
Cutaneous Manifestations of Human and Murine Leishmaniasis

1
Interdisciplinary Graduate Program in Immunology, University of Iowa, Iowa City, IA 52242, USA
2
Instituto de Pesquisa Gonçalo Moniz, Fundação Oswaldo Cruz (FIOCRUZ), Salvador, Bahia, 40296-710, Brazil
3
Federal University of Bahia, Hospital Universitário Prof. Edgard Santos, Salvador, BA, Brazil
4
Department of Internal Medicine, University of Iowa, Iowa City, IA 52242, USA
5
Department of Microbiology, University of Iowa, Iowa City, IA 52242, USA
6
Veterans’ Affairs Medical Center, Iowa City, IA 52242, USA
*
Author to whom correspondence should be addressed.
Int. J. Mol. Sci. 2017, 18(6), 1296; https://0-doi-org.brum.beds.ac.uk/10.3390/ijms18061296
Submission received: 28 January 2017
/
Revised: 23 May 2017
/
Accepted: 14 June 2017
/
Published: 18 June 2017
(This article belongs to the Special Issue Inflammatory Skin Conditions 2017)
Abstract
:The leishmaniases are diseases caused by pathogenic protozoan parasites of the genus Leishmania. Infections are initiated when a sand fly vector inoculates Leishmania parasites into the skin of a mammalian host. Leishmania causes a spectrum of inflammatory cutaneous disease manifestations. The type of cutaneous pathology is determined in part by the infecting Leishmania species, but also by a combination of inflammatory and anti-inflammatory host immune response factors resulting in different clinical outcomes. This review discusses the distinct cutaneous syndromes described in humans, and current knowledge of the inflammatory responses associated with divergent cutaneous pathologic responses to different Leishmania species. The contribution of key hematopoietic cells in experimental cutaneous leishmaniasis in mouse models are also reviewed and compared with those observed during human infection. We hypothesize that local skin events influence the ensuing adaptive immune response to Leishmania spp. infections, and that the balance between inflammatory and regulatory factors induced by infection are critical for determining cutaneous pathology and outcome of infection.
1. Introduction
Leishmaniasis is the group of diseases initiated through the bite of a female phlebotomine sand fly vector. Leishmaniasis is endemic in over 98 countries, and constitutes a widespread disease burden affecting many regions of the world [1]. With the exception of reported cases passed vertically from mother to child or, in dogs in the USA, from female dog to pups [2], Leishmania spp. protozoa are inoculated into mammalian skin during a sand fly blood meal at the onset of infection. Although most Leishmania spp. are introduced through the skin, the clinical syndrome caused by these protozoa can be widespread, influenced most heavily by the identity of the infecting species. Most fatalities are due to visceral leishmaniasis, in which Leishmania spp. parasites disseminate from the site of skin infection to visceral organs where they can cause pathology [3].
The most common form of leishmaniasis is localized cutaneous leishmaniasis (LCL), one type within a larger category of tegumentary leishmaniasis. During uncomplicated LCL, parasites remain localized in skin tissue and lead to chronic slowly healing skin ulcers. The lesions of LCL are painless and can self-heal without treatment. However, disease resolution can take several months and leave disfiguring scars [3]. In 2013, the World Health Organization (WHO) reported an estimated one million cases of LCL within the previous five years [1]. These estimates are likely vast underestimates [4]. Furthermore, the incidence is subject to fluctuation due to localized factors including population displacement events [5]. Soldiers and tourists traveling to endemic areas are at risk for acquiring leishmaniasis and major outbreaks have occurred in the US military [6].
A range of cutaneous manifestations occurs due to Leishmania infection. The species of parasite is the most important determinant of clinical outcome. Species of Leishmania that typically cause the different clinical forms of tegumentary leishmaniasis in humans are listed in Table 1.
There is considerable variety in disease manifestations beyond merely the species identity. Even within a species it has been difficult to discern the determinants that lead to different disease manifestations. Identification of parasite genetic determinants affecting phenotype has been hampered by the high degree of homology between Leishmania species. Genome sequences from multiple strains within species causing different forms of the disease have shown only a few differences [31]. Furthermore, gene synteny was conserved among 99% of genes and a high degree of amino acid conservation within coding regions (77–92%) was measured during a comparative analysis between three Leishmania species that cause divergent disease [31]. Most of the genomic variations between Leishmania species have been due to differences in gene copy number and chromosomal duplication events. Beyond that, post-transcriptional modifications have been identified between species [32,33].
In addition to the parasite, host immune response plays a key role in the clinical presentation of leishmaniasis. For instance, subjects with evidence of cell-mediated immunity against Leishmania antigen, when infected with L. (L.) amazonensis most often develop LCL. In contrast, patients with impairment of the T cell response often develop an anergic syndrome termed diffuse cutaneous leishmaniasis characterized by multiple nodular lesions full of parasites. Currently, it is not well defined what drives these differences in clinical outcome. It is known that parasite strain, genetically or environmentally determined host immune response, and vector components all contribute [3,34,35,36]. Further investigation is needed to define the contributions of host genetics and microbiota to the cutaneous manifestations of leishmaniasis [37,38,39,40].
Despite their different clinical manifestations, all forms of vector-borne leishmaniasis start with a similar series of events in host skin. These include the initial interactions among Leishmania promastigotes, vector components, and host skin cells, all serving to influence the innate immune activation cascade. It has become evident that the sand fly salivary components inoculated with the parasite have potent immunomodulatory properties, some of which are being exploited in vaccine development [34]. Furthermore, the parasite itself forms a gelatinous plug in the sand fly containing filamentous proteophosphoglycan, which has been called promastigote secretory gel (PSG). PSG is also included in the sand fly inoculum, and modulates local immune responses in the skin [41,42].
Following this, Leishmania parasites ultimately establish intracellular infection among macrophages, the primary Leishmania host cell housing a majority of parasites throughout chronic infection. These local events ultimately lead to presentation of Leishmania antigen to T lymphocytes to initiate a T cell-mediated immune response. Research in animal models has pointed towards a role for early tissue signals surrounding these events in polarization of the adaptive immune response, a response that guides the course of ensuing disease. A similar role of early events in human leishmaniasis remains difficult to discern [43,44,45,46,47]. Herein, we review the different types of tegumentary leishmaniasis during infection with different Leishmania species, and examine how host cutaneous responses contribute to or protect from disease.
2. Nomenclature
The terminologies that describe different clinical forms of leishmaniasis involving the skin can be confusing. Because “cutaneous leishmaniasis” (often abbreviated “CL”) often refers to a specific localized entity described below as Localized Cutaneous Leishmaniasis, “tegumentary leishmaniasis” will be used as an overall term for all forms of leishmaniasis skin involvement. Disseminated forms of tegumentary leishmaniasis called “disseminated cutaneous leishmaniasis” and “diffuse cutaneous leishmaniasis” are very different, but both have been abbreviated as DCL in the literature. Herein we will use the newer term “disseminated leishmaniasis” for disseminated cutaneous leishmaniasis, abbreviated DL. Diffuse cutaneous leishmaniasis is an anergic form sometimes called “anergic diffuse cutaneous leishmaniasis” or ADCL. Herein we will refer to this entity as ADCL, though diffuse cutaneous leishmaniasis is most often abbreviated DCL in the literature. Another disseminated form of leishmaniasis that manifests in mucus membranes is often called mucocutaneous leishmaniasis (abbreviated MCL) because it follows cutaneous lesions. The disease is also called “mucosal leishmaniasis” abbreviated ML, as the simultaneous occurrence of cutaneous and mucosal leishmaniasis is rare and because mucosal disease may develop in the absence of concomitant or previous LCL [48]. ML is the appropriate nomenclature for mucosal disease. Just as CL and LCL refer to the same clinical entity, MCL and ML are also interchangeable terms. Additionally, throughout the text (L.) denotes Leishmania species within the Leishmania subgenus and (V.) denotes Leishmania species within the Viannia subgenus.
3. Localized Cutaneous Leishmaniasis
LCL is the most prevalent clinical manifestation of leishmaniasis. It is generally non-life threatening but can be socially stigmatizing. LCL can result from infection with any one of a number of Leishmania species (Table 1). LCL symptoms develop an estimated 2–8 weeks after the bite of an infected sand fly, depending on the infecting species [49,50,51]. After the asymptomatic incubation period, a single or small number of nodular or papular lesions develop at the site of parasite deposition by the sand fly. Lymphadenopathy can develop as the earliest indication of disease caused by some species (notably L. (V.) braziliensis), and can precede lesion formation [52]. The lesions may progress into well-delimited ulcers with raised edges [3]. LCL can present with open lesions (wet lesions), which are subject to superficial secondary infection by bacterial or fungal species, with Staphylococcal species being most common [53]. For reasons that are still under debate, lesion ulceration has been associated lesion healing [54]. Indeed, patients infected with L. (V.) braziliensis who were treated before ulceration had a higher treatment fail rate compared to those treated after ulceration (75% vs. 25.8%) [54]. Lesions are capable of self-healing without treatment in many cases. However, resolution can take several months to years, and results in formation of a depressed, hypopigmented scar. Lesion resolution does not correspond to a sterile cure, as parasites or parasite DNA can be found in the scars of healed patients, years after successful treatment with clinical cure [55,56]. This propensity of parasites to persist in healthy tissue could have implications for many of the other forms of cutaneous leishmaniasis discussed later in this review.
Control of Leishmania replication is associated with development of Leishmania-specific T lymphocytes producing tumor necrosis factor (TNF) and interferon gamma (IFNγ). IFNγ promotes parasite control by activating infected macrophages, the primary host cell, to produce microbicidal effectors that can kill intracellular parasites [57]. Factors antagonizing the effects of these cytokines, such as interleukin (IL)-10 and transforming growth factor beta (TGFβ), are associated with lack of parasite control [57,58,59]. Active LCL in humans has been associated with a vigorous immune response, which can range from a predominantly type 1 immune response to a mixed type 1 and type 2 response [12,60,61]. Transcripts encoding innate mediators in lesional skin include IL-1β, IL-8, monocyte chemoattractant protein 1 (MCP1), and inducible nitric oxide synthase (iNOS) [8]. The T cell chemoattractants, chemokine (C-X-C motif) ligand 9 (CXCL9) and CXCL10, were the two most highly up regulated transcripts seen in LCL lesions compared to healthy skin in one study [62]. A T helper (TH)17-type signature was not observed in LCL lesions in this study, an observation relevant to the localized form of disease [63,64]. Activity of arginase, an enzyme expressed in both non-classically activated macrophages and in neutrophils [65], is also significantly up regulated in LCL lesions compared with healthy human skin [65,66]. After encountering parasites, monocyte-derived macrophages (MDMs) derived from LCL patients increase expression of CXCL8, CCL2, and CXCL9 in vitro, chemokines that recruit neutrophils, monocytes, and activated T cells, respectively; all of which may contribute to lesion formation [67]. Despite increased chemokine production and higher production of superoxide anion than cells from healthy subjects, MDMs from LCL subjects did not differ in their ability to control intracellular L. (V.) braziliensis infection, compared with healthy subject monocytes [67].
Subclinical infection is common in LCL and other forms of leishmaniasis, and can be detected by a lack of lesions or scars suggestive of healed CL, and lack of a history of chronic ulcer but a positive delayed type hypersensitivity (DTH) skin response to leishmanial antigen, or production of IFNγ by blood cells stimulated with Leishmania antigen, indicative of a functional cellular immune response [68,69,70]. However, the type 1 (TH1-type) response formed by asymptomatic hosts is not as robust as those who develop active disease, with smaller DTH induration, lower levels of antigen-induced IFNγ and TNF production by peripheral blood mononuclear cells (PBMCs), and higher IL-10 production [71]. What drives individuals to present with LCL or remain subclinically infected is not clear. Variation in basal T cell reactivity to Leishmania antigens may contribute. Naïve PBMCs from cohorts of healthy volunteers exposed to L. (V.) braziliensis or L. (L.) amazonensis antigen generate responses that can be stratified into high-IFNγ producing and low-IFNγ producing groups [72,73]. Monocyte-derived macrophages isolated from subclinically infected subjects controlled intracellular parasite replication better than macrophages from patients with active LCL, suggesting intrinsic differences in these innate immune cells even in the absence of T cells [67].
It has become clear that the outcome of tegumentary infections with the Leishmania spp. result from a balance between pro- and anti-inflammatory factors (Figure 1). In infected patients, tissue pathology is associated with a vigorous type 1 immune response to parasite antigens. Lesion size directly correlates with the magnitude Leishmania antigen-stimulated TNF production by PBMCs, and with the frequency of circulating TNF and IFNγ producing CD4+ lymphocytes [74,75]. During LCL, T cell-derived TNF and IFNγ production are observed by direct staining of early lesions and IL-10 and TGFβ transcripts increase in later stages, perhaps to curb the effects of these inflammatory cytokines once disease has been resolved [76,77]. Similarly, T regulatory cell (TREG) transcript forkhead box P3 (FOXP3) is increased intralesionally during chronic LCL due to L. (V.) guyanensis [78]. TREGS from these patients suppressed IFNγ production by autologous CD4+ T cells in vitro. The expression of these pro- and anti-inflammatory cytokines by LCL patient PBMCs is sensitive to the local cytokine environment [61]. In circulating monocytes of LCL patients, there is a positive correlation between TNF and IL-10, suggesting a balance between these co-regulatory mechanisms [79]. It may be that clinically overt disease results when the balance between these opposing factors becomes skewed, including LCL and the more extreme atypical manifestations discussed in detail in the following sections.
Recently, RNA-seq of LCL patient biopsies revealed lesional skin containing detectable L. (V.) braziliensis transcripts had a unique transcriptional signature compared with lesional skin that did not contain L. (V.) braziliensis transcripts [80]. Although inflammatory gene transcripts (IFNG and TNF) were increased in these lesions indicative of an active adaptive response, there was a concomitant increase in transcripts encoding inhibitory molecules (IL10, CTLA4, PD1, PDL1, and LAG3). Interestingly, parasite transcript-positive skin was also associated with a significant increase in B cell transcripts (CD79A, CD19, and CD20), suggesting B cell infiltration during active infection [80]. This is supported by another study showing an increase in B cells in L. (V.) braziliensis infected patient lymph nodes during the progression from early pre-lesional to lesional phases of diseases [81]. B cells are able to present Leishmania antigen to CD4+ T cells and elicit both pro-inflammatory and regulatory cytokine production [82]. Further, L. (V.) braziliensis specific IgG levels are high during the more severe Mucosal Leishmaniasis and decrease with time post treatment [83]. Thus, B cells may contribute to disease onset and persistence during CL.
The role of CD8+ T cells in the pathology of LCL has been well documented. While there is no association between CD8+ T cell activation and the inflammatory response in the pre-ulcerative phase of the disease, there is an association between the intensity of the inflammation and the frequency of CD8+ T cells expressing granzyme [84]. Although cytotoxic activity is higher in LCL than in healthy subjects or in subclinical individuals, CD8+ T cells kill L. (V.) braziliensis infected cells but fail to kill intracellular parasites [85,86].
4. Mucosal Leishmaniasis
Mucosal leishmaniasis [13] can occur as a complication of LCL caused by Leishmania species belonging to the Viannia subgenus. In regions where L. (V.) braziliensis is endemic, approximately 1–10% of LCL patients progress to ML. Known risk factors include sex (male > female), increased age, malnutrition, size and number of LCL lesions, lesions above the belt, and inadequate therapy [87,88,89]. ML can occur simultaneously or months–years after resolution of primary lesions, suggesting persistence of latent organisms in the interval [87,88,90]. In this form of tegumentary leishmaniasis, metastatic lesions appear in the mucosal surfaces of the upper respiratory and digestive tracts. Affected areas can include the nasal mucosa, soft palate, pharynx, larynx, lips or cheeks, and rarely, the trachea or genitalia [3]. ML is most prevalent in South America, typically caused by New World species of the Viannia subgenus, with L. (V.) braziliensis causing the highest number of cases [17,91,92]. ML is more difficult to treat than LCL, often requiring secondary therapy [93,94,95].
When it is allowed to progress, ML can be highly disfiguring and accompanied by extensive tissue destruction. Lesions contain active parasite-responsive T cells and other inflammatory cells, but the actual burden of detectable parasites is low [96]. Compared with LCL patients, Leishmania antigen-stimulated TNF and IFNγ responses of PBMCs are elevated, while IL-10 expression is reduced [97]. ML lesions tend to have higher IL-17 expression than LCL lesions, an inflammation-inducing cytokine, and TH17 cells, which may be involved in ML pathogenesis [63,64]. ML patients also have a higher percentage of activated CD4+ T cells expressing TNF and IFNγ in circulation compared with LCL patients and, contrary to LCL patients, ML patients do not show a positive correlation between the frequency of TNF+ monocytes and IL-10+ monocytes [79]. As a result, the ratio of inflammatory to regulatory cytokines is skewed towards the hypersensitivity pole during ML (Figure 2). Targeting this excess TNF for inhibition by pentoxifylline has shown promise in treating patients with refractory ML when combined with standard antimonial therapy [94]. PBMCs from ML patients are also less responsive to IL-10 regulation compared with LCL PBMCs [97] and ML patients have reduced intralesional IL-10 receptor expression [98]. The unregulated inflammatory response in the face of diminished regulatory activity is thought to drive the inflammatory responses leading to tissue destruction in ML. This highlights the importance of immunoregulation in determining the outcome of leishmaniasis.
5. Disseminated Leishmaniasis
Although ML was previously the most prevalent metastatic form of cutaneous leishmaniasis due to species of the Leishmania Viannia subgenus, disseminated leishmaniasis (DL, also called disseminated cutaneous leishmaniasis or DCL) has emerged as highly prevalent in some regions [12]. DL is characterized by high numbers of pleomorphic lesions, potentially numbering in the hundreds, in two or more noncontiguous anatomical regions [14,99]. Lesions are a mixture of acneiform, papular, nodular, and ulcerated types [25,27]. Parasite metastasis from the original infection site in DL is rapid, commonly occurring within weeks or even days of initial lesion formation [24,27]. The rapidity and breadth of dissemination, and the absence of lymph node enlargement, suggests bloodstream involvement in parasite spread during DL. Nasal mucosal lesions similar to the lesions of ML occur in up to 44% of DL cases [100]. Patients can respond to treatment [26], but may require multiple or slightly longer treatment regimens than LCL treatment recommendations [25,27,100]. Peripheral blood lymphocytes from DL patients produce lower levels of Th1 cytokines than CL patients [31,75,76]. However, at the lesion site, the immune response of DL patients is just as vigorous as lesion cells during LCL, as though a synchronous migration of antigen reactive cells to the multiple cutaneous lesions left a paucity of antigen-responsive cells in the blood [100]. One important characteristic of DL is the high inflammatory response that is mediated by the parasite strain itself. L. (V.) braziliensis isolates from DL patients induce higher production of TNF and IFNγ than L. (V.) braziliensis isolates from LCL patients [79]. Although the biochemical basis by which DL strains elicit a more vigorous cytokine response than LCL strains remains unknown, amplification of anonymous genetic markers from the genomes of different isolates shows evidence that L. (V.) braziliensis isolates causing different disease forms differ at a genomic level [101].
Some studies indicate DL is becoming more common in South America, and there is a site where it has surpassed ML in incidence in the state of Bahia, northeast Brazil [12]. However, in other regions, the occurrence of DL is still relatively rare [12,27,102]. Although the biochemical basis for differences between L. (V.) braziliensis isolates causing distinct syndromes is unknown, at least in one region genetic variability between strains within a species contributes to the outcome of disease. L. (V.) braziliensis strains isolated from DL vs. LCL patients in the same region of Brazil showed significant enrichment for distinct polymorphisms [102]. Furthermore, the expansion of strains was shown to parallel the geographical clustering of DL cases in areas of Brazil, indicating circulation of a particular parasite strain might account for the local increased incidence of DL [103].
Genetic variability in the human population has been reported as significantly associated with DL [104] or with ML in endemic regions [35,36,105,106,107]. Although some descriptions have confused DL with the anergic diffuse cutaneous leishmaniasis (ADCL), it is evident that DL is distinct clinically, immunologically and pathologically from LCL.
6. Anergic Diffuse Cutaneous Leishmaniasis
Patients with LCL, MCL or DL develop a vigorous type 1 immune response to the causative Leishmania species, which can be detected by a positive DTH response to Leishmania antigen. This contrasts with patients who contract anergic diffuse cutaneous leishmaniasis (ADCL), who have a conspicuous absence of a specific Leishmania DTH response (Figure 2).
ADCL (also called diffuse cutaneous leishmaniasis or DCL) is a rare but severe form of LCL, characterized by the development of multiple satellite lesions that can coalesce into plaques covering large areas of skin. Lesions are predominantly nodular or papular in nature, contain an abundance of amastigotes, and do not ulcerate [99]. Lesion histopathology shows numerous parasitized macrophages but few lymphocytes, and hyperkeratosis with epidermal hyperplasia [23]. The lack of ulceration and uncontrolled amastigote growth in ADCL are thought to be a consequence of an anergic cellular immune response. Supporting this hypothesis, ADCL patients have absent DTH and lymphocyte proliferative responses to Leishmania antigen [108,109]. Anergy is confined to Leishmania-specific cellular immune responses, as responses to unrelated antigens are preserved [25,110,111]. Very much like the responses in visceral leishmaniasis, circulating Leishmania specific antibodies are found at high levels in these patients [25,108,112]. Compared to LCL, ADCL lesions have significantly reduced numbers of IFNγ, iNOS, and IL-12 producing cells [113]. These infections are often highly resistant to treatment and exhibit frequent relapse. Cutaneous disease does not self-heal over time as is seen in LCL [108,114,115,116]. Additionally, patients with ADCL have significantly fewer circulating and lesional innate natural killer (NK) cells than LCL patients, and the NK cells present secrete less IFNγ and TNF than LCL NK cells following stimulation with Leishmania lipophosphoglycan, a parasite surface glycolipid that can ligate Toll-like receptor 2 [117]. In mouse models, high amounts of persistent antigen exposure have been shown to contribute to T cell anergy [118,119,120]. However, in ADCL it is not known whether immune hyposensitivity leads to the observed high parasite burden or vice versa.
ADCL is rare in areas endemic for transmission of other forms of CL, suggesting there may be a contribution of parasite strain to disease. However, when L. (V.) pifanoi parasites isolated from an ADCL lesion were inoculated into human volunteers, all developed classical LCL [20] suggesting this unusual disease form is caused by more than just the strain of parasite. Conventional chemotherapies affecting parasite growth have limited effects in these patients, perhaps because they do not correct the immune defects [121]. However, some success has been seen using an immunotherapy approach in ADCL patients that previously were unresponsive to treatment [122,123,124]. One goal of studies about mechanisms of human T cell anergy is to direct approaches of immunotherapy that might be used to improve treatment for ADCL patients.
7. Leishmaniasis Recidivans
Leishmaniasis recidivans (LR) is a rare, chronic relapsing form of tegumentary leishmaniasis that can occur following primary infection with L. (L.) tropica, L. (L.) major or L. (V.) braziliensis, in decreasing order of frequency [3,125,126]. About 5% of CL patients develop LR in Iran [127]. Disease manifests at variable time periods after resolution of a primary LCL infection, sometimes many years later. One case of late-onset LR occurred 43 years after initial infection, highlighting the ability of the parasite to persist in the human host for long periods of time without symptoms [125]. For unclear reasons, nascent lesions reactivate usually around the border, or adjacent to, the scars of previously healed Leishmania lesions [128]. LR lesions are usually painless and contain granulomatous inflammatory infiltrates but do not ulcerate. Amastigotes are rarely observed within lesions by microscopy, and the infecting parasite species can only be confirmed by PCR [126,127,128,129]. Similar to other recurrent or disseminated manifestations, this form of leishmaniasis can be difficult to treat [130].
The proximity of nascent LR lesions to primary lesion sites leads to the hypothesis that a small number of organisms survive silently within the skin at the site of initial infection, and can lead to reactivated inflammatory lesions years after initial cure, responding to an unknown stimulus. The cellular localization of parasites during periods of latency remains unknown. One hypothesis is that local trauma may play a role in reactivation of LR lesions, perhaps via inflammatory changes in the local microenvironment [125,131]. In mice, IL-10 and TGFβ have been implicated as factors promoting the persistence of L. (L.) tropica long after the initial infection subsides, but a role for these cytokines in human LR has not been documented [132].
8. Post Kala-Azar Dermal Leishmaniasis
Post Kala-Azar dermal leishmaniasis (PKDL) is a complication of visceral leishmaniasis caused by L. (L.) donovani, primarily occurring in east Africa (Sudan, Ethiopia, Kenya) and the Indian subcontinent (India, Bangladesh, Nepal) [29]. There are numerous clinical presentations of PKDL. In Africa, PKDL presents most commonly as a collection of dermatoses comprising a macular, maculopapular, or nodular rash [28]. In Indian PKDL, erythema and a combination of macular and papular lesions form and may coalesce into plaques [133,134]. PKDL lesions do not typically become ulcerated [135] and persist for several months [30]. PKDL can occur concurrently with visceral leishmaniasis (VL) or following resolution of visceral infection [28]. A subset of PKDL cases (15–20%) occur without any previous symptoms of VL, showing that for unknown reasons asymptomatic infections can convert to PKDL [134]. Fifty to sixty percent of Sudanese VL patients can develop PKDL, whereas the incidence in India is only 5–10% of VL cases [136]. In L. (L.) donovani endemic areas, the instance of PKDL is higher in HIV-positive patients [137]. The onset of PKDL in Africa also has different kinetics compared to Indian PKDL, as it manifests within months of VL treatment, while in India PKDL may take years to appear [29].
Although light microscopic studies show a low abundance of amastigotes within PKDL lesions [135,138], parasites can still be detected in affected skin using specific antibody staining [139,140]. This has lead scientists to hypothesize that subjects with PKDL serve as a reservoir maintaining continued parasite transmission in some regions, particularly in the Indian subcontinent [133]. This is consistent with the observation that VL in India is primarily an anthroponotic disease [141]. Following resolution of PKDL, there is apparent protective immunity against re-developing symptomatic disease, although there are rare cases of relapse [142,143,144]. Although PKDL can spontaneously heal without treatment (average 9.7 months in Sudan) [30], patients may contribute significantly to anthroponotic transmission of the infection. Therefore, it is considered an urgent priority to identify and treat PKDL cases to prevent continuation of VL in the Indian subcontinent [4].
Although most clinicians in India do not use the DTH skin test, studies in Sudan show that DTH skin tests are negative and PBMCs display absent proliferation and IFNγ responses to Leishmania antigen during active VL [6,136]. Successful treatment of VL precedes the recovery of T cell reactivity to Leishmania antigen, and in subjects who develop PKDL this recovery is coincident with PKDL development [140,145]. PKDL patients show an intermediate level of T cell reactivity between active VL patients and fully recovered patients. It is important to note, however, that only a subset of treated patients develop PKDL, despite restoration of skin test responses in both [28]. This discrepancy may be linked to IL-10 production in PKDL patients. Inadequate treatment of VL increases the probability of developing PKDL [28]. Unlike leishmaniasis recidivans, IL-10 is thought to play a major role in the etiology of PKDL. VL patients who develop PKDL have elevated serum IL-10 and antigen-induced IL-10 production by PBMCs compared to patients who do not develop PKDL [140]. PKDL lesions contain a range of infiltrating cells including numerous CD4+ and CD8+ cells, and granulomas are present at varying levels of maturity [146]. Concurrent IL-10 and IFNγ gene expression was observed in the lymph nodes, skin lesions [139,147] and skin keratinocytes of subjects with PKDL [140].
One theory is that PKDL develops in part due to the effects of UV light on vitamin D metabolism of local or circulating mononuclear cells. The localization of lesions in affected patients tends to be skewed toward sun-exposed skin surfaces [29,148]. Vitamin D3 synthesis is increased following UV exposure, and this in turn suppresses monocyte activation [149,150]. In addition, PKDL patients have higher amounts of the bioactive form of vitamin D3 in their plasma compared to other VL patients post-treatment [151]. PKDL circulating and intralesional monocyte/macrophages display a phenotype that is not classically activated, and has characteristics of M2-type cells with peroxisome proliferator-activated receptor gamma (PPARy), arginase 1 (ARG1), and CD206 expression [152]. This macrophage activation state was hypothesized to contribute to the persistence of this condition [151,153].
Reports of PKDL due to other visceralizing Leishmania species such as L. (L.) infantum are scarce. Some cases of non-ulcerating cutaneous lesions due to L. (L.) infantum infection have been reported, but these constitute a separate disease state from PKDL [154]. PKDL symptomology cannot be recapitulated using rodent models [155].
9. Cellular Determinants of the Immune Response in Murine Cutaneous Leishmaniasis
Mouse models have been used extensively to study cutaneous and visceral leishmaniasis [46]. As in human disease, clinical manifestations in mice vary after infection with different Leishmania species. Different mouse strains have genetically programmed variations in the severity of infection [156], an observation that has inspired many studies of human genetic susceptibility [157]. In particular, the mouse model of L. (L.) major infection has been widely used to study CL [46]. BALB/c mice are highly susceptible to L. (L.) major infection, developing a TH2-type CD4+ T cell response, large local ulcerating lesions, and eventually visceral dissemination of parasites. In contrast, C57BL/6, CBA, and C3H mice are relatively resistant to L. (L.) major, developing a TH1-type cellular immune response and small lesions that ultimately self-heal without dissemination [158,159,160]. Antigen specific TH1-type cells express IFNγ upon contact with antigen presenting cells [161], which in turn activates microbicidal defenses of infected macrophages to kill intracellular parasites [161,162]. Similar to humans, pathology during murine CL is caused by immune-mediated damage. As an illustration, CD4+ T cell deficient C57BL/6 mice experience minimal cutaneous pathology despite a high parasite burden in the ear [163]. Furthermore, during both L. (L.) major and L. (L.) infantum murine infection, there is a prolonged period of silent parasite replication during the first weeks of infection, during which pathology is not observed. After approximately four weeks, infiltration of immune cells coincides with the onset of pathologic changes [163,164].
Although murine models of L. (L.) major infection have been useful for elucidating mechanisms controlling type 1 vs. type 2 CD4+ T cell polarization, this dichotomy is not observed during murine infection with other Leishmania species. Murine infection with L. (L.) mexicana results in a chronic phenotype in most mouse strains [165]. L. (V.) braziliensis forms small, non-ulcerative and self-healing lesions in wild type mice [166]. There is a range of resistance to L. (L.) amazonensis in different mouse strains, with higher levels of antibody being associated with susceptibility [167]. In contrast to L. (L.) donovani and L. (L.) infantum, in which murine susceptibility maps to a single locus encoding solute carrier family 11 member 1 (Slc11a1, previously called Nramp1) on chromosome 1 [168], murine susceptibility to CL is associated with more than one chromosomal locus [168,169,170].
Mouse models of CL show that treated or self-resolved mice can achieve disease resolution, measured by decreased parasite burden and lack of cutaneous lesions, but that parasites persist in the host for long periods of time [171,172], as long as the production of IL-10 is intact [173]. Similar to humans, protective immunity in mouse models follows non-sterilizing cure [171,174]. Both CD4+ and CD8+ T cells contribute to this protection [175]. The cell types harboring latent parasites include macrophages, dendritic cells, and fibroblasts, although additional cell types may play similar roles [176,177,178]. In mice infected with L. (L.) major, IL-10 blockade at chronic time points leads to sterile cure [132,173]. Intriguingly, L. (L.) major-infected IL-10 deficient mice achieve a sterile cure, but these mice do not develop immunity to reinfection, suggesting that persistent parasites may be integral to achieving long-lasting immunity [179]. Nonetheless, IL-10 impairs cure of acute leishmaniasis, illustrated in IL-10 deficient mice infected with L. (L.) amazonensis or L. (L.) mexicana which are still unable to eliminate infection despite strong IFNγ production [180,181,182], at least in part through the action of IL-4 [181].
Early immune events within a critical 24–48 h post-infection have been implicated in instructing the polarization of the TH response in leishmaniasis [46]. The cytokine context within which dendritic cells (DCs) present antigen to CD4+ T cells is one important determinant of development of TH0 cells toward either TH1 vs. TH2 poles [183,184,185]. The signals that induce T cell polarizing cytokine expression are derived from the skin inflammatory milieu in which DCs encounter antigen. During experimental cutaneous leishmaniasis due to L. (L.) major, skin-infiltrating inflammatory monocytes differentiate locally into DCs and subsequently migrate to the draining lymph nodes, where they induce polarized CD4+ T cell responses [45]. Thus, the early skin microenvironment has an important role in influencing the adaptive T cell response and the progression or resolution of disease. Mice offer a unique opportunity to dissect early molecular and cellular mechanisms occurring within the site of infection.
9.1. Neutrophils
Neutrophils are the first immune cells recruited to the site of cutaneous infection after delivery of L. (L.) major to mouse ears [186,187]. There is conflicting evidence on whether neutrophils play a protective or pathogenic role in leishmaniasis. Neutrophils phagocytose parasites in vitro and within infected skin tissue [186,188]. It has been suggested that parasites remain viable within parasitophorous vacuoles after neutrophils undergo apoptosis. Apoptotic neutrophils are engulfed by macrophages in a process called efferocytosis (Figure 3), which induces an anti-inflammatory macrophage state with TGFβ release [189] and signaling through Mer tyrosine kinase receptors [190]. During leishmaniasis, macrophage clearance of apoptotic, infected neutrophils has been likened to a Trojan horse because microbicidal responses are suppressed during macrophage infection. Importantly, apoptotic infected neutrophils are also taken up by dendritic cells, with consequent signaling through the Mer kinase pathway and impaired ability to present antigen to T cells [190]. Dermal DCs that take up infected neutrophils have lowered expression of co-stimulatory receptors CD40 and CD80 [190]. Along these lines, one study showed higher activation of infected dermal DCs and enhanced CD4+ T cell priming in neutrophil depleted mice [187].
9.2. Langerhans Cells
Langerhans cells (LCs) are a subset of dendritic cells resident in the epidermis whose role during cutaneous leishmaniasis is unclear. It has been demonstrated that epidermal LCs can take up L. (L.) major parasites and migrate from the skin to the draining lymph nodes [191,192]. However, recent imaging studies of intradermally infected mouse skin revealed that L. (L.) major parasites are rapidly taken up by dermal and inflammatory DC subsets, but not by LCs [193,194]. Two studies depleted langerin-expressing cells (which are mostly LCs) using diphtheria toxin receptor transgenic mice, prior to L. (L.) major infection. The first showed no effect on CD4+ T cell priming or parasite burden [195], whereas the other showed reduced lesion size and reduced antigen-specific IL-10 production [196]. The data indicate that LCs interact with Leishmania spp. and participate in the immune response, although the biological relevance of these cells remains controversial.
9.3. Monocytes and Macrophages
Monocytes are recruited to the site of Leishmania infection and can internalize parasites (Figure 4). Monocytes differentiated into dendritic cells within the skin environment (termed dermal monocyte-derived DCs) are the major cell type responsible for inducing protective Leishmania-specific TH1 cells in skin draining lymph nodes [45].
Resident and recruited macrophages harbor the greatest burden of intracellular Leishmania spp. parasites throughout chronic leishmaniasis [186,197]. Macrophage infection can be acquired via efferocytosis, as summarized in Figure 3, and mouse and human macrophages internalize free parasites via receptor-mediated phagocytosis [198]. Both phagocytosis and inflammatory cytokines (IL-1, TNF, type 1 interferons) can stimulate macrophages to generate microbicidal reactive oxygen species and reactive nitrogen species, through assembly of the nicotinamide adenine dinucleotide phosphate (NADPH) oxidase or activation of iNOS, respectively [199,200,201,202,203]. Activation requires priming and activation signals, and TNF acts synergistically with IFNγ to induce NO and amastigote elimination by mouse macrophages (Figure 4) [204,205]. In both human and murine macrophages, the respiratory burst is subverted by Leishmania virulence factors [206,207]. Phagocytosis of amastigotes induces even less superoxide formation compared with promastigotes [208]. Mechanisms of subversion include inhibition of assembly and maturation of the NADPH oxidase machinery at the phagosomal membrane, antioxidant activity of Leishmania enzymes, and active macrophage deactivation through the SH2-domain containing inositol phosphatase 1 (SHIP-1) [203,209].
The relative importance of NADPH oxidase vs. iNOS for macrophage microbicidal responses was explored with knockout mouse models. Macrophages lacking a functional NADPH oxidase were able to kill intracellular L. (V.) braziliensis parasites when stimulated with IFNγ as well as wild-type macrophages, while iNOS-deficient macrophages were unresponsive to IFNγ activation [166]. Additionally, mice normally resistant to L. (L.) major infection lacking iNOS are susceptible, despite eliciting a robust TH1 immune response [210]. Macrophage killing mechanisms may be species dependent as both NO and ROS were required by C3H bone marrow-derived macrophages to kill intracellular L. (L.) amazonensis [211]. The extent to which human macrophages express and utilize NO production to combat Leishmania infection is controversial [202,212,213].
9.4. Natural Killer Cells
NK cells are innate immune effector cells that were shown to be protective against murine L. (L.) major infection [214,215]. NK cells are recruited to the skin infection site and draining lymph nodes within 24 h of L. (L.) major inoculation [216,217]. NK cell activity is higher during the early stages of L. (L.) major infection of resistant mice compared to susceptible mice, and the local parasite burden in the skin is inversely correlated with early NK cytotoxic activity in different mouse strains [215]. Activated NK cells augment IL-12p40 production by L. (L.) amazonensis promastigote-infected DCs [218]. These DC1 cells then preferentially induce TH1 polarization of naïve CD4+ T cells [215,219]. NK cell-derived IFNγ can also activate local Leishmania infected macrophages (Figure 4) but NK-derived IFNγ is insufficient to confer protection in the absence of TH1 cells [220,221]. Conversely, TGFβ suppresses NK cell IFNγ production, and blockade of TGFβ signaling in NK cells allows susceptible BALB/c mice to control L. (L.) major infection [222]. Overall, the literature supports a protective, but non-essential, role for NK cells during the early stages of experimental cutaneous leishmaniasis.
9.5. Keratinocytes
Other resident cell types critical to immune responses at the site of Leishmania spp. infection remain to be fully explored. Keratinocytes reside in the epidermis and participate a communicative immune network in the skin, mediated in part by cytokine exchange with the local cell populations [223]. In a model of L. (L.) major, expression of immunomodulatory mediators involved in TH1 induction were significantly highly induced in keratinocytes isolated from the infected skin of resistant mice and several were enriched in the epidermis upon RNA in situ hybridization, suggesting keratinocytes may be a rich source of skin derived cytokines in vivo [44]. Interferon-inducible T-cell alpha chemokine 1 (I-TAC), an early released cytokine that antagonized IL-12 production by DCs, is significantly increased in the epidermis and keratinocytes of susceptible compared to resistant mice infected with L. (L.) major [224]. Experiments performed in our lab suggest Leishmania species causing divergent clinical disease elicit distinct inflammatory responses by human keratinocytes in vitro. VL causing L. (L.) infantum-exposed human keratinocytes up-regulate pro-inflammatory cytokine gene expression with concurrent nuclear factor-κB (NF-κB) activation, while CL-causing L. (L.) major may inhibit keratinocyte activation [225]. Together, these studies suggest a role for keratinocyte-derived cytokines in shaping the immune response to Leishmania, warranting future investigation in this area.
10. Concluding Remarks
The different species of Leishmania cause characteristic clinical syndromes. However, there is nonetheless a spectrum of outcomes of infection with each Leishmania species in both human leishmaniasis and animal models. The range of clinical outcomes, when understood, seems to depend on key contributions from infecting parasite species and a delicate balance of pro- and anti-inflammatory host immune factors in response to infection. An interesting property of the Leishmania species infections is the persistence of parasites long after clinical cure of symptomatic infection, a property that can result in downstream cutaneous manifestations, but which also may be critical for developing immune protection against re-infection. The mechanisms and cell types involved in parasite persistence, and how they lead to disease re-emergence vs. immune protection, remain poorly understood and a subject of great interest for investigators.
Mouse models have revealed early host signals are important controllers of the clinical response to Leishmania spp. infection. This has led to the development of improved models that better mimic natural infection conditions. Our group and others have postulated a previously unappreciated role for inflammatory tissue signals derived from resident, non-hematopoietic cells such as keratinocytes, during early stages of Leishmania infection within the skin. Additional signals generated from sand fly derived factors also cannot be overlooked. Ongoing investigations aim to determine the cellular targets of these vector elicited effects. Mounting evidence suggests the skin microenvironment has a critical influence on the course of Leishmania infection. Therefore, elucidation of early cutaneous inflammatory mechanisms is an important subject of future research in the field of leishmaniasis.
Acknowledgments
This work was funded in part by the National Institutes of Health (NIH) grant AI076233, and by Merit Review grants 5I01BX001983-02 and 2I01BX000536 from the Department of Veterans’ Affairs (MEW). Studies were performed in part while B.M.S. was supported with funds from NIH T32 AI07511.
Author Contributions
Breanna M. Scorza and Mary E. Wilson designed and wrote the manuscript. Edgar M. Carvalho wrote the manuscript.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
| LCL | Localized cutaneous leishmaniasis (also called cutaneous leishmaniasis or CL) |
| ADCL | Anergic diffuse cutaneous leishmaniasis (also called diffuse cutaneous leishmaniasis or DCL) |
| DL | Disseminated leishmaniasis (also called disseminated cutaneous leishmaniasis) |
| LC | Langerhans’ cell |
| LR | Leishmaniasis recidivans |
| MDM | Monocyte derived macrophage |
| MCL | Mucocutaneous leishmaniasis (also called mucosal leishmaniasis or ML) |
| PBMC | Peripheral blood mononuclear cells |
| NADPH | Nicotinamide adenine dinucleotide phosphate |
References
- WHO. Weekly Epidemiological Record. Available online: http://www.who.int/wer/2016/wer9122.pdf?ua=1 (accessed on 31 August 2016).
- Grinnage-Pulley, T.; Scott, B.; Petersen, C.A. A mother’s gift: Congenital transmission of trypanosoma and leishmania species. PLoS Pathog. 2016, 12, e1005302. [Google Scholar] [CrossRef] [PubMed]
- Pearson, R.D.; Sousa, A.Q. Clinical spectrum of leishmaniasis. Clin. Infect. Dis. 1996, 22, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Malaviya, P.; Picado, A.; Singh, S.P.; Hasker, E.; Singh, R.P.; Boelaert, M.; Sundar, S. Visceral leishmaniasis in Muzaffarpur district, Bihar, India from 1990 to 2008. PLoS ONE 2011, 6, e14751. [Google Scholar]
- Al-Salem, W.S.; Pigott, D.M.; Subramaniam, K.; Haines, L.R.; Kelly-Hope, L.; Molyneux, D.H.; Hay, S.I.; Acosta-Serrano, A. Cutaneous leishmaniasis and conflict in Syria. Emerg. Infect. Dis. 2016, 22, 931–933. [Google Scholar] [CrossRef] [PubMed]
- Pavli, A.; Maltezou, H.C. Leishmaniasis, an emerging infection in travelers. Int. J. Infect. Dis. 2010, 14, e1032–e1039. [Google Scholar] [CrossRef] [PubMed]
- Grimaldi, G., Jr.; Tesh, R.B.; McMahon-Pratt, D. A review of the geographic distribution and epidemiology of leishmaniasis in the new world. Am. J. Trop. Med. Hyg. 1989, 41, 687–725. [Google Scholar] [CrossRef] [PubMed]
- Kumar, R.; Bumb, R.A.; Ansari, N.A.; Mehta, R.D.; Salotra, P. Cutaneous leishmaniasis caused by leishmania tropica in Bikaner, India: Parasite identification and characterization using molecular and immunologic tools. Am. J. Trop. Med. Hyg. 2007, 76, 896–901. [Google Scholar] [PubMed]
- Van Griensven, J.; Gadisa, E.; Aseffa, A.; Hailu, A.; Beshah, A.M.; Diro, E. Treatment of cutaneous leishmaniasis caused by Leishmania aethiopica: A systematic review. PLoS Negl. Trop. Dis. 2016, 10, e0004495. [Google Scholar] [CrossRef] [PubMed]
- Oliveira Neto, M.P.; Grimaldi, G., Jr.; Momen, H.; Pacheco, R.S.; Marzochi, M.C.; McMahon Pratt, D. Active cutaneous leishmaniasis in Brazil, induced by Leishmania donovani chagasi. Memorias do Instituto Oswaldo Cruz 1986, 81, 303–309. [Google Scholar] [CrossRef] [PubMed]
- Karunaweera, N.D.; Pratlong, F.; Siriwardane, H.V.; Ihalamulla, R.L.; Dedet, J.P. Sri lankan cutaneous leishmaniasis is caused by Leishmania donovani zymodeme mon-37. Trans. R. Soc. Trop. Med. Hyg. 2003, 97, 380–381. [Google Scholar] [CrossRef]
- Schriefer, A.; Wilson, M.E.; Carvalho, E.M. Recent developments leading toward a paradigm switch in the diagnostic and therapeutic approach to human leishmaniasis. Curr. Opin. Infect. Dis. 2008, 21, 483–488. [Google Scholar] [CrossRef] [PubMed]
- Constant, S.L.; Lee, K.S.; Bottomly, K. Site of antigen delivery can influence T cell priming: Pulmonary environment promotes preferential Th2-type differentiation. Eur. J. Immunol. 2000, 30, 840–847. [Google Scholar] [CrossRef]
- Costa, J.M.; Marsden, P.D.; Llanos-Cuentas, E.A.; Netto, E.M.; Carvalho, E.M.; Barral, A.; Rosa, A.C.; Cuba, C.C.; Magalhaes, A.V.; Barreto, A.C. Disseminated cutaneous leishmaniasis in a field clinic in Bahia, Brazil: A report of eight cases. J. Trop. Med. Hyg. 1986, 89, 319–323. [Google Scholar] [PubMed]
- Osorio, L.E.; Castillo, C.M.; Ochoa, M.T. Mucosal leishmaniasis due to Leishmania (Viannia) panamensis in Colombia: Clinical characteristics. Am. J. Trop. Med. Hyg. 1998, 59, 49–52. [Google Scholar] [CrossRef] [PubMed]
- Santrich, C.; Segura, I.; Arias, A.L.; Saravia, N.G. Mucosal disease caused by Leishmania braziliensis guyanensis. Am. J. Trop. Med. Hyg. 1990, 42, 51–55. [Google Scholar] [CrossRef] [PubMed]
- Lucas, C.M.; Franke, E.D.; Cachay, M.I.; Tejada, A.; Cruz, M.E.; Kreutzer, R.D.; Barker, D.C.; McCann, S.H.; Watts, D.M. Geographic distribution and clinical description of leishmaniasis cases in Peru. Am. J. Trop. Med. Hyg. 1998, 59, 312–317. [Google Scholar] [CrossRef] [PubMed]
- Barral, A.; Pedral-Sampaio, D.; Grimaldi Junior, G.; Momen, H.; McMahon-Pratt, D.; Ribeiro de Jesus, A.; Almeida, R.; Badaro, R.; Barral-Netto, M.; Carvalho, E.M.; et al. Leishmaniasis in Bahia, Brazil: Evidence that Leishmania amazonensis produces a wide spectrum of clinical disease. Am. J. Trop. Med. Hyg. 1991, 44, 536–546. [Google Scholar] [CrossRef] [PubMed]
- Velasco, O.; Savarino, S.J.; Walton, B.C.; Gam, A.A.; Neva, F.A. Diffuse cutaneous leishmaniasis in Mexico. Am. J. Trop. Med. Hyg. 1989, 41, 280–288. [Google Scholar] [CrossRef] [PubMed]
- Convit, J.; Pinardi, M.E.; Rondon, A.J. Diffuse cutaneous leishmaniasis: A disease due to an immunological defect of the host. Trans. R. Soc. Trop. Med. Hyg. 1972, 66, 603–610. [Google Scholar] [CrossRef]
- Akuffo, H.; Maasho, K.; Blostedt, M.; Hojeberg, B.; Britton, S.; Bakhiet, M. Leishmania aethiopica derived from diffuse leishmaniasis patients preferentially induce mRNA for interleukin-10 while those from localized leishmaniasis patients induce interferon-γ. J. Infect. Dis. 1997, 175, 737–741. [Google Scholar] [CrossRef] [PubMed]
- Develoux, M.; Diallo, S.; Dieng, Y.; Mane, I.; Huerre, M.; Pratlong, F.; Dedet, J.P.; Ndiaye, B. Diffuse cutaneous leishmaniasis due to leishmania major in Senegal. Trans. R. Soc. Trop. Med. Hyg. 1996, 90, 396–397. [Google Scholar] [CrossRef]
- Bryceson, A.D. Diffuse cutaneous leishmaniasis in Ethiopia. I. The clinical and histological features of the disease. Trans. R. Soc. Trop. Med. Hyg. 1969, 63, 708–737. [Google Scholar] [CrossRef]
- Carvalho, E.M.; Barral, A.; Costa, J.M.; Bittencourt, A.; Marsden, P. Clinical and immunopathological aspects of disseminated cutaneous leishmaniasis. Acta Trop. 1994, 56, 315–325. [Google Scholar] [CrossRef]
- Hashiguchi, Y.; Gomez, E.L.; Kato, H.; Martini, L.R.; Velez, L.N.; Uezato, H. Diffuse and disseminated cutaneous leishmaniasis: Clinical cases experienced in Ecuador and a brief review. Trop. Med. Health 2016, 44, 2. [Google Scholar] [CrossRef] [PubMed]
- Couppie, P.; Clyti, E.; Sainte-Marie, D.; Dedet, J.P.; Carme, B.; Pradinaud, R. Disseminated cutaneous leishmaniasis due to Leishmania guyanensis: Case of a patient with 425 lesions. Am. J. Trop. Med. Hyg. 2004, 71, 558–560. [Google Scholar] [PubMed]
- Turetz, M.L.; Machado, P.R.; Ko, A.I.; Alves, F.; Bittencourt, A.; Almeida, R.P.; Mobashery, N.; Johnson, W.D., Jr.; Carvalho, E.M. Disseminated leishmaniasis: A new and emerging form of leishmaniasis observed in Northeastern Brazil. J. Infect. Dis. 2002, 186, 1829–1834. [Google Scholar] [CrossRef] [PubMed]
- Zijlstra, E.E.; Khalil, E.A.; Kager, P.A.; El-Hassan, A.M. Post-kala-azar dermal leishmaniasis in the Sudan: Clinical presentation and differential diagnosis. Br. J. Dermatol. 2000, 143, 136–143. [Google Scholar] [CrossRef] [PubMed]
- Zijlstra, E.E.; Musa, A.M.; Khalil, E.A.; El-Hassan, I.M.; El-Hassan, A.M. Post-kala-azar dermal leishmaniasis. Lancet Infect. Dis. 2003, 3, 87–98. [Google Scholar] [CrossRef]
- Musa, A.M.; Khalil, E.A.; Raheem, M.A.; Zijlstra, E.E.; Ibrahim, M.E.; Elhassan, I.M.; Mukhtar, M.M.; El Hassan, A.M. The natural history of sudanese post-kala-azar dermal leishmaniasis: Clinical, immunological and prognostic features. Ann. Trop. Med. Parasitol. 2002, 96, 765–772. [Google Scholar] [CrossRef] [PubMed]
- Peacock, C.S.; Seeger, K.; Harris, D.; Murphy, L.; Ruiz, J.C.; Quail, M.A.; Peters, N.; Adlem, E.; Tivey, A.; Aslett, M.; et al. Comparative genomic analysis of three leishmania species that cause diverse human disease. Nat. Genet. 2007, 39, 839–847. [Google Scholar] [CrossRef] [PubMed]
- Rogers, M.B.; Hilley, J.D.; Dickens, N.J.; Wilkes, J.; Bates, P.A.; Depledge, D.P.; Harris, D.; Her, Y.; Herzyk, P.; Imamura, H.; et al. Chromosome and gene copy number variation allow major structural change between species and strains of leishmania. Genome Res. 2011, 21, 2129–2142. [Google Scholar] [CrossRef] [PubMed]
- Clayton, C.; Shapira, M. Post-transcriptional regulation of gene expression in trypanosomes and leishmanias. Mol. Biochem. Parasitol. 2007, 156, 93–101. [Google Scholar] [CrossRef] [PubMed]
- Abdeladhim, M.; Kamhawi, S.; Valenzuela, J.G. What’s behind a sand fly bite? The profound effect of sand fly saliva on host hemostasis, inflammation and immunity. Infect. Genet. Evol. 2014, 28, 691–703. [Google Scholar] [CrossRef] [PubMed]
- Castellucci, L.; Jamieson, S.E.; Almeida, L.; Oliveira, J.; Guimaraes, L.H.; Lessa, M.; Fakiola, M.; Jesus, A.R.; Nancy Miller, E.; Carvalho, E.M.; et al. Wound healing genes and susceptibility to cutaneous leishmaniasis in Brazil. Infect. Genet. Evol. 2012, 12, 1102–1110. [Google Scholar] [CrossRef] [PubMed]
- Castellucci, L.C.; Almeida, L.F.; Jamieson, S.E.; Fakiola, M.; Carvalho, E.M.; Blackwell, J.M. Host genetic factors in american cutaneous leishmaniasis: A critical appraisal of studies conducted in an endemic area of Brazil. Memorias do Instituto Oswaldo Cruz 2014, 109, 279–288. [Google Scholar] [CrossRef] [PubMed]
- Ribas-Silva, R.C.; Ribas, A.D.; Dos Santos, M.C.; da Silva, W.V.; Lonardoni, M.V.; Borelli, S.D.; Silveira, T.G. Association between HLA genes and american cutaneous leishmaniasis in endemic regions of southern Brazil. BMC Infect. Dis. 2013, 13, 198. [Google Scholar] [CrossRef] [PubMed]
- Samaranayake, N.; Fernando, S.D.; Neththikumara, N.F.; Rodrigo, C.; Karunaweera, N.D.; Dissanayake, V.H. Association of HLA class I and II genes with cutaneous leishmaniasis: A case control study from Sri Lanka and a systematic review. BMC Infect. Dis. 2016, 16, 292. [Google Scholar] [CrossRef] [PubMed]
- Lopes, M.E.; Carneiro, M.B.; Dos Santos, L.M.; Vieira, L.Q. Indigenous microbiota and leishmaniasis. Parasite Immunol. 2016, 38, 37–44. [Google Scholar] [CrossRef] [PubMed]
- Sakthianandeswaren, A.; Foote, S.J.; Handman, E. The role of host genetics in leishmaniasis. Trends Parasitol. 2009, 25, 383–391. [Google Scholar] [CrossRef] [PubMed]
- Rogers, M.; Kropf, P.; Choi, B.S.; Dillon, R.; Podinovskaia, M.; Bates, P.; Muller, I. Proteophosophoglycans regurgitated by leishmania-infected sand flies target the l-arginine metabolism of host macrophages to promote parasite survival. PLoS Pathog. 2009, 5, e1000555. [Google Scholar] [CrossRef] [PubMed]
- Rogers, M.E.; Corware, K.; Muller, I.; Bates, P.A. Leishmania infantum proteophosphoglycans regurgitated by the bite of its natural sand fly vector, lutzomyia longipalpis, promote parasite establishment in mouse skin and skin-distant tissues. Microbes Infect. Inst. Pasteur 2010, 12, 875–879. [Google Scholar] [CrossRef] [PubMed]
- Vannier-Santos, M.A.; Martiny, A.; de Souza, W. Cell biology of Leishmania spp.: Invading and evading. Curr. Pharm. Des. 2002, 8, 297–318. [Google Scholar] [CrossRef] [PubMed]
- Ehrchen, J.M.; Roebrock, K.; Foell, D.; Nippe, N.; von Stebut, E.; Weiss, J.M.; Munck, N.A.; Viemann, D.; Varga, G.; Muller-Tidow, C.; et al. Keratinocytes determine Th1 immunity during early experimental leishmaniasis. PLoS Pathog. 2010, 6, e1000871. [Google Scholar] [CrossRef] [PubMed]
- Leon, B.; Lopez-Bravo, M.; Ardavin, C. Monocyte-derived dendritic cells formed at the infection site control the induction of protective T helper 1 responses against leishmania. Immunity 2007, 26, 519–531. [Google Scholar] [CrossRef] [PubMed]
- Sacks, D.; Noben-Trauth, N. The immunology of susceptibility and resistance to leishmania major in mice. Nat. Rev. Immunol. 2002, 2, 845–858. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro-Gomes, F.L.; Sacks, D. The influence of early neutrophil-leishmania interactions on the host immune response to infection. Front. Cell. Infect. Microbiol. 2012, 2, 59. [Google Scholar] [CrossRef] [PubMed]
- Lessa, H.A.; Lessa, M.M.; Guimaraes, L.H.; Lima, C.M.; Arruda, S.; Machado, P.R.; Carvalho, E.M. A proposed new clinical staging system for patients with mucosal leishmaniasis. Trans. R. Soc. Trop. Med. Hyg. 2012, 106, 376–381. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, E.; Hatz, C.; Blum, J. New world cutaneous leishmaniasis in travellers. Lancet Infect. Dis. 2006, 6, 342–349. [Google Scholar] [CrossRef]
- Hochedez, P.; Caumes, E. Common skin infections in travelers. J. Travel Med. 2008, 15, 252–262. [Google Scholar] [CrossRef] [PubMed]
- El Hajj, L.; Thellier, M.; Carriere, J.; Bricaire, F.; Danis, M.; Caumes, E. Localized cutaneous leishmaniasis imported into Paris: A review of 39 cases. Int. J. Dermatol. 2004, 43, 120–125. [Google Scholar] [CrossRef] [PubMed]
- Barral, A.; Guerreiro, J.; Bomfim, G.; Correia, D.; Barral-Netto, M.; Carvalho, E.M. Lymphadenopathy as the first sign of human cutaneous infection by Leishmania braziliensis. Am. J. Trop. Med. Hyg. 1995, 53, 256–259. [Google Scholar] [CrossRef] [PubMed]
- Ziaei, H.; Sadeghian, G.; Hejazi, S.H. Distribution frequency of pathogenic bacteria isolated from cutaneus leishmaniasis lesions. Korean J. Parasitol. 2008, 46, 191–193. [Google Scholar] [CrossRef] [PubMed]
- Unger, A.; O’Neal, S.; Machado, P.R.; Guimaraes, L.H.; Morgan, D.J.; Schriefer, A.; Bacellar, O.; Glesby, M.J.; Carvalho, E.M. Association of treatment of american cutaneous leishmaniasis prior to ulcer development with high rate of failure in northeastern Brazil. Am. J. Trop. Med. Hyg. 2009, 80, 574–579. [Google Scholar] [PubMed]
- Schubach, A.; Haddad, F.; Oliveira-Neto, M.P.; Degrave, W.; Pirmez, C.; Grimaldi, G., Jr.; Fernandes, O. Detection of leishmania DNA by polymerase chain reaction in scars of treated human patients. J. Infect. Dis. 1998, 178, 911–914. [Google Scholar] [CrossRef] [PubMed]
- Schubach, A.; Marzochi, M.C.; Cuzzi-Maya, T.; Oliveira, A.V.; Araujo, M.L.; Oliveira, A.L.; Pacheco, R.S.; Momen, H.; Conceicao-Silva, F.; Coutinho, S.G.; et al. Cutaneous scars in american tegumentary leishmaniasis patients: A site of Leishmania (Viannia) braziliensis persistence and viability eleven years after antimonial therapy and clinical cure. Am. J. Trop. Med. Hyg. 1998, 58, 824–827. [Google Scholar] [CrossRef] [PubMed]
- Barral, A.; Teixeira, M.; Reis, P.; Vinhas, V.; Costa, J.; Lessa, H.; Bittencourt, A.L.; Reed, S.; Carvalho, E.M.; Barral-Netto, M. Transforming growth factor-β in human cutaneous leishmaniasis. Am. J. Pathol. 1995, 147, 947–954. [Google Scholar] [PubMed]
- Bourreau, E.; Prevot, G.; Gardon, J.; Pradinaud, R.; Launois, P. High intralesional interleukin-10 messenger RNA expression in localized cutaneous leishmaniasis is associated with unresponsiveness to treatment. J. Infect. Dis. 2001, 184, 1628–1630. [Google Scholar] [CrossRef] [PubMed]
- Salhi, A.; Rodrigues, V., Jr.; Santoro, F.; Dessein, H.; Romano, A.; Castellano, L.R.; Sertorio, M.; Rafati, S.; Chevillard, C.; Prata, A.; et al. Immunological and genetic evidence for a crucial role of IL-10 in cutaneous lesions in humans infected with Leishmania braziliensis. J. Immunol. 2008, 180, 6139–6148. [Google Scholar] [CrossRef] [PubMed]
- Castellano, L.R.; Filho, D.C.; Argiro, L.; Dessein, H.; Prata, A.; Dessein, A.; Rodrigues, V. Th1/Th2 immune responses are associated with active cutaneous leishmaniasis and clinical cure is associated with strong interferon-γ production. Hum. Immunol. 2009, 70, 383–390. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, W.N.; Ribeiro, L.E.; Schrieffer, A.; Machado, P.; Carvalho, E.M.; Bacellar, O. The role of inflammatory and anti-inflammatory cytokines in the pathogenesis of human tegumentary leishmaniasis. Cytokine 2014, 66, 127–132. [Google Scholar] [CrossRef] [PubMed]
- Novais, F.O.; Carvalho, L.P.; Passos, S.; Roos, D.S.; Carvalho, E.M.; Scott, P.; Beiting, D.P. Genomic profiling of human Leishmania braziliensis lesions identifies transcriptional modules associated with cutaneous immunopathology. J. Investig. Dermatol. 2015, 135, 94–101. [Google Scholar] [CrossRef] [PubMed]
- Bacellar, O.; Faria, D.; Nascimento, M.; Cardoso, T.M.; Gollob, K.J.; Dutra, W.O.; Scott, P.; Carvalho, E.M. Interleukin 17 production among patients with american cutaneous leishmaniasis. J. Infect. Dis. 2009, 200, 75–78. [Google Scholar] [CrossRef] [PubMed]
- Boaventura, V.S.; Santos, C.S.; Cardoso, C.R.; de Andrade, J.; Dos Santos, W.L.; Clarencio, J.; Silva, J.S.; Borges, V.M.; Barral-Netto, M.; Brodskyn, C.I.; et al. Human mucosal leishmaniasis: Neutrophils infiltrate areas of tissue damage that express high levels of Th17-related cytokines. Eur. J. Immunol. 2010, 40, 2830–2836. [Google Scholar] [CrossRef] [PubMed]
- Abebe, T.; Hailu, A.; Woldeyes, M.; Mekonen, W.; Bilcha, K.; Cloke, T.; Fry, L.; Seich Al Basatena, N.K.; Corware, K.; Modolell, M.; et al. Local increase of arginase activity in lesions of patients with cutaneous leishmaniasis in Ethiopia. PLoS Negl. Trop. Dis. 2012, 6, e1684. [Google Scholar] [CrossRef] [PubMed]
- Iniesta, V.; Carcelen, J.; Molano, L.; Peixoto, P.M.V.; Redondo, E.; Parra, P.; Mangas, M.; Monroy, I.; Campo, M.L.; Nieto, C.G.; et al. Arginase I induction during Leishmania major infection mediates the development of disease. Infect. Immun. 2005, 73, 6085–6090. [Google Scholar] [CrossRef] [PubMed]
- Giudice, A.; Vendrame, C.; Bezerra, C.; Carvalho, L.P.; Delavechia, T.; Carvalho, E.M.; Bacellar, O. Macrophages participate in host protection and the disease pathology associated with Leishmania braziliensis infection. BMC Infect. Dis. 2012, 12, 75. [Google Scholar] [CrossRef] [PubMed]
- Follador, I.; Araujo, C.; Bacellar, O.; Araujo, C.B.; Carvalho, L.P.; Almeida, R.P.; Carvalho, E.M. Epidemiologic and immunologic findings for the subclinical form of Leishmania braziliensis infection. Clin. Infect. Dis. 2002, 34, E54–E58. [Google Scholar] [CrossRef] [PubMed]
- Novoa, R.; Bacellar, O.; Nascimento, M.; Cardoso, T.M.; Ramasawmy, R.; Oliveira, W.N.; Schriefer, A.; Carvalho, E.M. IL-17 and regulatory cytokines (IL-10 and IL-27) in L. Braziliensis infection. Parasite Immunol. 2011, 33, 132–136. [Google Scholar] [CrossRef] [PubMed]
- Schnorr, D.; Muniz, A.C.; Passos, S.; Guimaraes, L.H.; Lago, E.L.; Bacellar, O.; Glesby, M.J.; Carvalho, E.M. IFN-γ production to leishmania antigen supplements the leishmania skin test in identifying exposure to L. Braziliensis infection. PLoS Negl. Trop. Dis. 2012, 6, e1947. [Google Scholar] [CrossRef] [PubMed]
- Bittar, R.C.; Nogueira, R.S.; Vieira-Goncalves, R.; Pinho-Ribeiro, V.; Mattos, M.S.; Oliveira-Neto, M.P.; Coutinho, S.G.; Da-Cruz, A.M. T-cell responses associated with resistance to leishmania infection in individuals from endemic areas for Leishmania (Viannia) braziliensis. Memorias do Instituto Oswaldo Cruz 2007, 102, 625–630. [Google Scholar] [CrossRef] [PubMed]
- Carneiro, M.W.; Fukutani, K.F.; Andrade, B.B.; Curvelo, R.P.; Cristal, J.R.; Carvalho, A.M.; Barral, A.; Van Weyenbergh, J.; Barral-Netto, M.; de Oliveira, C.I. Gene expression profile of high IFN-γ producers stimulated with Leishmania braziliensis identifies genes associated with cutaneous leishmaniasis. PLoS Negl. Trop. Dis. 2016, 10, e0005116. [Google Scholar] [CrossRef] [PubMed]
- Pompeu, M.M.; Brodskyn, C.; Teixeira, M.J.; Clarencio, J.; Van Weyenberg, J.; Coelho, I.C.; Cardoso, S.A.; Barral, A.; Barral-Netto, M. Differences in γ interferon production in vitro predict the pace of the in vivo response to Leishmania amazonensis in healthy volunteers. Infect. Immun. 2001, 69, 7453–7460. [Google Scholar] [CrossRef] [PubMed]
- Antonelli, L.R.; Dutra, W.O.; Almeida, R.P.; Bacellar, O.; Carvalho, E.M.; Gollob, K.J. Activated inflammatory T cells correlate with lesion size in human cutaneous leishmaniasis. Immunol. Lett. 2005, 101, 226–230. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, F.; Bafica, A.; Rosato, A.B.; Favali, C.B.; Costa, J.M.; Cafe, V.; Barral-Netto, M.; Barral, A. Lesion size correlates with leishmania antigen-stimulated TNF-levels in human cutaneous leishmaniasis. Am. J. Trop. Med. Hyg. 2011, 85, 70–73. [Google Scholar] [CrossRef] [PubMed]
- Melby, P.C.; Andrade-Narvaez, F.J.; Darnell, B.J.; Valencia-Pacheco, G.; Tryon, V.V.; Palomo-Cetina, A. Increased expression of proinflammatory cytokines in chronic lesions of human cutaneous leishmaniasis. Infect. Immun. 1994, 62, 837–842. [Google Scholar] [PubMed]
- Hejazi, S.; Hoseini, S.; Javanmard, S.; Zarkesh, S.; Khamesipour, A. Interleukin-10 and transforming growth factor-β in early and late lesions of patients with Leishmania major induced cutaneous leishmaniasis. Iran. J. Parasitol. 2012, 7, 16–23. [Google Scholar] [PubMed]
- Bourreau, E.; Ronet, C.; Darcissac, E.; Lise, M.C.; Sainte Marie, D.; Clity, E.; Tacchini-Cottier, F.; Couppie, P.; Launois, P. Intralesional regulatory T-cell suppressive function during human acute and chronic cutaneous leishmaniasis due to Leishmania guyanensis. Infect. Immun. 2009, 77, 1465–1474. [Google Scholar] [CrossRef] [PubMed]
- Gaze, S.T.; Dutra, W.O.; Lessa, M.; Lessa, H.; Guimaraes, L.H.; Jesus, A.R.; Carvalho, L.P.; Machado, P.; Carvalho, E.M.; Gollob, K.J. Mucosal leishmaniasis patients display an activated inflammatory T-cell phenotype associated with a nonbalanced monocyte population. Scand. J. Immunol. 2006, 63, 70–78. [Google Scholar] [CrossRef] [PubMed]
- Christensen, S.M.; Dillon, L.A.; Carvalho, L.P.; Passos, S.; Novais, F.O.; Hughitt, V.K.; Beiting, D.P.; Carvalho, E.M.; Scott, P.; El-Sayed, N.M.; et al. Meta-transcriptome profiling of the human Leishmania braziliensis cutaneous lesion. PLoS Negl. Trop. Dis. 2016, 10, e0004992. [Google Scholar] [CrossRef] [PubMed]
- Bomfim, G.; Andrade, B.B.; Santos, S.; Clarencio, J.; Barral-Netto, M.; Barral, A. Cellular analysis of cutaneous leishmaniasis lymphadenopathy: Insights into the early phases of human disease. Am. J. Trop. Med. Hyg. 2007, 77, 854–859. [Google Scholar] [PubMed]
- Rodriguez-Pinto, D.; Saravia, N.G.; McMahon-Pratt, D. CD4 T cell activation by B cells in human Leishmania (Viannia) infection. BMC Infect. Dis. 2014, 14, 108. [Google Scholar] [CrossRef] [PubMed]
- Junqueira Pedras, M.; Orsini, M.; Castro, M.; Passos, V.M.; Rabello, A. Antibody subclass profile against Leishmania braziliensis and Leishmania amazonensis in the diagnosis and follow-up of mucosal leishmaniasis. Diagn. Microbiol. Infect. Dis. 2003, 47, 477–485. [Google Scholar] [CrossRef]
- Faria, D.R.; Souza, P.E.; Duraes, F.V.; Carvalho, E.M.; Gollob, K.J.; Machado, P.R.; Dutra, W.O. Recruitment of CD8+ T cells expressing granzyme a is associated with lesion progression in human cutaneous leishmaniasis. Parasite Immunol. 2009, 31, 432–439. [Google Scholar] [CrossRef] [PubMed]
- Cardoso, T.M.; Machado, A.; Costa, D.L.; Carvalho, L.P.; Queiroz, A.; Machado, P.; Scott, P.; Carvalho, E.M.; Bacellar, O. Protective and pathological functions of CD8+ T cells in Leishmania braziliensis infection. Infect. Immun. 2015, 83, 898–906. [Google Scholar] [CrossRef] [PubMed]
- Da Silva Santos, C.; Brodskyn, C.I. The role of CD4 and CD8 T cells in human cutaneous leishmaniasis. Front. Public Health 2014, 2, 165. [Google Scholar] [CrossRef] [PubMed]
- Marsden, P.D. Mucosal leishmaniasis (“espundia” Escomel, 1911). Trans. R. Soc. Trop. Med. Hyg. 1986, 80, 859–876. [Google Scholar] [CrossRef]
- Jones, T.C.; Johnson, W.D., Jr.; Barretto, A.C.; Lago, E.; Badaro, R.; Cerf, B.; Reed, S.G.; Netto, E.M.; Tada, M.S.; Franca, T.F.; et al. Epidemiology of american cutaneous leishmaniasis due to Leishmania braziliensis braziliensis. J. Infect. Dis. 1987, 156, 73–83. [Google Scholar] [CrossRef] [PubMed]
- Machado-Coelho, G.L.; Caiaffa, W.T.; Genaro, O.; Magalhaes, P.A.; Mayrink, W. Risk factors for mucosal manifestation of American cutaneous leishmaniasis. Trans. R. Soc. Trop. Med. Hyg. 2005, 99, 55–61. [Google Scholar] [CrossRef] [PubMed]
- Marsden, P.D. Mucocutaneous leishmaniasis. BMJ 1990, 301, 656–657. [Google Scholar] [CrossRef] [PubMed]
- Saravia, N.G.; Segura, I.; Holguin, A.F.; Santrich, C.; Valderrama, L.; Ocampo, C. Epidemiologic, genetic, and clinical associations among phenotypically distinct populations of Leishmania (Viannia) in Colombia. Am. J. Trop. Med. Hyg. 1998, 59, 86–94. [Google Scholar] [CrossRef] [PubMed]
- Amato, V.S.; Tuon, F.F.; Bacha, H.A.; Neto, V.A.; Nicodemo, A.C. Mucosal leishmaniasis. Current scenario and prospects for treatment. Acta Trop. 2008, 105, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Franke, E.D.; Wignall, F.S.; Cruz, M.E.; Rosales, E.; Tovar, A.A.; Lucas, C.M.; Llanos-Cuentas, A.; Berman, J.D. Efficacy and toxicity of sodium stibogluconate for mucosal leishmaniasis. Ann. Intern. Med. 1990, 113, 934–940. [Google Scholar] [CrossRef] [PubMed]
- Lessa, H.A.; Machado, P.; Lima, F.; Cruz, A.A.; Bacellar, O.; Guerreiro, J.; Carvalho, E.M. Successful treatment of refractory mucosal leishmaniasis with pentoxifylline plus antimony. Am. J. Trop. Med. Hyg. 2001, 65, 87–89. [Google Scholar] [CrossRef] [PubMed]
- Amato, V.S.; Tuon, F.F.; Siqueira, A.M.; Nicodemo, A.C.; Neto, V.A. Treatment of mucosal leishmaniasis in Latin America: Systematic review. Am. J. Trop. Med. Hyg. 2007, 77, 266–274. [Google Scholar] [PubMed]
- Ridley, D.S.; De Magalhaes, A.V.; Marsden, P.D. Histological analysis and the pathogenesis of mucocutaneous leishmaniasis. J. Pathol. 1989, 159, 293–299. [Google Scholar] [CrossRef] [PubMed]
- Bacellar, O.; Lessa, H.; Schriefer, A.; Machado, P.; Ribeiro de Jesus, A.; Dutra, W.O.; Gollob, K.J.; Carvalho, E.M. Up-regulation of Th1-type responses in mucosal leishmaniasis patients. Infect. Immun. 2002, 70, 6734–6740. [Google Scholar] [CrossRef] [PubMed]
- Faria, D.R.; Gollob, K.J.; Barbosa, J., Jr.; Schriefer, A.; Machado, P.R.; Lessa, H.; Carvalho, L.P.; Romano-Silva, M.A.; de Jesus, A.R.; Carvalho, E.M.; et al. Decreased in situ expression of interleukin-10 receptor is correlated with the exacerbated inflammatory and cytotoxic responses observed in mucosal leishmaniasis. Infect. Immun. 2005, 73, 7853–7859. [Google Scholar] [CrossRef] [PubMed]
- Goto, H.; Lindoso, J.A. Current diagnosis and treatment of cutaneous and mucocutaneous leishmaniasis. Expert Rev. Anti-Infect. Ther. 2010, 8, 419–433. [Google Scholar] [CrossRef] [PubMed]
- Machado, P.R.; Rosa, M.E.; Costa, D.; Mignac, M.; Silva, J.S.; Schriefer, A.; Teixeira, M.M.; Bacellar, O.; Carvalho, E.M. Reappraisal of the immunopathogenesis of disseminated leishmaniasis: In situ and systemic immune response. Trans. R. Soc. Trop. Med. Hyg. 2011, 105, 438–444. [Google Scholar] [CrossRef] [PubMed]
- Schriefer, A.L.F.; Goes Neto, A.; Guimaraes, L.H.; Carvalho, L.P.; Almeida, R.P.; Machado, P.R.; Lessa, H.A.; Jesus, A.R.; Riley, L.W.; Carvalho, E.M. Multiclonal Leishmania braziliensis population structure and its clinical implication in a region of endemicity for american tegumentary leishmaniasis. Infect. Immun. 2004, 72, 508–514. [Google Scholar] [CrossRef] [PubMed]
- Queiroz, A.; Sousa, R.; Heine, C.; Cardoso, M.; Guimaraes, L.H.; Machado, P.R.; Carvalho, E.M.; Riley, L.W.; Wilson, M.E.; Schriefer, A. Association between an emerging disseminated form of leishmaniasis and Leishmania (Viannia) braziliensis strain polymorphisms. J. Clin. Microbiol. 2012, 50, 4028–4034. [Google Scholar] [CrossRef] [PubMed]
- Schriefer, A.; Guimaraes, L.H.; Machado, P.R.; Lessa, M.; Lessa, H.A.; Lago, E.; Ritt, G.; Goes-Neto, A.; Schriefer, A.L.; Riley, L.W.; et al. Geographic clustering of leishmaniasis in northeastern Brazil. Emerg. Infect. Dis. 2009, 15, 871–876. [Google Scholar] [CrossRef] [PubMed]
- Leopoldo, P.T.; Machado, P.R.; Almeida, R.P.; Schriefer, A.; Giudice, A.; de Jesus, A.R.; Ho, J.L.; Guimaraes, L.H.; Bacellar, O.; Carvalho, E.M. Differential effects of antigens from l. Braziliensis isolates from disseminated and cutaneous leishmaniasis on in vitro cytokine production. BMC Infect. Dis. 2006, 6, 75. [Google Scholar] [CrossRef] [PubMed]
- Castellucci, L.; Menezes, E.; Oliveira, J.; Magalhaes, A.; Guimaraes, L.H.; Lessa, M.; Ribeiro, S.; Reale, J.; Noronha, E.F.; Wilson, M.E.; et al. IL6-174 G/C promoter polymorphism influences susceptibility to mucosal but not localized cutaneous leishmaniasis in Brazil. J. Infect. Dis. 2006, 194, 519–527. [Google Scholar] [CrossRef] [PubMed]
- Castellucci, L.; Jamieson, S.E.; Miller, E.N.; Menezes, E.; Oliveira, J.; Magalhaes, A.; Guimaraes, L.H.; Lessa, M.; de Jesus, A.R.; Carvalho, E.M.; et al. CXCR1 and SLC11A1 polymorphisms affect susceptibility to cutaneous leishmaniasis in Brazil: A case-control and family-based study. BMC Med. Genet. 2010, 11, 10. [Google Scholar] [CrossRef] [PubMed]
- Castellucci, L.; Jamieson, S.E.; Miller, E.N.; de Almeida, L.F.; Oliveira, J.; Magalhaes, A.; Guimaraes, L.H.; Lessa, M.; Lago, E.; de Jesus, A.R.; et al. FLI1 polymorphism affects susceptibility to cutaneous leishmaniasis in Brazil. Genes Immun. 2011, 12, 589–594. [Google Scholar] [CrossRef] [PubMed]
- Bomfim, G.; Nascimento, C.; Costa, J.; Carvalho, E.M.; Barral-Netto, M.; Barral, A. Variation of cytokine patterns related to therapeutic response in diffuse cutaneous leishmaniasis. Exp. Parasitol. 1996, 84, 188–194. [Google Scholar] [CrossRef] [PubMed]
- Silveira, F.T.; Lainson, R.; Corbett, C.E. Clinical and immunopathological spectrum of american cutaneous leishmaniasis with special reference to the disease in amazonian Brazil: A review. Memorias do Instituto Oswaldo Cruz 2004, 99, 239–251. [Google Scholar] [CrossRef] [PubMed]
- Convit, J.; Kerdel-Vegas, F.; Gordon, B. Disseminated anergic cutaneous leishmaniasis. Br. J. Dermatol. 1962, 74, 132–135. [Google Scholar] [CrossRef] [PubMed]
- Petersen, E.A.; Neva, F.A.; Oster, C.N.; Bogaert Diaz, H. Specific inhibition of lymphocyte-proliferation responses by adherent suppressor cells in diffuse cutaneous leishmaniasis. N. Engl. J. Med. 1982, 306, 387–392. [Google Scholar] [CrossRef] [PubMed]
- Barral, A.; Costa, J.M.; Bittencourt, A.L.; Barral-Netto, M.; Carvalho, E.M. Polar and subpolar diffuse cutaneous leishmaniasis in Brazil: Clinical and immunopathologic aspects. Int. J. Dermatol. 1995, 34, 474–479. [Google Scholar] [CrossRef] [PubMed]
- Diaz, N.L.; Arvelaez, F.A.; Zerpa, O.; Tapia, F.J. Inducible nitric oxide synthase and cytokine pattern in lesions of patients with american cutaneous leishmaniasis. Clin. Exp. Dermatol. 2006, 31, 114–117. [Google Scholar] [CrossRef] [PubMed]
- Zerpa, O.; Ulrich, M.; Blanco, B.; Polegre, M.; Avila, A.; Matos, N.; Mendoza, I.; Pratlong, F.; Ravel, C.; Convit, J. Diffuse cutaneous leishmaniasis responds to miltefosine but then relapses. Br. J. Dermatol. 2007, 156, 1328–1335. [Google Scholar] [CrossRef] [PubMed]
- Calvopina, M.; Gomez, E.A.; Sindermann, H.; Cooper, P.J.; Hashiguchi, Y. Relapse of new world diffuse cutaneous leishmaniasis caused by Leishmania (Leishmania) mexicana after miltefosine treatment. Am. J. Trop. Med. Hyg. 2006, 75, 1074–1077. [Google Scholar] [PubMed]
- Salaiza-Suazo, N.; Volkow, P.; Tamayo, R.; Moll, H.; Gillitzer, R.; Perez-Torres, A.; Perez-Montfort, R.; Dominguez, J.D.; Velasco-Castrejon, O.; Crippa, M.; et al. Treatment of two patients with diffuse cutaneous leishmaniasis caused by Leishmania mexicana modifies the immunohistological profile but not the disease outcome. Trop. Med. Int. Health 1999, 4, 801–811. [Google Scholar] [CrossRef] [PubMed]
- Caneda-Guzman, I.C.; Salaiza-Suazo, N.; Fernandez-Figueroa, E.A.; Carrada-Figueroa, G.; Aguirre-Garcia, M.; Becker, I. NK cell activity differs between patients with localized and diffuse cutaneous leishmaniasis infected with Leishmania mexicana: A comparative study of TLRs and cytokines. PLoS ONE 2014, 9, e112410. [Google Scholar] [CrossRef] [PubMed]
- Han, S.; Asoyan, A.; Rabenstein, H.; Nakano, N.; Obst, R. Role of antigen persistence and dose for CD4+ T-cell exhaustion and recovery. Proc. Natl. Acad. Sci. USA 2010, 107, 20453–20458. [Google Scholar] [CrossRef] [PubMed]
- Kahan, S.M.; Wherry, E.J.; Zajac, A.J. T cell exhaustion during persistent viral infections. Virology 2015, 479–480, 180–193. [Google Scholar] [CrossRef] [PubMed]
- Wherry, E.J.; Kurachi, M. Molecular and cellular insights into T cell exhaustion. Nat. Rev. Immunol. 2015, 15, 486–499. [Google Scholar] [CrossRef] [PubMed]
- Franke, E.D.; Lucas, C.M.; Tovar, A.A.; Kruger, J.H.; De Rivera, M.V.; Wignall, F.S. Diffuse cutaneous leishmaniasis acquired in Peru. Am. J. Trop. Med. Hyg. 1990, 43, 260–262. [Google Scholar] [CrossRef] [PubMed]
- Convit, J.; Castellanos, P.L.; Ulrich, M.; Castes, M.; Rondon, A.; Pinardi, M.E.; Rodriquez, N.; Bloom, B.R.; Formica, S.; Valecillos, L.; et al. Immunotherapy of localized, intermediate, and diffuse forms of american cutaneous leishmaniasis. J. Infect. Dis. 1989, 160, 104–115. [Google Scholar] [CrossRef] [PubMed]
- Pereira, L.I.; Dorta, M.L.; Pereira, A.J.; Bastos, R.P.; Oliveira, M.A.; Pinto, S.A.; Galdino, H., Jr.; Mayrink, W.; Barcelos, W.; Toledo, V.P.; et al. Increase of NK cells and proinflammatory monocytes are associated with the clinical improvement of diffuse cutaneous leishmaniasis after immunochemotherapy with BCG/leishmania antigens. Am. J. Trop. Med. Hyg. 2009, 81, 378–383. [Google Scholar] [PubMed]
- Badaro, R.; Johnson, W.D., Jr. The role of interferon-γ in the treatment of visceral and diffuse cutaneous leishmaniasis. J. Infect. Dis. 1993, 167 (Suppl. 1), S13–S17. [Google Scholar] [CrossRef] [PubMed]
- Marovich, M.A.; Lira, R.; Shepard, M.; Fuchs, G.H.; Kruetzer, R.; Nutman, T.B.; Neva, F.A. Leishmaniasis recidivans recurrence after 43 years: A clinical and immunologic report after successful treatment. Clin. Infect. Dis. 2001, 33, 1076–1079. [Google Scholar] [CrossRef] [PubMed]
- Sharifi, I.; Fekri, A.R.; Aflatoonian, M.R.; Khamesipour, A.; Mahboudi, F.; Dowlati, Y.; Nadim, A.; Modabber, F. Leishmaniasis recidivans among school children in bam, south-east Iran, 1994–2006. Int. J. Dermatol. 2010, 49, 557–561. [Google Scholar] [CrossRef] [PubMed]
- Momeni, A.Z.; Aminjavaheri, M. Treatment of recurrent cutaneous leishmaniasis. Int. J. Dermatol. 1995, 34, 129–133. [Google Scholar] [CrossRef] [PubMed]
- Berlin, C. Leishmaniasis recidiva cutis; leishmanid. Arch. Dermatol. Syphilol. 1940, 41, 874–886. [Google Scholar] [CrossRef]
- Oliveira-Neto, M.P.; Mattos, M.; Souza, C.S.; Fernandes, O.; Pirmez, C. Leishmaniasis recidiva cutis in new world cutaneous leishmaniasis. Int. J. Dermatol. 1998, 37, 846–849. [Google Scholar] [CrossRef] [PubMed]
- Strick, R.A.; Borok, M.; Gasiorowski, H.C. Recurrent cutaneous leishmaniasis. J. Am. Acad. Dermatol. 1983, 9, 437–443. [Google Scholar] [CrossRef]
- Wortmann, G.W.; Aronson, N.E.; Miller, R.S.; Blazes, D.; Oster, C.N. Cutaneous leishmaniasis following local trauma: A clinical pearl. Clin. Infect. Dis. 2000, 31, 199–201. [Google Scholar] [CrossRef] [PubMed]
- Anderson, C.F.; Lira, R.; Kamhawi, S.; Belkaid, Y.; Wynn, T.A.; Sacks, D. Il-10 and TGF-β control the establishment of persistent and transmissible infections produced by leishmania tropica in C57BL/6 mice. J. Immunol. 2008, 180, 4090–4097. [Google Scholar] [CrossRef] [PubMed]
- Girgla, H.S.; Marsden, R.A.; Singh, G.M.; Ryan, T.J. Post-kala-azar dermal leishmaniasis. Br. J. Dermatol. 1977, 97, 307–311. [Google Scholar] [CrossRef] [PubMed]
- Ramesh, V.; Mukherjee, A. Post-kala-azar dermal leishmaniasis. Int. J. Dermatol. 1995, 34, 85–91. [Google Scholar] [CrossRef] [PubMed]
- El Hassan, A.M.; Ghalib, H.W.; Zijlstra, E.E.; Eltoum, I.A.; Satti, M.; Ali, M.S.; Ali, H.M. Post kala-azar dermal leishmaniasis in the Sudan: Clinical features, pathology and treatment. Trans. R. Soc. Trop. Med. Hyg. 1992, 86, 245–248. [Google Scholar] [CrossRef]
- Zijlstra, E.E.; El-Hassan, A.M. Leishmanin and tuberculin sensitivity in leishmaniasis in the Sudan, with special reference to kala-azar. Trans. R. Soc. Trop. Med. Hyg. 1993, 87, 425–427. [Google Scholar] [CrossRef]
- Ritmeijer, K.; Veeken, H.; Melaku, Y.; Leal, G.; Amsalu, R.; Seaman, J.; Davidson, R.N. Ethiopian visceral leishmaniasis: Generic and proprietary sodium stibogluconate are equivalent; HIV co-infected patients have a poor outcome. Trans. R. Soc. Trop. Med. Hyg. 2001, 95, 668–672. [Google Scholar] [CrossRef]
- Ramesh, V.; Kaushal, H.; Mishra, A.K.; Singh, R.; Salotra, P. Clinico-epidemiological analysis of post kala-azar dermal leishmaniasis (PKDL) cases in india over last two decades: A hospital based retrospective study. BMC Public Health 2015, 15, 1092. [Google Scholar] [CrossRef] [PubMed]
- Ismail, A.; Kharazmi, A.; Permin, H.; El Hassan, A.M. Detection and characterization of leishmania in tissues of patients with post kala-azar dermal leishmaniasis using a specific monoclonal antibody. Trans. R. Soc. Trop. Med. Hyg. 1997, 91, 283–285. [Google Scholar] [CrossRef]
- Gasim, S.; Elhassan, A.M.; Khalil, E.A.; Ismail, A.; Kadaru, A.M.; Kharazmi, A.; Theander, T.G. High levels of plasma IL-10 and expression of IL-10 by keratinocytes during visceral leishmaniasis predict subsequent development of post-kala-azar dermal leishmaniasis. Clin. Exp. Immunol. 1998, 111, 64–69. [Google Scholar] [CrossRef] [PubMed]
- Singh, N.; Mishra, J.; Singh, R.; Singh, S. Animal reservoirs of visceral leishmaniasis in India. J. Parasitol. 2013, 99, 64–67. [Google Scholar] [CrossRef] [PubMed]
- Sinha, R.; Roychoudhury, J.; Palit, P.; Ali, N. Cationic liposomal sodium stibogluconate (SSG), a potent therapeutic tool for treatment of infection by SSG-sensitive and -resistant Leishmania donovani. Antimicrob. Agents Chemother. 2015, 59, 344–355. [Google Scholar] [CrossRef] [PubMed]
- Sen Gupta, P.C.; Mukherjee, A.M. Recurrence of kala-azar associated with post-kala-azar dermal leishmaniasis. J. Indian Med. Assoc. 1968, 50, 1–5. [Google Scholar] [PubMed]
- Nandy, A.; Addy, M.; Maji, A.K.; Guha, S.K.; Banerjee, D.; Chaudhuri, D. Recurrence of kala-azar after PKDL: Role of co-factors. Trop. Med. Int. Health 1998, 3, 76–78. [Google Scholar] [CrossRef] [PubMed]
- Haldar, J.P.; Ghose, S.; Saha, K.C.; Ghose, A.C. Cell-mediated immune response in Indian kala-azar and post-kala-azar dermal leishmaniasis. Infect. Immun. 1983, 42, 702–707. [Google Scholar] [PubMed]
- Ghosh, M.K.; Nandy, A.; Addy, M.; Maitra, T.K.; Ghose, A.C. Subpopulations of T lymphocytes in the peripheral blood, dermal lesions and lymph nodes of post kala-azar dermal leishmaniasis patients. Scand. J. Immunol. 1995, 41, 11–17. [Google Scholar] [CrossRef] [PubMed]
- Ghalib, H.W.; Piuvezam, M.R.; Skeiky, Y.A.; Siddig, M.; Hashim, F.A.; El-Hassan, A.M.; Russo, D.M.; Reed, S.G. Interleukin 10 production correlates with pathology in human Leishmania donovani infections. J. Clin. Investig. 1993, 92, 324–329. [Google Scholar] [CrossRef] [PubMed]
- Ismail, A.; Khalil, E.A.; Musa, A.M.; El Hassan, I.M.; Ibrahim, M.E.; Theander, T.G.; El Hassan, A.M. The pathogenesis of post kala-azar dermal leishmaniasis from the field to the molecule: Does ultraviolet light (UVB) radiation play a role? Med. Hypotheses 2006, 66, 993–999. [Google Scholar] [CrossRef] [PubMed]
- Sadeghi, K.; Wessner, B.; Laggner, U.; Ploder, M.; Tamandl, D.; Friedl, J.; Zugel, U.; Steinmeyer, A.; Pollak, A.; Roth, E.; et al. Vitamin D3 down-regulates monocyte TLR expression and triggers hyporesponsiveness to pathogen-associated molecular patterns. Eur. J. Immunol. 2006, 36, 361–370. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Leung, D.Y.; Richers, B.N.; Liu, Y.; Remigio, L.K.; Riches, D.W.; Goleva, E. Vitamin D inhibits monocyte/macrophage proinflammatory cytokine production by targeting MAPK phosphatase-1. J. Immunol. 2012, 188, 2127–2135. [Google Scholar] [CrossRef] [PubMed]
- Mukhopadhyay, D.; Mukherjee, S.; Roy, S.; Dalton, J.E.; Kundu, S.; Sarkar, A.; Das, N.K.; Kaye, P.M.; Chatterjee, M. M2 polarization of monocytes-macrophages is a hallmark of Indian post kala-azar dermal leishmaniasis. PLoS Negl. Trop. Dis. 2015, 9, e0004145. [Google Scholar] [CrossRef] [PubMed]
- Mosser, D.M.; Edwards, J.P. Exploring the full spectrum of macrophage activation. Nat. Rev. Immunol. 2008, 8, 958–969. [Google Scholar] [CrossRef] [PubMed]
- Whitcomb, J.P.; Deagostino, M.; Ballentine, M.; Fu, J.; Tenniswood, M.; Welsh, J.; Cantorna, M.; McDowell, M.A. The role of vitamin D and vitamin D receptor in immunity to Leishmania major infection. J. Parasitol. Res. 2012, 2012, 134645. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.W.; Ramasamy, G.; McCall, L.I.; Haydock, A.; Ranasinghe, S.; Abeygunasekara, P.; Sirimanna, G.; Wickremasinghe, R.; Myler, P.; Matlashewski, G. Genetic analysis of leishmania donovani tropism using a naturally attenuated cutaneous strain. PLoS Pathog. 2014, 10, e1004244. [Google Scholar] [CrossRef] [PubMed]
- Nuwayri-Salti, N.; Matta, M.; Shbaklo, Z.; Lakkis, M.; Kabbani, Z.E. Behavior in a mouse model of isolates of leishmania donovani sensu lato cultured from the blood of patients with chronic cutaneous lesions. Am. J. Trop. Med. Hyg. 1998, 58, 710–714. [Google Scholar] [CrossRef] [PubMed]
- Wilson, M.E.; Jeronimo, S.M.; Pearson, R.D. Immunopathogenesis of infection with the visceralizing Leishmania species. Microb. Pathog. 2005, 38, 147–160. [Google Scholar] [CrossRef] [PubMed]
- Blackwell, J.M.; Fakiola, M.; Ibrahim, M.E.; Jamieson, S.E.; Jeronimo, S.B.; Miller, E.N.; Mishra, A.; Mohamed, H.S.; Peacock, C.S.; Raju, M.; et al. Genetics and visceral leishmaniasis: Of mice and man. Parasite Immunol. 2009, 31, 254–266. [Google Scholar] [CrossRef] [PubMed]
- Scott, P.; Novais, F.O. Cutaneous leishmaniasis: Immune responses in protection and pathogenesis. Nat. Rev. Immunol. 2016, 16, 581–592. [Google Scholar] [CrossRef] [PubMed]
- Scott, P.; Natovitz, P.; Coffman, R.L.; Pearce, E.; Sher, A. CD4+ T cell subsets in experimental cutaneous leishmaniasis. Memorias do Instituto Oswaldo Cruz 1988, 83 (Suppl. 1), 256–259. [Google Scholar] [CrossRef] [PubMed]
- Heinzel, F.P.; Sadick, M.D.; Holaday, B.J.; Coffman, R.L.; Locksley, R.M. Reciprocal expression of interferon γ or interleukin 4 during the resolution or progression of murine leishmaniasis. Evidence for expansion of distinct helper T cell subsets. J. Exp. Med. 1989, 169, 59–72. [Google Scholar] [CrossRef] [PubMed]
- Muller, A.J.; Filipe-Santos, O.; Eberl, G.; Aebischer, T.; Spath, G.F.; Bousso, P. CD4+ T cells rely on a cytokine gradient to control intracellular pathogens beyond sites of antigen presentation. Immunity 2012, 37, 147–157. [Google Scholar] [CrossRef] [PubMed]
- Filipe-Santos, O.; Pescher, P.; Breart, B.; Lippuner, C.; Aebischer, T.; Glaichenhaus, N.; Spath, G.F.; Bousso, P. A dynamic map of antigen recognition by CD4 T cells at the site of Leishmania major infection. Cell Host Microbe 2009, 6, 23–33. [Google Scholar] [CrossRef] [PubMed]
- Belkaid, Y.; Mendez, S.; Lira, R.; Kadambi, N.; Milon, G.; Sacks, D. A natural model of Leishmania major infection reveals a prolonged “silent” phase of parasite amplification in the skin before the onset of lesion formation and immunity. J. Immunol. 2000, 165, 969–977. [Google Scholar] [CrossRef] [PubMed]
- Wilson, M.E.; Recker, T.J.; Rodriguez, N.E.; Young, B.M.; Burnell, K.K.; Streit, J.A.; Kline, J.N. The TGF-β response to leishmania chagasi in the absence of IL-12. Eur. J. Immunol. 2002, 32, 3556–3565. [Google Scholar] [CrossRef]
- Perez, H.; Malave, I.; Arredondo, B. The effects of protein malnutrition on the course of leishmania mexicana infection in C57BL/6 mice: Nutrition and susceptibility to leishmaniasis. Clin. Exp. Immunol. 1979, 38, 453–460. [Google Scholar] [PubMed]
- Rocha, F.J.; Schleicher, U.; Mattner, J.; Alber, G.; Bogdan, C. Cytokines, signaling pathways, and effector molecules required for the control of Leishmania (Viannia) braziliensis in mice. Infect. Immun. 2007, 75, 3823–3832. [Google Scholar] [CrossRef] [PubMed]
- De Oliveira Cardoso, F.; de Souza Cda, S.; Mendes, V.G.; Abreu-Silva, A.L.; Goncalves da Costa, S.C.; Calabrese, K.S. Immunopathological studies of Leishmania amazonensis infection in resistant and in susceptible mice. J. Infect. Dis. 2010, 201, 1933–1940. [Google Scholar] [CrossRef] [PubMed]
- Lipoldova, M.; Demant, P. Genetic susceptibility to infectious disease: Lessons from mouse models of leishmaniasis. Nat. Rev. Genet. 2006, 7, 294–305. [Google Scholar] [CrossRef] [PubMed]
- Loeuillet, C.; Banuls, A.L.; Hide, M. Study of leishmania pathogenesis in mice: Experimental considerations. Parasit. Vectors 2016, 9, 144. [Google Scholar] [CrossRef] [PubMed]
- Blackwell, J.M. Genetic susceptibility to leishmanial infections: Studies in mice and man. Parasitology 1996, 112, S67–S74. [Google Scholar] [PubMed]
- Aebischer, T.; Moody, S.F.; Handman, E. Persistence of virulent Leishmania major in murine cutaneous leishmaniasis: A possible hazard for the host. Infect. Immun. 1993, 61, 220–226. [Google Scholar] [PubMed]
- Aebischer, T.; Morris, L.; Handman, E. Intravenous injection of irradiated leishmania major into susceptible BALB/c mice: Immunization or protective tolerance. Int. Immunol. 1994, 6, 1535–1543. [Google Scholar] [CrossRef] [PubMed]
- Belkaid, Y.; Hoffmann, K.F.; Mendez, S.; Kamhawi, S.; Udey, M.C.; Wynn, T.A.; Sacks, D.L. The role of interleukin (IL)-10 in the persistence of Leishmania major in the skin after healing and the therapeutic potential of anti-IL-10 receptor antibody for sterile cure. J. Exp. Med. 2001, 194, 1497–1506. [Google Scholar] [CrossRef] [PubMed]
- Liew, F.Y.; Hale, C.; Howard, J.G. Immunologic regulation of experimental cutaneous leishmaniasis. V. Characterization of effector and specific suppressor T cells. J. Immunol. 1982, 128, 1917–1922. [Google Scholar] [PubMed]
- Muller, I. Role of T cell subsets during the recall of immunologic memory to Leishmania major. Eur. J. Immunol. 1992, 22, 3063–3069. [Google Scholar] [CrossRef] [PubMed]
- Alexander, J.; Russell, D.G. The interaction of leishmania species with macrophages. Adv. Parasitol. 1992, 31, 175–254. [Google Scholar] [PubMed]
- Moll, H.; Flohe, S.; Rollinghoff, M. Dendritic cells in leishmania major-immune mice harbor persistent parasites and mediate an antigen-specific T cell immune response. Eur. J. Immunol. 1995, 25, 693–699. [Google Scholar] [CrossRef] [PubMed]
- Bogdan, C.; Donhauser, N.; Doring, R.; Rollinghoff, M.; Diefenbach, A.; Rittig, M.G. Fibroblasts as host cells in latent leishmaniosis. J. Exp. Med. 2000, 191, 2121–2130. [Google Scholar] [CrossRef] [PubMed]
- Belkaid, Y.; Piccirillo, C.A.; Mendez, S.; Shevach, E.M.; Sacks, D.L. CD4+CD25+ regulatory T cells control Leishmania major persistence and immunity. Nature 2002, 420, 502–507. [Google Scholar] [CrossRef] [PubMed]
- Jones, D.E.; Ackermann, M.R.; Wille, U.; Hunter, C.A.; Scott, P. Early enhanced th1 response after Leishmania amazonensis infection of C57BL/6 interleukin-10-deficient mice does not lead to resolution of infection. Infect. Immun. 2002, 70, 2151–2158. [Google Scholar] [CrossRef] [PubMed]
- Padigel, U.M.; Alexander, J.; Farrell, J.P. The role of interleukin-10 in susceptibility of BALB/c mice to infection with Leishmania mexicana and Leishmania amazonensis. J. Immunol. 2003, 171, 3705–3710. [Google Scholar] [CrossRef] [PubMed]
- Ji, J.; Sun, J.; Soong, L. Impaired expression of inflammatory cytokines and chemokines at early stages of infection with Leishmania amazonensis. Infect. Immun. 2003, 71, 4278–4288. [Google Scholar] [CrossRef] [PubMed]
- Heinzel, F.P.; Schoenhaut, D.S.; Rerko, R.M.; Rosser, L.E.; Gately, M.K. Recombinant interleukin 12 cures mice infected with Leishmania major. J. Exp. Med. 1993, 177, 1505–1509. [Google Scholar] [CrossRef] [PubMed]
- Sypek, J.P.; Chung, C.L.; Mayor, S.E.; Subramanyam, J.M.; Goldman, S.J.; Sieburth, D.S.; Wolf, S.F.; Schaub, R.G. Resolution of cutaneous leishmaniasis: Interleukin 12 initiates a protective T helper type 1 immune response. J. Exp. Med. 1993, 177, 1797–1802. [Google Scholar] [CrossRef] [PubMed]
- Chatelain, R.; Varkila, K.; Coffman, R.L. IL-4 induces a Th2 response in leishmania major-infected mice. J. Immunol. 1992, 148, 1182–1187. [Google Scholar] [PubMed]
- Peters, N.C.; Egen, J.G.; Secundino, N.; Debrabant, A.; Kimblin, N.; Kamhawi, S.; Lawyer, P.; Fay, M.P.; Germain, R.N.; Sacks, D. In vivo imaging reveals an essential role for neutrophils in leishmaniasis transmitted by sand flies. Science 2008, 321, 970–974. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro-Gomes, F.L.; Peters, N.C.; Debrabant, A.; Sacks, D.L. Efficient capture of infected neutrophils by dendritic cells in the skin inhibits the early anti-leishmania response. PLoS Pathog. 2012, 8, e1002536. [Google Scholar] [CrossRef] [PubMed]
- Conceicao, J.; Davis, R.; Carneiro, P.P.; Giudice, A.; Muniz, A.C.; Wilson, M.E.; Carvalho, E.M.; Bacellar, O. Characterization of neutrophil function in human cutaneous leishmaniasis caused by Leishmania braziliensis. PLoS Negl. Trop. Dis. 2016, 10, e0004715. [Google Scholar] [CrossRef] [PubMed]
- Van Zandbergen, G.; Klinger, M.; Mueller, A.; Dannenberg, S.; Gebert, A.; Solbach, W.; Laskay, T. Cutting edge: Neutrophil granulocyte serves as a vector for leishmania entry into macrophages. J. Immunol. 2004, 173, 6521–6525. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro-Gomes, F.L.; Romano, A.; Lee, S.; Roffe, E.; Peters, N.C.; Debrabant, A.; Sacks, D. Apoptotic cell clearance of Leishmania major-infected neutrophils by dendritic cells inhibits cCD8+ T-cell priming in vitro by MER tyrosine kinase-dependent signaling. Cell Death Dis. 2015, 6, e2018. [Google Scholar] [CrossRef] [PubMed]
- Blank, C.; Fuchs, H.; Rappersberger, K.; Rollinghoff, M.; Moll, H. Parasitism of epidermal Langerhans cells in experimental cutaneous leishmaniasis with Leishmania major. J. Infect. Dis. 1993, 167, 418–425. [Google Scholar] [CrossRef] [PubMed]
- Moll, H.; Fuchs, H.; Blank, C.; Rollinghoff, M. Langerhans cells transport leishmania major from the infected skin to the draining lymph node for presentation to antigen-specific T cells. Eur. J. Immunol. 1993, 23, 1595–1601. [Google Scholar] [CrossRef] [PubMed]
- Ritter, U.; Meissner, A.; Scheidig, C.; Korner, H. CD8 α- and langerin-negative dendritic cells, but not Langerhans cells, act as principal antigen-presenting cells in leishmaniasis. Eur. J. Immunol. 2004, 34, 1542–1550. [Google Scholar] [CrossRef] [PubMed]
- Ng, L.G.; Hsu, A.; Mandell, M.A.; Roediger, B.; Hoeller, C.; Mrass, P.; Iparraguirre, A.; Cavanagh, L.L.; Triccas, J.A.; Beverley, S.M.; et al. Migratory dermal dendritic cells act as rapid sensors of protozoan parasites. PLoS Pathog. 2008, 4, e1000222. [Google Scholar] [CrossRef] [PubMed]
- Brewig, N.; Kissenpfennig, A.; Malissen, B.; Veit, A.; Bickert, T.; Fleischer, B.; Mostbock, S.; Ritter, U. Priming of CD8+ and CD4+ T cells in experimental leishmaniasis is initiated by different dendritic cell subtypes. J. Immunol. 2009, 182, 774–783. [Google Scholar] [CrossRef] [PubMed]
- Kautz-Neu, K.; Noordegraaf, M.; Dinges, S.; Bennett, C.L.; John, D.; Clausen, B.E.; von Stebut, E. Langerhans cells are negative regulators of the anti-leishmania response. J. Exp. Med. 2011, 208, 885–891. [Google Scholar] [CrossRef] [PubMed]
- Hsiao, C.H.; Ueno, N.; Shao, J.Q.; Schroeder, K.R.; Moore, K.C.; Donelson, J.E.; Wilson, M.E. The effects of macrophage source on the mechanism of phagocytosis and intracellular survival of leishmania. Microbes Infect. Inst. Pasteur 2011, 13, 1033–1044. [Google Scholar] [CrossRef] [PubMed]
- Ueno, N.; Wilson, M.E. Receptor-mediated phagocytosis of leishmania: Implications for intracellular survival. Trends Parasitol. 2012, 28, 335–344. [Google Scholar] [CrossRef] [PubMed]
- Stafford, J.L.; Neumann, N.F.; Belosevic, M. Macrophage-mediated innate host defense against protozoan parasites. Crit. Rev. Microbiol. 2002, 28, 187–248. [Google Scholar] [CrossRef] [PubMed]
- Bronte, V.; Murray, P.J. Understanding local macrophage phenotypes in disease: Modulating macrophage function to treat cancer. Nat. Med. 2015, 21, 117–119. [Google Scholar] [CrossRef] [PubMed]
- Minakami, R.; Sumimotoa, H. Phagocytosis-coupled activation of the superoxide-producing phagocyte oxidase, a member of the NADPH oxidase (NOX) family. Int. J. Hematol. 2006, 84, 193–198. [Google Scholar] [CrossRef] [PubMed]
- Gantt, K.R.; Goldman, T.L.; McCormick, M.L.; Miller, M.A.; Jeronimo, S.M.; Nascimento, E.T.; Britigan, B.E.; Wilson, M.E. Oxidative responses of human and murine macrophages during phagocytosis of Leishmania chagasi. J. Immunol. 2001, 167, 893–901. [Google Scholar] [CrossRef] [PubMed]
- Van Assche, T.; Deschacht, M.; da Luz, R.A.; Maes, L.; Cos, P. Leishmania-macrophage interactions: Insights into the redox biology. Free Radic. Biol. Med. 2011, 51, 337–351. [Google Scholar] [CrossRef] [PubMed]
- Liew, F.Y.; Millott, S.; Parkinson, C.; Palmer, R.M.; Moncada, S. Macrophage killing of leishmania parasite in vivo is mediated by nitric oxide from l-arginine. J. Immunol. 1990, 144, 4794–4797. [Google Scholar] [PubMed]
- Bogdan, C.; Moll, H.; Solbach, W.; Rollinghoff, M. Tumor necrosis factor-α in combination with interferon-γ, but not with interleukin 4 activates murine macrophages for elimination of Leishmania major amastigotes. Eur. J. Immunol. 1990, 20, 1131–1135. [Google Scholar] [CrossRef] [PubMed]
- Almeida, T.F.; Palma, L.C.; Mendez, L.C.; Noronha-Dutra, A.A.; Veras, P.S. Leishmania amazonensis fails to induce the release of reactive oxygen intermediates by CBA macrophages. Parasite Immunol. 2012, 34, 492–498. [Google Scholar] [CrossRef] [PubMed]
- Matheoud, D.; Moradin, N.; Bellemare-Pelletier, A.; Shio, M.T.; Hong, W.J.; Olivier, M.; Gagnon, E.; Desjardins, M.; Descoteaux, A. Leishmania evades host immunity by inhibiting antigen cross-presentation through direct cleavage of the SNARE VAMP8. Cell Host Microbe 2013, 14, 15–25. [Google Scholar] [CrossRef] [PubMed]
- Channon, J.Y.; Roberts, M.B.; Blackwell, J.M. A study of the differential respiratory burst activity elicited by promastigotes and amastigotes of Leishmania donovani in murine resident peritoneal macrophages. Immunology 1984, 53, 345–355. [Google Scholar] [PubMed]
- Nandan, D.; Reiner, N.E. Leishmania donovani engages in regulatory interference by targeting macrophage protein tyrosine phosphatase SHP-1. Clin. Immunol. 2005, 114, 266–277. [Google Scholar] [CrossRef] [PubMed]
- Wei, X.Q.; Charles, I.G.; Smith, A.; Ure, J.; Feng, G.J.; Huang, F.P.; Xu, D.; Muller, W.; Moncada, S.; Liew, F.Y. Altered immune responses in mice lacking inducible nitric oxide synthase. Nature 1995, 375, 408–411. [Google Scholar] [CrossRef] [PubMed]
- Mukbel, R.M.; Patten, C., Jr.; Gibson, K.; Ghosh, M.; Petersen, C.; Jones, D.E. Macrophage killing of Leishmania amazonensis amastigotes requires both nitric oxide and superoxide. Am. J. Trop. Med. Hyg. 2007, 76, 669–675. [Google Scholar] [PubMed]
- Novais, F.O.; Nguyen, B.T.; Beiting, D.P.; Carvalho, L.P.; Glennie, N.D.; Passos, S.; Carvalho, E.M.; Scott, P. Human classical monocytes control the intracellular stage of Leishmania braziliensis by reactive oxygen species. J. Infect. Dis. 2014, 209, 1288–1296. [Google Scholar] [CrossRef] [PubMed]
- Weinberg, J.B.; Misukonis, M.A.; Shami, P.J.; Mason, S.N.; Sauls, D.L.; Dittman, W.A.; Wood, E.R.; Smith, G.K.; McDonald, B.; Bachus, K.E.; et al. Human mononuclear phagocyte inducible nitric oxide synthase (iNOS): Analysis of iNOS mRNA, iNOS protein, biopterin, and nitric oxide production by blood monocytes and peritoneal macrophages. Blood 1995, 86, 1184–1195. [Google Scholar] [PubMed]
- Bogdan, C. Natural killer cells in experimental and human leishmaniasis. Front. Cell. Infect. Microbiol. 2012, 2, 69. [Google Scholar] [CrossRef] [PubMed]
- Scharton, T.M.; Scott, P. Natural killer cells are a source of interferon γ that drives differentiation of CD4+ T cell subsets and induces early resistance to Leishmania major in mice. J. Exp. Med. 1993, 178, 567–577. [Google Scholar] [CrossRef] [PubMed]
- Muller, K.; van Zandbergen, G.; Hansen, B.; Laufs, H.; Jahnke, N.; Solbach, W.; Laskay, T. Chemokines, natural killer cells and granulocytes in the early course of Leishmania major infection in mice. Med. Microbiol. Immunol. 2001, 190, 73–76. [Google Scholar] [CrossRef] [PubMed]
- Bajenoff, M.; Breart, B.; Huang, A.Y.; Qi, H.; Cazareth, J.; Braud, V.M.; Germain, R.N.; Glaichenhaus, N. Natural killer cell behavior in lymph nodes revealed by static and real-time imaging. J. Exp. Med. 2006, 203, 619–631. [Google Scholar] [CrossRef] [PubMed]
- Sanabria, M.X.; Vargas-Inchaustegui, D.A.; Xin, L.; Soong, L. Role of natural killer cells in modulating dendritic cell responses to leishmania amazonensis infection. Infect. Immun. 2008, 76, 5100–5109. [Google Scholar] [CrossRef] [PubMed]
- Mailliard, R.B.; Son, Y.I.; Redlinger, R.; Coates, P.T.; Giermasz, A.; Morel, P.A.; Storkus, W.J.; Kalinski, P. Dendritic cells mediate NK cell help for Th1 and CTL responses: Two-signal requirement for the induction of NK cell helper function. J. Immunol. 2003, 171, 2366–2373. [Google Scholar] [CrossRef] [PubMed]
- Prajeeth, C.K.; Haeberlein, S.; Sebald, H.; Schleicher, U.; Bogdan, C. Leishmania-infected macrophages are targets of NK cell-derived cytokines but not of NK cell cytotoxicity. Infect. Immun. 2011, 79, 2699–2708. [Google Scholar] [CrossRef] [PubMed]
- Wakil, A.E.; Wang, Z.E.; Ryan, J.C.; Fowell, D.J.; Locksley, R.M. Interferon γ derived from CD4+ T cells is sufficient to mediate T helper cell type 1 development. J. Exp. Med. 1998, 188, 1651–1656. [Google Scholar] [CrossRef] [PubMed]
- Laouar, Y.; Sutterwala, F.S.; Gorelik, L.; Flavell, R.A. Transforming growth factor-β controls T helper type 1 cell development through regulation of natural killer cell interferon-γ. Nat. Immunol. 2005, 6, 600–607. [Google Scholar] [CrossRef] [PubMed]
- Takashima, A.; Bergstresser, P.R. Cytokine-mediated communication by keratinocytes and Langerhans cells with dendritic epidermal T cells. Semin. Immunol. 1996, 8, 333–339. [Google Scholar] [CrossRef] [PubMed]
- Roebrock, K.; Sunderkotter, C.; Munck, N.A.; Wolf, M.; Nippe, N.; Barczyk, K.; Varga, G.; Vogl, T.; Roth, J.; Ehrchen, J. Epidermal expression of I-TAC (CXCL11) instructs adaptive Th2-type immunity. FASEB J. 2014, 28, 1724–1734. [Google Scholar] [CrossRef] [PubMed]
- Scorza, B.M.; Wacker, M.A.; Messingham, K.; Fairley, J.; Kim, P.; Klingelhutz, A.; Wilson, M.E. Differential activation of human keratinocytes by Leishmania spp. causing localized or disseminated disease. J. Investig. Dermatol. 2017, accepted/in press. [Google Scholar]
Figure 1.
Immune regulation of human macrophages contributing to different pathologic states during Leishmania infection. Human macrophages parasitized by Leishmania spp. are subject to regulation by cytokines present within the skin at the infection site. Inflammatory Type 1 cytokines, IFNγ and TNF, can synergistically induce ROS production by macrophages, thereby inhibiting Leishmania replication within the phagolysosome. TH17 cells are increased in inflammatory MCL lesions. Chemoattractant transcripts (IL8, MCP1, CXCL9, and CXCL10) and transcripts associated with classical (type 1) macrophage activation (iNOS, IL1β) have been measured in inflammatory CL lesions. In a regulatory environment (left), there are low levels of type 1 cytokines with lack of microbicidal effectors. T cell or macrophage-derived cytokines including type 2 cytokines, IL-10 and TGFβ antagonize the effects of IFNγ and TNF, and enhanced polyamines can result in parasite proliferation. TREG cells can suppress the effects of IFNγ. Inhibitory receptors on CD4+ or CD8+ T cells (CTLA4, PD1, and LAG3) and their counter-ligands (CD80, CD86, and PDL1) are associated with T cell exhaustion. Arginase activity is associated with M2-type non-classical macrophage activation. IFNγ: Interferon γ; TNF: Tumor necrosis factor; IL10: Interleukin 10; TGFβ: Transforming growth factor β; ROS: Reactive oxygen species; ADCL: Anergic diffuse cutaneous leishmaniasis; PKDL: Post Kala-Azar dermal leishmaniasis; LCL: Localized cutaneous leishmaniasis; LR: Leishmania recidivans; DL: Disseminated leishmaniasis; MCL: Mucocutaneous leishmaniasis; DTH: Delayed type 1 hypersensitivity; TREG: T regulatory cell; TH: T helper cell; MCP: Monocyte chemoattractant protein; CXCL: Chemokine (C-X-C motif) ligand; CTLA4: Cytotoxic T-lymphocyte associated protein 4; PD1: Programmed cell death protein 1; LAG3: Lymphocyte activation gene 3; PDL1: Programmed death ligand 1.
Figure 1.
Immune regulation of human macrophages contributing to different pathologic states during Leishmania infection. Human macrophages parasitized by Leishmania spp. are subject to regulation by cytokines present within the skin at the infection site. Inflammatory Type 1 cytokines, IFNγ and TNF, can synergistically induce ROS production by macrophages, thereby inhibiting Leishmania replication within the phagolysosome. TH17 cells are increased in inflammatory MCL lesions. Chemoattractant transcripts (IL8, MCP1, CXCL9, and CXCL10) and transcripts associated with classical (type 1) macrophage activation (iNOS, IL1β) have been measured in inflammatory CL lesions. In a regulatory environment (left), there are low levels of type 1 cytokines with lack of microbicidal effectors. T cell or macrophage-derived cytokines including type 2 cytokines, IL-10 and TGFβ antagonize the effects of IFNγ and TNF, and enhanced polyamines can result in parasite proliferation. TREG cells can suppress the effects of IFNγ. Inhibitory receptors on CD4+ or CD8+ T cells (CTLA4, PD1, and LAG3) and their counter-ligands (CD80, CD86, and PDL1) are associated with T cell exhaustion. Arginase activity is associated with M2-type non-classical macrophage activation. IFNγ: Interferon γ; TNF: Tumor necrosis factor; IL10: Interleukin 10; TGFβ: Transforming growth factor β; ROS: Reactive oxygen species; ADCL: Anergic diffuse cutaneous leishmaniasis; PKDL: Post Kala-Azar dermal leishmaniasis; LCL: Localized cutaneous leishmaniasis; LR: Leishmania recidivans; DL: Disseminated leishmaniasis; MCL: Mucocutaneous leishmaniasis; DTH: Delayed type 1 hypersensitivity; TREG: T regulatory cell; TH: T helper cell; MCP: Monocyte chemoattractant protein; CXCL: Chemokine (C-X-C motif) ligand; CTLA4: Cytotoxic T-lymphocyte associated protein 4; PD1: Programmed cell death protein 1; LAG3: Lymphocyte activation gene 3; PDL1: Programmed death ligand 1.

Figure 2.
Schematic representation of the spectrum of human cutaneous leishmaniasis manifestations. The localized disease LCL results in the presence of an IFNγ-dominated response, detected by DTH response and T cells. In MCL, T cell responsiveness to Leishmania antigen further increases towards the hypersensitivity pole, resulting in tissue damage but low parasite burden. The opposite polar responses with low or absent T cell responsiveness, high parasite burden and antibody titers culminate in ADCL. ADCL: Anergic diffuse cutaneous leishmaniasis; PKDL: Post Kala-Azar dermal leishmaniasis; LCL: Localized cutaneous leishmaniasis; LR: Leishmania recidivans; DL: Disseminated leishmaniasis; MCL: Mucocutaneous leishmaniasis; DTH: Delayed type 1 hypersensitivity; TH: T helper cell; TH1: TH1-type immune response; TH2: TH2-type immune response; IFNγ: Interferon gamma; IL-10: Interleukin 10.
Figure 2.
Schematic representation of the spectrum of human cutaneous leishmaniasis manifestations. The localized disease LCL results in the presence of an IFNγ-dominated response, detected by DTH response and T cells. In MCL, T cell responsiveness to Leishmania antigen further increases towards the hypersensitivity pole, resulting in tissue damage but low parasite burden. The opposite polar responses with low or absent T cell responsiveness, high parasite burden and antibody titers culminate in ADCL. ADCL: Anergic diffuse cutaneous leishmaniasis; PKDL: Post Kala-Azar dermal leishmaniasis; LCL: Localized cutaneous leishmaniasis; LR: Leishmania recidivans; DL: Disseminated leishmaniasis; MCL: Mucocutaneous leishmaniasis; DTH: Delayed type 1 hypersensitivity; TH: T helper cell; TH1: TH1-type immune response; TH2: TH2-type immune response; IFNγ: Interferon gamma; IL-10: Interleukin 10.

Figure 3.
Neutrophils at the cutaneous site of Leishmania spp. inoculation in mice. Neutrophils are rapidly recruited to the site of Leishmania infection, initiated by either sand fly or needle inoculation, where they phagocytose parasites. Although some Leishmania are killed by neutrophils, some survive within neutrophils until they undergo apoptosis. Uptake of apoptotic infected neutrophils by macrophages leads to MERTK signaling and increased TGFβ release, promoting an anti-inflammatory macrophage state and intracellular parasite replication. DCs that take up apoptotic infected neutrophils show a decreased capacity to present antigen to naïve CD4+ T cells. MERTK: Mer tyrosine kinase; TGFβ: Transforming growth factor β, TH0: naïve CD4+ T cell.
Figure 3.
Neutrophils at the cutaneous site of Leishmania spp. inoculation in mice. Neutrophils are rapidly recruited to the site of Leishmania infection, initiated by either sand fly or needle inoculation, where they phagocytose parasites. Although some Leishmania are killed by neutrophils, some survive within neutrophils until they undergo apoptosis. Uptake of apoptotic infected neutrophils by macrophages leads to MERTK signaling and increased TGFβ release, promoting an anti-inflammatory macrophage state and intracellular parasite replication. DCs that take up apoptotic infected neutrophils show a decreased capacity to present antigen to naïve CD4+ T cells. MERTK: Mer tyrosine kinase; TGFβ: Transforming growth factor β, TH0: naïve CD4+ T cell.

Figure 4.
Monocytic cells in control of Leishmania replication in murine skin. Epidermal cytokines can modulate this process. Monocyte derived dendritic cells (Mo-DCs) transport antigen to draining lymph nodes for presentation to naïve CD4+ T cells and resultant TH1 cells traffic back to the site of infection where they secrete IFNγ. Recruited inflammatory monocytes are the primary host cell allowing intracellular survival and replication of Leishmania spp. Monocytes recruited to the site of intradermal Leishmania inoculation can differentiate locally into dendritic cells or macrophages. Once phagocytosed by macrophages, Leishmania use a variety of virulence mechanisms to suppress the host cell microbicidal response and replicate intracellularly. Infected macrophages can either facilitate intracellular survival and/or replication, or if exposed to activating signals such as IFNγ from TH1-type cells or NK cells, can upregulate iNOS. The combination of reactive oxygen and reactive nitrogen species can contribute to killing of intracellular parasites. Mo-DC: Monocyte derived dendritic cell; NK cell: Natural killer cell; IFNγ: Interferon γ; IL-12: Interleukin-12; iNOS: Inducible nitric oxide synthase; TH1: T helper 1 cell, TH0: Naïve CD4+ T cell.
Figure 4.
Monocytic cells in control of Leishmania replication in murine skin. Epidermal cytokines can modulate this process. Monocyte derived dendritic cells (Mo-DCs) transport antigen to draining lymph nodes for presentation to naïve CD4+ T cells and resultant TH1 cells traffic back to the site of infection where they secrete IFNγ. Recruited inflammatory monocytes are the primary host cell allowing intracellular survival and replication of Leishmania spp. Monocytes recruited to the site of intradermal Leishmania inoculation can differentiate locally into dendritic cells or macrophages. Once phagocytosed by macrophages, Leishmania use a variety of virulence mechanisms to suppress the host cell microbicidal response and replicate intracellularly. Infected macrophages can either facilitate intracellular survival and/or replication, or if exposed to activating signals such as IFNγ from TH1-type cells or NK cells, can upregulate iNOS. The combination of reactive oxygen and reactive nitrogen species can contribute to killing of intracellular parasites. Mo-DC: Monocyte derived dendritic cell; NK cell: Natural killer cell; IFNγ: Interferon γ; IL-12: Interleukin-12; iNOS: Inducible nitric oxide synthase; TH1: T helper 1 cell, TH0: Naïve CD4+ T cell.


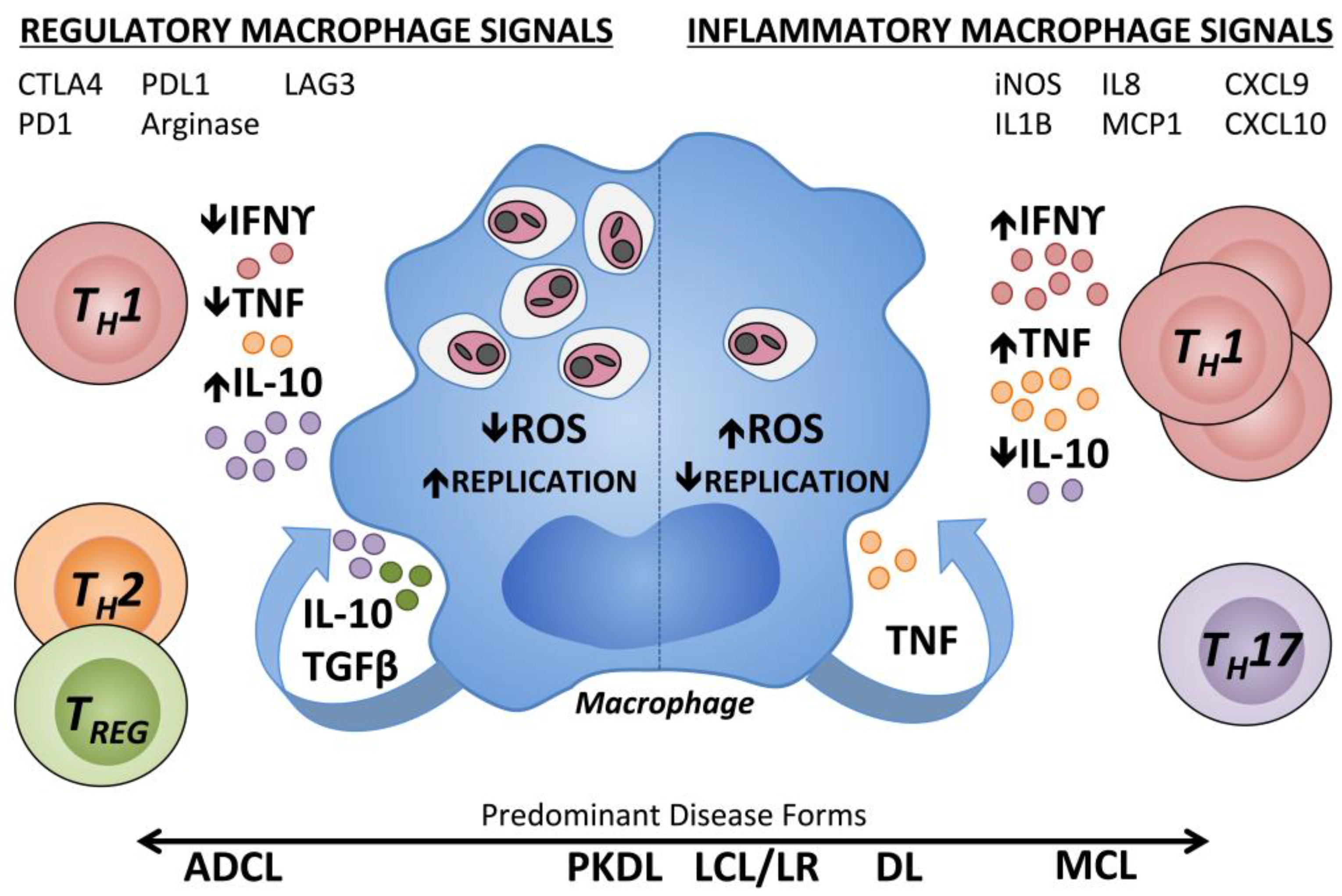{kind=link}
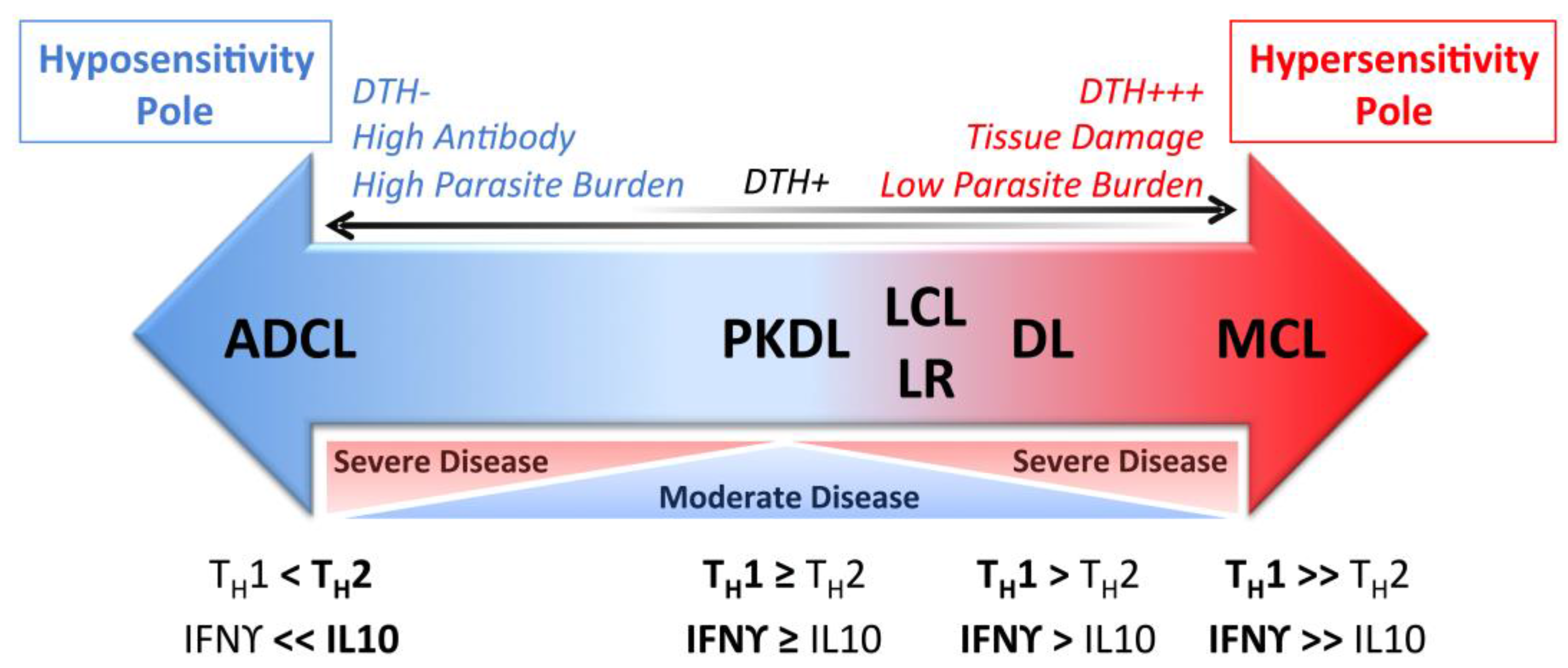{kind=link}
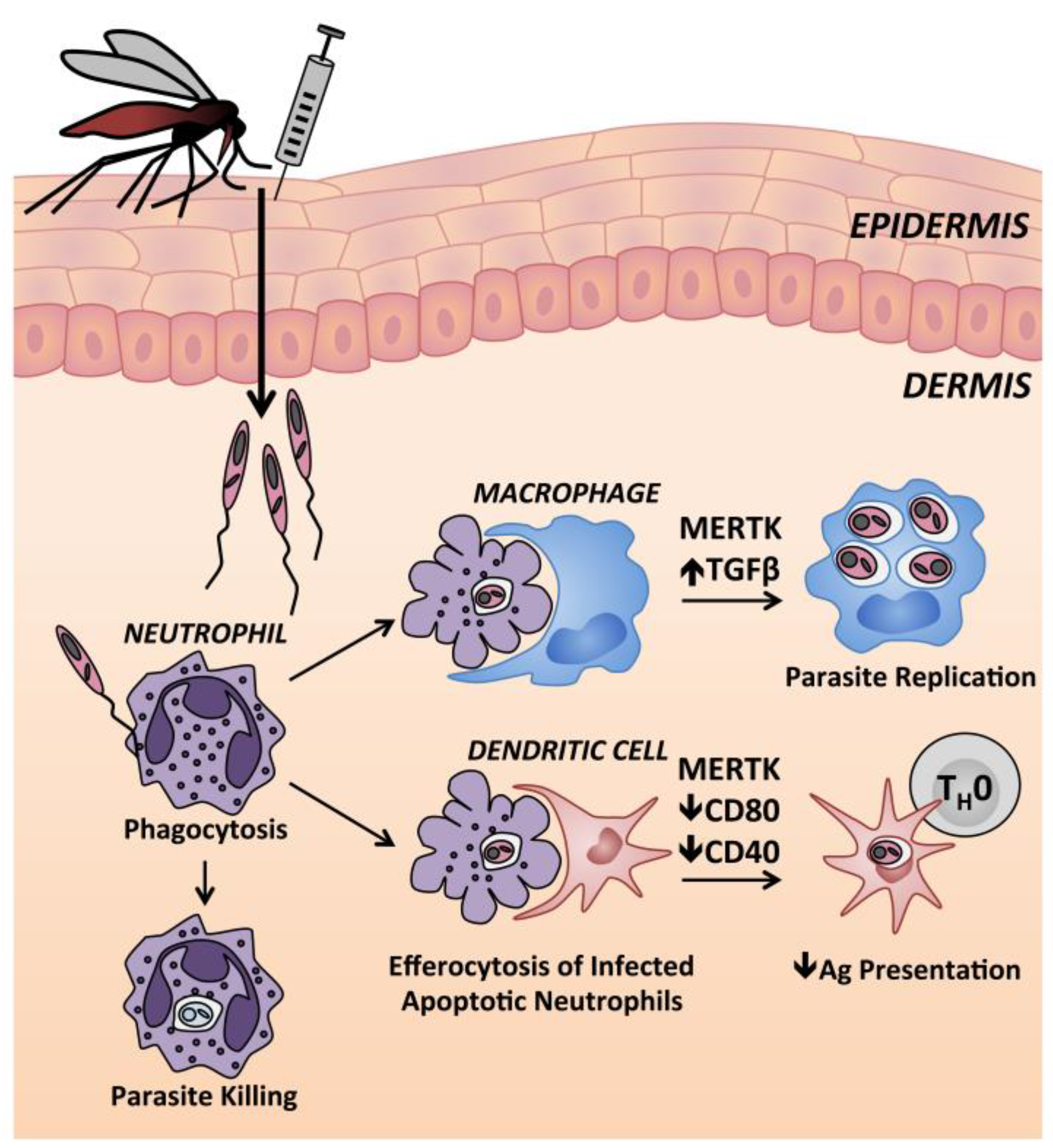{kind=link}
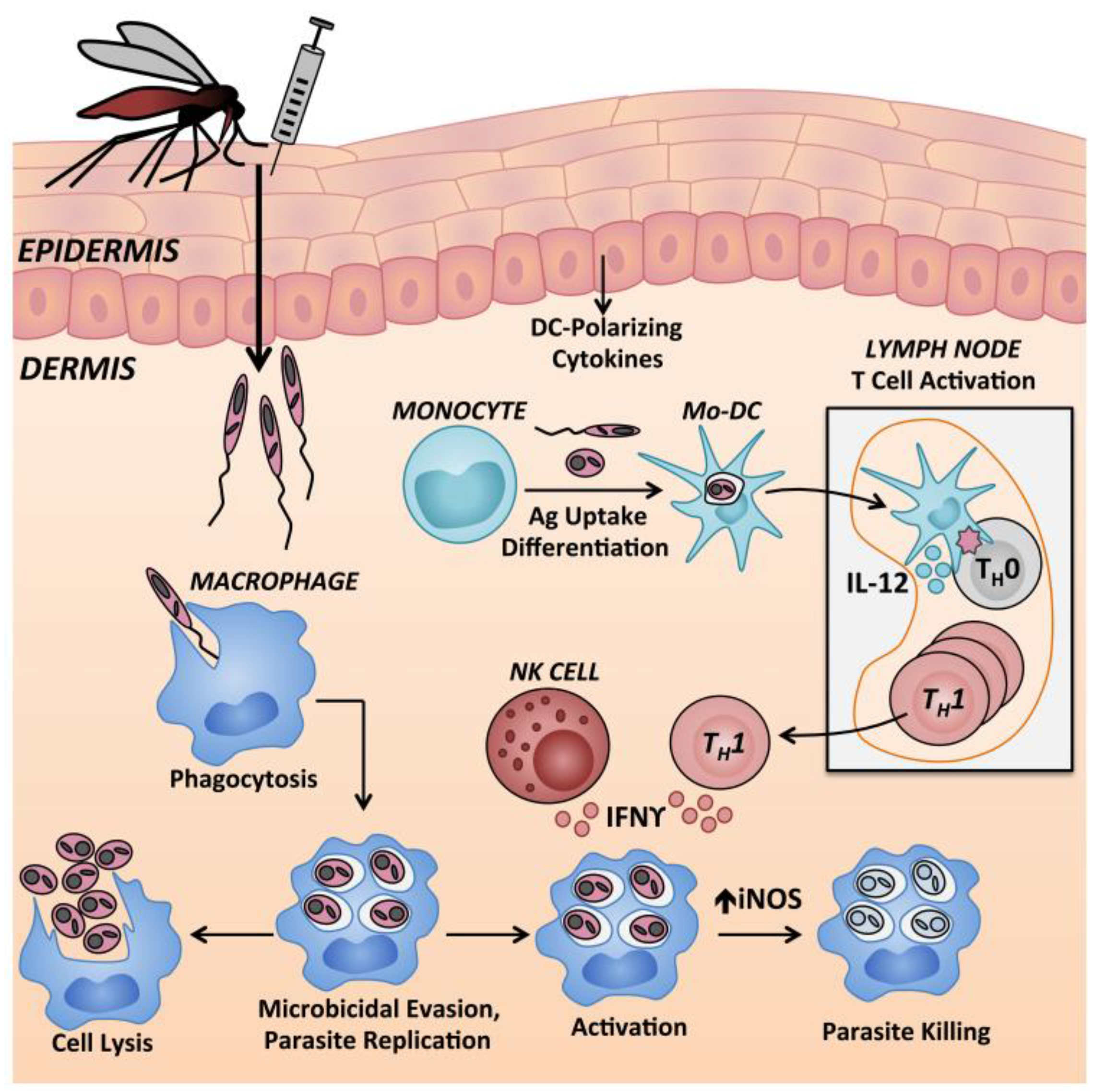{kind=link}
Table 1.
Major clinical forms of tegumentary leishmaniasis and causative Leishmania species.
| Clinical Syndrome | Causative Species 1 | Clinical Manifestation |
|---|---|---|
| Localized Cutaneous Leishmaniasis (LCL) | L. (L.) major [3] L. (L.) mexicana [7] L. (L.) amazonensis [7] L. (V.) braziliensis [7] L. (L.) tropica [8] L. (L.) aethiopica [9] L. (V.) panamanensis [7] L. (L.) infantum [10] L. (L.) donovani [11] | Single or limited number of lesions; ulcers formed can be wet or dry with raised crateriform border. Moderate parasite loads in biopsies of the ulcer border; positive DTH 2 response [12] |
| Mucosal Leishmaniasis [13] | L. (V.) braziliensis [14] L. (V.) panamanensis [15] L. (V.) guyanensis [16] L. (L.) amazonensis [14,17] | Highly inflammatory lesions involving mucosal membranes; can be disfiguring. Rare parasite forms present in biopsies; strong DTH response [12] |
| Anergic Diffuse Cutaneous Leishmaniasis (ADCL) | L. (L.) amazonensis [18] L. (L.) mexicana [19] L. (V.) pifanoi [20] L. (L.) aethiopica [21] L. (L.) major [22] | Multiple, disseminated, non-ulcerative nodular lesions; many parasites in lesions; absent DTH response (anergy) [20,23] |
| Disseminated Leishmaniasis (DL) | L. (V.) braziliensis [24] L. (V.) panamanensis [25] L. (V.) guyanensis [25,26] L. (L.) amazonensis [24] | Numerous papular/acneiform lesions in ≥2 non-contiguous areas of the body, commonly involving mucosal membranes. Few parasites in lesions; strong DTH response [27] |
| Post-Kala Azar Dermal Leishmaniasis (PKDL) | L. (L.) donovani [1] | Hypopigmented macular, maculopapular, or nodular rash. Interferon γ (IFNγ) response to Leishmania antigens. Parasites are present in lesions [28,29,30] |
1 (L.) denotes Leishmania subgenus. (V.) denotes Viannia subgenus. 2 DTH refers to a delayed type hypersensitivity response to Leishmania antigen, a test that is also called the Leishmania skin test (LST) or Montenegro test.
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Scorza, B.M.; Carvalho, E.M.; Wilson, M.E. Cutaneous Manifestations of Human and Murine Leishmaniasis. Int. J. Mol. Sci. 2017, 18, 1296. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms18061296
AMA Style
Scorza BM, Carvalho EM, Wilson ME. Cutaneous Manifestations of Human and Murine Leishmaniasis. International Journal of Molecular Sciences. 2017; 18(6):1296. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms18061296
Chicago/Turabian StyleScorza, Breanna M., Edgar M. Carvalho, and Mary E. Wilson. 2017. "Cutaneous Manifestations of Human and Murine Leishmaniasis" International Journal of Molecular Sciences 18, no. 6: 1296. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms18061296
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.