
Genomic Variations in Pancreatic Cancer and Potential Opportunities for Development of New Approaches for Diagnosis and Treatment

Abstract
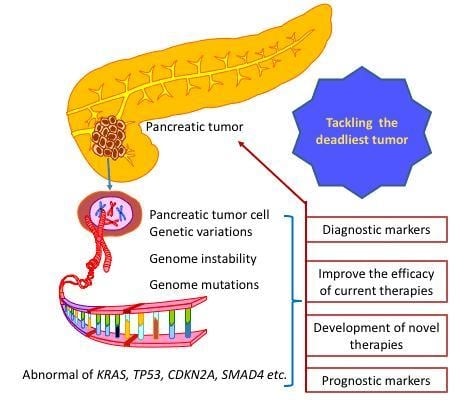
:
1. Introduction
2. Types of Genomic Variations
2.1. Chromosomal Aberrations
2.2. Copy Number Variations
2.3. Point Mutations and Indels
2.4. Epigenetic Changes
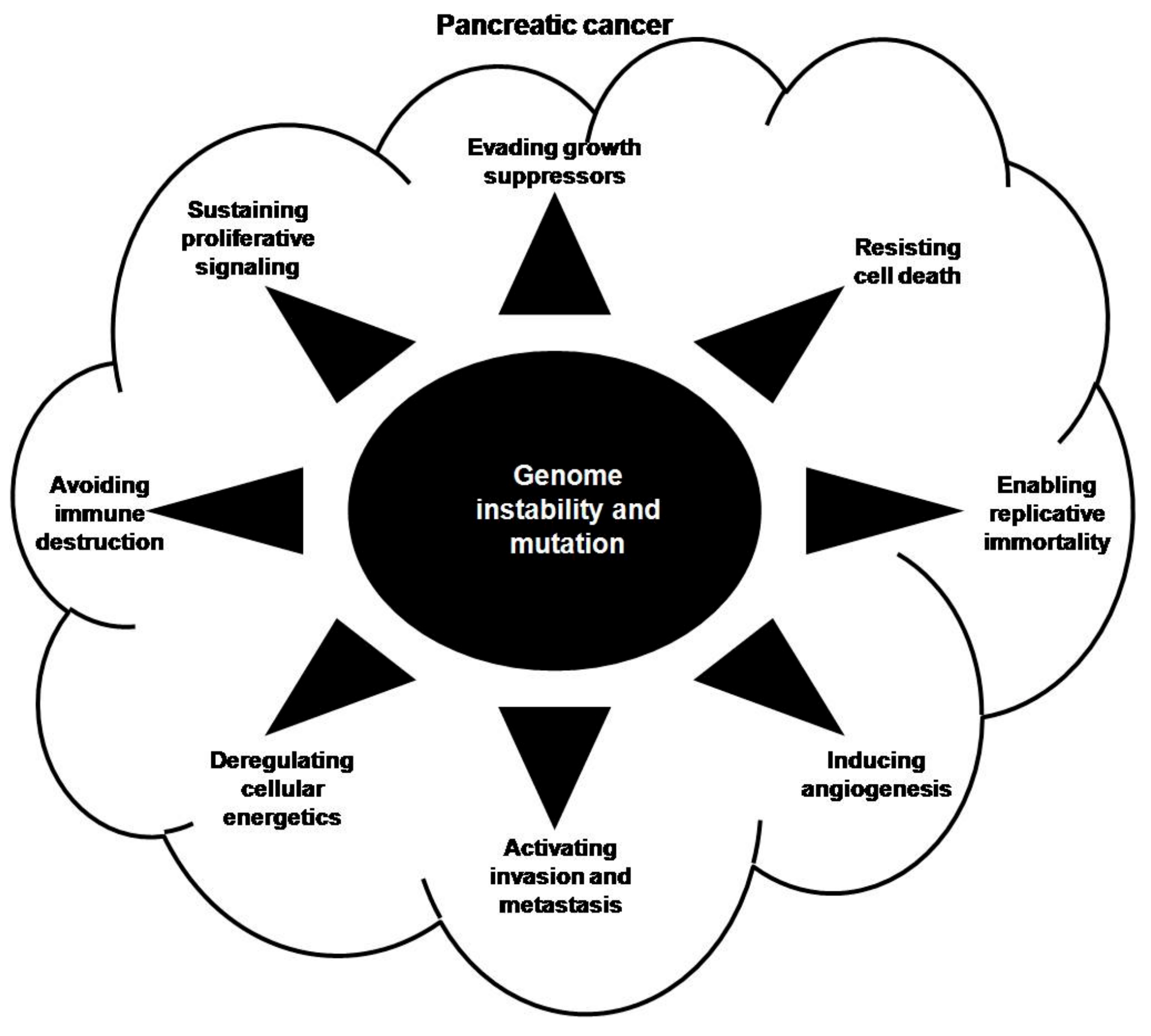
3. Contribution of Genomic Instability to Pancreatic Carcinogenesis
3.1. Sustained Proliferation
3.2. Evading Growth Suppression
3.3. Resisting Cell Death
3.4. Enabling Replicative Immortality
3.5. Inducing Angiogenesis
3.6. Activating Invasion and Metastasis
3.7. Deregulating Cellular Energetics
3.8. Avoiding Immune Destruction
4. Effects of Pancreatic Cancer Genetics on Host Immunity
5. Implications of Genomic Variations on Pancreatic Cancer Diagnosis and Treatment
5.1. Diagnostic Biomarkers
5.2. Prognostic Biomarkers
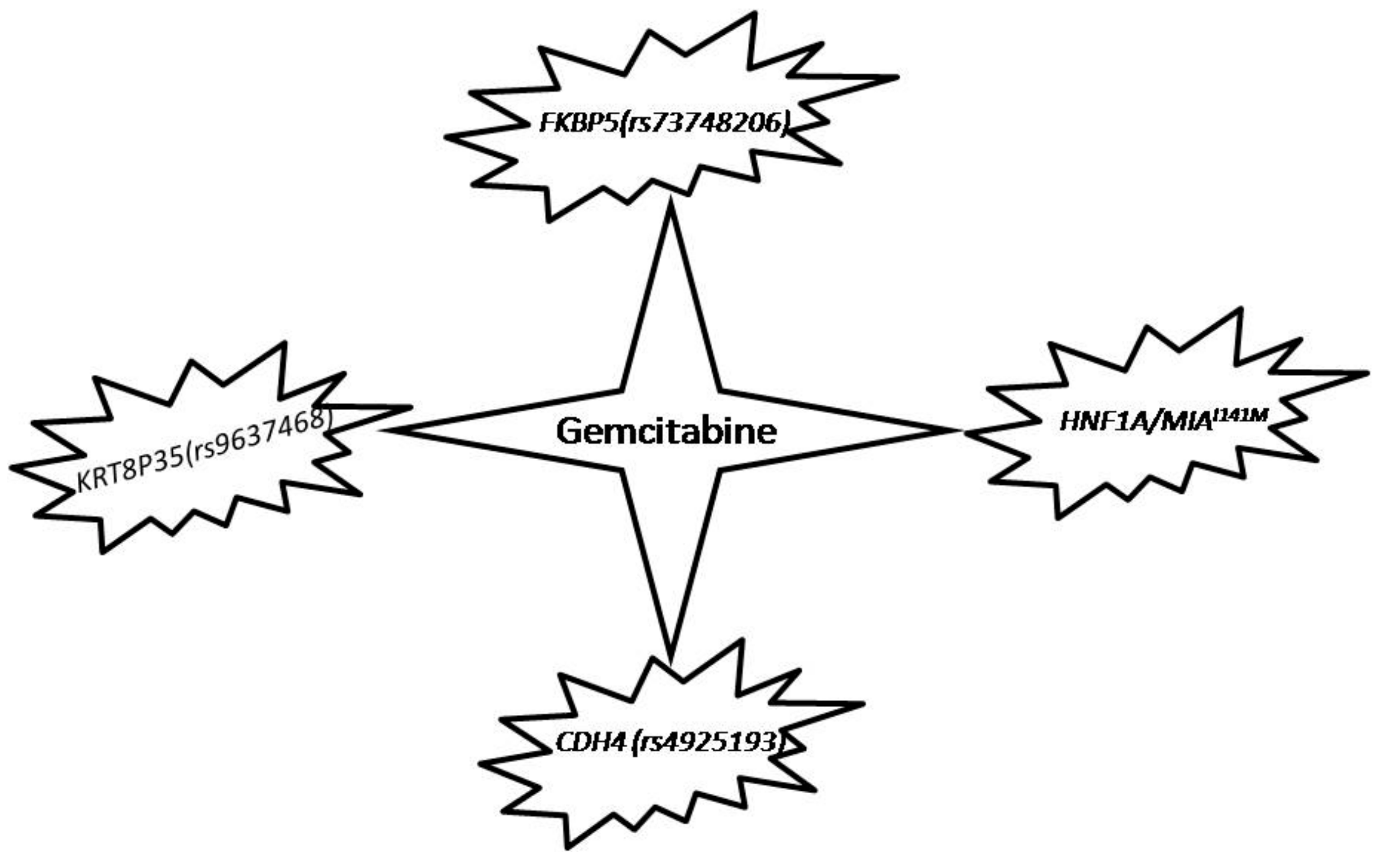
5.3. Exploiting Genetic Variations to Improve Patient Responses to Gemcitabine
5.4. Therapeutic Inhibition of KRAS
5.5. Exploring Immunotherapy for Pancreatic Cancer Based on Genetic Variations
6. Conclusions
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2016. CA Cancer J. Clin. 2016, 66, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Zheng, R.; Baade, P.D.; Zhang, S.; Zeng, H.; Bray, F.; Jemal, A.; Yu, X.Q.; He, J. Cancer statistics in China, 2015. CA Cancer J. Clin. 2016, 66, 115–132. [Google Scholar] [CrossRef] [PubMed]
- Ryan, D.P.; Hong, T.S.; Bardeesy, N. Pancreatic adenocarcinoma. N. Engl. J. Med. 2014, 371, 1039–1049. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.S.; Park, S.B.; Kim, S.A.; Kwon, S.K.; Cha, H.; Lee, D.Y.; Ro, S.; Cho, J.M.; Song, S.Y. A novel HDAC inhibitor, CG200745, inhibits pancreatic cancer cell growth and overcomes gemcitabine resistance. Sci. Rep. 2017, 7. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, A.M.; Wang, Y.H. Viro-immune therapy: A new strategy for treatment of pancreatic cancer. World J. Gastroenterol. 2016, 22, 748–763. [Google Scholar] [CrossRef] [PubMed]
- Stathis, A.; Moore, M.J. Advanced pancreatic carcinoma: Current treatment and future challenges. Nat. Rev. Clin. Oncol. 2010, 7, 163–172. [Google Scholar] [CrossRef] [PubMed]
- Sausen, M.; Phallen, J.; Adleff, V.; Jones, S.; Leary, R.J.; Barrett, M.T.; Anagnostou, V.; Parpart-Li, S.; Murphy, D.; Li, Q.K.; et al. Clinical implications of genomic alterations in the tumour and circulation of pancreatic cancer patients. Nat. Commun. 2015, 6, 7686. [Google Scholar] [CrossRef] [PubMed]
- Garrido-Laguna, I.; Hidalgo, M. Pancreatic cancer: From state-of-the-art treatments to promising novel therapies. Nat. Rev. Clin. Oncol. 2015, 12, 319–334. [Google Scholar] [CrossRef] [PubMed]
- Ottenhof, N.A.; Milne, A.N.A.; Morsink, F.H.M.; Drillenburg, P.; ten Kate, F.J.W.; Maitra, A.; Offerhaus, G.J. Pancreatic Intraepithelial Neoplasia and Pancreatic Tumorigenesis—Of Mice and Men. Arch. Pathol. Lab. Med. 2009, 133, 375–381. [Google Scholar] [CrossRef] [PubMed]
- Kanda, M.; Matthaei, H.; Wu, J.; Hong, S.M.; Yu, J.; Borges, M.; Hruban, R.H.; Maitra, A.; Kinzler, K.; Vogelstein, B.; et al. Presence of Somatic Mutations in Most Early-Stage Pancreatic Intraepithelial Neoplasia. Gastroenterology 2012, 142, 730–733. [Google Scholar] [CrossRef] [PubMed]
- Wood, L.D.; Parsons, D.W.; Jones, S.; Lin, J.; Sjoblom, T.; Leary, R.J.; Shen, D.; Boca, S.M.; Barber, T.; Ptak, J.; et al. The genomic landscapes of human breast and colorectal cancers. Science 2007, 318, 1108–1113. [Google Scholar] [CrossRef] [PubMed]
- Kowalski, J.; Morsberger, L.A.; Blackford, A.; Hawkins, A.; Yeo, C.J.; Hruban, R.H.; Griffin, C.A. Chromosomal abnormalities of adenocarcinoma of the pancreas: Identifying early and late changes. Cancer Genet. Cytogenet. 2007, 178, 26–35. [Google Scholar] [CrossRef] [PubMed]
- Bailey, P.; Chang, D.K.; Nones, K.; Johns, A.L.; Patch, A.M.; Gingras, M.C.; Miller, D.K.; Christ, A.N.; Bruxner, T.J.; Quinn, M.C.; et al. Genomic analyses identify molecular subtypes of pancreatic cancer. Nature 2016, 531, 47–52. [Google Scholar] [CrossRef] [PubMed]
- Waddell, N.; Pajic, M.; Patch, A.M.; Chang, D.K.; Kassahn, K.S.; Bailey, P.; Johns, A.L.; Miller, D.; Nones, K.; Quek, K.; et al. Whole genomes redefine the mutational landscape of pancreatic cancer. Nature 2015, 518, 495–501. [Google Scholar] [CrossRef] [PubMed]
- Donahue, T.R.; Tran, L.M.; Hill, R.; Li, Y.F.; Kovochich, A.; Calvopina, J.H.; Patel, S.G.; Wu, N.P.; Hindoyan, A.; Farrell, J.J.; et al. Integrative Survival-Based Molecular Profiling of Human Pancreatic Cancer. Clin. Cancer Res. 2012, 18, 1352–1363. [Google Scholar] [CrossRef] [PubMed]
- Ranganathan, P.; Harsha, H.C.; Pandey, A. Molecular alterations in exocrine neoplasms of the pancreas. Arch. Pathol. Lab. Med. 2009, 133, 405–412. [Google Scholar] [PubMed]
- Rustgi, A.K. The molecular pathogenesis of pancreatic cancer: Clarifying a complex circuitry. Genes Dev. 2006, 20, 3049–3053. [Google Scholar] [CrossRef] [PubMed]
- Liang, J.W.; Shi, Z.Z.; Shen, T.Y.; Che, X.; Wang, Z.; Shi, S.S.; Xu, X.; Cai, Y.; Zhao, P.; Wang, C.F.; et al. Identification of genomic alterations in pancreatic cancer using array-based comparative genomic hybridization. PLoS ONE 2014, 9, e114616. [Google Scholar] [CrossRef] [PubMed]
- Matthaios, D.; Zarogoulidis, P.; Balgouranidou, I.; Chatzaki, E.; Kakolyris, S. Molecular pathogenesis of pancreatic cancer and clinical perspectives. Oncology 2011, 81, 259–272. [Google Scholar] [CrossRef] [PubMed]
- Asting, A.G.; Ljungman, D.; Caren, H.; Dambrauskas, Z.; Iresjo, B.M.; Hyltander, A.; Naredi, P.; Lundholm, K. Alterations in Tumor DNA Are Related to Short Postoperative Survival in Patients Resected for Pancreatic Carcinoma Aimed at Cure. Pancreas 2016, 45, 900–907. [Google Scholar] [CrossRef] [PubMed]
- Jones, S.; Zhang, X.; Parsons, D.W.; Lin, J.C.; Leary, R.J.; Angenendt, P.; Mankoo, P.; Carter, H.; Kamiyama, H.; Jimeno, A.; et al. Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science 2008, 321, 1801–1806. [Google Scholar] [CrossRef] [PubMed]
- Sahin, I.H.; Lowery, M.A.; Stadler, Z.K.; Salo-Mullen, E.; Iacobuzio-Donahue, C.A.; Kelsen, D.P.; O′Reilly, E.M. Genomic instability in pancreatic adenocarcinoma: A new step towards precision medicine and novel therapeutic approaches. Expert Rev. Gastroenterol. Hepatol. 2016, 10, 893–905. [Google Scholar] [CrossRef] [PubMed]
- Welsch, T.; Kleeff, J.; Friess, H. Molecular pathogenesis of pancreatic cancer: Advances and challenges. Curr. Mol. Med. 2007, 7, 504–521. [Google Scholar] [CrossRef] [PubMed]
- Hu, C.; Hart, S.N.; Bamlet, W.R.; Moore, R.M.; Nandakumar, K.; Eckloff, B.W.; Lee, Y.K.; Petersen, G.M.; McWilliams, R.R.; Couch, F.J. Prevalence of Pathogenic Mutations in Cancer Predisposition Genes among Pancreatic Cancer Patients. Cancer Epidemiol. Biomark. Prev. 2016, 25, 207–211. [Google Scholar] [CrossRef] [PubMed]
- Skoulidis, F.; Cassidy, L.D.; Pisupati, V.; Jonasson, J.G.; Bjarnason, H.; Eyfjord, J.E.; Karreth, F.A.; Lim, M.; Barber, L.M.; Clatworthy, S.A.; et al. Germline Brca2 heterozygosity promotes Kras(G12D)—Driven carcinogenesis in a murine model of familial pancreatic cancer. Cancer Cell 2010, 18, 499–509. [Google Scholar] [CrossRef] [PubMed]
- Slater, E.P.; Langer, P.; Fendrich, V.; Habbe, N.; Chaloupka, B.; Matthai, E.; Sina, M.; Hahn, S.A.; Bartsch, D.K. Prevalence of BRCA2 and CDKN2a mutations in German familial pancreatic cancer families. Fam. Cancer 2010, 9, 335–343. [Google Scholar] [CrossRef] [PubMed]
- Feldmann, G.; Karikari, C.; dal Molin, M.; Duringer, S.; Volkmann, P.; Bartsch, D.K.; Bisht, S.; Koorstra, J. B.; Brossart, P.; Maitra, A.; et al. Inactivation of Brca2 cooperates with Trp53(R172H) to induce invasive pancreatic ductal adenocarcinomas in mice: A mouse model of familial pancreatic cancer. Cancer Biol. Ther. 2011, 11, 959–968. [Google Scholar] [CrossRef] [PubMed]
- Harinck, F.; Kluijt, I.; van der Stoep, N.; Oldenburg, R.A.; Wagner, A.; Aalfs, C.M.; Sijmons, R.H.; Poley, J.W.; Kuipers, E.J.; Fockens, P.; et al. Indication for CDKN2A-mutation analysis in familial pancreatic cancer families without melanomas. J. Med. Genet. 2012, 49, 362–365. [Google Scholar] [CrossRef] [PubMed]
- Bournet, B.; Buscail, C.; Muscari, F.; Cordelier, P.; Buscail, L. Targeting KRAS for diagnosis, prognosis, and treatment of pancreatic cancer: Hopes and realities. Eur. J. Cancer 2016, 54, 75–83. [Google Scholar] [CrossRef] [PubMed]
- Biankin, A.V.; Waddell, N.; Kassahn, K.S.; Gingras, M.C.; Muthuswamy, L.B.; Johns, A.L.; Miller, D.K.; Wilson, P.J.; Patch, A.M.; Wu, J.; et al. Pancreatic cancer genomes reveal aberrations in axon guidance pathway genes. Nature 2012, 491, 399–405. [Google Scholar] [CrossRef] [PubMed]
- Zhen, D.B.; Rabe, K.G.; Gallinger, S.; Syngal, S.; Schwartz, A.G.; Goggins, M.G.; Hruban, R.H.; Cote, M.L.; McWilliams, R.R.; Roberts, N.J.; et al. BRCA1, BRCA2, PALB2, and CDKN2A mutations in familial pancreatic cancer: A PACGENE study. Genet. Med. 2015, 17, 569–577. [Google Scholar] [CrossRef] [PubMed]
- Ueki, T.; Toyota, M.; Sohn, T.; Yeo, C.J.; Issa, J.P.; Hruban, R.H.; Goggins, M. Hypermethylation of multiple genes in pancreatic adenocarcinoma. Cancer Res. 2000, 60, 1835–1839. [Google Scholar] [PubMed]
- Esteller, M.; Sparks, A.; Toyota, M.; Sanchez-Cespedes, M.; Capella, G.; Peinado, M.A.; Gonzalez, S.; Tarafa, G.; Sidransky, D.; Meltzer, S.J.; et al. Analysis of adenomatous polyposis coli promoter hypermethylation in human cancer. Cancer Res. 2000, 60, 4366–4371. [Google Scholar] [PubMed]
- Jansen, M.; Fukushima, N.; Rosty, C.; Walter, K.; Altink, R.; Heek, T.V.; Hruban, R.; Offerhaus, J.G.; Goggins, M. Aberrant methylation of the 5′ CpG island of TSLC1 is common in pancreatic ductal adenocarcinoma and is first manifest in high-grade PanlNs. Cancer Biol. Ther. 2002, 1, 293–296. [Google Scholar] [CrossRef] [PubMed]
- Fukushima, N.; Sato, N.; Sahin, F.; Su, G.H.; Hruban, R.H.; Goggins, M. Aberrant methylation of suppressor of cytokine signalling-1 (SOCS-1) gene in pancreatic ductal neoplasms. Br. J. Cancer 2003, 89, 338–343. [Google Scholar] [CrossRef] [PubMed]
- Matsubayashi, H.; Sato, N.; Fukushima, N.; Yeo, C.J.; Walter, K.M.; Brune, K.; Sahin, F.; Hruban, R.H.; Goggins, M. Methylation of cyclin D2 is observed frequently in pancreatic cancer but is also an age-related phenomenon in gastrointestinal tissues. Clin. Cancer Res. 2003, 9, 1446–1452. [Google Scholar] [PubMed]
- Dammann, R.; Schagdarsurengin, U.; Liu, L.; Otto, N.; Gimm, O.; Dralle, H.; Boehm, B.O.; Pfeifer, G.P.; Hoang-Vu, C. Frequent RASSF1A promoter hypermethylation and K-ras mutations in pancreatic carcinoma. Oncogene 2003, 22, 3806–3812. [Google Scholar] [CrossRef] [PubMed]
- Kuroki, T.; Yendamuri, S.; Trapasso, F.; Matsuyama, A.; Aqeilan, R.I.; Alder, H.; Rattan, S.; Cesari, R.; Nolli, M.L.; Williams, N.N.; et al. The tumor suppressor gene WWOX at FRA16D is involved in pancreatic carcinogenesis. Clin. Cancer Res. 2004, 10, 2459–2465. [Google Scholar] [CrossRef] [PubMed]
- Wada, M.; Yazumi, S.; Takaishi, S.; Hasegawa, K.; Sawada, M.; Tanaka, H.; Ida, H.; Sakakura, C.; Ito, K.; Ito, Y.; et al. Frequent loss of RUNX3 gene expression in human bile duct and pancreatic cancer cell lines. Oncogene 2004, 23, 2401–2407. [Google Scholar] [CrossRef] [PubMed]
- Sakai, M.; Hibi, K.; Koshikawa, K.; Inoue, S.; Takeda, S.; Kaneko, T.; Nakao, A. Frequent promoter methylation and gene silencing of CDH13 in pancreatic cancer. Cancer Sci. 2004, 95, 588–591. [Google Scholar] [CrossRef] [PubMed]
- Martin, S.T.; Sato, N.; Dhara, S.; Chang, R.; Hustinx, S.R.; Abe, T.; Maitra, A.; Goggins, M. Aberrant methylation of the Human Hedgehog interacting protein (HHIP) gene in pancreatic neoplasms. Cancer Biol. Ther. 2005, 4, 728–733. [Google Scholar] [CrossRef] [PubMed]
- Park, J.Y.; Helm, J.F.; Zheng, W.; Ly, Q.P.; Hodul, P.J.; Centeno, B.A.; Malafa, M.P. Silencing of the candidate tumor suppressor gene solute carrier family 5 member 8 (SLC5A8) in human pancreatic cancer. Pancreas 2008, 36, e32–e39. [Google Scholar] [CrossRef] [PubMed]
- Weber, M.; Davies, J.J.; Wittig, D.; Oakeley, E.J.; Haase, M.; Lam, W.L.; Schubeler, D. Chromosome-wide and promoter-specific analyses identify sites of differential DNA methylation in normal and transformed human cells. Nat. Genet. 2005, 37, 853–862. [Google Scholar] [CrossRef] [PubMed]
- Schutte, M.; Hruban, R.H.; Geradts, J.; Maynard, R.; Hilgers, W.; Rabindran, S.K.; Moskaluk, C.A.; Hahn, S.A.; Schwarte-Waldhoff, I.; Schmiegel, W.; et al. Abrogation of the Rb/p16 tumor-suppressive pathway in virtually all pancreatic carcinomas. Cancer Res. 1997, 57, 3126–3130. [Google Scholar] [PubMed]
- Hwang, H.W.; Mendell, J.T. MicroRNAs in cell proliferation, cell death, and tumorigenesis. Br. J. Cancer 2006, 94, 776–780. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Getz, G.; Miska, E.A.; Alvarez-Saavedra, E.; Lamb, J.; Peck, D.; Sweet-Cordero, A.; Ebert, B.L.; Mak, R.H.; Ferrando, A.A.; et al. MicroRNA expression profiles classify human cancers. Nature 2005, 435, 834–838. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.J.; Gusev, Y.; Jiang, J.M.; Nuovo, G.J.; Lerner, M.R.; Frankel, W.L.; Morgan, D.L.; Postier, R.G.; Brackett, D.J.; Schmittgen, T.D. Expression profiling identifies microRNA signature in pancreatic cancer. Int. J. Cancer 2007, 120, 1046–1054. [Google Scholar] [CrossRef] [PubMed]
- Szafranska, A.E.; Davison, T.S.; John, J.; Cannon, T.; Sipos, B.; Maghnouj, A.; Labourier, E.; Hahn, S.A. MicroRNA expression alterations are linked to tumorigenesis and non-neoplastic processes in pancreatic ductal adenocarcinoma. Oncogene 2007, 26, 4442–4452. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Z.; Gao, W.; Qian, Z.; Miao, Y. Genetic variation of miRNA sequence in pancreatic cancer. Acta Biochim. Biophys. Sin. Shanghai 2009, 41, 407–413. [Google Scholar] [CrossRef] [PubMed]
- Bloomston, M.; Frankel, W.L.; Petrocca, F.; Volinia, S.; Alder, H.; Hagan, J.P.; Liu, C.G.; Bhatt, D.; Taccioli, C.; Croce, C.M. MicroRNA expression patterns to differentiate pancreatic adenocarcinoma from normal pancreas and chronic pancreatitis. J. Am. Med. Assoc. 2007, 297, 1901–1908. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Jiang, B.H.; Liu, L.Z. PI3K/PTEN signaling in angiogenesis and tumorigenesis. Adv. Cancer Res. 2009, 102, 19–65. [Google Scholar] [CrossRef] [PubMed]
- Yuan, T.L.; Cantley, L.C. PI3K pathway alterations in cancer: Variations on a theme. Oncogene 2008, 27, 5497–5510. [Google Scholar] [CrossRef] [PubMed]
- Bournet, B.; Muscari, F.; Buscail, C.; Assenat, E.; Barthet, M.; Hammel, P.; Selves, J.; Guimbaud, R.; Cordelier, P.; Buscail, L. KRAS G12D Mutation Subtype Is A Prognostic Factor for Advanced Pancreatic Adenocarcinoma. Clin. Transl. Gastroenterol. 2016, 7, e157. [Google Scholar] [CrossRef] [PubMed]
- Sherr, C.J.; McCormick, F. The RB and p53 pathways in cancer. Cancer Cell 2002, 2, 103–112. [Google Scholar] [CrossRef]
- Burkhart, D.L.; Sage, J. Cellular mechanisms of tumour suppression by the retinoblastoma gene. Nat. Rev. Cancer 2008, 8, 671–682. [Google Scholar] [CrossRef] [PubMed]
- Deshpande, A.; Sicinski, P.; Hinds, P.W. Cyclins and cdks in development and cancer: A perspective. Oncogene 2005, 24, 2909–2915. [Google Scholar] [CrossRef] [PubMed]
- Curto, M.; Cole, B.K.; Lallemand, D.; Liu, C.H.; McClatchey, A.I. Contact-dependent inhibition of EGFR signaling by Nf2/Merlin. J. Cell Biol. 2007, 177, 893–903. [Google Scholar] [CrossRef] [PubMed]
- Okada, T.; Lopez-Lago, M.; Giancotti, F.G. Merlin/NF-2 mediates contact inhibition of growth by suppressing recruitment of Rac to the plasma membrane. J. Cell Biol. 2005, 171, 361–371. [Google Scholar] [CrossRef] [PubMed]
- Partanen, J.I.; Nieminen, A.I.; Klefstrom, J. 3D view to tumor suppression: Lkb1, polarity and the arrest of oncogenic c-Myc. Cell Cycle 2009, 8, 716–724. [Google Scholar] [CrossRef] [PubMed]
- Hezel, A.F.; Bardeesy, N. LKB1; linking cell structure and tumor suppression. Oncogene 2008, 27, 6908–6919. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.C.; Wang, H.C.; Hou, Y.C.; Tung, H.L.; Chiu, T.J.; Shan, Y.S. Blockade of autophagy reduces pancreatic cancer stem cell activity and potentiates the tumoricidal effect of gemcitabine. Mol. Cancer 2015, 14, 179. [Google Scholar] [CrossRef] [PubMed]
- Quan, M.; Cui, J.; Xia, T.; Jia, Z.; Xie, D.; Wei, D.; Huang, S.; Huang, Q.; Zheng, S.; Xie, K. Merlin/NF2 Suppresses Pancreatic Tumor Growth and Metastasis by Attenuating the FOXM1-Mediated Wnt/β-Catenin Signaling. Cancer Res. 2015, 75, 4778–4789. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.J.; Wei, Z.M.; Meng, Y.X.; Ji, X.R. β-catenin up-regulates the expression of cyclinD1, c-myc and MMP-7 in human pancreatic cancer: Relationships with carcinogenesis and metastasis. World J. Gastroenterol. 2005, 11, 2117–2123. [Google Scholar] [CrossRef] [PubMed]
- Adams, J.M.; Cory, S. The Bcl-2 apoptotic switch in cancer development and therapy. Oncogene 2007, 26, 1324–1337. [Google Scholar] [CrossRef] [PubMed]
- Pandharipande, P.V.; Jeon, A.; Heberle, C.R.; Dowling, E.C.; Kong, C.Y.; Chung, D.C.; Brugge, W.R.; Hur, C. Screening for Pancreatic Adenocarcinoma in BRCA2 Mutation Carriers: Results of a Disease Simulation Model. EBioMedicine 2015, 2, 1980–1986. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Fan, Z. The epidermal growth factor receptor antibody cetuximab induces autophagy in cancer cells by downregulating HIF-1α and Bcl-2 and activating the beclin 1/hVps34 complex. Cancer Res. 2010, 70, 5942–5952. [Google Scholar] [CrossRef] [PubMed]
- Levine, B.; Kroemer, G. Autophagy in the pathogenesis of disease. Cell 2008, 132, 27–42. [Google Scholar] [CrossRef] [PubMed]
- White, E.; DiPaola, R.S. The double-edged sword of autophagy modulation in cancer. Clin. Cancer Res. 2009, 15, 5308–5316. [Google Scholar] [CrossRef] [PubMed]
- Mujumdar, N.; Saluja, A.K. Autophagy in pancreatic cancer: An emerging mechanism of cell death. Autophagy 2010, 6, 997–998. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Shi, H.; Lin, S.; Ba, M.; Cui, S. MicroRNA-216a enhances the radiosensitivity of pancreatic cancer cells by inhibiting beclin-1-mediated autophagy. Oncol. Rep. 2015, 34, 1557–1564. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Chen, X.; Tang, M. MicroRNA-216a inhibits pancreatic cancer by directly targeting Janus kinase 2. Oncol. Rep. 2014, 32, 2824–2830. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.N.; Hua, X.; Deng, W.Q.; Wu, Q.N.; Mei, H.; Chen, B. PCDH10 Interacts with hTERT and Negatively Regulates Telomerase Activity. Medicine 2015, 94, e2230. [Google Scholar] [CrossRef] [PubMed]
- Yu, B.; Yang, H.; Zhang, C.; Wu, Q.; Shao, Y.; Zhang, J.; Guan, M.; Wan, J.; Zhang, W. High-resolution melting analysis of PCDH10 methylation levels in gastric, colorectal and pancreatic cancers. Neoplasma 2010, 57, 247–252. [Google Scholar] [CrossRef] [PubMed]
- Hata, T.; Ishida, M.; Motoi, F.; Yamaguchi, T.; Naitoh, T.; Katayose, Y.; Egawa, S.; Unno, M. Telomerase activity in pancreatic juice differentiates pancreatic cancer from chronic pancreatitis: A meta-analysis. Pancreatology 2016, 6, 372–381. [Google Scholar] [CrossRef] [PubMed]
- Cong, Y.; Shay, J.W. Actions of human telomerase beyond telomeres. Cell Res. 2008, 18, 725–732. [Google Scholar] [CrossRef] [PubMed]
- Lefter, L.P.; Dima, S.; Sunamura, M.; Furukawa, T.; Sato, Y.; Abe, M.; Chivu, M.; Popescu, I.; Horii, A. Transcriptional silencing of ETS-1 efficiently suppresses angiogenesis of pancreatic cancer. Cancer Gene Ther. 2009, 16, 137–148. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.Y.; Wu, Y.; Tsuneyama, K.; Baba, T.; Mukaida, N. Essential contribution of Ets-1 to constitutive Pim-3 expression in human pancreatic cancer cells. Cancer Sci. 2009, 100, 396–404. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.Y.; Popivanova, B.K.; Nagai, Y.; Ishikura, H.; Fujii, C.; Mukaida, N. Pim-3, a proto-oncogene with serine/threonine kinase activity, is aberrantly expressed in human pancreatic cancer and phosphorylates bad to block bad-mediated apoptosis in human pancreatic cancer cell lines. Cancer Res. 2006, 66, 6741–6747. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Wang, Z.; Li, H.Y.; Zhang, B.; Ping, B.; Li, Y.Y. Pim-3 promotes human pancreatic cancer growth by regulating tumor vasculogenesis. Oncol. Rep. 2014, 31, 2625–2634. [Google Scholar] [CrossRef] [PubMed]
- Micalizzi, D.S.; Farabaugh, S.M.; Ford, H.L. Epithelial-mesenchymal transition in cancer: Parallels between normal development and tumor progression. J. Mammary Gland Biol. Neoplasia 2010, 15, 117–134. [Google Scholar] [CrossRef] [PubMed]
- Taube, J.H.; Herschkowitz, J.I.; Komurov, K.; Zhou, A.Y.; Gupta, S.; Yang, J.; Hartwell, K.; Onder, T.T.; Gupta, P.B.; Evans, K.W.; et al. Core epithelial-to-mesenchymal transition interactome gene-expression signature is associated with claudin-low and metaplastic breast cancer subtypes. Proc. Natl. Acad. Sci. USA 2010, 107, 15449–15454. [Google Scholar] [CrossRef] [PubMed]
- Schmalhofer, O.; Brabletz, S.; Brabletz, T. E-cadherin, β-catenin, and ZEB1 in malignant progression of cancer. Cancer Metastasis Rev. 2009, 28, 151–166. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Weinberg, R.A. Epithelial-mesenchymal transition: At the crossroads of development and tumor metastasis. Dev. Cell 2008, 14, 818–829. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Wang, H.; Liu, X.; Yu, T. miR-1271 inhibits migration, invasion and epithelial-mesenchymal transition by targeting ZEB1 and TWIST1 in pancreatic cancer cells. Biochem. Biophys. Res. Commun. 2016, 472, 346–352. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Wang, Z.; Chen, Y.; Zhou, M.; Zhang, H.; Chen, R.; Shi, F.; Wang, C.; Rui, Z. Transcriptional silencing of ETS-1 abrogates epithelial-mesenchymal transition resulting in reduced motility of pancreatic cancer cells. Oncol. Rep. 2015, 33, 559–565. [Google Scholar] [CrossRef] [PubMed]
- Katoh, M. Integrative genomic analyses of ZEB2: Transcriptional regulation of ZEB2 based on SMADs, ETS1, HIF1α, POU/OCT, and NF-kappaB. Int. J. Oncol. 2009, 34, 1737–1742. [Google Scholar] [CrossRef] [PubMed]
- Dong, X.; Li, Y.; Chang, P.; Tang, H.; Hess, K.R.; Abbruzzese, J.L.; Li, D. Glucose metabolism gene variants modulate the risk of pancreatic cancer. Cancer Prev. Res. Phila. 2011, 4, 758–766. [Google Scholar] [CrossRef] [PubMed]
- Cairns, R.A.; Harris, I.S.; Mak, T.W. Regulation of cancer cell metabolism. Nat. Rev. Cancer 2011, 11, 85–95. [Google Scholar] [CrossRef] [PubMed]
- Dong, X.; Tang, H.; Hess, K.R.; Abbruzzese, J.L.; Li, D. Glucose metabolism gene polymorphisms and clinical outcome in pancreatic cancer. Cancer 2011, 117, 480–491. [Google Scholar] [CrossRef] [PubMed]
- Chaika, N.V.; Yu, F.; Purohit, V.; Mehla, K.; Lazenby, A.J.; DiMaio, D.; Anderson, J.M.; Yeh, J.J.; Johnson, K.R.; Hollingsworth, M.A.; et al. Differential expression of metabolic genes in tumor and stromal components of primary and metastatic loci in pancreatic adenocarcinoma. PLoS ONE 2012, 7, e32996. [Google Scholar] [CrossRef] [PubMed]
- Paschka, P.; Schlenk, R.F.; Gaidzik, V.I.; Habdank, M.; Kronke, J.; Bullinger, L.; Spath, D.; Kayser, S.; Zucknick, M.; Gotze, K.; et al. IDH1 and IDH2 mutations are frequent genetic alterations in acute myeloid leukemia and confer adverse prognosis in cytogenetically normal acute myeloid leukemia with NPM1 mutation without FLT3 internal tandem duplication. J. Clin. Oncol. 2010, 28, 3636–3643. [Google Scholar] [CrossRef] [PubMed]
- Yan, H.; Parsons, D.W.; Jin, G.; McLendon, R.; Rasheed, B.A.; Yuan, W.; Kos, I.; Batinic-Haberle, I.; Jones, S.; Riggins, G.J.; et al. IDH1 and IDH2 mutations in gliomas. N. Engl. J. Med. 2009, 360, 765–773. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Hu, J.; Sun, W.; Duan, X.; Chen, X. Hypoxia-mediated immune evasion of pancreatic carcinoma cells. Mol. Med. Rep. 2015, 11, 3666–3672. [Google Scholar] [CrossRef] [PubMed]
- Peng, Y.P.; Zhang, J.J.; Liang, W.B.; Tu, M.; Lu, Z.P.; Wei, J.S.; Jiang, K.R.; Gao, W.T.; Wu, J.L.; Xu, Z.K.; et al. Elevation of MMP-9 and IDO induced by pancreatic cancer cells mediates natural killer cell dysfunction. BMC Cancer 2014, 14, 738. [Google Scholar] [CrossRef] [PubMed]
- Inman, K.S.; Francis, A.A.; Murray, N.R. Complex role for the immune system in initiation and progression of pancreatic cancer. World J. Gastroenterol. 2014, 20, 11160–11181. [Google Scholar] [CrossRef] [PubMed]
- Makohon-Moore, A.; Iacobuzio-Donahue, C.A. Pancreatic cancer biology and genetics from an evolutionary perspective. Nat. Rev. Cancer 2016, 16, 553–565. [Google Scholar] [CrossRef] [PubMed]
- Kunk, P.R.; Bauer, T.W.; Slingluff, C.L.; Rahma, O.E. From bench to bedside a comprehensive review of pancreatic cancer immunotherapy. J. Immunother. Cancer 2016, 4, 14. [Google Scholar] [CrossRef] [PubMed]
- Goldszmid, R.S.; Dzutsev, A.; Trinchieri, G. Host Immune Response to Infection and Cancer: Unexpected Commonalities. Cell Host Microbe 2014, 15, 295–305. [Google Scholar] [CrossRef] [PubMed]
- Vonderheide, R.H. Tumor-Promoting Inflammatory Networks in Pancreatic Neoplasia: Another Reason to Loathe Kras. Cancer Cell 2014, 25, 553–554. [Google Scholar] [CrossRef] [PubMed]
- Pylayeva-Gupta, Y.; Lee, K.E.; Hajdu, C.H.; Miller, G.; Bar-Sagi, D. Oncogenic Kras-Induced GM-CSF Production Promotes the Development of Pancreatic Neoplasia. Cancer Cell 2012, 21, 836–847. [Google Scholar] [CrossRef] [PubMed]
- Bayne, L.J.; Beatty, G.L.; Jhala, N.; Clark, C.E.; Rhim, A.D.; Stanger, B.Z.; Vonderheide, R.H. Tumor-Derived Granulocyte-Macrophage Colony-Stimulating Factor Regulates Myeloid Inflammation and T Cell Immunity in Pancreatic Cancer. Cancer Cell 2012, 21, 822–835. [Google Scholar] [CrossRef] [PubMed]
- McAllister, F.; Bailey, J.M.; Alsina, J.; Nirschl, C.J.; Sharma, R.; Fan, H.N.; Rattigan, Y.; Roeser, J.C.; Lankapalli, R.H.; Zhang, H.; et al. Oncogenic Kras Activates a Hematopoietic-to-Epithelial IL-17 Signaling Axis in Preinvasive Pancreatic Neoplasia. Cancer Cell 2014, 25, 621–637. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Niu, Z.Y.; Liang, Z.Y.; Zhou, W.X.; You, L.; Wang, M.Y.; Yao, L.T.; Liao, Q.; Zhao, Y.P. HLA-G impairs host immune response and predicts poor prognosis in pancreatic cancer. Am. J. Transl. Res. 2015, 7, 2036–2044. [Google Scholar] [PubMed]
- Rossi, M.L.; Rehman, A.A.; Gondi, C.S. Therapeutic options for the management of pancreatic cancer. World J. Gastroenterol. 2014, 20, 11142–11159. [Google Scholar] [CrossRef] [PubMed]
- Takai, E.; Yachida, S. Genomic alterations in pancreatic cancer and their relevance to therapy. World J. Gastrointest. Oncol. 2015, 7, 250–258. [Google Scholar] [PubMed]
- Hayashi, H.; Nishihara, H. A Novel Treatment Strategy for Pancreatic Cancer Based on Gene Profiles. Gan Kagaku Ryoho 2016, 43, 1326–1331. [Google Scholar]
- Goonetilleke, K.S.; Siriwardena, A.K. Systematic review of carbohydrate antigen (CA 19-9) as a biochemical marker in the diagnosis of pancreatic cancer. Eur. J. Surg. Oncol. 2007, 33, 266–270. [Google Scholar] [CrossRef] [PubMed]
- Chang, J.C.; Kundranda, M. Novel Diagnostic and Predictive Biomarkers in Pancreatic Adenocarcinoma. Int. J. Mol. Sci. 2017, 18, 667. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, Y.; Murakami, Y.; Uemura, K.; Hayashidani, Y.; Sudo, T.; Ohge, H.; Fukuda, E.; Sueda, T.; Hiyama, E. Detection of human telomerase reverse transcriptase (hTERT) expression in tissue and pancreatic juice from pancreatic cancer. Surgery 2008, 143, 113–125. [Google Scholar] [CrossRef] [PubMed]
- Yip-Schneider, M.T.; Wu, H.; Dumas, R.P.; Hancock, B.A.; Agaram, N.; Radovich, M.; Schmidt, C.M. Vascular endothelial growth factor, a novel and highly accurate pancreatic fluid biomarker for serous pancreatic cysts. J. Am. Coll. Surg. 2014, 218, 608–617. [Google Scholar] [CrossRef] [PubMed]
- Collisson, E.A.; Sadanandam, A.; Olson, P.; Gibb, W.J.; Truitt, M.; Gu, S.; Cooc, J.; Weinkle, J.; Kim, G. E.; Jakkula, L.; et al. Subtypes of pancreatic ductal adenocarcinoma and their differing responses to therapy. Nat. Med. 2011, 17, 500–503. [Google Scholar] [CrossRef] [PubMed]
- Yachida, S.; White, C.M.; Naito, Y.; Zhong, Y.; Brosnan, J.A.; Macgregor-Das, A.M.; Morgan, R.A.; Saunders, T.; Laheru, D.A.; Herman, J.M.; et al. Clinical significance of the genetic landscape of pancreatic cancer and implications for identification of potential long-term survivors. Clin. Cancer Res. 2012, 18, 6339–6347. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Tu, H.; Meng, Z.Q.; Chen, Z.; Wang, P.; Liu, L.M. K-ras mutational status predicts poor prognosis in unresectable pancreatic cancer. Eur. J. Surg. Oncol. 2010, 36, 657–662. [Google Scholar] [CrossRef] [PubMed]
- Xiang, J.F.; Wang, W.Q.; Liu, L.; Xu, H.X.; Wu, C.T.; Yang, J.X.; Qi, Z.H.; Wang, Y.Q.; Xu, J.; Liu, C.; et al. Mutant p53 determines pancreatic cancer poor prognosis to pancreatectomy through upregulation of cavin-1 in patients with preoperative serum CA19-9 ≥ 1000 U/mL. Sci. Rep. 2016, 6, 19222. [Google Scholar] [CrossRef] [PubMed]
- Chang, D.T.; Chapman, C.H.; Norton, J.A.; Visser, B.; Fisher, G.A.; Kunz, P.; Ford, J.M.; Koong, A.C.; Pai, R.K. Expression of p16(INK4A) but not hypoxia markers or poly adenosine diphosphate-ribose polymerase is associated with improved survival in patients with pancreatic adenocarcinoma. Cancer 2010, 116, 5179–5187. [Google Scholar] [CrossRef] [PubMed]
- Kang, C.M.; Hwang, H.K.; Park, J.; Kim, C.; Cho, S.K.; Yun, M.; Lee, W.J. Maximum Standard Uptake Value as a Clinical Biomarker for Detecting Loss of SMAD4 Expression and Early Systemic Tumor Recurrence in Resected Left-Sided Pancreatic Cancer. Medicine 2016, 95, e3452. [Google Scholar] [CrossRef] [PubMed]
- Kornmann, M.; Ishiwata, T.; Itakura, J.; Tangvoranuntakul, P.; Beger, H.G.; Korc, M. Increased cyclin D1 in human pancreatic cancer is associated with decreased postoperative survival. Oncology 1998, 55, 363–369. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Giovannetti, E.; Hwang, J.H.; Petrini, I.; Wang, Q.; Voortman, J.; Wang, Y.; Steinberg, S.M.; Funel, N.; Meltzer, P.S.; et al. Loss of 18q22.3 involving the carboxypeptidase of glutamate-like gene is associated with poor prognosis in resected pancreatic cancer. Clin. Cancer Res. 2012, 18, 524–533. [Google Scholar] [CrossRef] [PubMed]
- Dong, X.; Jiao, L.; Li, Y.; Evans, D.B.; Wang, H.; Hess, K.R.; Abbruzzese, J.L.; Li, D. Significant associations of mismatch repair gene polymorphisms with clinical outcome of pancreatic cancer. J. Clin. Oncol. 2009, 27, 1592–1599. [Google Scholar] [CrossRef] [PubMed]
- Walker, E.J.; Ko, A.H. Beyond first-line chemotherapy for advanced pancreatic cancer: An expanding array of therapeutic options? World J. Gastroenterol. 2014, 20, 2224–2236. [Google Scholar] [CrossRef] [PubMed]
- Ellsworth, K.A.; Eckloff, B.W.; Li, L.; Moon, I.; Fridley, B.L.; Jenkins, G.D.; Carlson, E.; Brisbin, A.; Abo, R.; Bamlet, W.; et al. Contribution of FKBP5 Genetic Variation to Gemcitabine Treatment and Survival in Pancreatic Adenocarcinoma. PLoS ONE 2013, 8, e70216. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Zhang, J.W.; Jenkins, G.; Xie, F.; Carlson, E.E.; Fridley, B.L.; Bamlet, W.R.; Petersen, G.M.; McWilliams, R.R.; Wang, L.W. Genetic variations associated with gemcitabine treatment outcome in pancreatic cancer. Pharmacogenet. Genom. 2016, 26, 527–537. [Google Scholar] [CrossRef] [PubMed]
- Kong, B.; Wu, W.W.; Valkovska, N.; Jager, C.; Hong, X.; Nitsche, U.; Friess, H.; Esposito, I.; Erkan, M.; Kleeff, J.; et al. A common genetic variation of melanoma inhibitory activity-2 labels a subtype of pancreatic adenocarcinoma with high endoplasmic reticulum stress levels. Sci. Rep. 2015, 5, 8109. [Google Scholar] [CrossRef] [PubMed]
- Collins, M.A.; di Magliano, M.P. Kras as a key oncogene and therapeutic target in pancreatic cancer. Front. Physiol. 2014, 4, 407. [Google Scholar] [CrossRef] [PubMed]
- Fang, B. RAS signaling and anti-RAS therapy: Lessons learned from genetically engineered mouse models, human cancer cells, and patient-related studies. Acta Biochim. Biophys. Sin. Shanghai 2016, 48, 27–38. [Google Scholar] [CrossRef] [PubMed]
- Laheru, D.; Shah, P.; Rajeshkumar, N.V.; McAllister, F.; Taylor, G.; Goldsweig, H.; Le, D.T.; Donehower, R.; Jimeno, A.; Linden, S.; et al. Integrated preclinical and clinical development of S-trans, trans-Farnesylthiosalicylic Acid (FTS, Salirasib) in pancreatic cancer. Investig. New Drugs 2012, 30, 2391–2399. [Google Scholar] [CrossRef] [PubMed]
- Bryant, K.L.; Mancias, J.D.; Kimmelman, A.C.; Der, C.J. KRAS: Feeding pancreatic cancer proliferation. Trends Biochem. Sci. 2014, 39, 91–100. [Google Scholar] [CrossRef] [PubMed]
- Eser, S.; Schnieke, A.; Schneider, G.; Saur, D. Oncogenic KRAS signalling in pancreatic cancer. Br. J. Cancer 2014, 111, 817–822. [Google Scholar] [CrossRef] [PubMed]
- Fang, Y.T.; Yao, Q.Z.; Chen, Z.Y.; Xiang, J.B.; William, F.E.; Gibbs, R.A.; Chen, C.Y. Genetic and molecular alterations in pancreatic cancer: Implications for personalized medicine. Med. Sci. Monit. 2013, 19, 916–926. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Hegde, S.; Knolhoff, B.L.; Zhu, Y.; Herndon, J.M.; Meyer, M.A.; Nywening, T.M.; Hawkins, W.G.; Shapiro, I.M.; Weaver, D.T.; et al. Targeting focal adhesion kinase renders pancreatic cancers responsive to checkpoint immunotherapy. Nat. Med. 2016, 22, 851–860. [Google Scholar] [CrossRef] [PubMed]
- Seton-Rogers, S. Pancreatic cancer: Dodging immunosuppression. Nat. Rev. Cancer 2016, 16, 480–481. [Google Scholar] [CrossRef] [PubMed]
- Soares, K.C.; Zheng, L.; Edil, B.; Jaffee, E.M. Vaccines for Pancreatic Cancer. Cancer J. 2012, 18, 642–652. [Google Scholar] [CrossRef] [PubMed]
- Kawaoka, T.; Oka, M.; Takashima, M.; Ueno, T.; Yamamoto, K.; Yahara, N.; Yoshino, S.; Hazama, S. Adoptive immunotherapy for pancreatic cancer: Cytotoxic T lymphocytes stimulated by the MUC1-expressing human pancreatic cancer cell line YPK-1. Oncol. Rep. 2008, 20, 155–163. [Google Scholar] [CrossRef] [PubMed]
- Fesnak, A.D.; June, C.H.; Levine, B.L. Engineered T cells: The promise and challenges of cancer immunotherapy. Nat. Rev. Cancer 2016, 16, 566–581. [Google Scholar] [CrossRef] [PubMed]
- Weiner, G.J. Building better monoclonal antibody-based therapeutics. Nat. Rev. Cancer 2015, 15, 361–370. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.H.; Hwang-Verslues, W.W.; Lee, W.H.; Huang, C.K.; Wei, P.C.; Chen, C.L.; Shew, J.Y.; Lee, E.Y.H.P.; Jeng, Y.M.; Tien, Y.W.; et al. Targeting IL-17B-IL-17RB signaling with an anti-IL-17RB antibody blocks pancreatic cancer metastasis by silencing multiple chemokines. J. Exp. Med. 2015, 212, 333–349. [Google Scholar] [CrossRef] [PubMed]
- Leach, S.D. Interleukin interrupted: A new strategy for the treatment of pancreatic cancer. J. Exp. Med. 2015, 212, 284. [Google Scholar] [CrossRef] [PubMed]
- Steele, C.W.; Karim, S.A.; Leach, J.D.G.; Bailey, P.; Upstill-Goddard, R.; Rishi, L.; Foth, M.; Bryson, S.; McDaid, K.; Wilson, Z.; et al. CXCR2 Inhibition Profoundly Suppresses Metastases and Augments Immunotherapy in Pancreatic Ductal Adenocarcinoma. Cancer Cell 2016, 29, 832–845. [Google Scholar] [CrossRef] [PubMed]
- Dart, A. Metastasis: CXCR2-targeted therapy for pancreatic cancer. Nat. Rev. Cancer 2016, 16, 411. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
| Pancreatic Tumor Traits | Genomic Variation Pathways |
|---|---|
| Sustained proliferation signaling | PI3K/AKT; KRAS; PTEN |
| Growth suppressor evasion | TP53; RB; NF2/MERLIN; LKB1 |
| Cell death resistance | CCND1; BCL2; TP53; BRCA2; miRNA216a/BECLIN1 |
| Enabling of replicative immortality | PCDH10/hTERT |
| Induction of angiogenesis | ETS1/PIM3 |
| Activation of invasion and metastasis | SNAIL; SLUG; miRNA-1271/TWIST; EST1/ZEB2 |
| Evasion of immune destruction | HIF1α/MIC; MMP9 |
| Deregulated cellular energetics | KRAS/IDH; FH; SDH |
| Drugs | Mechanism | Efficacy |
|---|---|---|
| FTIs (Lonafarnib and Tipifarnib) | Inhibiting farnesylation of KRAS | Not promising |
| FTS, salirasib | Preventing KRAS from reaching cell membranes | Promising |
| Deltarasin | Enabling KRAS to be farnesylated but halting it from reaching the membrane | Decreasing the proliferation of KRAS-driven PDAC cell lines |
| CI-1040 and PD0325901 | Inhibiting MEK/MAPK pathway downstream of KRAS | Not significant |
| LY294002 | Inhibiting PI3K pathway downstream of KRAS | Promoting apoptosis in vitro and preventing tumor proliferation in vivo |
| Immunotherapies | Examples | Mechanism |
|---|---|---|
| Tumor antigens identification | MUC1; KRAS; Mesothelin, etc. | Development of more specific and potent cancer vaccines |
| Adoptive T-cell therapy | Tumor infiltrating lymphocytes (TILs) | Expanding and activating of the patient’s T-cells ex-vivo and then re-infusing them back into the patient to kill tumor cells |
| Engineered T-cells which express a specific cancer T-cell receptor (TCR) | ||
| T-cells which express a chimeric antigen receptor (CAR) | ||
| Tumor-targeted oncolytic viruses (TOVs) | Reolysin, etc. | Selectively eliminating cancer cells and producing systemic anti-tumor effects such as promoting long lasting anti-tumor immunity |
| Monoclonal antibody | IL17RB; IL17RA, etc. | Direct targeting of the cancer cells; altering the host immune response; redirecting host immunity towards the cancerous cells; and delivering cytotoxic materials |
| Immune checkpoint therapy | CTLA4; PD1, etc. | Enhancing T cells function |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lu, S.; Ahmed, T.; Du, P.; Wang, Y. Genomic Variations in Pancreatic Cancer and Potential Opportunities for Development of New Approaches for Diagnosis and Treatment. Int. J. Mol. Sci. 2017, 18, 1201. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms18061201
Lu S, Ahmed T, Du P, Wang Y. Genomic Variations in Pancreatic Cancer and Potential Opportunities for Development of New Approaches for Diagnosis and Treatment. International Journal of Molecular Sciences. 2017; 18(6):1201. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms18061201
Chicago/Turabian StyleLu, Shuangshuang, Tasqeen Ahmed, Pan Du, and Yaohe Wang. 2017. "Genomic Variations in Pancreatic Cancer and Potential Opportunities for Development of New Approaches for Diagnosis and Treatment" International Journal of Molecular Sciences 18, no. 6: 1201. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms18061201