Izzo, P.; Duraturo, F. Incomplete Segregation of MSH6 Frameshift Variants with Phenotype of Lynch Syndrome. Int. J. Mol. Sci. 2017, 18, 999. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms18050999
Liccardo R, De Rosa M, Rossi GB, Carlomagno N, Izzo P, Duraturo F. Incomplete Segregation of MSH6 Frameshift Variants with Phenotype of Lynch Syndrome. International Journal of Molecular Sciences. 2017; 18(5):999. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms18050999
Chicago/Turabian StyleLiccardo, Raffaella, Marina De Rosa, Giovanni Battista Rossi, Nicola Carlomagno, Paola Izzo, and Francesca Duraturo. 2017. "Incomplete Segregation of MSH6 Frameshift Variants with Phenotype of Lynch Syndrome" International Journal of Molecular Sciences 18, no. 5: 999. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms18050999