
Association of Smoking, Alcohol Use, and Betel Quid Chewing with Epigenetic Aberrations in Cancers



Abstract
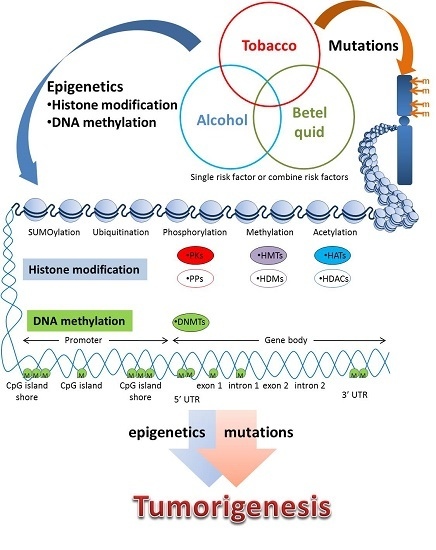
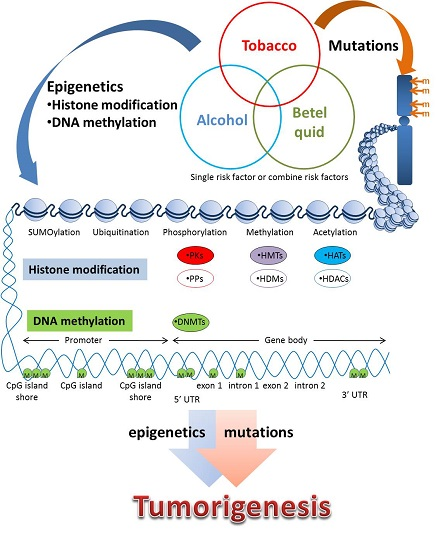
:
1. Introduction
2. Epigenetic Modification of DNA
2.1. DNA Methylation
2.2. Epigenetic Modification of Histones
2.2.1. Histone Modification
2.2.2. Acetylation
2.2.3. Methylation
2.2.4. Phosphorylation
2.2.5. Ubiquitination
2.2.6. SUMOylation
3. Smoking Modulates the Epigenome during Carcinogenesis
3.1. Smoking Induces DNA Methylation-Associated Enzymes Activity
3.2. DNA Methylation in Smoking-Related Cancers
3.3. Smoking Induces DNA Methylation
3.4. Smoking Induces Histone Modification
4. Alcohol Use Known to Induce DNA Methylation during Carcinogenesis
5. Betel Quid Chewing Modulates the Epigenome during Carcinogenesis
6. Therapeutic Drugs Targeting the Epigenome
7. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Hardy, T.M.; Tollefsbol, T.O. Epigenetic diet: Impact on the epigenome and cancer. Epigenomics 2011, 3, 503–518. [Google Scholar] [CrossRef] [PubMed]
- Bishop, K.S.; Ferguson, L.R. The interaction between epigenetics, nutrition and the development of cancer. Nutrients 2015, 7, 922–947. [Google Scholar] [CrossRef] [PubMed]
- Legaki, E.; Gazouli, M. Influence of environmental factors in the development of inflammatory bowel diseases. World J. Gastrointest. Pharmacol. Ther. 2016, 7, 112–125. [Google Scholar] [CrossRef] [PubMed]
- Leung, A.; Trac, C.; Du, J.; Natarajan, R.; Schones, D.E. Persistent chromatin modifications induced by high fat diet. J. Biol. Chem. 2016, 291, 10446–10455. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.S. Impact of maternal diet on the epigenome during in utero life and the developmental programming of diseases in childhood and adulthood. Nutrients 2015, 7, 9492–9507. [Google Scholar] [CrossRef] [PubMed]
- Krautkramer, K.A.; Kreznar, J.H.; Romano, K.A.; Vivas, E.I.; Barrett-Wilt, G.A.; Rabaglia, M.E.; Keller, M.P.; Attie, A.D.; Rey, F.E.; Denu, J.M. Diet-microbiota interactions mediate global epigenetic programming in multiple host tissues. Mol. Cell 2016, 64, 982–992. [Google Scholar] [CrossRef] [PubMed]
- Maury, E.; Hashizume, R. Epigenetic modification in chromatin machinery and its deregulation in pediatric brain tumors: Insight into epigenetic therapies. Epigenetics 2017, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Maroni, P.; Matteucci, E.; Bendinelli, P.; Desiderio, M.A. Functions and epigenetic regulation of WWOX in bone metastasis from breast carcinoma: Comparison with primary tumors. Int. J. Mol. Sci. 2017, 18. [Google Scholar] [CrossRef] [PubMed]
- Koschmann, C.; Nunez, F.J.; Mendez, F.; Brosnan-Cashman, J.A.; Meeker, A.K.; Lowenstein, P.R.; Castro, M.G. Mutated chromatin regulatory factors as tumor drivers in cancer. Cancer Res. 2017, 77, 227–233. [Google Scholar] [CrossRef] [PubMed]
- O’Keefe, S.J. Diet, microorganisms and their metabolites, and colon cancer. Nat. Rev. Gastroenterol. Hepatol. 2016, 13, 691–706. [Google Scholar] [CrossRef] [PubMed]
- Laufer, B.I.; Kapalanga, J.; Castellani, C.A.; Diehl, E.J.; Yan, L.; Singh, S.M. Associative DNA methylation changes in children with prenatal alcohol exposure. Epigenomics 2015, 7, 1259–1274. [Google Scholar] [CrossRef] [PubMed]
- Finegersh, A.; Homanics, G.E. Paternal alcohol exposure reduces alcohol drinking and increases behavioral sensitivity to alcohol selectively in male offspring. PLoS ONE 2014, 9, e99078. [Google Scholar] [CrossRef] [PubMed]
- Grandjean, P.; Barouki, R.; Bellinger, D.C.; Casteleyn, L.; Chadwick, L.H.; Cordier, S.; Etzel, R.A.; Gray, K.A.; Ha, E.H.; Junien, C.; et al. Life-long implications of developmental exposure to environmental stressors: New perspectives. Endocrinology 2015, 156, 3408–3415. [Google Scholar] [CrossRef] [PubMed]
- Shiu, M.N.; Chen, T.H.; Chang, S.H.; Hahn, L.J. Risk factors for leukoplakia and malignant transformation to oral carcinoma: A leukoplakia cohort in Taiwan. Br. J. Cancer 2000, 82, 1871–1874. [Google Scholar] [CrossRef] [PubMed]
- Moutasim, K.A.; Jenei, V.; Sapienza, K.; Marsh, D.; Weinreb, P.H.; Violette, S.M.; Lewis, M.P.; Marshall, J.F.; Fortune, F.; Tilakaratne, W.M.; et al. Betel-derived alkaloid up-regulates keratinocyte αvβ6 integrin expression and promotes oral submucous fibrosis. J. Pathol. 2011, 223, 366–377. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.T.; Lan, S.J.; Hsieh, C.C.; Yang, M.J.; Ko, Y.C.; Tsai, C.C.; Yen, Y.Y. Prevalence and characteristics of areca nut chewers among junior high school students in Changhua county, Taiwan. Community Dent. Oral Epidemiol. 1993, 21, 370–373. [Google Scholar] [CrossRef] [PubMed]
- Wong, Y.K.; Liu, T.Y.; Chang, K.W.; Lin, S.C.; Chao, T.W.; Li, P.L.; Chang, C.S. P53 alterations in betel quid- and tobacco-associated oral squamous cell carcinomas from Taiwan. J. Oral Pathol. Med. 1998, 27, 243–248. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Gimenez-Conti, I.B.; Cunningham, J.E.; Collet, A.M.; Luna, M.A.; Lanfranchi, H.E.; Spitz, M.R.; Conti, C.J. Alterations of p53, cyclin D1, rb, and H-ras in human oral carcinomas related to tobacco use. Cancer 1998, 83, 204–212. [Google Scholar] [CrossRef]
- Chiu, C.F.; Tsai, M.H.; Tseng, H.C.; Wang, C.L.; Wang, C.H.; Wu, C.N.; Lin, C.C.; Bau, D.T. A novel single nucleotide polymorphism in XRCC4 gene is associated with oral cancer susceptibility in taiwanese patients. Oral Oncol. 2008, 44, 898–902. [Google Scholar] [CrossRef] [PubMed]
- Tseng, H.C.; Tsai, M.H.; Chiu, C.F.; Wang, C.H.; Chang, N.W.; Huang, C.Y.; Tsai, C.W.; Liang, S.Y.; Wang, C.L.; Bau, D.T. Association of XRCC4 codon 247 polymorphism with oral cancer susceptibility in Taiwan. Anticancer Res. 2008, 28, 1687–1691. [Google Scholar] [PubMed]
- Yang, C.H.; Lin, Y.D.; Yen, C.Y.; Chuang, L.Y.; Chang, H.W. A systematic gene-gene and gene-environment interaction analysis of DNA repair genes XRCC1, XRCC2, XRCC3, XRCC4, and oral cancer risk. Omics 2015, 19, 238–247. [Google Scholar] [CrossRef] [PubMed]
- Laird, P.W. The power and the promise of DNA methylation markers. Nat. Rev. Cancer 2003, 3, 253–266. [Google Scholar] [CrossRef] [PubMed]
- Ramsahoye, B.H.; Biniszkiewicz, D.; Lyko, F.; Clark, V.; Bird, A.P.; Jaenisch, R. Non-CpG methylation is prevalent in embryonic stem cells and may be mediated by DNA methyltransferase 3a. Proc. Natl. Acad. Sci. USA 2000, 97, 5237–5242. [Google Scholar] [CrossRef] [PubMed]
- Venhoranta, H.; Li, S.; Salamon, S.; Flisikowska, T.; Andersson, M.; Switonski, M.; Kind, A.; Schnieke, A.; Flisikowski, K. Non-CpG hypermethylation in placenta of mutation-induced intrauterine growth restricted bovine foetuses. Biochem. Biophys. Res. Commun. 2014, 444, 391–394. [Google Scholar] [CrossRef] [PubMed]
- Han, L.; Su, B.; Li, W.H.; Zhao, Z. Cpg island density and its correlations with genomic features in mammalian genomes. Genome. Biol. 2008, 9, R79. [Google Scholar] [CrossRef] [PubMed]
- Rakyan, V.K.; Down, T.A.; Balding, D.J.; Beck, S. Epigenome-wide association studies for common human diseases. Nat. Rev. Genet. 2011, 12, 529–541. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Su, S.; Barnes, V.A.; de Miguel, C.; Pollock, J.; Ownby, D.; Shi, H.; Zhu, H.; Snieder, H.; Wang, X. A genome-wide methylation study on obesity: Differential variability and differential methylation. Epigenetics 2013, 8, 522–533. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.W.; Pausova, Z. Cigarette smoking and DNA methylation. Front. Genet. 2013, 4, 132. [Google Scholar] [CrossRef] [PubMed]
- Fatemi, M.; Pao, M.M.; Jeong, S.; Gal-Yam, E.N.; Egger, G.; Weisenberger, D.J.; Jones, P.A. Footprinting of mammalian promoters: Use of a CpG DNA methyltransferase revealing nucleosome positions at a single molecule level. Nucleic Acids Res. 2005, 33, e176. [Google Scholar] [CrossRef] [PubMed]
- Sabounchi, S.; Bollyky, J.; Nadeau, K. Review of environmental impact on the epigenetic regulation of atopic diseases. Curr. Allergy Asthma Rep. 2015, 15, 33. [Google Scholar] [CrossRef] [PubMed]
- Bird, A. DNA methylation patterns and epigenetic memory. Genes Dev. 2002, 16, 6–21. [Google Scholar] [CrossRef] [PubMed]
- Graff, J.; Kim, D.; Dobbin, M.M.; Tsai, L.H. Epigenetic regulation of gene expression in physiological and pathological brain processes. Physiol. Rev. 2011, 91, 603–649. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, M.; Daujat, S.; Schneider, R. Lateral thinking: How histone modifications regulate gene expression. Trends Genet. 2016, 32, 42–56. [Google Scholar] [CrossRef] [PubMed]
- Zou, C.; Mallampalli, R.K. Regulation of histone modifying enzymes by the ubiquitin-proteasome system. Biochim. Biophys. Acta 2014, 1843, 694–702. [Google Scholar] [CrossRef] [PubMed]
- Tessarz, P.; Kouzarides, T. Histone core modifications regulating nucleosome structure and dynamics. Nat. Rev. Mol. Cell Biol. 2014, 15, 703–708. [Google Scholar] [CrossRef] [PubMed]
- Verdin, E.; Ott, M. 50 years of protein acetylation: From gene regulation to epigenetics, metabolism and beyond. Nat. Rev. Mol. Cell Biol. 2015, 16, 258–264. [Google Scholar] [CrossRef] [PubMed]
- Greer, E.L.; Shi, Y. Histone methylation: A dynamic mark in health, disease and inheritance. Nat. Rev. Genet. 2012, 13, 343–357. [Google Scholar] [CrossRef] [PubMed]
- Tan, M.; Luo, H.; Lee, S.; Jin, F.; Yang, J.S.; Montellier, E.; Buchou, T.; Cheng, Z.; Rousseaux, S.; Rajagopal, N.; et al. Identification of 67 histone marks and histone lysine crotonylation as a new type of histone modification. Cell 2011, 146, 1016–1028. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Carey, M.; Workman, J.L. The role of chromatin during transcription. Cell 2007, 128, 707–719. [Google Scholar] [CrossRef] [PubMed]
- Shilatifard, A. Chromatin modifications by methylation and ubiquitination: Implications in the regulation of gene expression. Annu. Rev. Biochem. 2006, 75, 243–269. [Google Scholar] [CrossRef] [PubMed]
- Weake, V.M.; Workman, J.L. Histone ubiquitination: Triggering gene activity. Mol. Cell 2008, 29, 653–663. [Google Scholar] [CrossRef] [PubMed]
- Gill, G. Sumo and ubiquitin in the nucleus: Different functions, similar mechanisms? Genes Dev. 2004, 18, 2046–2059. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.W.; Zhang, J.; Heine, G.F.; Arora, M.; Gulcin Ozer, H.; Onti-Srinivasan, R.; Huang, K.; Parvin, J.D. Chromatin modification by sumo-1 stimulates the promoters of translation machinery genes. Nucleic Acids Res. 2012, 40, 10172–10186. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Killian, J.K.; Yang, M.; Walker, R.L.; Hong, J.A.; Zhang, M.; Davis, S.; Zhang, Y.; Hussain, M.; Xi, S.; et al. Epigenomic alterations and gene expression profiles in respiratory epithelia exposed to cigarette smoke condensate. Oncogene 2010, 29, 3650–3664. [Google Scholar] [CrossRef] [PubMed]
- Izzotti, A.; Pulliero, A. Molecular damage and lung tumors in cigarette smoke-exposed mice. Ann. N. Y. Acad. Sci. 2015, 1340, 75–83. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.J.; Chang, L.W.; Lin, P.; Wang, Y.J. Epigenetic effects and molecular mechanisms of tumorigenesis induced by cigarette smoke: An overview. J. Oncol. 2011, 2011, 654931. [Google Scholar] [CrossRef] [PubMed]
- Moritsugu, K.P. The 2006 report of the surgeon general: The health consequences of involuntary exposure to tobacco smoke. Am. J. Prev. Med. 2007, 32, 542–543. [Google Scholar] [CrossRef] [PubMed]
- Peterson, L.A. Context matters: Contribution of specific DNA adducts to the genotoxic properties of the tobacco-specific nitrosamine NNK. Chem. Res. Toxicol. 2017, 30, 420–433. [Google Scholar] [CrossRef] [PubMed]
- Leanderson, P.; Tagesson, C. Cigarette smoke-induced DNA damage in cultured human lung cells: Role of hydroxyl radicals and endonuclease activation. Chem. Biol. Interact. 1992, 81, 197–208. [Google Scholar] [CrossRef]
- Risch, A.; Plass, C. Lung cancer epigenetics and genetics. Int. J. Cancer 2008, 123, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Vineis, P.; Caporaso, N. Tobacco and cancer: Epidemiology and the laboratory. Environ. Health Perspect. 1995, 103, 156–160. [Google Scholar] [CrossRef] [PubMed]
- Mukhopadhyay, P.; Greene, R.M.; Pisano, M.M. Cigarette smoke induces proteasomal-mediated degradation of DNA methyltransferases and methyl CpG-/CpG domain-binding proteins in embryonic orofacial cells. Reprod. Toxicol. 2015, 58, 140–148. [Google Scholar] [CrossRef] [PubMed]
- Pace, E.; di Vincenzo, S.; Ferraro, M.; Siena, L.; Chiappara, G.; Dino, P.; Vitulo, P.; Bertani, A.; Saibene, F.; Lanata, L.; et al. Effects of carbocysteine and beclomethasone on histone acetylation/deacetylation processes in cigarette smoke exposed bronchial epithelial cells. J. Cell. Physiol. 2016. [Google Scholar] [CrossRef]
- Sundar, I.K.; Rahman, I. Gene expression profiling of epigenetic chromatin modification enzymes and histone marks by cigarette smoke: Implications for COPD and lung cancer. Am. J. Physiol. Lung Cell. Mol. Physiol. 2016, 311, L1245–L1258. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Deng, J.; Wang, S.S.; Ma, L.; Pei, J.; Zeng, X.X.; Tang, J.X. Association of methylation of the RAR-β gene with cigarette smoking in non-small cell lung cancer with southern-central Chinese population. Asian Pac. J. Cancer Prev. 2014, 15, 10937–10941. [Google Scholar] [CrossRef] [PubMed]
- Freeman, J.R.; Chu, S.; Hsu, T.; Huang, Y.T. Epigenome-wide association study of smoking and DNA methylation in non-small cell lung neoplasms. Oncotarget 2016, 7, 69579–69591. [Google Scholar] [CrossRef] [PubMed]
- Philibert, R.; Hollenbeck, N.; Andersen, E.; McElroy, S.; Wilson, S.; Vercande, K.; Beach, S.R.; Osborn, T.; Gerrard, M.; Gibbons, F.X.; et al. Reversion of AHRR demethylation is a quantitative biomarker of smoking cessation. Front. Psychiatry 2016, 7, 55. [Google Scholar] [CrossRef] [PubMed]
- Marsit, C.J.; Karagas, M.R.; Schned, A.; Kelsey, K.T. Carcinogen exposure and epigenetic silencing in bladder cancer. Ann. N. Y. Acad. Sci. 2006, 1076, 810–821. [Google Scholar] [CrossRef] [PubMed]
- Marsit, C.J.; Karagas, M.R.; Danaee, H.; Liu, M.; Andrew, A.; Schned, A.; Nelson, H.H.; Kelsey, K.T. Carcinogen exposure and gene promoter hypermethylation in bladder cancer. Carcinogenesis 2006, 27, 112–116. [Google Scholar] [CrossRef] [PubMed]
- Shui, I.M.; Wong, C.J.; Zhao, S.; Kolb, S.; Ebot, E.M.; Geybels, M.S.; Rubicz, R.; Wright, J.L.; Lin, D.W.; Klotzle, B.; et al. Prostate tumor DNA methylation is associated with cigarette smoking and adverse prostate cancer outcomes. Cancer 2016, 122, 2168–2177. [Google Scholar] [CrossRef] [PubMed]
- Brait, M.; Munari, E.; LeBron, C.; Noordhuis, M.G.; Begum, S.; Michailidi, C.; Gonzalez-Roibon, N.; Maldonado, L.; Sen, T.; Guerrero-Preston, R.; et al. Genome-wide methylation profiling and the PI3K-AKT pathway analysis associated with smoking in urothelial cell carcinoma. Cell Cycle 2013, 12, 1058–1070. [Google Scholar] [CrossRef] [PubMed]
- Marsit, C.J.; Houseman, E.A.; Schned, A.R.; Karagas, M.R.; Kelsey, K.T. Promoter hypermethylation is associated with current smoking, age, gender and survival in bladder cancer. Carcinogenesis 2007, 28, 1745–1751. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Cui, S.; Ma, J.; Lu, Q.; Kong, C.; Liu, T.; Sun, Z. Cigarette smoking extract causes hypermethylation and inactivation of WWOX gene in T-24 human bladder cancer cells. Neoplasma 2012, 59, 216–223. [Google Scholar] [CrossRef] [PubMed]
- Pulling, L.C.; Vuillemenot, B.R.; Hutt, J.A.; Devereux, T.R.; Belinsky, S.A. Aberrant promoter hypermethylation of the death-associated protein kinase gene is early and frequent in murine lung tumors induced by cigarette smoke and tobacco carcinogens. Cancer Res. 2004, 64, 3844–3848. [Google Scholar] [CrossRef] [PubMed]
- Beleford, D.; Liu, Z.; Rattan, R.; Quagliuolo, L.; Boccellino, M.; Baldi, A.; Maguire, J.; Staub, J.; Molina, J.; Shridhar, V. Methylation induced gene silencing of HtrA3 in smoking-related lung cancer. Clin. Cancer Res. 2010, 16, 398–409. [Google Scholar] [CrossRef] [PubMed]
- Cheng, G.; Li, J.; Zheng, M.; Zhao, Y.; Zhou, J.; Li, W. NNK, a tobacco-specific carcinogen, inhibits the expression of lysyl oxidase, a tumor suppressor. Int. J. Environ. Res. Public Health 2014, 12, 64–82. [Google Scholar] [CrossRef] [PubMed]
- Knopik, V.S.; Maccani, M.A.; Francazio, S.; McGeary, J.E. The epigenetics of maternal cigarette smoking during pregnancy and effects on child development. Dev. Psychopathol. 2012, 24, 1377–1390. [Google Scholar] [CrossRef] [PubMed]
- Xi, S.; Xu, H.; Shan, J.; Tao, Y.; Hong, J.A.; Inchauste, S.; Zhang, M.; Kunst, T.F.; Mercedes, L.; Schrump, D.S. Cigarette smoke mediates epigenetic repression of miR-487b during pulmonary carcinogenesis. J. Clin. Investig. 2013, 123, 1241–1261. [Google Scholar] [CrossRef] [PubMed]
- Xi, S.; Inchauste, S.; Guo, H.; Shan, J.; Xiao, Z.; Xu, H.; Miettenen, M.; Zhang, M.R.; Hong, J.A.; Raiji, M.T.; et al. Cigarette smoke mediates epigenetic repression of miR-217 during esophageal adenocarcinogenesis. J. Clin. Investig. 2015, 34, 5548–5559. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Liu, Y.; Luo, F.; Xu, Y.; Qin, Y.; Lu, X.; Xu, W.; Shi, L.; Liu, Q.; Xiang, Q. Epigenetic silencing of microRNA-218 via EZH2-mediated H3K27 trimethylation is involved in malignant transformation of HBE cells induced by cigarette smoke extract. Arch. Toxicol. 2016, 90, 449–461. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Wang, B.; Liu, X.; Lu, L.; Luo, F.; Lu, X.; Shi, L.; Xu, W.; Liu, Q. Epigenetic silencing of p21 by long non-coding RNA hotair is involved in the cell cycle disorder induced by cigarette smoke extract. Toxicol. Lett. 2016, 240, 60–67. [Google Scholar] [CrossRef] [PubMed]
- Sundar, I.K.; Nevid, M.Z.; Friedman, A.E.; Rahman, I. Cigarette smoke induces distinct histone modifications in lung cells: Implications for the pathogenesis of COPD and lung cancer. J. Proteome Res. 2014, 13, 982–996. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Fang, L.; Li, H.; Tang, M.S.; Jin, C. Cigarette smoke component acrolein modulates chromatin assembly by inhibiting histone acetylation. J. Biol. Chem. 2013, 288, 21678–21687. [Google Scholar] [CrossRef] [PubMed]
- Ibuki, Y.; Toyooka, T.; Zhao, X.; Yoshida, I. Cigarette sidestream smoke induces histone H3 phosphorylation via JNK and PI3K/Akt pathways, leading to the expression of proto-oncogenes. Carcinogenesis 2014, 35, 1228–1237. [Google Scholar] [CrossRef] [PubMed]
- Dalton, A.M.; Jones, A.P.; Sharp, S.J.; Cooper, A.J.; Griffin, S.; Wareham, N.J. Residential neighbourhood greenspace is associated with reduced risk of incident diabetes in older people: A prospective cohort study. BMC Public Health 2016, 16, 1171. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Willett, W.C.; Rimm, E.B.; Stampfer, M.J.; Giovannucci, E.L. Light to moderate intake of alcohol, drinking patterns, and risk of cancer: Results from two prospective us cohort studies. BMJ 2015, 351, h4238. [Google Scholar] [CrossRef] [PubMed]
- Mahnke, A.H.; Miranda, R.C.; Homanics, G.E. Epigenetic mediators and consequences of excessive alcohol consumption. Alcohol 2017, 60, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Pandey, S.C.; Kyzar, E.J.; Zhang, H. Epigenetic basis of the dark side of alcohol addiction. Neuropharmacology 2017. [Google Scholar] [CrossRef] [PubMed]
- Bohacek, J.; Mansuy, I.M. Epigenetic inheritance of disease and disease risk. Neuropsychopharmacology 2013, 38, 220–236. [Google Scholar] [CrossRef] [PubMed]
- Finegersh, A.; Homanics, G.E. Acute ethanol alters multiple histone modifications at model gene promoters in the cerebral cortex. Alcohol. Clin. Exp. Res. 2014, 38, 1865–1873. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Kusumo, H.; Sakharkar, A.J.; Pandey, S.C.; Guizzetti, M. Regulation of DNA methylation by ethanol induces tissue plasminogen activator expression in astrocytes. J. Neurochem. 2014, 128, 344–349. [Google Scholar] [CrossRef] [PubMed]
- Kaur, J.; Demokan, S.; Tripathi, S.C.; Macha, M.A.; Begum, S.; Califano, J.A.; Ralhan, R. Promoter hypermethylation in Indian primary oral squamous cell carcinoma. Int. J. Cancer 2010, 127, 2367–2373. [Google Scholar] [CrossRef] [PubMed]
- Pattani, K.M.; Zhang, Z.; Demokan, S.; Glazer, C.; Loyo, M.; Goodman, S.; Sidransky, D.; Bermudez, F.; Jean-Charles, G.; McCaffrey, T.; et al. Endothelin receptor type B gene promoter hypermethylation in salivary rinses is independently associated with risk of oral cavity cancer and premalignancy. Cancer Prev. Res. 2010, 3, 1093–1103. [Google Scholar] [CrossRef] [PubMed]
- Arjungi, K.N. Areca nut: A review. Arzneimittelforschung 1976, 26, 951–956. [Google Scholar] [PubMed]
- Thomas, S.J.; MacLennan, R. Slaked lime and betel nut cancer in papua new guinea. Lancet 1992, 340, 577–578. [Google Scholar] [CrossRef]
- Ko, Y.C.; Huang, Y.L.; Lee, C.H.; Chen, M.J.; Lin, L.M.; Tsai, C.C. Betel quid chewing, cigarette smoking and alcohol consumption related to oral cancer in Taiwan. J. Oral Pathol. Med. 1995, 24, 450–453. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.H.; Lee, H.S.; Mar, K.; Ji, D.D.; Huang, M.S.; Hsia, K.T. Loss expression of O6-methylguanine DNA methyltransferase by promoter hypermethylation and its relationship to betel quid chewing in oral squamous cell carcinoma. Oral Surg. Oral Med. Oral Pathol. Oral Radiol. Endod 2010, 109, 883–889. [Google Scholar] [CrossRef] [PubMed]
- Rai, A.K.; Freddy, A.J.; Banerjee, A.; Kurkalang, S.; Rangad, G.M.; Islam, M.; Nongrum, H.B.; Dkhar, H.; Chatterjee, A. Distinct involvement of 9p21–24 and 13q14.1–14.3 chromosomal regions in raw betel-nut induced esophageal cancers in the state of meghalaya, India. Asian Pac. J. Cancer Prev. 2012, 13, 2629–2633. [Google Scholar] [CrossRef] [PubMed]
- Takeshima, M.; Saitoh, M.; Kusano, K.; Nagayasu, H.; Kurashige, Y.; Malsantha, M.; Arakawa, T.; Takuma, T.; Chiba, I.; Kaku, T.; et al. High frequency of hypermethylation of p14, p15 and p16 in oral pre-cancerous lesions associated with betel-quid chewing in Sri Lanka. J. Oral Pathol. Med. 2008, 37, 475–479. [Google Scholar] [CrossRef] [PubMed]
- Singh, V.; Singh, L.C.; Singh, A.P.; Sharma, J.; Borthakur, B.B.; Debnath, A.; Rai, A.K.; Phukan, R.K.; Mahanta, J.; Kataki, A.C.; et al. Status of epigenetic chromatin modification enzymes and esophageal squamous cell carcinoma risk in northeast Indian population. Am. J. Cancer Res. 2015, 5, 979–999. [Google Scholar] [PubMed]
- Chen, J.H.; Yeh, K.T.; Yang, Y.M.; Chang, J.G.; Lee, H.E.; Hung, S.Y. High expressions of histone methylation- and phosphorylation-related proteins are associated with prognosis of oral squamous cell carcinoma in male population of Taiwan. Med. Oncol. 2013, 30, 513. [Google Scholar] [CrossRef] [PubMed]
- Chang, H.H.; Chiang, C.P.; Hung, H.C.; Lin, C.Y.; Deng, Y.T.; Kuo, M.Y. Histone deacetylase 2 expression predicts poorer prognosis in oral cancer patients. Oral Oncol. 2009, 45, 610–614. [Google Scholar] [CrossRef] [PubMed]
- Prokopczyk, B.; Rivenson, A.; Bertinato, P.; Brunnemann, K.D.; Hoffmann, D. 3-(methylnitrosamino) propionitrile: Occurrence in saliva of betel quid chewers, carcinogenicity, and DNA methylation in f344 rats. Cancer Res. 1987, 47, 467–471. [Google Scholar] [PubMed]
- Lin, K.H.; Lin, C.Y.; Liu, C.C.; Chou, M.Y.; Lin, J.K. Arecoline n-oxide: Its mutagenicity and possible role as ultimate carcinogen in areca oral carcinogenesis. J. Agric. Food Chem. 2011, 59, 3420–3428. [Google Scholar] [CrossRef] [PubMed]
- Tsai, Y.S.; Lee, K.W.; Huang, J.L.; Liu, Y.S.; Juo, S.H.; Kuo, W.R.; Chang, J.G.; Lin, C.S.; Jong, Y.J. Arecoline, a major alkaloid of areca nut, inhibits p53, represses DNA repair, and triggers DNA damage response in human epithelial cells. Toxicology 2008, 249, 230–237. [Google Scholar] [CrossRef] [PubMed]
- Jeng, J.H.; Chang, M.C.; Hahn, L.J. Role of areca nut in betel quid-associated chemical carcinogenesis: Current awareness and future perspectives. Oral Oncol. 2001, 37, 477–492. [Google Scholar] [CrossRef]
- Lin, P.C.; Chang, W.H.; Chen, Y.H.; Lee, C.C.; Lin, Y.H.; Chang, J.G. Cytotoxic effects produced by arecoline correlated to epigenetic regulation in human K-562 cells. J. Toxicol. Environ. Health 2011, 74, 737–745. [Google Scholar] [CrossRef] [PubMed]
- Lai, Z.L.; Tsou, Y.A.; Fan, S.R.; Tsai, M.H.; Chen, H.L.; Chang, N.W.; Cheng, J.C.; Chen, C.M. Methylation-associated gene silencing of RARβ in areca carcinogens induced mouse oral squamous cell carcinoma. BioMed Res. Int. 2014, 2014, 378358. [Google Scholar] [CrossRef] [PubMed]
- Qi, Y.; Wang, D.; Jin, T.; Yang, L.; Wu, H.; Li, Y.; Zhao, J.; Du, F.; Song, M.; Wang, R. Hedd: The human epigenetic drug database. Database J. Biol. Databases Curation 2016, 2016, baw159. [Google Scholar] [CrossRef] [PubMed]
- Figueroa, M.E.; Skrabanek, L.; Li, Y.; Jiemjit, A.; Fandy, T.E.; Paietta, E.; Fernandez, H.; Tallman, M.S.; Greally, J.M.; Carraway, H.; et al. Mds and secondary aml display unique patterns and abundance of aberrant DNA methylation. Blood 2009, 114, 3448–3458. [Google Scholar] [CrossRef] [PubMed]
- Minkovsky, A.; Sahakyan, A.; Bonora, G.; Damoiseaux, R.; Dimitrova, E.; Rubbi, L.; Pellegrini, M.; Radu, C.G.; Plath, K. A high-throughput screen of inactive X chromosome reactivation identifies the enhancement of DNA demethylation by 5-Aza-2′-dC upon inhibition of ribonucleotide reductase. Epigenet. Chromatin 2015, 8, 42. [Google Scholar] [CrossRef] [PubMed]
- Sun, F.; Chan, E.; Wu, Z.; Yang, X.; Marquez, V.E.; Yu, Q. Combinatorial pharmacologic approaches target EZH2-mediated gene repression in breast cancer cells. Mol. Cancer Ther. 2009, 8, 3191–3202. [Google Scholar] [CrossRef] [PubMed]
- Miranda, T.B.; Cortez, C.C.; Yoo, C.B.; Liang, G.; Abe, M.; Kelly, T.K.; Marquez, V.E.; Jones, P.A. Dznep is a global histone methylation inhibitor that reactivates developmental genes not silenced by DNA methylation. Mol. Cancer Ther. 2009, 8, 1579–1588. [Google Scholar] [CrossRef] [PubMed]
- Hascher, A.; Haase, A.K.; Hebestreit, K.; Rohde, C.; Klein, H.U.; Rius, M.; Jungen, D.; Witten, A.; Stoll, M.; Schulze, I.; et al. DNA methyltransferase inhibition reverses epigenetically embedded phenotypes in lung cancer preferentially affecting polycomb target genes. Clin. Cancer Res. 2014, 20, 814–826. [Google Scholar] [CrossRef] [PubMed]
- Heller, G.; Weinzierl, M.; Noll, C.; Babinsky, V.; Ziegler, B.; Altenberger, C.; Minichsdorfer, C.; Lang, G.; Dome, B.; End-Pfutzenreuter, A.; et al. Genome-wide miRNA expression profiling identifies miR-9–3 and miR-193a as targets for DNA methylation in non-small cell lung cancers. Clin. Cancer Res. 2012, 18, 1619–1629. [Google Scholar] [CrossRef] [PubMed]
- Hudson, R.S.; Yi, M.; Esposito, D.; Watkins, S.K.; Hurwitz, A.A.; Yfantis, H.G.; Lee, D.H.; Borin, J.F.; Naslund, M.J.; Alexander, R.B.; et al. Microrna-1 is a candidate tumor suppressor and prognostic marker in human prostate cancer. Nucleic Acids Res. 2012, 40, 3689–3703. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.R.; Bryan, J.N.; Esebua, M.; Amos-Landgraf, J.; May, T.J. Testis specific y-like 5: Gene expression, methylation and implications for drug sensitivity in prostate carcinoma. BMC Cancer 2017, 17, 158. [Google Scholar] [CrossRef] [PubMed]
- Duenas-Gonzalez, A.; Coronel, J.; Cetina, L.; Gonzalez-Fierro, A.; Chavez-Blanco, A.; Taja-Chayeb, L. Hydralazine-valproate: A repositioned drug combination for the epigenetic therapy of cancer. Expert Opin. Drug Metab. Toxicol. 2014, 10, 1433–1444. [Google Scholar] [CrossRef] [PubMed]
- Hilakivi-Clarke, L.; Warri, A.; Bouker, K.B.; Zhang, X.; Cook, K.L.; Jin, L.; Zwart, A.; Nguyen, N.; Hu, R.; Cruz, M.I.; et al. Effects of in utero exposure to ethinyl estradiol on tamoxifen resistance and breast cancer recurrence in a preclinical model. J. Natl. Cancer Inst. 2017, 109. [Google Scholar] [CrossRef] [PubMed]
- Pinkerneil, M.; Hoffmann, M.J.; Deenen, R.; Kohrer, K.; Arent, T.; Schulz, W.A.; Niegisch, G. Inhibition of class I histone deacetylases 1 and 2 promotes urothelial carcinoma cell death by various mechanisms. Mol. Cancer Ther. 2016, 15, 299–312. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Salvador, L.A.; Byeon, S.; Ying, Y.; Kwan, J.C.; Law, B.K.; Hong, J.; Luesch, H. Anticolon cancer activity of largazole, a marine-derived tunable histone deacetylase inhibitor. J. Pharmacol. Exp. Ther. 2010, 335, 351–361. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, Y.; Yamauchi, T.; Hosono, N.; Uzui, K.; Negoro, E.; Morinaga, K.; Nishi, R.; Yoshida, A.; Kimura, S.; Maekawa, T.; et al. Combination of panobinostat with ponatinib synergistically overcomes imatinib-resistant cml cells. Cancer Sci. 2016, 107, 1029–1038. [Google Scholar] [CrossRef] [PubMed]
- Van Maldegem, A.M.; Bovee, J.V.; Gelderblom, H. Panobinostat-a potential treatment for metastasized ewing sarcoma? A case report. Pediatr. Blood Cancer 2016, 63, 1840–1843. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Jia, Y.; Liu, X.; Winters, C.; Wang, X.; Zhang, Y.; Devor, E.J.; Hovey, A.M.; Reyes, H.D.; Xiao, X.; et al. Systematic dissection of the mechanisms underlying progesterone receptor downregulation in endometrial cancer. Oncotarget 2014, 5, 9783–9797. [Google Scholar] [CrossRef] [PubMed]
- Welsbie, D.S.; Xu, J.; Chen, Y.; Borsu, L.; Scher, H.I.; Rosen, N.; Sawyers, C.L. Histone deacetylases are required for androgen receptor function in hormone-sensitive and castrate-resistant prostate cancer. Cancer Res. 2009, 69, 958–966. [Google Scholar] [CrossRef] [PubMed]
- Kong, L.R.; Tan, T.Z.; Ong, W.R.; Bi, C.; Huynh, H.; Lee, S.C.; Chng, W.J.; Eichhorn, P.J.; Goh, B.C. Belinostat exerts anti-tumor cytotoxicity through the ubiquitin-proteasome pathway in lung squamous cell carcinoma. Mol. Oncol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Feng, R.; Oton, A.; Mapara, M.Y.; Anderson, G.; Belani, C.; Lentzsch, S. The histone deacetylase inhibitor, pxd101, potentiates bortezomib-induced anti-multiple myeloma effect by induction of oxidative stress and DNA damage. Br. J. Haematol. 2007, 139, 385–397. [Google Scholar] [CrossRef] [PubMed]
- Han, H.; Yang, X.; Pandiyan, K.; Liang, G. Synergistic re-activation of epigenetically silenced genes by combinatorial inhibition of dnmts and lsd1 in cancer cells. PLoS ONE 2013, 8, e75136. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Flamand, V.; Peehl, D.M. Anti-oncogenic and pro-differentiation effects of clorgyline, a monoamine oxidase a inhibitor, on high grade prostate cancer cells. BMC Med. Genom. 2009, 2, 55. [Google Scholar] [CrossRef] [PubMed]
- Valiuliene, G.; Stirblyte, I.; Jasnauskaite, M.; Borutinskaite, V.; Navakauskiene, R. Anti-leukemic effects of hdaci belinostat and hmti 3-deazaneplanocin a on human acute promyelocytic leukemia cells. Eur. J. Pharmacol. 2017, 799, 143–153. [Google Scholar] [CrossRef] [PubMed]
- Arif, M.; Vedamurthy, B.M.; Choudhari, R.; Ostwal, Y.B.; Mantelingu, K.; Kodaganur, G.S.; Kundu, T.K. Nitric oxide-mediated histone hyperacetylation in oral cancer: Target for a water-soluble HAT inhibitor, CTK7A. Chem. Biol. 2010, 17, 903–913. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.; Kim, S.H.; Datta, I.; Levin, A.; Dyson, G.; Li, J.; Kaypee, S.; Swamy, M.M.; Gupta, N.; Kwon, H.J.; et al. Hydrazinobenzoylcurcumin inhibits androgen receptor activity and growth of castration-resistant prostate cancer in mice. Oncotarget 2015, 6, 6136–6150. [Google Scholar] [CrossRef] [PubMed]
- Rath, S.; Das, L.; Kokate, S.B.; Ghosh, N.; Dixit, P.; Rout, N.; Singh, S.P.; Chattopadhyay, S.; Ashktorab, H.; Smoot, D.T.; et al. Inhibition of histone/lysine acetyltransferase activity kills CoCL2-treated and hypoxia-exposed gastric cancer cells and reduces their invasiveness. Int. J. Biochem. Cell Biol. 2017, 82, 28–40. [Google Scholar] [CrossRef] [PubMed]
- Venturelli, S.; Berger, A.; Bocker, A.; Busch, C.; Weiland, T.; Noor, S.; Leischner, C.; Schleicher, S.; Mayer, M.; Weiss, T.S.; et al. Resveratrol as a pan-HDAC inhibitor alters the acetylation status of histone [corrected] proteins in human-derived hepatoblastoma cells. PLoS ONE 2013, 8, e73097. [Google Scholar] [CrossRef]
- Hassan, W.A.; Takebayashi, S.I.; Abdalla, M.O.A.; Fujino, K.; Kudoh, S.; Motooka, Y.; Sato, Y.; Naito, Y.; Higaki, K.; Wakimoto, J.; et al. Correlation between histone acetylation and expression of notch1 in human lung carcinoma and its possible role in combined small-cell lung carcinoma. Lab. Investig. 2017. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.F.; Yan, Q.; Shen, W.; Gurunathan, S. Trichostatin a enhances the apoptotic potential of palladium nanoparticles in human cervical cancer cells. Int. J. Mol. Sci. 2016, 17, 1354. [Google Scholar] [CrossRef] [PubMed]
- Boyanapalli, S.S.; Li, W.; Fuentes, F.; Guo, Y.; Ramirez, C.N.; Gonzalez, X.P.; Pung, D.; Kong, A.N. Epigenetic reactivation of rassf1a by phenethyl isothiocyanate (peitc) and promotion of apoptosis in lncap cells. Pharmacol. Res. 2016, 114, 175–184. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Jin, T.; Zhu, K.; Piao, Y.; Quan, T.; Quan, C.; Lin, Z. Pi3k/mtor dual inhibitor bez235 and histone deacetylase inhibitor trichostatin a synergistically exert anti-tumor activity in breast cancer. Oncotarget 2017, 8, 11937–11949. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
| Category | Synonyms | Cas Number | Disease | FDA Approval Number |
|---|---|---|---|---|
| DNMTi | 5-Azacytidine | 320-67-2 | Myelodysplastic syndromes | 50794 |
| 5-Aza-2′-deoxycytidine | 2353-33-5 | Myelodysplastic syndromes | 21790 | |
| Hydralazine | 86-54-4 | Hypertension | 8303 | |
| HDACi | Suberoylanilide hydroxamic acid | 149647-78-9 | Cutaneous T-cell lymphoma | 21991 |
| Panobinostat | 404950-80-7 | Multiple myeloma | 205353 | |
| Belinostat | 414864-00-9 | Peripheral T-cell lymphoma | 206256 | |
| Romidepsin | 128517-07-7 | Cutaneous T-cell lymphoma | 22393 |
| Risk Factor | Epigenetic Aberrations | Cancer Types | References |
|---|---|---|---|
| Smoke | DNA methylation | Lung cancer | [55,56,57,64,65,66] [68,69,70] |
| Bladder cancers | [58,59] | ||
| Prostate cancer | [60,62,63] | ||
| Histone modification | Lung cancer | [72,73,74] | |
| Alcohol | DNA methylation | Liver cancer | [77] |
| Oral cancer | [82] | ||
| Histone modification | Oral cancer | [77] | |
| Betel nut | DNA methylation | Esophageal cancers | [88] |
| Oral pre-cancerous lesions | [89] | ||
| Histone modification | Esophageal squamous cell carcinoma | [90] | |
| Oral squamous cell carcinoma | [91] | ||
| Oral cancer | [92] |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, T.-H.; Hsia, S.-M.; Shih, Y.-H.; Shieh, T.-M. Association of Smoking, Alcohol Use, and Betel Quid Chewing with Epigenetic Aberrations in Cancers. Int. J. Mol. Sci. 2017, 18, 1210. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms18061210
Wang T-H, Hsia S-M, Shih Y-H, Shieh T-M. Association of Smoking, Alcohol Use, and Betel Quid Chewing with Epigenetic Aberrations in Cancers. International Journal of Molecular Sciences. 2017; 18(6):1210. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms18061210
Chicago/Turabian StyleWang, Tong-Hong, Shih-Min Hsia, Yin-Hwa Shih, and Tzong-Ming Shieh. 2017. "Association of Smoking, Alcohol Use, and Betel Quid Chewing with Epigenetic Aberrations in Cancers" International Journal of Molecular Sciences 18, no. 6: 1210. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms18061210