Pharmacogenomic Variants May Influence the Urinary Excretion of Novel Kidney Injury Biomarkers in Patients Receiving Cisplatin

Abstract
:1. Introduction
2. Results
2.1. Patient Characteristics
2.2. Associations between SLC22A2 and SLC31A1 Variants and Estimated Glomerular Filtration Rate (eGFR) in Patients Receiving Cisplatin
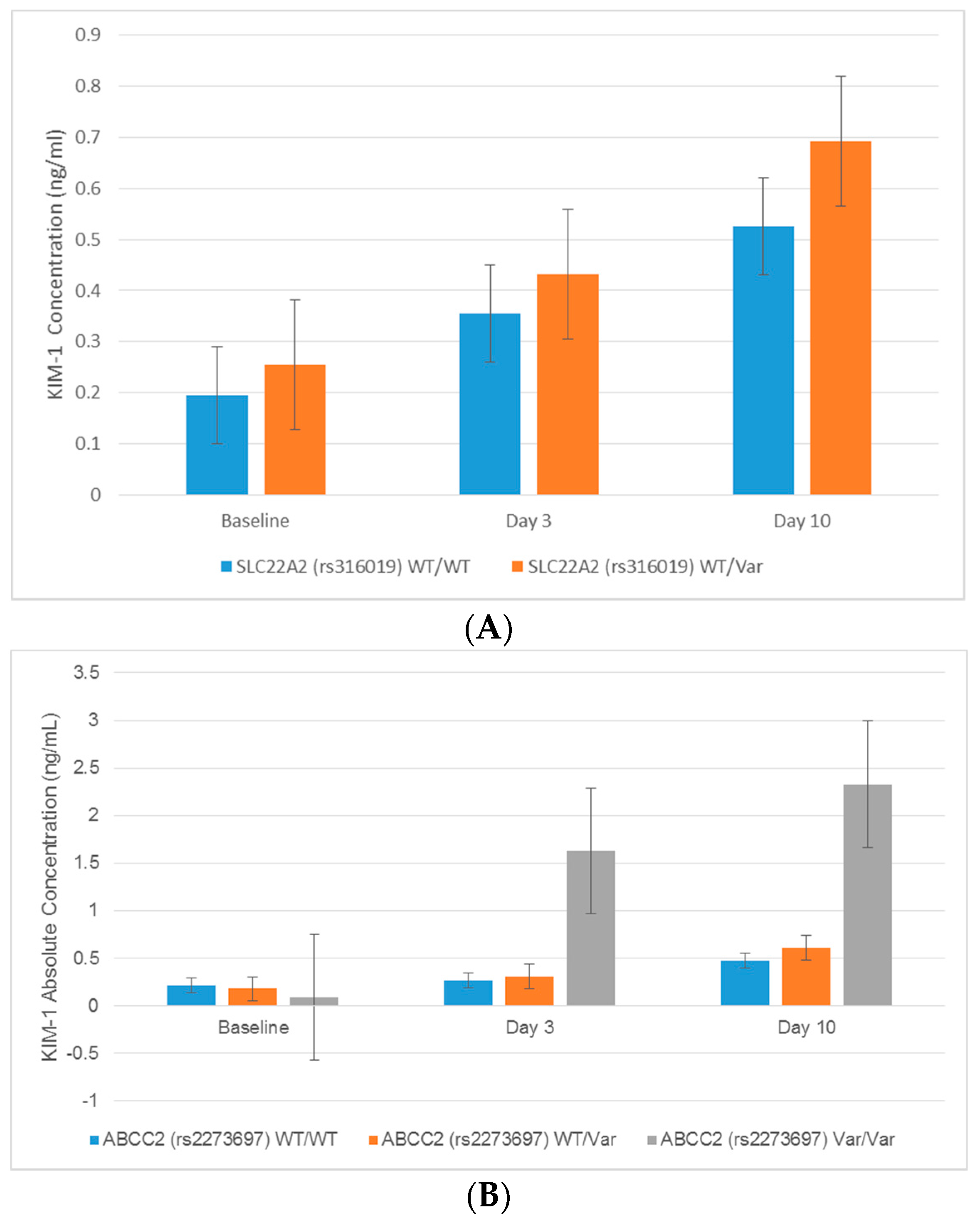
2.3. Associations between Transporter Gene Variants and Novel Urinary Biomarkers of Kidney Injury in Patients Receiving Cisplatin
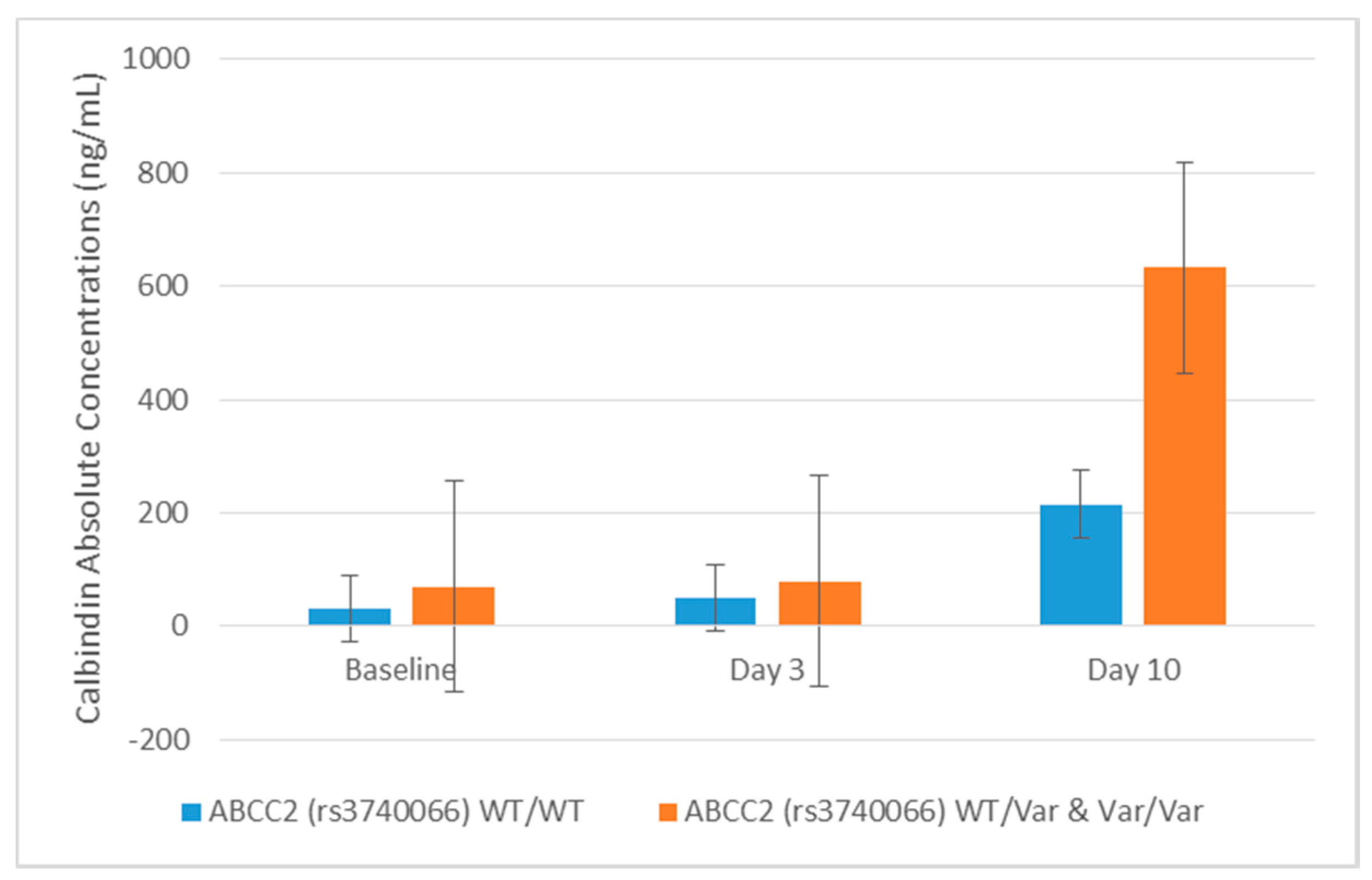
2.4. Association between Cisplatin Metabolism Genes and Novel Urinary Biomarkers of Kidney Injury in Patients Receiving Cisplatin
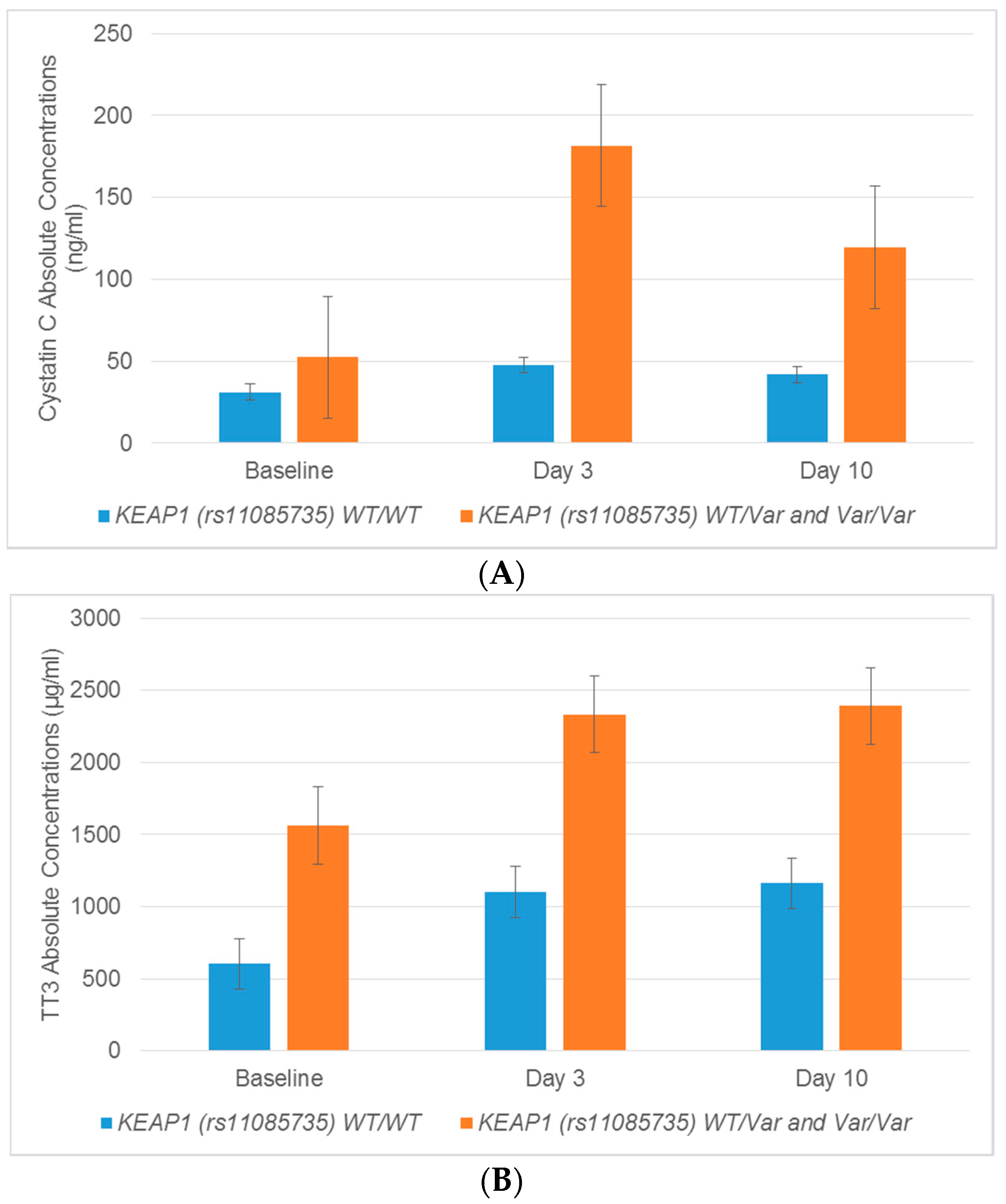
2.5. Association between Regulatory Genes and Novel Urinary Biomarkers of Kidney Injury in Patients Receiving Cisplatin
3. Discussion
4. Materials and Methods
4.1. General Reagents
4.2. Study Population
4.3. DNA Isolation
4.4. Genotyping
4.5. Collection of Urine Samples
4.6. Assessment of Urinary Biomarkers
4.7. Statistical Analysis
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Bolis, G.; Favalli, G.; Danese, S.; Zanaboni, F.; Mangili, G.; Scarabelli, C.; Tateo, S.; Valsecchi, M.G.; Scarfone, G.; Richiardi, G.; et al. Weekly cisplatin given for 2 months versus cisplatin plus cyclophosphamide given for 5 months after cytoreductive surgery for advanced ovarian cancer. J. Clin. Oncol. 1997, 15, 1938–1944. [Google Scholar] [CrossRef] [PubMed]
- Coppin, C.M.; Gospodarowicz, M.K.; James, K.; Tannock, I.F.; Zee, B.; Carson, J.; Pater, J.; Sullivan, L.D. Improved local control of invasive bladder cancer by concurrent cisplatin and preoperative or definitive radiation. The National Cancer Institute of Canada Clinical Trials Group. J. Clin. Oncol. 1996, 14, 2901–2907. [Google Scholar] [CrossRef] [PubMed]
- Gatzemeier, U.; von Pawel, J.; Gottfried, M.; ten Velde, G.P.; Mattson, K.; de Marinis, F.; Harper, P.; Salvati, F.; Robinet, G.; Lucenti, A.; et al. Phase III comparative study of high-dose cisplatin versus a combination of paclitaxel and cisplatin in patients with advanced non-small-cell lung cancer. J. Clin. Oncol. 2000, 18, 3390–3399. [Google Scholar] [CrossRef] [PubMed]
- Hoskins, P.; Eisenhauer, E.; Vergote, I.; Dubuc-Lissoir, J.; Fisher, B.; Grimshaw, R.; Oza, A.; Plante, M.; Stuart, G.; Vermorken, J. Phase II feasibility study of sequential couplets of Cisplatin/Topotecan followed by paclitaxel/cisplatin as primary treatment for advanced epithelial ovarian cancer: A National Cancer Institute of Canada Clinical Trials Group Study. J. Clin. Oncol. 2000, 18, 4038–4044. [Google Scholar] [CrossRef] [PubMed]
- Pabla, N.; Dong, Z. Cisplatin nephrotoxicity: Mechanisms and renoprotective strategies. Kidney Int. 2008, 73, 994–1007. [Google Scholar] [CrossRef] [PubMed]
- Planting, A.S.; Catimel, G.; de Mulder, P.H.; de Graeff, A.; Hoppener, F.; Verweij, J.; Oster, W.; Vermorken, J.B. Randomized study of a short course of weekly cisplatin with or without amifostine in advanced head and neck cancer. EORTC Head and Neck Cooperative Group. Ann. Oncol. 1999, 10, 693–700. [Google Scholar] [CrossRef] [PubMed]
- Rose, P.G.; Bundy, B.N.; Watkins, E.B.; Thigpen, J.T.; Deppe, G.; Maiman, M.A.; Clarke-Pearson, D.L.; Insalaco, S. Concurrent cisplatin-based radiotherapy and chemotherapy for locally advanced cervical cancer. N. Engl. J. Med. 1999, 340, 1144–1153. [Google Scholar] [CrossRef] [PubMed]
- Lebwohl, D.; Canetta, R. Clinical development of platinum complexes in cancer therapy: An historical perspective and an update. Eur. J. Cancer 1998, 34, 1522–1534. [Google Scholar] [CrossRef]
- Shiraishi, F.; Curtis, L.M.; Truong, L.; Poss, K.; Visner, G.A.; Madsen, K.; Nick, H.S.; Agarwal, A. Heme oxygenase-1 gene ablation or expression modulates cisplatin-induced renal tubular apoptosis. Am. J. Physiol. Renal. Physiol. 2000, 278, F726–F736. [Google Scholar] [PubMed]
- Ozkok, A.; Edelstein, C.L. Pathophysiology of cisplatin-induced acute kidney injury. BioMed Res. Int. 2014, 2014, 967826. [Google Scholar] [CrossRef] [PubMed]
- Bag, A.; Jyala, N.S.; Bag, N. Cytochrome P450 1A1 genetic polymorphisms as cancer biomarkers. Indian J. Cancer 2015, 52, 479–489. [Google Scholar] [CrossRef] [PubMed]
- Townsend, D.M.; Deng, M.; Zhang, L.; Lapus, M.G.; Hanigan, M.H. Metabolism of Cisplatin to a nephrotoxin in proximal tubule cells. J. Am. Soc. Nephrol. 2003, 14, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Ciarimboli, G.; Deuster, D.; Knief, A.; Sperling, M.; Holtkamp, M.; Edemir, B.; Pavenstadt, H.; Lanvers-Kaminsky, C.; am Zehnhoff-Dinnesen, A.; Schinkel, A.H.; et al. Organic cation transporter 2 mediates cisplatin-induced oto- and nephrotoxicity and is a target for protective interventions. Am. J. Pathol. 2010, 176, 1169–1180. [Google Scholar] [CrossRef] [PubMed]
- Ishida, S.; Lee, J.; Thiele, D.J.; Herskowitz, I. Uptake of the anticancer drug cisplatin mediated by the copper transporter Ctr1 in yeast and mammals. Proc. Natl. Acad. Sci. USA 2002, 99, 14298–14302. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Gonzalez, P.D.; Lopez-Hernandez, F.J.; Lopez-Novoa, J.M.; Morales, A.I. An integrative view of the pathophysiological events leading to cisplatin nephrotoxicity. Crit. Rev. Toxicol. 2011, 41, 803–821. [Google Scholar] [CrossRef] [PubMed]
- Hanigan, M.H.; Lykissa, E.D.; Townsend, D.M.; Ou, C.N.; Barrios, R.; Lieberman, M.W. γ-Glutamyl transpeptidase-deficient mice are resistant to the nephrotoxic effects of cisplatin. Am. J. Pathol. 2001, 159, 1889–1894. [Google Scholar] [CrossRef]
- Ishikawa, T.; Wright, C.D.; Ishizuka, H. GS-X pump is functionally overexpressed in cis-diamminedichloroplatinum (II)-resistant human leukemia HL-60 cells and down-regulated by cell differentiation. J. Biol. Chem. 1994, 269, 29085–29093. [Google Scholar] [PubMed]
- Nakamura, T.; Yonezawa, A.; Hashimoto, S.; Katsura, T.; Inui, K. Disruption of multidrug and toxin extrusion MATE1 potentiates cisplatin-induced nephrotoxicity. Biochem. Pharmacol. 2010, 80, 1762–1767. [Google Scholar] [CrossRef] [PubMed]
- Wen, X.; Buckley, B.; McCandlish, E.; Goedken, M.J.; Syed, S.; Pelis, R.; Manautou, J.E.; Aleksunes, L.M. Transgenic expression of the human MRP2 transporter reduces cisplatin accumulation and nephrotoxicity in Mrp2-null mice. Am. J. Pathol. 2014, 184, 1299–1308. [Google Scholar] [CrossRef] [PubMed]
- Filipski, K.K.; Mathijssen, R.H.; Mikkelsen, T.S.; Schinkel, A.H.; Sparreboom, A. Contribution of organic cation transporter 2 (OCT2) to cisplatin-induced nephrotoxicity. Clin. Pharmacol. Ther. 2009, 86, 396–402. [Google Scholar] [CrossRef] [PubMed]
- Friling, R.S.; Bensimon, A.; Tichauer, Y.; Daniel, V. Xenobiotic-inducible expression of murine glutathione S-transferase Ya subunit gene is controlled by an electrophile-responsive element. Proc. Natl. Acad. Sci. USA 1990, 87, 6258–6262. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.; Sherratt, P.J.; Pickett, C.B. Regulatory mechanisms controlling gene expression mediated by the antioxidant response element. Annu. Rev. Pharmacol. Toxicol. 2003, 43, 233–260. [Google Scholar] [CrossRef] [PubMed]
- Rushmore, T.H.; Morton, M.R.; Pickett, C.B. The antioxidant responsive element. Activation by oxidative stress and identification of the DNA consensus sequence required for functional activity. J. Biol. Chem. 1991, 266, 11632–11639. [Google Scholar] [PubMed]
- Kansanen, E.; Kuosmanen, S.M.; Leinonen, H.; Levonen, A.L. The κ1-Nrf2 pathway: Mechanisms of activation and dysregulation in cancer. Redox Biol. 2013, 1, 45–49. [Google Scholar] [CrossRef] [PubMed]
- Aleksunes, L.M.; Goedken, M.J.; Rockwell, C.E.; Thomale, J.; Manautou, J.E.; Klaassen, C.D. Transcriptional regulation of renal cytoprotective genes by Nrf2 and its potential use as a therapeutic target to mitigate cisplatin-induced nephrotoxicity. J. Pharmacol. Exp. Ther. 2010, 335, 2–12. [Google Scholar] [CrossRef] [PubMed]
- Star, R.A. Treatment of acute renal failure. Kidney Int. 1998, 54, 1817–1831. [Google Scholar] [CrossRef] [PubMed]
- Khwaja, A. KDIGO clinical practice guidelines for acute kidney injury. Nephron Clin. Pract. 2012, 120, c179–c184. [Google Scholar] [CrossRef] [PubMed]
- Group, K.A.W. KDIGO clinical practice guideline for acute kidney injury. Kidney Int. 2012, 17, 1–138. [Google Scholar]
- Han, W.K.; Bailly, V.; Abichandani, R.; Thadhani, R.; Bonventre, J.V. Kidney Injury Molecule-1 (KIM-1): A novel biomarker for human renal proximal tubule injury. Kidney Int. 2002, 62, 237–244. [Google Scholar] [CrossRef] [PubMed]
- Takashi, M.; Zhu, Y.; Miyake, K.; Kato, K. Urinary 28-kD calbindin-D as a new marker for damage to distal renal tubules caused by cisplatin-based chemotherapy. Urol. Int. 1996, 56, 174–179. [Google Scholar] [CrossRef] [PubMed]
- Waring, W.S.; Moonie, A. Earlier recognition of nephrotoxicity using novel biomarkers of acute kidney injury. Clin. Toxicol. 2011, 49, 720–728. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Jin, H.; Holder, D.; Ozer, J.S.; Villarreal, S.; Shughrue, P.; Shi, S.; Figueroa, D.J.; Clouse, H.; Su, M.; et al. Urinary biomarkers trefoil factor 3 and albumin enable early detection of kidney tubular injury. Nat. Biotechnol. 2010, 28, 470–477. [Google Scholar] [CrossRef] [PubMed]
- Levey, A.S.; Coresh, J.; Balk, E.; Kausz, A.T.; Levin, A.; Steffes, M.W.; Hogg, R.J.; Perrone, R.D.; Lau, J.; Eknoyan, G. National Kidney Foundation practice guidelines for chronic kidney disease: Evaluation, classification, and stratification. Ann. Intern. Med. 2003, 139, 137–147. [Google Scholar] [CrossRef] [PubMed]
- Bai, X.; Chen, Y.; Hou, X.; Huang, M.; Jin, J. Emerging role of NRF2 in chemoresistance by regulating drug-metabolizing enzymes and efflux transporters. Drug Metab. Rev. 2016, 48, 541–567. [Google Scholar] [CrossRef] [PubMed]
- Atilano-Roque, A.; Aleksunes, L.M.; Joy, M.S. Bardoxolone methyl modulates efflux transporter and detoxifying enzyme expression in cisplatin-induced kidney cell injury. Toxicol. Lett. 2016, 259, 52–59. [Google Scholar] [CrossRef] [PubMed]
- Lanvers-Kaminsky, C.; Sprowl, J.A.; Malath, I.; Deuster, D.; Eveslage, M.; Schlatter, E.; Mathijssen, R.H.; Boos, J.; Jurgens, H.; Am Zehnhoff-Dinnesen, A.G.; et al. Human OCT2 variant c.808G>T confers protection effect against cisplatin-induced ototoxicity. Pharmacogenomics 2015, 16, 323–332. [Google Scholar] [CrossRef] [PubMed]
- Qian, C.Y.; Zheng, Y.; Wang, Y.; Chen, J.; Liu, J.Y.; Zhou, H.H.; Yin, J.Y.; Liu, Z.Q. Associations of genetic polymorphisms of the transporters organic cation transporter 2 (OCT2), multidrug and toxin extrusion 1 (MATE1), and ATP-binding cassette subfamily C member 2 (ABCC2) with platinum-based chemotherapy response and toxicity in non-small cell lung cancer patients. Chin. J. Cancer 2016, 35, 85. [Google Scholar] [PubMed]
- Christensen, M.M.; Pedersen, R.S.; Stage, T.B.; Brasch-Andersen, C.; Nielsen, F.; Damkier, P.; Beck-Nielsen, H.; Brosen, K. A gene-gene interaction between polymorphisms in the OCT2 and MATE1 genes influences the renal clearance of metformin. Pharmacogenet. Genom. 2013, 23, 526–534. [Google Scholar] [CrossRef] [PubMed]
- Wanga, V.; Venuto, C.; Morse, G.D.; Acosta, E.P.; Daar, E.S.; Haas, D.W.; Li, C.; Shepherd, B.E. Genomewide association study of tenofovir pharmacokinetics and creatinine clearance in AIDS Clinical Trials Group protocol A5202. Pharmacogenet. Genom. 2015, 25, 450–461. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Duan, L.; Zhou, B.; Ma, R.; Zhou, H.; Liu, Z. Prediction of copper transport protein 1 (CTR1) genotype on severe cisplatin induced toxicity in non-small cell lung cancer (NSCLC) patients. Lung Cancer 2012, 77, 438–442. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Duan, L.; Zhou, B.; Ma, R.; Zhou, H.; Liu, Z. Genetic polymorphism of copper transporter protein 1 is related to platinum resistance in Chinese non-small cell lung carcinoma patients. Clin. Exp. Pharmacol. Physiol. 2012, 39, 786–792. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Teranishi, K.; Li, S.; Yee, S.W.; Hesselson, S.; Stryke, D.; Johns, S.J.; Ferrin, T.E.; Kwok, P.; Giacomini, K.M. Genetic variants in multidrug and toxic compound extrusion-1, hMATE1, alter transport function. Pharmacogenom. J. 2009, 9, 127–136. [Google Scholar] [CrossRef] [PubMed]
- Becker, M.L.; Visser, L.E.; van Schaik, R.H.; Hofman, A.; Uitterlinden, A.G.; Stricker, B.H. Interaction between polymorphisms in the OCT1 and MATE1 transporter and metformin response. Pharmacogenet. Genom. 2010, 20, 38–44. [Google Scholar] [CrossRef] [PubMed]
- He, R.; Zhang, D.; Lu, W.; Zheng, T.; Wan, L.; Liu, F.; Jia, W. SLC47A1 gene rs2289669 G>A variants enhance the glucose-lowering effect of metformin via delaying its excretion in Chinese type 2 diabetes patients. Diabetes Res. Clin. Pract. 2015, 109, 57–63. [Google Scholar] [CrossRef] [PubMed]
- Stocker, S.L.; Morrissey, K.M.; Yee, S.W.; Castro, R.A.; Xu, L.; Dahlin, A.; Ramirez, A.H.; Roden, D.M.; Wilke, R.A.; McCarty, C.A.; et al. The effect of novel promoter variants in MATE1 and MATE2 on the pharmacokinetics and pharmacodynamics of metformin. Clin. Pharmacol. Ther. 2013, 93, 186–194. [Google Scholar] [CrossRef] [PubMed]
- Xiao, D.; Guo, Y.; Li, X.; Yin, J.Y.; Zheng, W.; Qiu, X.W.; Xiao, L.; Liu, R.R.; Wang, S.Y.; Gong, W.J.; et al. The Impacts of SLC22A1 rs594709 and SLC47A1 rs2289669 Polymorphisms on Metformin Therapeutic Efficacy in Chinese Type 2 Diabetes Patients. Int. J. Endocrinol. 2016, 2016, 4350712. [Google Scholar] [CrossRef] [PubMed]
- Ma, C.L.; Wu, X.Y.; Jiao, Z.; Hong, Z.; Wu, Z.Y.; Zhong, M.K. SCN1A, ABCC2 and UGT2B7 gene polymorphisms in association with individualized oxcarbazepine therapy. Pharmacogenomics 2015, 16, 347–360. [Google Scholar] [CrossRef] [PubMed]
- Qu, J.; Zhou, B.T.; Yin, J.Y.; Xu, X.J.; Zhao, Y.C.; Lei, G.H.; Tang, Q.; Zhou, H.H.; Liu, Z.Q. ABCC2 polymorphisms and haplotype are associated with drug resistance in Chinese epileptic patients. CNS Neurosci. Ther. 2012, 18, 647–651. [Google Scholar] [CrossRef] [PubMed]
- Sha’ari, H.M.; Haerian, B.S.; Baum, L.; Saruwatari, J.; Tan, H.J.; Rafia, M.H.; Raymond, A.A.; Kwan, P.; Ishitsu, T.; Nakagawa, K.; et al. ABCC2 rs2273697 and rs3740066 polymorphisms and resistance to antiepileptic drugs in Asia Pacific epilepsy cohorts. Pharmacogenomics 2014, 15, 459–466. [Google Scholar] [CrossRef] [PubMed]
- Ufer, M.; von Stulpnagel, C.; Muhle, H.; Haenisch, S.; Remmler, C.; Majed, A.; Plischke, H.; Stephani, U.; Kluger, G.; Cascorbi, I. Impact of ABCC2 genotype on antiepileptic drug response in Caucasian patients with childhood epilepsy. Pharmacogenet. Genom. 2011, 21, 624–630. [Google Scholar] [CrossRef] [PubMed]
- Nishijima, T.; Komatsu, H.; Higasa, K.; Takano, M.; Tsuchiya, K.; Hayashida, T.; Oka, S.; Gatanaga, H. Single Nucleotide Polymorphisms in ABCC2 Associate With Tenofovir-Induced Kidney Tubular Dysfunction in Japanese Patients with HIV-1 Infection: A Pharmacogenetic Study. Clin. Infect. Dis. 2012, 55, 1558–1567. [Google Scholar] [CrossRef] [PubMed]
- Sprowl, J.A.; Gregorc, V.; Lazzari, C.; Mathijssen, R.H.; Loos, W.J.; Sparreboom, A. Associations between ABCC2 polymorphisms and cisplatin disposition and efficacy. Clin. Pharmacol. Ther. 2012, 91, 1022–1026. [Google Scholar] [CrossRef] [PubMed]
- Lecomte, T.; Landi, B.; Beaune, P.; Laurent-Puig, P.; Loriot, M.A. Glutathione S-transferase P1 polymorphism (Ile105Val) predicts cumulative neuropathy in patients receiving oxaliplatin-based chemotherapy. Clin. Cancer Res. 2006, 12, 3050–3066. [Google Scholar] [CrossRef] [PubMed]
- Stoehlmacher, J.; Park, D.J.; Zhang, W.; Yang, D.; Groshen, S.; Zahedy, S.; Lenz, H.J. A multivariate analysis of genomic polymorphisms: Prediction of clinical outcome to 5-FU/oxaliplatin combination chemotherapy in refractory colorectal cancer. Br. J. Cancer 2004, 91, 344–354. [Google Scholar] [CrossRef] [PubMed]
- Spracklen, T.F.; Vorster, A.A.; Ramma, L.; Dalvie, S.; Ramesar, R.S. Promoter region variation in NFE2L2 influences susceptibility to ototoxicity in patients exposed to high cumulative doses of cisplatin. Pharmacogenomics J. 2016. [Google Scholar] [CrossRef] [PubMed]
- World Medical Association Declaration of Helsinki. Ethical principles for medical research involving human subjects. JAMA 2013, 310, 2191–2194. [Google Scholar]
- George, B.; Wen, X.; Mercke, N.; Gomez, M.; O’Bryant, C.; Bowles, D.W.; Hu, Y.; Hogan, S.L.; Joy, M.S.; Aleksunes, L.M. Profiling of kidney injury biomarkers in patients receiving cisplatin: Time-dependent changes in absence of clinical nephrotoxicity. Clin. Pharmacol. Ther. 2017, 101, 510–518. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Data Presented as Mean ± Standard Deviation | |
|---|---|
| Age | 53 ± 14 years |
| BSA | 1.9 ± 0.3 m2 |
| Gender | 51% male: 49% female |
| Weight | 80 ± 20 kg |
| Race | 92% White: 8% Other |
| First Cisplatin Dose | 59 ± 25 mg/m2 |
| Total Dose | 479 ± 219 mg |
| Baseline Serum Creatinine | 0.9 ± 0.2 mg/dL |
| Baseline eGFR | 91 ± 21 mL/min/1.73 m2 |
| Cancer Etiologies (number, %) | Genital (54, 26%) |
| Head and Neck (41, 20%) | |
| Melanoma (31, 15%) | |
| Lung (25, 12%) | |
| Digestive (21, 10%) | |
| Urinary (18, 9%) | |
| Other (16, 8%) | |
| Gene | Variant | Homozygous Wildtype Frequency Observed | Heterozygous Frequency Observed | Homozygous Variant Frequency Observed | Undeter-Mined | Major Allele Frequency Observed (Expected) | Minor Allele Frequency Observed (Expected) | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| SLC22A2 (OCT2) | rs316019 * | C/C | 0.679 | A/C | 0.187 | A/A | 0.014 | 0.120 | C | 0.824 (0.79) | A | 0.118 (0.21) |
| rs3127573 | A/A | 0.737 | A/G | 0.211 | G/G | 0.010 | 0.043 | A | 0.858 (0.88) | G | 0.10 (0.12) | |
| rs2279463 | A/A | 0.665 | A/G | 0.201 | G/G | 0.005 | 0.014 | A | 0.869 (0.88) | G | 0.097 (0.12) | |
| rs596881 | C/C | 0.741 | C/T | 0.230 | T/T | 0.010 | 0.019 | C | 0.861 (0.89) | T | 0.098 (0.11) | |
| SLC31A1 (CTR1) | rs7851395 | A/A | 0.306 | A/G | 0.431 | G/G | 0.187 | 0.077 | A | 0.553 (0.53) | G | 0.431 (0.47) |
| rs12686377 | C/C | 0.718 | A/C | 0.158 | A/A | 0.038 | 0.086 | C | 0.847 (0.92) | A | 0.194 (0.08) | |
| SLC47A1 (MATE1) | rs2289669 | G/G | 0.278 | A/G | 0.464 | A/A | 0.196 | 0.062 | G | 0.527 (0.54) | A | 0.443 (0.46) |
| ABCC2 (MRP2) | rs717620 | C/C | 0.603 | C/T | 0.258 | T/T | 0.043 | 0.096 | C | 0.776 (0.81) | T | 0.207 (0.19) |
| rs2273697 * | G/G | 0.531 | A/G | 0.325 | A/A | 0.053 | 0.091 | G | 0.729 (0.82) | A | 0.23 (0.18) | |
| rs3740066 | C/C | 0.397 | C/T | 0.368 | T/T | 0.144 | 0.091 | C | 0.63(0.62) | T | 0.379 (0.38) | |
| GGT1 | rs4820599 | A/A | 0.464 | A/G | 0.349 | G/G | 0.100 | 0.086 | A | 0.681 (0.73) | G | 0.316 (0.27) |
| GSTP1 | rs1695 * | A/A | 0.354 | A/G | 0.402 | G/G | 0.129 | 0.033 | A | 0.63(0.59) | G | 0.372 (0.41) |
| KEAP1 | rs11085735 | C/C | 0.746 | A/C | 0.139 | A/A | 0.014 | 0.100 | C | 0.864 (0.91) | A | 0.118 (0.09) |
| rs1048290 | C/C | 0.282 | C/G | 0.407 | G/G | 0.129 | 0.182 | C | 0.531 (0.68) | G | 0.359 (0.32) | |
| NFE2L2 (NRF2) | rs2886162 | A/A | 0.239 | A/G | 0.459 | G/G | 0.220 | 0.081 | A | 0.489 (0.58) | G | 0.469 (0.42) |
| rs1806649 | C/C | 0.512 | C/T | 0.311 | T/T | 0.053 | 0.124 | C | 0.716 (0.77) | T | 0.23 (0.23) | |
| rs1962142 | G/G | 0.000 | A/G | 0.670 | A/A | 0.244 | 0.086 | G | 0(0.92) | A | 0.494 (0.08) | |
| rs2706110 | C/C | 0.560 | C/T | 0.344 | T/T | 0.086 | 0.010 | C | 0.748 (0.8) | T | 0.293 (0.2) | |
| rs6721961 | G/G | 0.665 | G/T | 0.225 | T/T | 0.024 | 0.091 | G | 0.815 (0.8) | T | 0.155 (0.2) | |
| Gene | Variant | Protein Biomarker | Time | Fold Change (WT/WT vs. WT/Var) | p-value | Fold Change (WT/WT vs. Var/Var) | p-value |
|---|---|---|---|---|---|---|---|
| SLC22A2 (OCT2) | rs596881 | B2M | Day 3 | −2.134 | 0.039 | 2.474 | 0.057 |
| Osteopontin | Day 3 | 1.918 | 0.049 | −1.052 | 0.972 | ||
| rs316019 | KIM-1 | Day 3 | 1.77 × 10171 | 0.038 | N/A | N/A | |
| KIM-1 | Day 10 | −1.38 × 1084 | 0.046 | N/A | N/A | ||
| SLC31A1 (CTR1) | rs7851395 | Osteopontin | Day 3 | 1.341 | 0.488 | 2.509 | 0.015 |
| SLC47A1 (MATE1) | rs2289669 | KIM-1 | Day 3 | 1.379 | 0.636 | 3.605 | 0.007 |
| MCP-1 | Day 3 | 1.322 | 0.629 | 2.952 | 0.015 | ||
| ABCC2 (MRP2) | rs717620 | Clusterin | Day 3 | 3.534 | 0.024 | −2.981 | 0.854 |
| Cystatin C | Day 3 | 2.627 | 0.034 | 1.279 | 0.910 | ||
| rs3740066 | Calbindin | Day 3 | 1.900 | 0.017 | 1.068 | 0.883 | |
| Calbindin | Day 10 | 2.732 | 0.023 | −1.269 | 0.822 | ||
| Clusterin | Day 3 | 1.170 | 0.870 | 4.384 | 0.012 | ||
| Cystatin C | Day 3 | 1.601 | 0.447 | 3.094 | 0.038 | ||
| NGAL | Day 3 | 2.110 | 0.030 | 1.011 | 0.986 | ||
| rs2273697 | KIM-1 | Day 3 | 1.153 | 0.730 | 5.966 | 4.29 × 10−5 | |
| KIM-1 | Day 10 | 1.163 | 0.636 | 2.808 | 0.042 | ||
| Calbindin | Day 3 | 1.124 | 0.687 | 2.648 | 0.038 | ||
| MCP-1 | Day 3 | −1.215 | 0.635 | 3.500 | 0.010 | ||
| GSTP1 | rs1695 | KIM-1 | Day 3 | 1.195 | 0.732 | 2.690 | 0.029 |
| Calbindin | Day 3 | 1.369 | 0.354 | 2.371 | 0.012 | ||
| IL-18 | Day 10 | −1.566 | 0.337 | 2.287 | 0.012 | ||
| NGAL | Day 3 | 1.294 | 0.572 | 2.569 | 0.027 | ||
| KEAP1 | rs1048290 | NGAL | Day 3 | −1.849 | 0.211 | 2.051 | 0.035 |
| rs11085735 | Calbindin | Day 10 | 2.944 | 0.019 | −17.366 | 0.661 | |
| Cystatin C | Day 3 | 3.275 | 0.011 | 1.207 | 0.928 | ||
| Cystatin C | Day 10 | 2.600 | 0.027 | −2.481 | 0.751 | ||
| TFF3 | Day 3 | 1.747 | 0.038 | 1.466 | 0.619 | ||
| TFF3 | Day 10 | 1.776 | 0.034 | −1.629 | 0.685 | ||
| NFE2L2 | rs2886162 | Calbindin | Day 10 | 3.486 | 0.029 | 1.53 | 0.710 |
| rs1806649 | IL-18 | Day 3 | −1.298 | 0.692 | 3.744 | 0.002 | |
| TFF3 | Day 3 | −1.133 | 0.709 | 2.337 | 0.004 | ||
| rs1962142 | TFF3 | Day 10 | −1.638 | 0.028 | N/A | N/A | |
| rs2706110 | MCP-1 | Day 10 | 1.236 | 0.621 | 3.182 | 0.002 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chang, C.; Hu, Y.; Hogan, S.L.; Mercke, N.; Gomez, M.; O’Bryant, C.; Bowles, D.W.; George, B.; Wen, X.; Aleksunes, L.M.; et al. Pharmacogenomic Variants May Influence the Urinary Excretion of Novel Kidney Injury Biomarkers in Patients Receiving Cisplatin. Int. J. Mol. Sci. 2017, 18, 1333. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms18071333
Chang C, Hu Y, Hogan SL, Mercke N, Gomez M, O’Bryant C, Bowles DW, George B, Wen X, Aleksunes LM, et al. Pharmacogenomic Variants May Influence the Urinary Excretion of Novel Kidney Injury Biomarkers in Patients Receiving Cisplatin. International Journal of Molecular Sciences. 2017; 18(7):1333. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms18071333
Chicago/Turabian StyleChang, Cara, Yichun Hu, Susan L. Hogan, Nickie Mercke, Madeleine Gomez, Cindy O’Bryant, Daniel W. Bowles, Blessy George, Xia Wen, Lauren M. Aleksunes, and et al. 2017. "Pharmacogenomic Variants May Influence the Urinary Excretion of Novel Kidney Injury Biomarkers in Patients Receiving Cisplatin" International Journal of Molecular Sciences 18, no. 7: 1333. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms18071333