The Role of Tumor Microenvironment in Chemoresistance: To Survive, Keep Your Enemies Closer

 ,
,  , , and
, , and
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
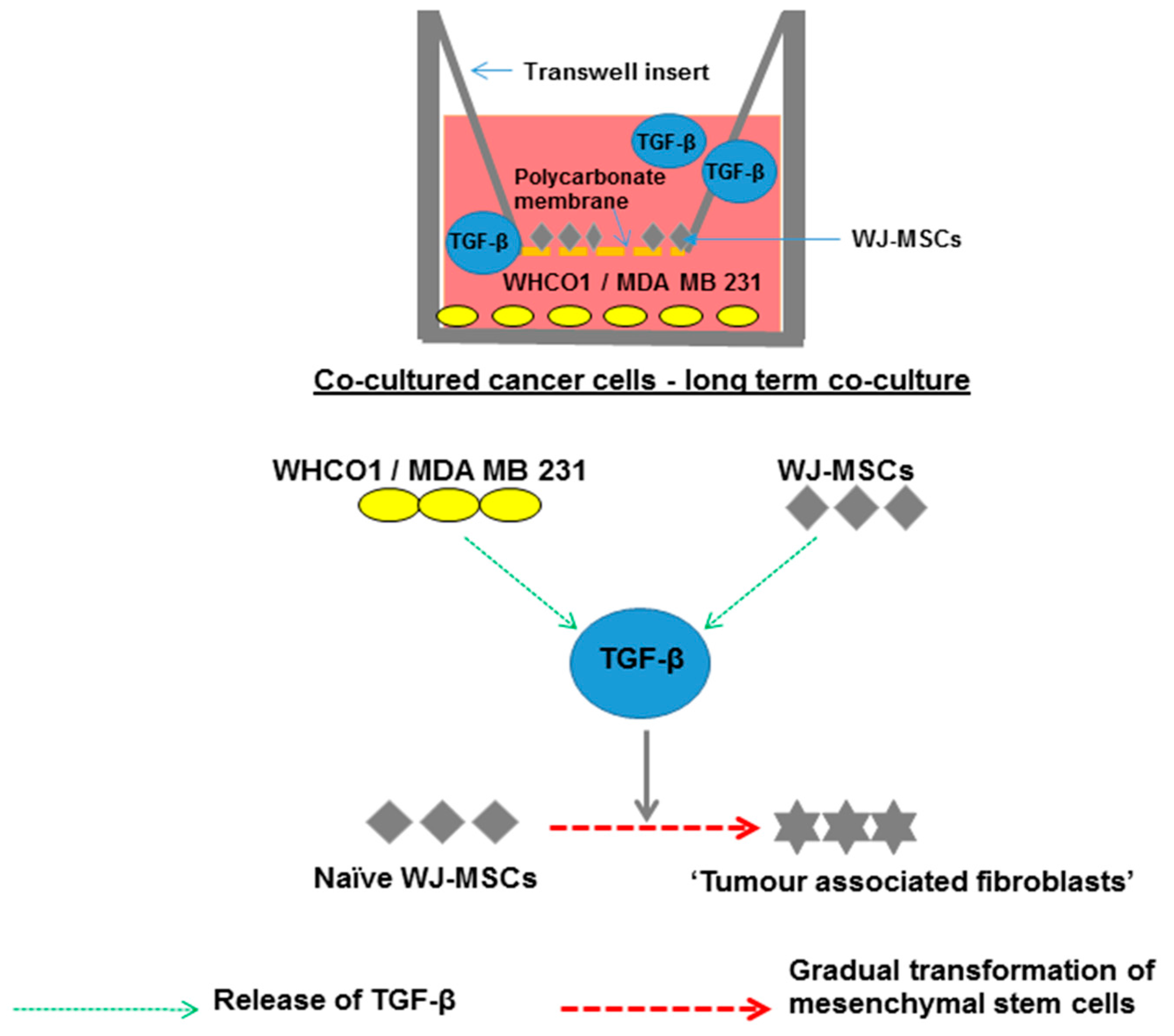{kind=link}
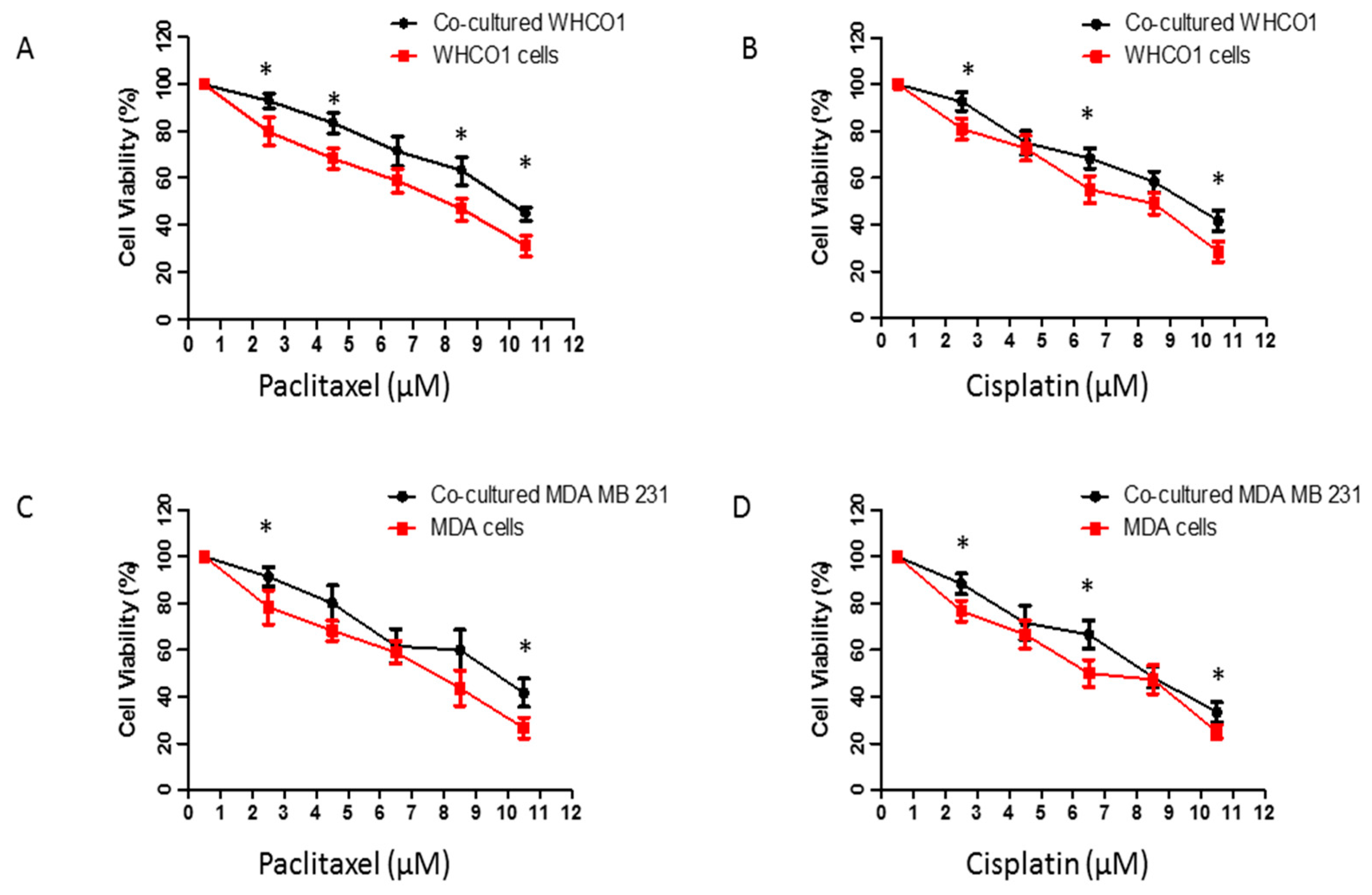{kind=link}
1. Introduction
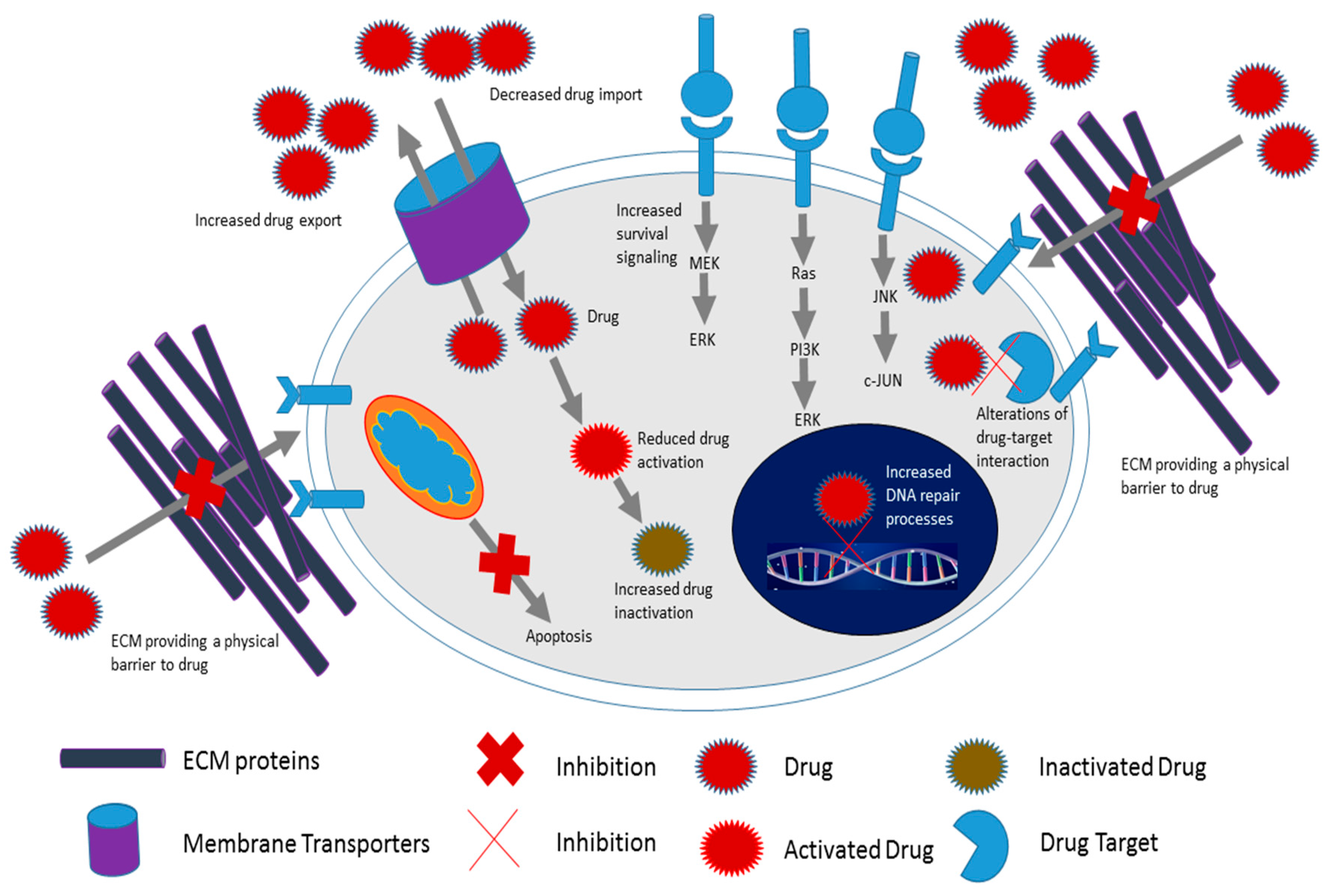
2. Cancer Cell Chemoresistance
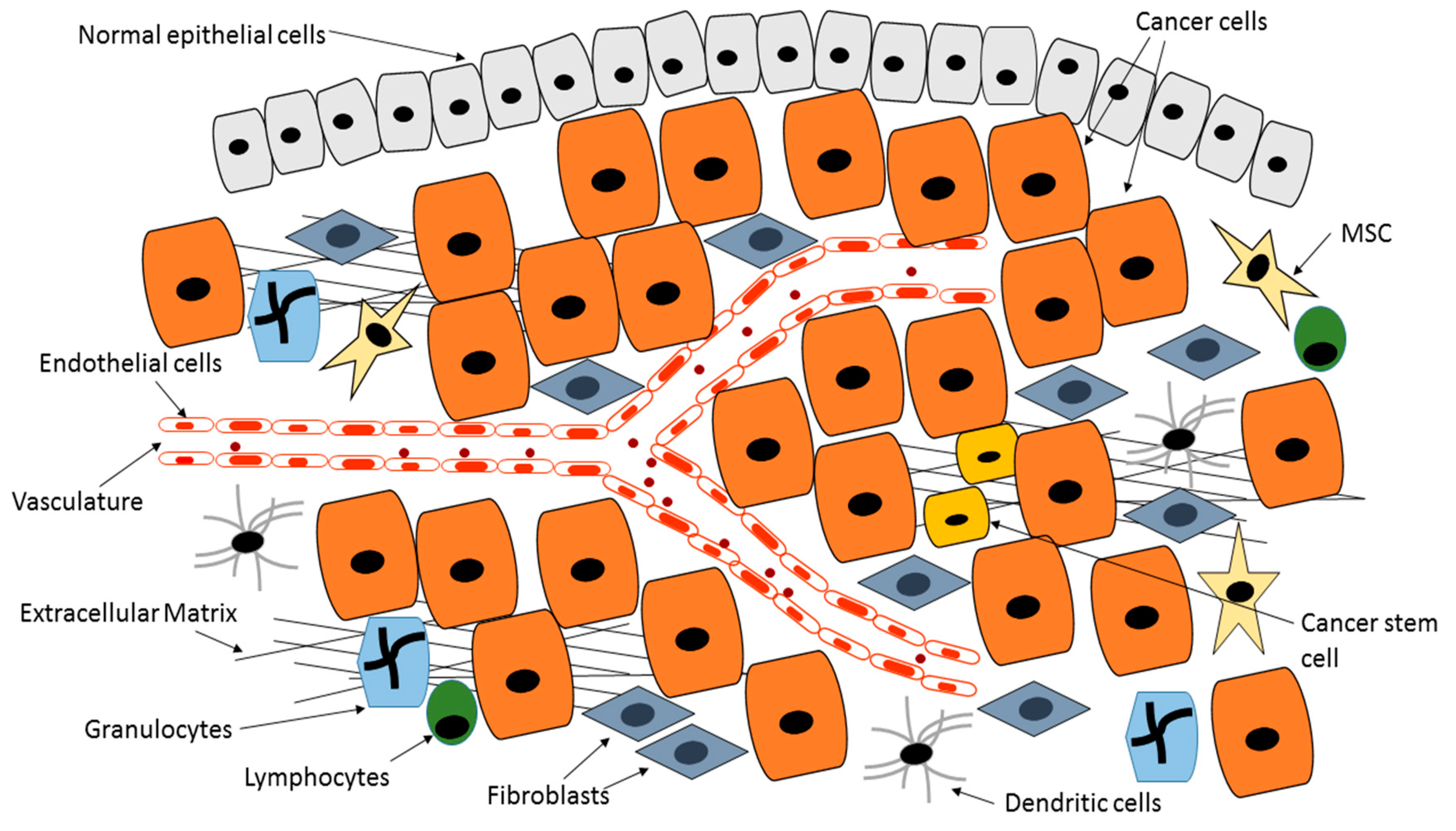
3. Tumor Microenvironment
3.1. Cancer-Associated Fibroblasts (CAFs)
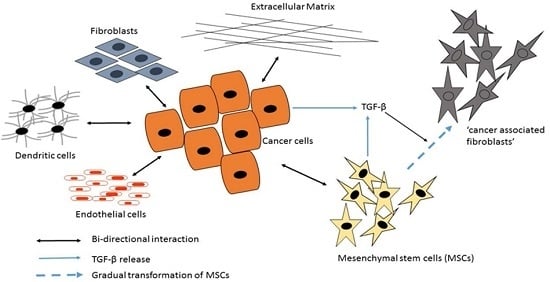
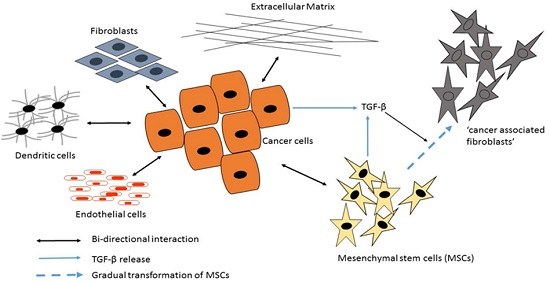
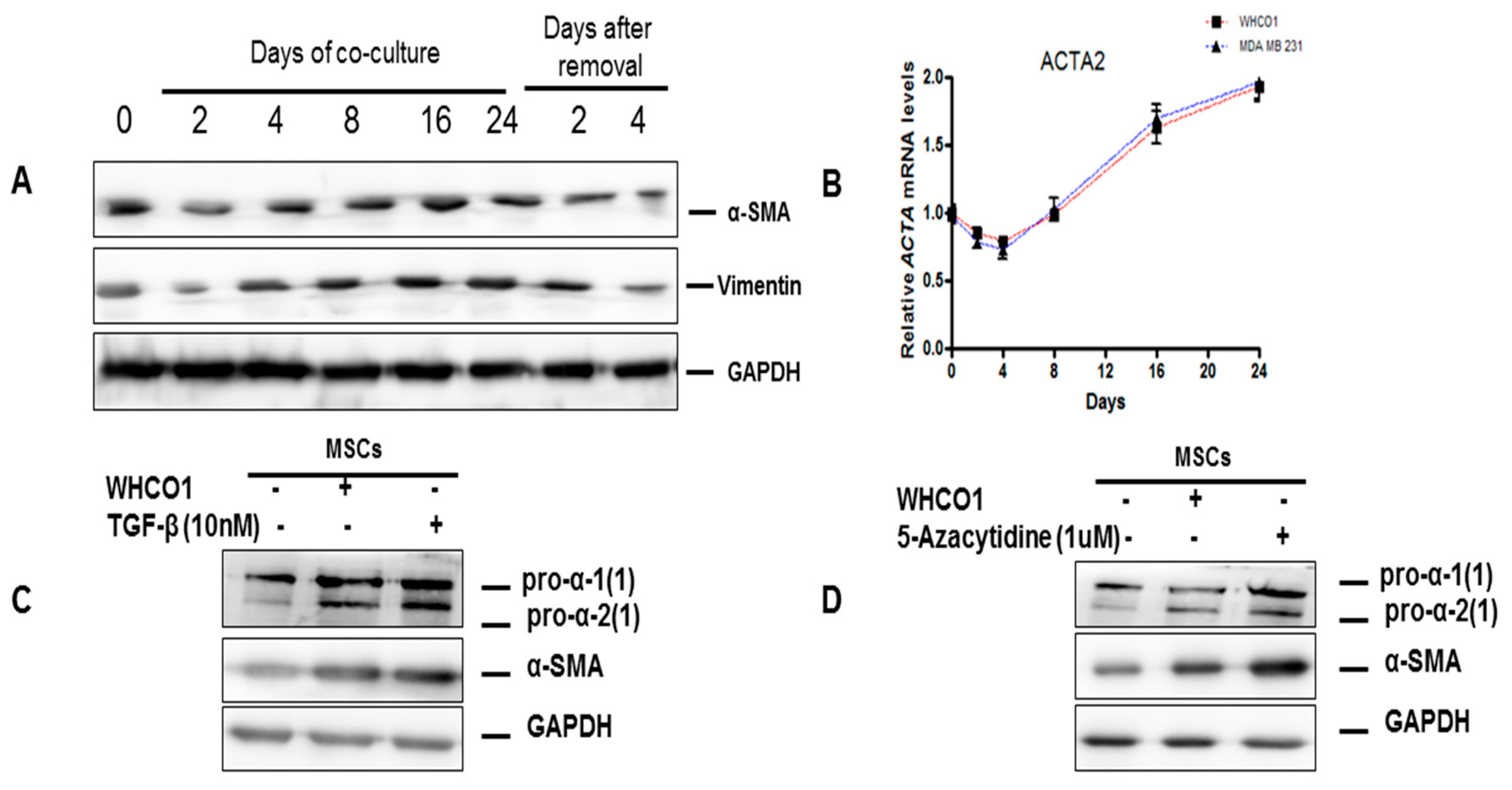
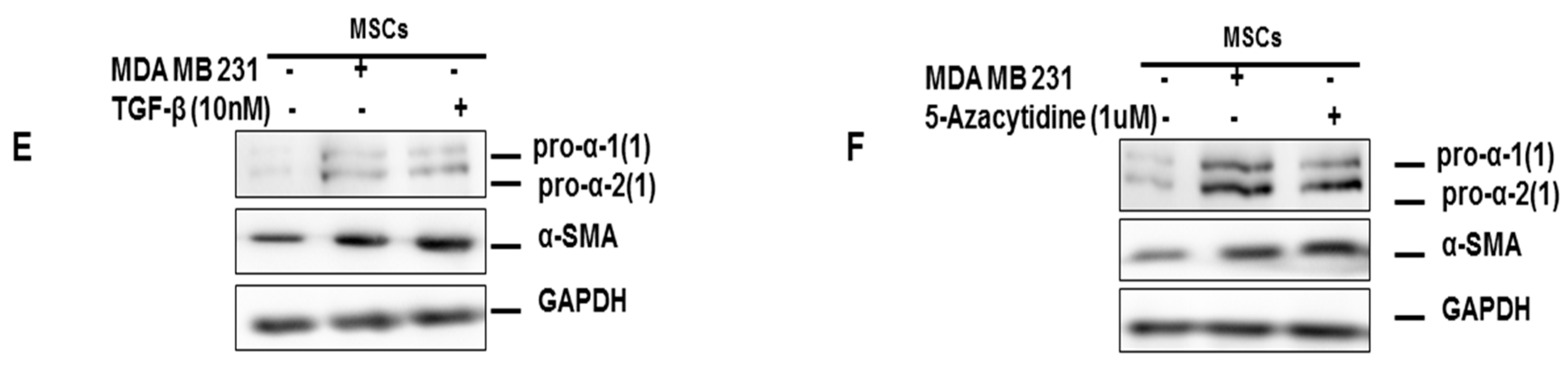
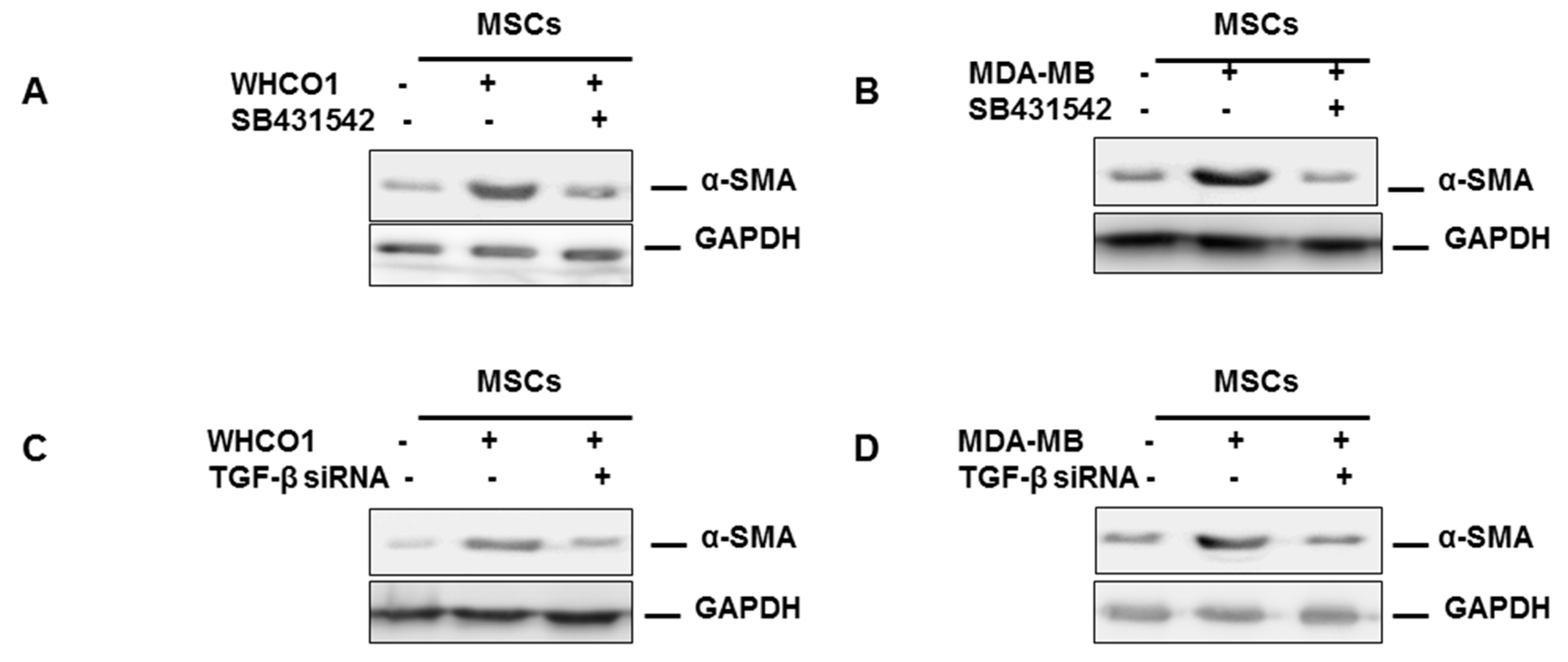
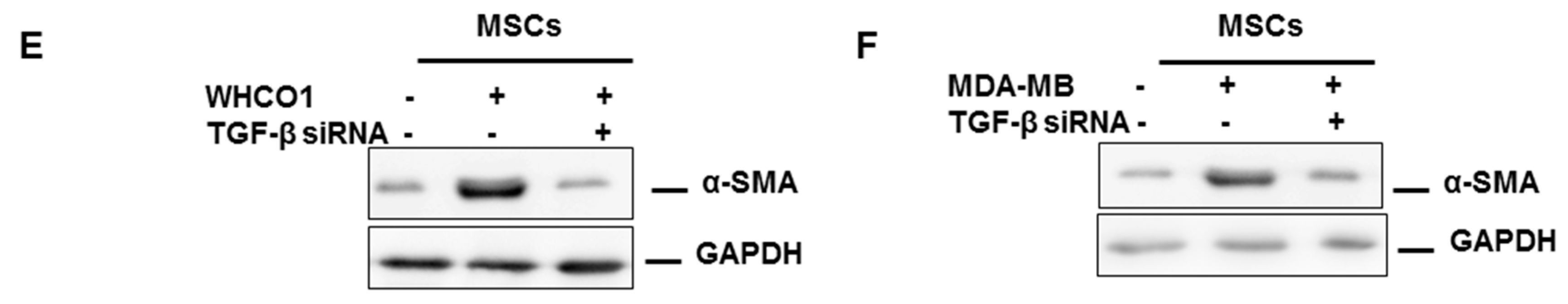
3.2. Mesenchymal Stromal/Stem Cells (MSCs)
3.3. The Role of the Extracellular Matrix in Chemotherapeutic Resistance
3.3.1. Collagen
3.3.2. Laminin
3.3.3. Fibronectin
3.3.4. Periostin
4. Strategies to Overcome Chemoresistance
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Lemoine, M.; Girard, P.M.; Thursz, M.; Raguin, G. In the shadow of hiv/aids: Forgotten diseases in sub-Saharan Africa: Global health issues and funding agency responsibilities. J. Public Health Policy 2012, 33, 430–438. [Google Scholar] [CrossRef] [PubMed]
- Levitt, N.S.; Steyn, K.; Dave, J.; Bradshaw, D. Chronic noncommunicable diseases and hiv-aids on a collision course: Relevance for health care delivery, particularly in low-resource settings—Insights from South Africa. Am. J. Clin. Nutr. 2011, 94, 1690S–1696S. [Google Scholar] [CrossRef] [PubMed]
- Lipson, E.J.; Sharfman, W.H.; Drake, C.G.; Wollner, I.; Taube, J.M.; Anders, R.A.; Xu, H.; Yao, S.; Pons, A.; Chen, L.; et al. Durable cancer regression off-treatment and effective reinduction therapy with an anti-pd-1 antibody. Clin. Cancer Res. 2013, 19, 462–468. [Google Scholar] [CrossRef] [PubMed]
- Philips, G.K.; Atkins, M. Therapeutic uses of anti-pd-1 and anti-pd-l1 antibodies. Int. Immunol. 2015, 27, 39–46. [Google Scholar] [CrossRef] [PubMed]
- Wilson, T.R.; Longley, D.B.; Johnston, P.G. Chemoresistance in solid tumours. Ann. Oncol. 2006, 17, x315–x324. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- You, Y.N.; Lakhani, V.T.; Wells, S.A. The role of prophylactic surgery in cancer prevention. World J. Surg. 2007, 31, 450–464. [Google Scholar] [CrossRef] [PubMed]
- Luqmani, Y.A. Mechanisms of drug resistance in cancer chemotherapy. Med. Princ. Pract. Int. J. Kuwait Univ. Health Sci. Cent. 2005, 14, 35–48. [Google Scholar] [CrossRef] [PubMed]
- Thomas, M.; Coyle, K.; Sultan, M.; Vaghar-Kashani, A.; Marcato, P. Chemoresistance in cancer stem cells and strategies to overcome resistance. Chemotherapy 2014, 3, 2. [Google Scholar]
- Bilen, M.A.; Hess, K.R.; Campbell, M.T.; Wang, J.; Broaddus, R.R.; Karam, J.A.; Ward, J.F.; Wood, C.G.; Choi, S.L.; Rao, P.; et al. Intratumoral heterogeneity and chemoresistance in nonseminomatous germ cell tumor of the testis. Oncotarget 2016, 7, 86280–86289. [Google Scholar] [PubMed]
- Brown, F.C.; Cifani, P.; Drill, E.; He, J.; Still, E.; Zhong, S.; Balasubramanian, S.; Pavlick, D.; Yilmazel, B.; Knapp, K.M.; et al. Genomics of primary chemoresistance and remission induction failure in paediatric and adult acute myeloid leukaemia. Br. J. Haematol. 2017, 176, 86–91. [Google Scholar] [CrossRef] [PubMed]
- Fletcher, N.M.; Belotte, J.; Saed, M.G.; Memaj, I.; Diamond, M.P.; Morris, R.T.; Saed, G.M. Specific point mutations in key redox enzymes are associated with chemoresistance in epithelial ovarian cancer. Free Radic. Biol. Med. 2017, 102, 122–132. [Google Scholar] [CrossRef] [PubMed]
- Guryanova, O.A.; Shank, K.; Spitzer, B.; Luciani, L.; Koche, R.P.; Garrett-Bakelman, F.E.; Ganzel, C.; Durham, B.H.; Mohanty, A.; Hoermann, G.; et al. DNMT3A mutations promote anthracycline resistance in acute myeloid leukemia via impaired nucleosome remodeling. Nat. Med. 2016, 22, 1488–1495. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Baytak, E.; Li, J.; Akman, B.; Okay, K.; Hu, G.; Scuto, A.; Zhang, W.; Kucuk, C. The relationship of rel proto-oncogene to pathobiology and chemoresistance in follicular and transformed follicular lymphoma. Leuk. Res. 2017, 54, 30–38. [Google Scholar] [CrossRef] [PubMed]
- Janczar, S.; Janczar, K.; Pastorczak, A.; Harb, H.; Paige, A.J.; Zalewska-Szewczyk, B.; Danilewicz, M.; Mlynarski, W. The role of histone protein modifications and mutations in histone modifiers in pediatric b-cell progenitor acute lymphoblastic leukemia. Cancers 2017, 9, 2. [Google Scholar] [CrossRef] [PubMed]
- Takada, M.; Nagai, S.; Haruta, M.; Sugino, R.P.; Tozuka, K.; Takei, H.; Ohkubo, F.; Inoue, K.; Kurosumi, M.; Miyazaki, M.; et al. BRCA1 alterations with additional defects in DNA damage response genes may confer chemoresistance to BRCA-like breast cancers treated with neoadjuvant chemotherapy. Genes Chromosomes Cancer 2017, 56, 405–420. [Google Scholar] [CrossRef] [PubMed]
- Taylor-Weiner, A.; Zack, T.; O’Donnell, E.; Guerriero, J.L.; Bernard, B.; Reddy, A.; Han, G.C.; AlDubayan, S.; Amin-Mansour, A.; Schumacher, S.E.; et al. Genomic evolution and chemoresistance in germ-cell tumours. Nature 2016, 540, 114–118. [Google Scholar] [CrossRef] [PubMed]
- Chan, R.; Sethi, P.; Jyoti, A.; McGarry, R.; Upreti, M. Investigating the radioresistant properties of lung cancer stem cells in the context of the tumor microenvironment. Radiat. Res. 2016, 185, 169–181. [Google Scholar] [CrossRef] [PubMed]
- Conrad, C.A.; Fueyo, J.; Gomez-Manzano, C. Intratumoral heterogeneity and intraclonal plasticity: From warburg to oxygen and back again. Neuro Oncol. 2014, 16, 1025–1026. [Google Scholar] [CrossRef] [PubMed]
- Gentric, G.; Mieulet, V.; Mechta-Grigoriou, F. Heterogeneity in cancer metabolism: New concepts in an old field. Antioxid. Redox Signal. 2016, 26, 462–485. [Google Scholar] [CrossRef] [PubMed]
- Hida, K.; Maishi, N.; Torii, C.; Hida, Y. Tumor angiogenesis—Characteristics of tumor endothelial cells. Int. J. Clin. Oncol. 2016, 21, 206–212. [Google Scholar] [CrossRef] [PubMed]
- Martin, J.D.; Fukumura, D.; Duda, D.G.; Boucher, Y.; Jain, R.K. Reengineering the tumor microenvironment to alleviate hypoxia and overcome cancer heterogeneity. Cold Spring Harb. Perspect. Med. 2016, 6, a027094. [Google Scholar] [CrossRef] [PubMed]
- Mumenthaler, S.M.; Foo, J.; Choi, N.C.; Heise, N.; Leder, K.; Agus, D.B.; Pao, W.; Michor, F.; Mallick, P. The impact of microenvironmental heterogeneity on the evolution of drug resistance in cancer cells. Cancer Inform. 2015, 14, 19–31. [Google Scholar] [PubMed]
- Pucciarelli, D.; Lengger, N.; Takacova, M.; Csaderova, L.; Bartosova, M.; Breiteneder, H.; Pastorekova, S.; Hafner, C. Hypoxia increases the heterogeneity of melanoma cell populations and affects the response to vemurafenib. Mol. Med. Rep. 2016, 13, 3281–3288. [Google Scholar] [CrossRef] [PubMed]
- Guerra, L.; Odorisio, T.; Zambruno, G.; Castiglia, D. Stromal microenvironment in type VII collagen-deficient skin: The ground for squamous cell carcinoma development. Matrix Biol. J. Int. Soc. Matrix Biol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Fuzer, A.M.; Lee, S.Y.; Mott, J.D.; Cominetti, M.R. [10]-Gingerol reverts malignant phenotype of breast cancer cells in 3d culture. J. Cell. Biochem. 2017, 118, 2693–2699. [Google Scholar] [CrossRef] [PubMed]
- Tadeo, I.; Berbegall, A.P.; Navarro, S.; Castel, V.; Noguera, R. A stiff extracellular matrix is associated with malignancy in peripheral neuroblastic tumors. Pediatr. Blood Cancer 2017. [Google Scholar] [CrossRef] [PubMed]
- Affo, S.; Yu, L.; Schwabe, R.F. The role of cancer-associated fibroblasts and fibrosis in liver cancer. Annu. Rev. Pathol. 2017, 24, 153–186. [Google Scholar] [CrossRef] [PubMed]
- Gjorevski, N.; Sachs, N.; Manfrin, A.; Giger, S.; Bragina, M.E.; Ordonez-Moran, P.; Clevers, H.; Lutolf, M.P. Designer matrices for intestinal stem cell and organoid culture. Nature 2016, 539, 560–564. [Google Scholar] [CrossRef] [PubMed]
- Kopanska, K.S.; Alcheikh, Y.; Staneva, R.; Vignjevic, D.; Betz, T. Tensile forces originating from cancer spheroids facilitate tumor invasion. PLoS ONE 2016, 11, e0156442. [Google Scholar] [CrossRef] [PubMed]
- McLane, J.S.; Ligon, L.A. Stiffened extracellular matrix and signaling from stromal fibroblasts via osteoprotegerin regulate tumor cell invasion in a 3-d tumor in situ model. Cancer Microenviron. 2016, 9, 127–139. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Kim, D.H.; Kim, H.N.; Wang, C.J.; Kwak, M.K.; Hur, E.; Suh, K.Y.; An, S.S.; Levchenko, A. Directed migration of cancer cells guided by the graded texture of the underlying matrix. Nat. Mater. 2016, 15, 792–801. [Google Scholar] [CrossRef] [PubMed]
- Romero-Lopez, M.; Trinh, A.L.; Sobrino, A.; Hatch, M.M.; Keating, M.T.; Fimbres, C.; Lewis, D.E.; Gershon, P.D.; Botvinick, E.L.; Digman, M.; et al. Recapitulating the human tumor microenvironment: Colon tumor-derived extracellular matrix promotes angiogenesis and tumor cell growth. Biomaterials 2017, 116, 118–129. [Google Scholar] [CrossRef] [PubMed]
- Shin, J.W.; Mooney, D.J. Extracellular matrix stiffness causes systematic variations in proliferation and chemosensitivity in myeloid leukemias. Proc. Natl. Acad. Sci. USA 2016, 113, 12126–12131. [Google Scholar] [CrossRef] [PubMed]
- Dave, B.; Gonzalez, D.D.; Liu, Z.B.; Li, X.; Wong, H.; Granados, S.; Ezzedine, N.E.; Sieglaff, D.H.; Ensor, J.E.; Miller, K.D.; et al. Role of RPL39 in metaplastic breast cancer. J. Natl. Cancer Inst. 2017. [Google Scholar] [CrossRef] [PubMed]
- Jahani, M.; Azadbakht, M.; Norooznezhad, F.; Mansouri, K. L-arginine alters the effect of 5-fluorouracil on breast cancer cells in favor of apoptosis. Biomed. Pharmacother. Biomed. Pharmacother. 2017, 88, 114–123. [Google Scholar] [CrossRef] [PubMed]
- Spitschak, A.; Meier, C.; Kowtharapu, B.; Engelmann, D.; Putzer, B.M. MiR-182 promotes cancer invasion by linking ret oncogene activated NF-κB to loss of the hes1/notch1 regulatory circuit. Mol. Cancer 2017, 16, 24. [Google Scholar] [CrossRef] [PubMed]
- Avnet, S.; Di Pompo, G.; Chano, T.; Errani, C.; Ibrahim-Hashim, A.; Gillies, R.J.; Donati, D.M.; Baldini, N. Cancer-associated mesenchymal stroma fosters the stemness of osteosarcoma cells in response to intratumoral acidosis via nf-kappab activation. Int. J. Cancer 2017, 140, 1331–1345. [Google Scholar] [CrossRef] [PubMed]
- Cortini, M.; Massa, A.; Avnet, S.; Bonuccelli, G.; Baldini, N. Tumor-activated mesenchymal stromal cells promote osteosarcoma stemness and migratory potential via IL-6 secretion. PLoS ONE 2016, 11, e0166500. [Google Scholar] [CrossRef] [PubMed]
- Cramer, G.M.; Jones, D.P.; El-Hamidi, H.; Celli, J.P. Ecm composition and rheology regulate growth, motility, and response to photodynamic therapy in 3d models of pancreatic ductal adenocarcinoma. Mol. Cancer Res. MCR 2017, 15, 15–25. [Google Scholar] [CrossRef] [PubMed]
- Dauer, P.; Nomura, A.; Saluja, A.; Banerjee, S. Microenvironment in determining chemo-resistance in pancreatic cancer: Neighborhood matters. Pancreatology 2016. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Li, F.; Gao, F.; Xing, L.; Qin, P.; Liang, X.; Zhang, J.; Qiao, X.; Lin, L.; Zhao, Q.; et al. Periostin promotes the chemotherapy resistance to gemcitabine in pancreatic cancer. Tumour Biol. 2016, 37, 15283–15291. [Google Scholar] [CrossRef] [PubMed]
- Majidinia, M.; Yousefi, B. Breast tumor stroma: A driving force in the development of resistance to therapies. Chem. Biol. Drug Des. 2017, 89, 309–318. [Google Scholar] [CrossRef] [PubMed]
- Rao, C.V.; Janakiram, N.B.; Mohammed, A. Molecular pathways: Mucins and drug delivery in cancer. Clin. Cancer Res. 2017, 23, 1373–1378. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Kim, S.H.; Kim, K.M.; Choi, E.K.; Kim, J.; Seo, H.R. Activated hepatic stellate cells play pivotal roles in hepatocellular carcinoma cell chemoresistance and migration in multicellular tumor spheroids. Sci. Rep. 2016, 6, 36750. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Xie, C.; Yue, J.; Jiang, Z.; Zhou, R.; Xie, R.; Wang, Y.; Wu, S. Cancer-associated fibroblasts mediated chemoresistance by a FOXO1/TGFβ1 signaling loop in esophageal squamous cell carcinoma. Mol. Carcinog. 2017, 56, 1150–1164. [Google Scholar] [CrossRef] [PubMed]
- Afik, R.; Zigmond, E.; Vugman, M.; Klepfish, M.; Shimshoni, E.; Pasmanik-Chor, M.; Shenoy, A.; Bassat, E.; Halpern, Z.; Geiger, T.; et al. Tumor macrophages are pivotal constructors of tumor collagenous matrix. J. Exp. Med. 2016, 213, 2315–2331. [Google Scholar] [CrossRef] [PubMed]
- Kaushik, S.; Pickup, M.W.; Weaver, V.M. From transformation to metastasis: Deconstructing the extracellular matrix in breast cancer. Cancer Metastasis Rev. 2016, 35, 655–667. [Google Scholar] [CrossRef] [PubMed]
- Lim, E.J.; Suh, Y.; Yoo, K.C.; Lee, J.H.; Kim, I.G.; Kim, M.J.; Chang, J.H.; Kang, S.G.; Lee, S.J. Tumor-associated mesenchymal stem-like cells provide extracellular signaling cue for invasiveness of glioblastoma cells. Oncotarget 2017, 8, 1438–1448. [Google Scholar] [CrossRef] [PubMed]
- Mellone, M.; Hanley, C.J.; Thirdborough, S.; Mellows, T.; Garcia, E.; Woo, J.; Tod, J.; Frampton, S.; Jenei, V.; Moutasim, K.A.; et al. Induction of fibroblast senescence generates a non-fibrogenic myofibroblast phenotype that differentially impacts on cancer prognosis. Aging 2016, 9, 114–132. [Google Scholar] [CrossRef] [PubMed]
- Miroshnikova, Y.A.; Mouw, J.K.; Barnes, J.M.; Pickup, M.W.; Lakins, J.N.; Kim, Y.; Lobo, K.; Persson, A.I.; Reis, G.F.; McKnight, T.R.; et al. Tissue mechanics promote IDH1-dependent HIF1α-tenascin c feedback to regulate glioblastoma aggression. Nat. Cell Biol. 2016, 18, 1336–1345. [Google Scholar] [CrossRef] [PubMed]
- Mongiat, M.; Andreuzzi, E.; Tarticchio, G.; Paulitti, A. Extracellular matrix, a hard player in angiogenesis. Int. J. Mol. Sci. 2016, 17, 1822. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, S.; Itakura, S.; Matsui, R.; Nakayama, K.; Nishi, T.; Nishimoto, A.; Hama, S.; Kogure, K. Tumor microenvironment-sensitive liposomes penetrate tumor tissue via attenuated interaction of the extracellular matrix and tumor cells and accompanying actin depolymerization. Biomacromolecules 2017, 18, 535–547. [Google Scholar] [CrossRef] [PubMed]
- Affolter, A.; Hess, J. Preclinical models in head and neck tumors: Evaluation of cellular and molecular resistance mechanisms in the tumor microenvironment. HNO 2016, 64, 860–869. [Google Scholar] [CrossRef] [PubMed]
- Eiro, N.; Fernandez-Gomez, J.; Sacristan, R.; Fernandez-Garcia, B.; Lobo, B.; Gonzalez-Suarez, J.; Quintas, A.; Escaf, S.; Vizoso, F.J. Stromal factors involved in human prostate cancer development, progression and castration resistance. J. Cancer Res. Clin. Oncol. 2017, 143, 351–359. [Google Scholar] [CrossRef] [PubMed]
- Fujimura, T.; Kakizaki, A.; Furudate, S.; Kambayashi, Y.; Aiba, S. Tumor-associated macrophages in skin: How to treat their heterogeneity and plasticity. J. Dermatol. Sci. 2016, 83, 167–173. [Google Scholar] [CrossRef] [PubMed]
- Mitrofanova, I.; Zavyalova, M.; Telegina, N.; Buldakov, M.; Riabov, V.; Cherdyntseva, N.; Kzhyshkowska, J. Tumor-associated macrophages in human breast cancer parenchyma negatively correlate with lymphatic metastasis after neoadjuvant chemotherapy. Immunobiology 2017, 222, 101–109. [Google Scholar] [CrossRef] [PubMed]
- Parajuli, H.; Teh, M.T.; Abrahamsen, S.; Christoffersen, I.; Neppelberg, E.; Lybak, S.; Osman, T.; Johannessen, A.C.; Gullberg, D.; Skarstein, K.; et al. Integrin α11 is overexpressed by tumour stroma of head and neck squamous cell carcinoma and correlates positively with α smooth muscle actin expression. J. Oral Pathol. Med. 2017, 46, 267–275. [Google Scholar] [CrossRef] [PubMed]
- Prime, S.S.; Cirillo, N.; Hassona, Y.; Lambert, D.W.; Paterson, I.C.; Mellone, M.; Thomas, G.J.; James, E.N.; Parkinson, E.K. Fibroblast activation and senescence in oral cancer. J. Oral Pathol. Med. 2017, 46, 82–88. [Google Scholar] [CrossRef] [PubMed]
- Bissell, M.J.; Kenny, P.A.; Radisky, D.C. Microenvironmental regulators of tissue structure and function also regulate tumor induction and progression: The role of extracellular matrix and its degrading enzymes. Cold Spring Harb. Symp. Quant. Biol. 2005, 70, 343–356. [Google Scholar] [CrossRef] [PubMed]
- Bizzarri, M.; Cucina, A.; Conti, F.; D’Anselmi, F. Beyond the oncogene paradigm: Understanding complexity in cancerogenesis. Acta Biotheor. 2008, 56, 173–196. [Google Scholar] [CrossRef] [PubMed]
- Bizzarri, M.; Cucina, A.; Proietti, S. Tumor reversion: Mesenchymal-epithelial transition as a critical step in managing the tumor-microenvironment cross-talk. Curr. Pharm. Des. 2017. [Google Scholar] [CrossRef]
- Aihara, K.; Mukasa, A.; Nagae, G.; Nomura, M.; Yamamoto, S.; Ueda, H.; Tatsuno, K.; Shibahara, J.; Takahashi, M.; Momose, T.; et al. Genetic and epigenetic stability of oligodendrogliomas at recurrence. Acta Neuropathol. Commun. 2017, 5, 18. [Google Scholar] [CrossRef] [PubMed]
- Antonucci, I.; Provenzano, M.; Sorino, L.; Rodrigues, M.; Palka, G.; Stuppia, L. A new case of “de novo” brca1 mutation in a patient with early-onset breast cancer. Clin. Case Rep. 2017, 5, 238–240. [Google Scholar] [CrossRef] [PubMed]
- Cheema, P.K.; Raphael, S.; El-Maraghi, R.; Li, J.; McClure, R.; Zibdawi, L.; Chan, A.; Victor, J.C.; Dolley, A.; Dziarmaga, A. Rate of EGFR mutation testing for patients with nonsquamous non-small-cell lung cancer with implementation of reflex testing by pathologists. Curr. Oncol. 2017, 24, 16–22. [Google Scholar] [CrossRef] [PubMed]
- Dolatkhah, R.; Somi, M.H.; Kermani, I.A.; Farassati, F.; Dastgiri, S. A novel kras gene mutation report in sporadic colorectal cancer, from northwest of Iran. Clin. Case Rep. 2017, 5, 338–341. [Google Scholar] [CrossRef] [PubMed]
- Jiangdian, S.; Di, D.; Yanqi, H.; Yali, Z.; Zaiyi, L.; Jie, T. Association between tumor heterogeneity and progression-free survival in non-small cell lung cancer patients with EGFR mutations undergoing tyrosine kinase inhibitors therapy. In Proceedings of the 2016 IEEE 38th Annual International Conference of the Engineering in Medicine and Biology Society (EMBC), Orlando, FL, USA, 16–20 August 2016; pp. 1268–1271. [Google Scholar]
- Mahalakshmi, R.; Husayn Ahmed, P.; Mahadevan, V. HDAC inhibitors show differential epigenetic regulation and cell survival strategies on p53 mutant colon cancer cells. J. Biomol. Struct. Dyn. 2017. [Google Scholar] [CrossRef]
- Tan, R.Y.; Walsh, M.; Howard, A.; Winship, I. Multiple cutaneous leiomyomas leading to discovery of novel splice mutation in the fumarate hydratase gene associated with HLRCC. Australas. J. Dermatol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Walton, S.J.; Frayling, I.M.; Clark, S.K.; Latchford, A. Gastric tumours in FAP. Fam. Cancer 2017, 16, 363–369. [Google Scholar] [CrossRef] [PubMed]
- Alderton, G.K. Tumour evolution: Epigenetic and genetic heterogeneity in metastasis. Nat. Rev. Cancer 2017, 17, 141. [Google Scholar] [CrossRef] [PubMed]
- Brown, D.V.; Filiz, G.; Daniel, P.M.; Hollande, F.; Dworkin, S.; Amiridis, S.; Kountouri, N.; Ng, W.; Morokoff, A.P.; Mantamadiotis, T. Expression of cd133 and cd44 in glioblastoma stem cells correlates with cell proliferation, phenotype stability and intra-tumor heterogeneity. PLoS ONE 2017, 12, e0172791. [Google Scholar] [CrossRef] [PubMed]
- Carmona-Fontaine, C.; Deforet, M.; Akkari, L.; Thompson, C.B.; Joyce, J.A.; Xavier, J.B. Metabolic origins of spatial organization in the tumor microenvironment. Proc. Natl. Acad. Sci. USA 2017, 114, 2934–2939. [Google Scholar] [CrossRef] [PubMed]
- Lapa, C.; Schirbel, A.; Samnick, S.; Luckerath, K.; Kortum, K.M.; Knop, S.; Wester, H.J.; Buck, A.K.; Schreder, M. The gross picture: Intraindividual tumour heterogeneity in a patient with nonsecretory multiple myeloma. Eur. J. Nucl. Med. Mol. Imaging 2017, 44, 1097–1098. [Google Scholar] [CrossRef] [PubMed]
- Mehta, R.S.; Song, M.; Nishihara, R.; Drew, D.A.; Wu, K.; Qian, Z.R.; Fung, T.T.; Hamada, T.; Masugi, Y.; da Silva, A.; et al. Dietary patterns and risk of colorectal cancer: Analysis by tumor location and molecular subtypes. Gastroenterology 2017, 152, 1944–1952. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Sun, Y.; Xu, X.; Zhang, Y.; Zhang, J.; Xue, J.; Wang, M.; Yuan, H.; Hu, S.; Shi, W.; et al. The assessment of estrogen receptor status and its intratumoral heterogeneity in breast cancer patients by using 18 f-fluoroestradiol pet/ct. Clin. Nucl. Med. 2017, 42, 421–427. [Google Scholar] [CrossRef] [PubMed]
- Zhai, W.; Lim, T.K.; Zhang, T.; Phang, S.T.; Tiang, Z.; Guan, P.; Ng, M.H.; Lim, J.Q.; Yao, F.; Li, Z.; et al. The spatial organization of intra-tumour heterogeneity and evolutionary trajectories of metastases in hepatocellular carcinoma. Nat. Commun. 2017, 8, 4565. [Google Scholar] [CrossRef] [PubMed]
- Saunders, N.A.; Simpson, F.; Thompson, E.W.; Hill, M.M.; Endo-Munoz, L.; Leggatt, G.; Minchin, R.F.; Guminski, A. Role of intratumoural heterogeneity in cancer drug resistance: Molecular and clinical perspectives. EMBO Mol. Med. 2012, 4, 675–684. [Google Scholar] [CrossRef] [PubMed]
- Gerlinger, M.; Horswell, S.; Larkin, J.; Rowan, A.J.; Salm, M.P.; Varela, I.; Fisher, R.; McGranahan, N.; Matthews, N.; Santos, C.R.; et al. Genomic architecture and evolution of clear cell renal cell carcinomas defined by multiregion sequencing. Nat. Genet. 2014, 46, 225–233. [Google Scholar] [CrossRef] [PubMed]
- Burmakin, M.; van Wieringen, T.; Olsson, P.O.; Stuhr, L.; Ahgren, A.; Heldin, C.H.; Reed, R.K.; Rubin, K.; Hellberg, C. Imatinib increases oxygen delivery in extracellular matrix-rich but not in matrix-poor experimental carcinoma. J. Transl. Med. 2017, 15, 47. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Chou, S.; Swidnicka-Siergiejko, A.; Badi, N.; Chavez-Tomar, M.; Lesinski, G.B.; Bekaii-Saab, T.; Farren, M.R.; Mace, T.A.; Schmidt, C.; Liu, Y.; et al. Lipocalin-2 promotes pancreatic ductal adenocarcinoma by regulating inflammation in the tumor microenvironment. Cancer Res. 2017, 77, 2647–2660. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.S.; Yoo, J.E.; Kim, H.; Rhee, H.; Koh, M.J.; Nahm, J.H.; Choi, J.S.; Lee, K.H.; Park, Y.N. Tumor stroma with senescence-associated secretory phenotype in steatohepatitic hepatocellular carcinoma. PLoS ONE 2017, 12, e0171922. [Google Scholar] [CrossRef] [PubMed]
- Nordby, Y.; Richardsen, E.; Rakaee, M.; Ness, N.; Donnem, T.; Patel, H.R.; Busund, L.T.; Bremnes, R.M.; Andersen, S. High expression of pdgfr-β in prostate cancer stroma is independently associated with clinical and biochemical prostate cancer recurrence. Sci. Rep. 2017, 7, 43378. [Google Scholar] [CrossRef] [PubMed]
- Ramamonjisoa, N.; Ackerstaff, E. Characterization of the tumor microenvironment and tumor-stroma interaction by non-invasive preclinical imaging. Front. Oncol. 2017, 7, 3. [Google Scholar] [CrossRef] [PubMed]
- Szebeni, G.J.; Vizler, C.; Kitajka, K.; Puskas, L.G. Inflammation and cancer: Extra- and intracellular determinants of tumor-associated macrophages as tumor promoters. Mediat. Inflamm. 2017, 2017, 9294018. [Google Scholar] [CrossRef] [PubMed]
- Chang, A. Chemotherapy, chemoresistance and the changing treatment landscape for nsclc. Lung Cancer 2011, 71, 3–10. [Google Scholar] [CrossRef] [PubMed]
- Zahreddine, H.; Borden, K.L. Mechanisms and insights into drug resistance in cancer. Front. Pharmacol. 2013, 4, 28. [Google Scholar] [CrossRef] [PubMed]
- Tredan, O.; Galmarini, C.M.; Patel, K.; Tannock, I.F. Drug resistance and the solid tumor microenvironment. J. Natl. Cancer Inst. 2007, 99, 1441–1454. [Google Scholar] [CrossRef] [PubMed]
- Acerbi, I.; Cassereau, L.; Dean, I.; Shi, Q.; Au, A.; Park, C.; Chen, Y.Y.; Liphardt, J.; Hwang, E.S.; Weaver, V.M. Human breast cancer invasion and aggression correlates with ECM stiffening and immune cell infiltration. Integr. Biol. 2015, 7, 1120–1134. [Google Scholar] [CrossRef] [PubMed]
- Dzobo, K.; Vogelsang, M.; Thomford, N.E.; Dandara, C.; Kallmeyer, K.; Pepper, M.S.; Parker, M.I. Wharton’s jelly-derived mesenchymal stromal cells and fibroblast-derived extracellular matrix synergistically activate apoptosis in a p21-dependent mechanism in whco1 and MDA MB 231 cancer cells in vitro. Stem Cells Int. 2016, 2016, 4842134. [Google Scholar] [CrossRef] [PubMed]
- Kerbel, R.S.; Rak, J.; Kobayashi, H.; Man, M.S.; St Croix, B.; Graham, C.H. Multicellular resistance: A new paradigm to explain aspects of acquired drug resistance of solid tumors. Cold Spring Harb. Symp. Quant. Biol. 1994, 59, 661–672. [Google Scholar] [CrossRef] [PubMed]
- Fodale, V.; Pierobon, M.; Liotta, L.; Petricoin, E. Mechanism of cell adaptation: When and how do cancer cells develop chemoresistance? Cancer J. 2011, 17, 89–95. [Google Scholar] [CrossRef] [PubMed]
- Asimakopoulos, F.; Hope, C.; Johnson, M.G.; Pagenkopf, A.; Gromek, K.; Nagel, B. Extracellular matrix and the myeloid-in-myeloma compartment: Balancing tolerogenic and immunogenic inflammation in the myeloma niche. J. Leukoc. Biol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.; Dai, W.; He, B.; Zhang, H.; Wang, X.; Wang, Y.; Zhang, Q. Current multistage drug delivery systems based on the tumor microenvironment. Theranostics 2017, 7, 538–558. [Google Scholar] [CrossRef] [PubMed]
- La Porta, C.A.; Zapperi, S. Complexity in cancer stem cells and tumor evolution: Toward precision medicine. Semin. Cancer Biol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Nettersheim, D.; Schorle, H. The plasticity of germ cell cancers and its dependence on the cellular microenvironment. J. Cell. Mol. Med. 2017. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Zhang, Y. Tumor-associated macrophages: From basic research to clinical application. J. Hematol. Oncol. 2017, 10, 58. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.S.; Duchamp, M.; Oklu, R.; Ellisen, L.W.; Langer, R.; Khademhosseini, A. Bioprinting the cancer microenvironment. ACS Biomater. Sci. Eng. 2016, 2, 1710–1721. [Google Scholar] [CrossRef] [PubMed]
- Kabeer, M.H.; Loudon, W.G.; Dethlefs, B.A.; Li, Z.; Zhong, J.F.; Luo, J.J.; Vu, L.T.; Li, S.C. Tissue elasticity bridges cancer stem cells to the tumor microenvironment through micrornas: Implications for a “watch-and-wait” approach to cancer. Curr. Stem Cell Res. Ther. 2017. [Google Scholar] [CrossRef] [PubMed]
- Maturu, P.; Jones, D.; Ruteshouser, E.C.; Hu, Q.; Reynolds, J.M.; Hicks, J.; Putluri, N.; Ekmekcioglu, S.; Grimm, E.A.; Dong, C.; et al. Role of cyclooxygenase-2 pathway in creating an immunosuppressive microenvironment and in initiation and progression of wilms’ tumor. Neoplasia 2017, 19, 237–249. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.Y.; Wood, C.N.; Dolorito, J.A.; Libove, E.; Epstein, E.H., Jr. Differing tumor-suppressor functions of arf and p53 in murine basal cell carcinoma initiation and progression. Oncogene 2017, 36, 3772–3780. [Google Scholar] [CrossRef] [PubMed]
- Fuhrmann, A.; Banisadr, A.; Beri, P.; Tlsty, T.D.; Engler, A.J. Metastatic state of cancer cells may be indicated by adhesion strength. Biophys. J. 2017, 112, 736–745. [Google Scholar] [CrossRef] [PubMed]
- Ring, K.L.; Yemelyanova, A.V.; Soliman, P.T.; Frumovitz, M.M.; Jazaeri, A.A. Potential immunotherapy targets in recurrent cervical cancer. Gynecol. Oncol. 2017, 143, 462–468. [Google Scholar] [CrossRef] [PubMed]
- Dzobo, K.; Senthebane, D.A.; Rowe, A.; Thomford, N.E.; Mwapagha, L.M.; Al-Awwad, N.; Dandara, C.; Parker, M.I. Cancer stem cell hypothesis for therapeutic innovation in clinical oncology? Taking the root out, not chopping the leaf. Omics 2016, 20, 681–691. [Google Scholar] [CrossRef] [PubMed]
- Castells, M.; Thibault, B.; Delord, J.-P.; Couderc, B. Implication of tumor microenvironment in chemoresistance: Tumor-associated stromal cells protect tumor cells from cell death. Int. J. Mol. Sci. 2012, 13, 9545–9571. [Google Scholar] [CrossRef] [PubMed]
- Sung, S.Y.; Hsieh, C.L.; Wu, D.; Chung, L.W.; Johnstone, P.A. Tumor microenvironment promotes cancer progression, metastasis, and therapeutic resistance. Curr. Probl. Cancer 2007, 31, 36–100. [Google Scholar] [CrossRef] [PubMed]
- Whatcott, C.J.; Han, H.; Posner, R.G.; Hostetter, G.; Von Hoff, D.D. Targeting the tumor microenvironment in cancer: Why hyaluronidase deserves a second look. Cancer Discov. 2011, 1, 291–296. [Google Scholar] [CrossRef] [PubMed]
- Whiteside, T.L. The tumor microenvironment and its role in promoting tumor growth. Oncogene 2008, 27, 5904–5912. [Google Scholar] [CrossRef] [PubMed]
- Ahmadzadeh, H.; Webster, M.R.; Behera, R.; Jimenez Valencia, A.M.; Wirtz, D.; Weeraratna, A.T.; Shenoy, V.B. Modeling the two-way feedback between contractility and matrix realignment reveals a nonlinear mode of cancer cell invasion. Proc. Natl. Acad. Sci. USA 2017, 114, E1617–E1626. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Han, H.; Koo, H.; Na, J.H.; Yoon, H.Y.; Lee, K.E.; Lee, H.; Kim, H.; Kwon, I.C.; Kim, K. Extracellular matrix remodeling in vivo for enhancing tumor-targeting efficiency of nanoparticle drug carriers using the pulsed high intensity focused ultrasound. J. Control. Release 2017. [Google Scholar] [CrossRef] [PubMed]
- Logun, M.T.; Bisel, N.S.; Tanasse, E.A.; Zhao, W.; Gunasekera, B.; Mao, L.; Karumbaiah, L. Glioma cell invasion is significantly enhanced in composite hydrogel matrices composed of chondroitin 4- and 4,6-sulfated glycosaminoglycans. J. Mater. Chem. B 2016, 4, 6052–6064. [Google Scholar] [CrossRef] [PubMed]
- Maddaly, R.; Subramaniyan, A.; Balasubramanian, H. Cancer cytokines and the relevance of 3d cultures for studying those implicated in human cancers. J. Cell. Biochem. 2017, 118, 2544–2558. [Google Scholar] [CrossRef] [PubMed]
- Pinto, M.L.; Rios, E.; Silva, A.C.; Neves, S.C.; Caires, H.R.; Pinto, A.T.; Duraes, C.; Carvalho, F.A.; Cardoso, A.P.; Santos, N.C.; et al. Decellularized human colorectal cancer matrices polarize macrophages towards an anti-inflammatory phenotype promoting cancer cell invasion via ccl18. Biomaterials 2017, 124, 211–224. [Google Scholar] [CrossRef] [PubMed]
- Tourell, M.C.; Shokoohmand, A.; Landgraf, M.; Holzapfel, N.P.; Poh, P.S.; Loessner, D.; Momot, K.I. The distribution of the apparent diffusion coefficient as an indicator of the response to chemotherapeutics in ovarian tumour xenografts. Sci. Rep. 2017, 7, 42905. [Google Scholar] [CrossRef] [PubMed]
- Binder, M.J.; McCoombe, S.; Williams, E.D.; McCulloch, D.R.; Ward, A.C. The extracellular matrix in cancer progression: Role of hyalectan proteoglycans and adamts enzymes. Cancer Lett. 2017, 385, 55–64. [Google Scholar] [CrossRef] [PubMed]
- Di Marzo, L.; Desantis, V.; Solimando, A.G.; Ruggieri, S.; Annese, T.; Nico, B.; Fumarulo, R.; Vacca, A.; Frassanito, M.A. Microenvironment drug resistance in multiple myeloma: Emerging new players. Oncotarget 2016, 7, 60698–60711. [Google Scholar] [CrossRef] [PubMed]
- Ribatti, D. Epithelial-mesenchymal transition in morphogenesis, cancer progression and angiogenesis. Exp. Cell Res. 2017, 353, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Riechelmann, R.; Grothey, A. Antiangiogenic therapy for refractory colorectal cancer: Current options and future strategies. Ther. Adv. Med. Oncol. 2017, 9, 106–126. [Google Scholar] [CrossRef] [PubMed]
- Simone, V.; Brunetti, O.; Lupo, L.; Testini, M.; Maiorano, E.; Simone, M.; Longo, V.; Rolfo, C.; Peeters, M.; Scarpa, A.; et al. Targeting angiogenesis in biliary tract cancers: An open option. Int. J. Mol. Sci. 2017, 18, 418. [Google Scholar] [CrossRef] [PubMed]
- Epshtein, M.; Korin, N. Shear targeted drug delivery to stenotic blood vessels. J. Biomech. 2017, 50, 217–221. [Google Scholar] [CrossRef] [PubMed]
- Saber, M.M.; Bahrainian, S.; Dinarvand, R.; Atyabi, F. Targeted drug delivery of sunitinib malate to tumor blood vessels by crgd-chiotosan-gold nanoparticles. Int. J. Pharm. 2017, 517, 269–278. [Google Scholar] [CrossRef] [PubMed]
- Wenes, M.; Shang, M.; Di Matteo, M.; Goveia, J.; Martin-Perez, R.; Serneels, J.; Prenen, H.; Ghesquiere, B.; Carmeliet, P.; Mazzone, M. Macrophage metabolism controls tumor blood vessel morphogenesis and metastasis. Cell Metab. 2016, 24, 701–715. [Google Scholar] [CrossRef] [PubMed]
- Wong, P.P.; Bodrug, N.; Hodivala-Dilke, K.M. Exploring novel methods for modulating tumor blood vessels in cancer treatment. Curr. Biol. 2016, 26, R1161–R1166. [Google Scholar] [CrossRef] [PubMed]
- Haldorsen, I.S.; Stefansson, I.; Gruner, R.; Husby, J.A.; Magnussen, I.J.; Werner, H.M.; Salvesen, O.O.; Bjorge, L.; Trovik, J.; Taxt, T.; et al. Increased microvascular proliferation is negatively correlated to tumour blood flow and is associated with unfavourable outcome in endometrial carcinomas. Br. J. Cancer 2014, 110, 107–114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsafnat, N.; Tsafnat, G.; Lambert, T.D. A three-dimensional fractal model of tumour vasculature. In Proceedings of the 26th Annual International Conference of the IEEE Engineering in Medicine and Biology Society, San Francisco, CA, USA, 1–5 September 2004; pp. 683–686. [Google Scholar]
- Choi, S.H.; Park, J.Y. Regulation of the hypoxic tumor environment in hepatocellular carcinoma using RNA interference. Cancer Cell Int. 2017, 17, 3. [Google Scholar] [CrossRef] [PubMed]
- Daniell, K.; Nucera, C. Effect of the micronutrient iodine in thyroid carcinoma angiogenesis. Aging 2016, 8, 3180–3184. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Gao, F.; Song, W. Periostin contributes to arsenic trioxide resistance in hepatocellular carcinoma cells under hypoxia. Biomed. Pharmacother. 2017, 88, 342–348. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Ji, N.; Wei, W.; Sun, W.; Gong, X.; Wang, X. Mir-142 modulates human pancreatic cancer proliferation and invasion by targeting hypoxia-inducible factor 1 (HIF-1α) in the tumor microenvironments. Biol. Open 2017, 6, 252–259. [Google Scholar] [CrossRef] [PubMed]
- Semenza, G.L. Hypoxia-inducible factors: Coupling glucose metabolism and redox regulation with induction of the breast cancer stem cell phenotype. EMBO J. 2017, 36, 252–259. [Google Scholar] [CrossRef] [PubMed]
- Tarrado-Castellarnau, M.; de Atauri, P.; Cascante, M. Oncogenic regulation of tumor metabolic reprogramming. Oncotarget 2016, 7, 62726–62753. [Google Scholar] [CrossRef] [PubMed]
- Demaria, M.; O’Leary, M.N.; Chang, J.; Shao, L.; Liu, S.; Alimirah, F.; Koenig, K.; Le, C.; Mitin, N.; Deal, A.M.; et al. Cellular senescence promotes adverse effects of chemotherapy and cancer relapse. Cancer Discov. 2016. [Google Scholar] [CrossRef] [PubMed]
- Luna, J.I.; Grossenbacher, S.K.; Murphy, W.J.; Canter, R.J. Targeting cancer stem cells with natural killer cell immunotherapy. Expert Opin. Biol. Ther. 2016. [Google Scholar] [CrossRef] [PubMed]
- Pearl Mizrahi, S.; Gefen, O.; Simon, I.; Balaban, N.Q. Persistence to anti-cancer treatments in the stationary to proliferating transition. Cell Cycle 2016, 15, 3442–3453. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Wu, M.Y.; Jiang, M.; Zhi, Q.; Bian, X.; Xu, M.D.; Gong, F.R.; Hou, J.; Tao, M.; Shou, L.M.; et al. TNF-α sensitizes chemotherapy and radiotherapy against breast cancer cells. Cancer Cell Int. 2017, 17, 13. [Google Scholar] [CrossRef] [PubMed]
- Dart, A. Tumour metabolism: Packed full of protein! Nat. Rev. Cancer 2017, 17, 77. [Google Scholar] [CrossRef] [PubMed]
- Kremer, J.C.; Prudner, B.C.; Lange, S.E.; Bean, G.R.; Schultze, M.B.; Brashears, C.B.; Radyk, M.D.; Redlich, N.; Tzeng, S.C.; Kami, K.; et al. Arginine deprivation inhibits the warburg effect and upregulates glutamine anaplerosis and serine biosynthesis in ass1-deficient cancers. Cell Rep. 2017, 18, 991–1004. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, L.; Seyfried, T.; Alfarouk, K.O.; Da Veiga Moreira, J.; Fais, S. Out of warburg effect: An effective cancer treatment targeting the tumor specific metabolism and dysregulated ph. Semin. Cancer Biol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Cui, L.; Tse, K.; Zahedi, P.; Harding, S.M.; Zafarana, G.; Jaffray, D.A.; Bristow, R.G.; Allen, C. Hypoxia and cellular localization influence the radiosensitizing effect of gold nanoparticles (aunps) in breast cancer cells. Radiat. Res. 2014, 182, 475–488. [Google Scholar] [CrossRef] [PubMed]
- Michiels, C.; Tellier, C.; Feron, O. Cycling hypoxia: A key feature of the tumor microenvironment. Biochim. Biophys. Acta 2016, 1866, 76–86. [Google Scholar] [CrossRef] [PubMed]
- Muller-Edenborn, K.; Leger, K.; Glaus Garzon, J.F.; Oertli, C.; Mirsaidi, A.; Richards, P.J.; Rehrauer, H.; Spielmann, P.; Hoogewijs, D.; Borsig, L.; et al. Hypoxia attenuates the proinflammatory response in colon cancer cells by regulating ikappab. Oncotarget 2015, 6, 20288–20301. [Google Scholar] [CrossRef] [PubMed]
- Sun, Q.; Li, X. Targeting cyclic hypoxia to prevent malignant progression and therapeutic resistance of cancers. Histol. Histopathol. 2015, 30, 51–60. [Google Scholar] [PubMed]
- Vaupel, P.; Mayer, A. Hypoxia in tumors: Pathogenesis-related classification, characterization of hypoxia subtypes, and associated biological and clinical implications. Adv. Exp. Med. Biol. 2014, 812, 19–24. [Google Scholar] [PubMed]
- Vaupel, P.; Mayer, A. Tumor hypoxia: Causative mechanisms, microregional heterogeneities, and the role of tissue-based hypoxia markers. Adv. Exp. Med. Biol. 2016, 923, 77–86. [Google Scholar] [PubMed]
- Zhang, C.; Cao, S.; Toole, B.P.; Xu, Y. Cancer may be a pathway to cell survival under persistent hypoxia and elevated ros: A model for solid-cancer initiation and early development. Int. J. Cancer 2015, 136, 2001–2011. [Google Scholar] [CrossRef] [PubMed]
- Hu, Z.; Dong, N.; Lu, D.; Jiang, X.; Xu, J.; Wu, Z.; Zheng, D.; Wechsler, D.S. A positive feedback loop between ros and mxi1–0 promotes hypoxia-induced vegf expression in human hepatocellular carcinoma cells. Cell. Signal. 2017, 31, 79–86. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Yu, S.S.; Zong, M.; Fan, S.S.; Lu, T.B.; Gong, R.H.; Sun, L.S.; Fan, L.Y. Glucose-6-phosphate isomerase (G6PI) mediates hypoxia-induced angiogenesis in rheumatoid arthritis. Sci. Rep. 2017, 7, 40274. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Xu, Y.; Ma, J.; Pang, X.; Dong, M. Adrenomedullin promotes angiogenesis in epithelial ovarian cancer through upregulating hypoxia-inducible factor-1α and vascular endothelial growth factor. Sci. Rep. 2017, 7, 40524. [Google Scholar] [CrossRef] [PubMed]
- Da Silva, E.F.; Krause, G.C.; Lima, K.G.; Haute, G.V.; Pedrazza, L.; Mesquita, F.C.; Basso, B.S.; Velasquez, A.C.; Nunes, F.B.; de Oliveira, J.R. Rapamycin and fructose-1,6-bisphosphate reduce the HEPG2 cell proliferation via increase of free radicals and apoptosis. Oncol. Rep. 2016, 36, 2647–2652. [Google Scholar] [CrossRef] [PubMed]
- Fong, C.W. Platinum based radiochemotherapies: Free radical mechanisms and radiotherapy sensitizers. Free Radic. Biol. Med. 2016, 99, 99–109. [Google Scholar] [CrossRef] [PubMed]
- Guo, P.; Wang, S.; Liang, W.; Wang, W.; Wang, H.; Zhao, M.; Liu, X. Salvianolic acid b reverses multidrug resistance in HCT8/VCR human colorectal cancer cells by increasing ROS levels. Mol. Med. Rep. 2017, 15, 724–730. [Google Scholar] [PubMed]
- Cao, Z.; Scandura, J.M.; Inghirami, G.G.; Shido, K.; Ding, B.S.; Rafii, S. Molecular checkpoint decisions made by subverted vascular niche transform indolent tumor cells into chemoresistant cancer stem cells. Cancer Cell 2017, 31, 110–126. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Mazas, C.; Csaba, N.; Garcia-Fuentes, M. Biomaterials to suppress cancer stem cells and disrupt their tumoral niche. Int. J. Pharm. 2017, 523, 490–505. [Google Scholar] [CrossRef] [PubMed]
- Lee, G.; Hall, R.R., 3rd; Ahmed, A.U. Cancer stem cells: Cellular plasticity, niche, and its clinical relevance. J. Stem Cell Res. Ther. 2016, 6, 363. [Google Scholar] [CrossRef] [PubMed]
- Oei, A.L.; Vriend, L.E.; Krawczyk, P.M.; Horsman, M.R.; Franken, N.A.; Crezee, J. Targeting therapy-resistant cancer stem cells by hyperthermia. Int. J. Hyperth. 2017. [Google Scholar] [CrossRef] [PubMed]
- Picco, N.; Gatenby, R.A.; Anderson, A.R. Stem cell plasticity and niche dynamics in cancer progression. IEEE Trans. Biomed. Eng. 2016. [Google Scholar] [CrossRef] [PubMed]
- Shahriyari, L.; Mahdipour Shirayeh, A. Modeling dynamics of mutants in heterogeneous stem cell niche. Phys. Biol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Comerford, K.M.; Wallace, T.J.; Karhausen, J.; Louis, N.A.; Montalto, M.C.; Colgan, S.P. Hypoxia-inducible factor-1-dependent regulation of the multidrug resistance (mdr1) gene. Cancer Res. 2002, 62, 3387–3394. [Google Scholar] [PubMed]
- Cowan, D.S.; Tannock, I.F. Factors that influence the penetration of methotrexate through solid tissue. Int. J. Cancer 2001, 91, 120–125. [Google Scholar] [CrossRef]
- Mahoney, B.P.; Raghunand, N.; Baggett, B.; Gillies, R.J. Tumor acidity, ion trapping and chemotherapeutics. I. Acid pH affects the distribution of chemotherapeutic agents in vitro. Biochem. Pharmacol. 2003, 66, 1207–1218. [Google Scholar] [CrossRef]
- Abulaiti, A.; Shintani, Y.; Funaki, S.; Nakagiri, T.; Inoue, M.; Sawabata, N.; Minami, M.; Okumura, M. Interaction between non-small-cell lung cancer cells and fibroblasts via enhancement of tgf-β signaling by il-6. Lung Cancer 2013, 82, 204–213. [Google Scholar] [CrossRef] [PubMed]
- Mukaida, N.; Sasaki, S. Fibroblasts, an inconspicuous but essential player in colon cancer development and progression. World J. Gastroenterol. 2016, 22, 5301–5316. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Li, Q.; Yamada, T.; Matsumoto, K.; Matsumoto, I.; Oda, M.; Watanabe, G.; Kayano, Y.; Nishioka, Y.; Sone, S.; et al. Crosstalk to stromal fibroblasts induces resistance of lung cancer to epidermal growth factor receptor tyrosine kinase inhibitors. Clin. Cancer Res. 2009, 15, 6630–6638. [Google Scholar] [CrossRef] [PubMed]
- De Veirman, K.; Rao, L.; De Bruyne, E.; Menu, E.; Van Valckenborgh, E.; Van Riet, I.; Frassanito, M.A.; Di Marzo, L.; Vacca, A.; Vanderkerken, K. Cancer associated fibroblasts and tumor growth: Focus on multiple myeloma. Cancers 2014, 6, 1363–1381. [Google Scholar] [CrossRef] [PubMed]
- Matsuo, Y.; Ochi, N.; Sawai, H.; Yasuda, A.; Takahashi, H.; Funahashi, H.; Takeyama, H.; Tong, Z.; Guha, S. CXCL8/Il-8 and CXCL12/SDF-1α co-operatively promote invasiveness and angiogenesis in pancreatic cancer. Int. J. Cancer. J. Int. Cancer 2009, 124, 853–861. [Google Scholar] [CrossRef] [PubMed]
- Park, J.E.; Lenter, M.C.; Zimmermann, R.N.; Garin-Chesa, P.; Old, L.J.; Rettig, W.J. Fibroblast activation protein, a dual specificity serine protease expressed in reactive human tumor stromal fibroblasts. J. Biol. Chem. 1999, 274, 36505–36512. [Google Scholar] [CrossRef] [PubMed]
- Bharti, R.; Dey, G.; Mandal, M. Cancer development, chemoresistance, epithelial to mesenchymal transition and stem cells: A snapshot of il-6 mediated involvement. Cancer Lett. 2016, 375, 51–61. [Google Scholar] [CrossRef] [PubMed]
- Conze, D.; Weiss, L.; Regen, P.S.; Bhushan, A.; Weaver, D.; Johnson, P.; Rincon, M. Autocrine production of interleukin 6 causes multidrug resistance in breast cancer cells. Cancer Res. 2001, 61, 8851–8858. [Google Scholar] [PubMed]
- Sun, X.; Mao, Y.; Wang, J.; Zu, L.; Hao, M.; Cheng, G.; Qu, Q.; Cui, D.; Keller, E.T.; Chen, X.; et al. Il-6 secreted by cancer-associated fibroblasts induces tamoxifen resistance in luminal breast cancer. Oncogene 2014. [Google Scholar] [CrossRef] [PubMed]
- Houthuijzen, J.M.; Daenen, L.G.; Roodhart, J.M.; Voest, E.E. The role of mesenchymal stem cells in anti-cancer drug resistance and tumour progression. Br. J. Cancer 2012, 106, 1901–1906. [Google Scholar] [CrossRef] [PubMed]
- Sun, Z.; Wang, S.; Zhao, R.C. The roles of mesenchymal stem cells in tumor inflammatory microenvironment. J. Hematol. Oncol. 2014, 7, 14. [Google Scholar] [CrossRef] [PubMed]
- Roodhart, J.M.; Daenen, L.G.; Stigter, E.C.; Prins, H.J.; Gerrits, J.; Houthuijzen, J.M.; Gerritsen, M.G.; Schipper, H.S.; Backer, M.J.; van Amersfoort, M.; et al. Mesenchymal stem cells induce resistance to chemotherapy through the release of platinum-induced fatty acids. Cancer Cell 2011, 20, 370–383. [Google Scholar] [CrossRef] [PubMed]
- Erdogan, B.; Webb, D.J. Cancer-associated fibroblasts modulate growth factor signaling and extracellular matrix remodeling to regulate tumor metastasis. Biochem. Soc. Trans. 2017, 45, 229–236. [Google Scholar] [CrossRef] [PubMed]
- Gascard, P.; Tlsty, T.D. Carcinoma-associated fibroblasts: Orchestrating the composition of malignancy. Genes Dev. 2016, 30, 1002–1019. [Google Scholar] [CrossRef] [PubMed]
- Kalaszczynska, I.; Ferdyn, K. Wharton’s jelly derived mesenchymal stem cells: Future of regenerative medicine? Recent findings and clinical significance. BioMed Res. Int. 2015, 2015, 430847. [Google Scholar] [CrossRef] [PubMed]
- Underwood, T.J.; Hayden, A.L.; Derouet, M.; Garcia, E.; Noble, F.; White, M.J.; Thirdborough, S.; Mead, A.; Clemons, N.; Mellone, M.; et al. Cancer-associated fibroblasts predict poor outcome and promote periostin-dependent invasion in oesophageal adenocarcinoma. J. Pathol. 2015, 235, 466–477. [Google Scholar] [CrossRef] [PubMed]
- Chong, I.W.; Chang, M.Y.; Chang, H.C.; Yu, Y.P.; Sheu, C.C.; Tsai, J.R.; Hung, J.Y.; Chou, S.H.; Tsai, M.S.; Hwang, J.J.; et al. Great potential of a panel of multiple hMTH1, SPD, ITGA11 and COL11A1 markers for diagnosis of patients with non-small cell lung cancer. Oncol. Rep. 2006, 16, 981–988. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.H.; Chang, T.H.; Huang, Y.F.; Huang, H.D.; Chou, C.Y. Col11a1 promotes tumor progression and predicts poor clinical outcome in ovarian cancer. Oncogene 2014, 33, 3432–3440. [Google Scholar] [CrossRef] [PubMed]
- Lu, P.; Weaver, V.M.; Werb, Z. The extracellular matrix: A dynamic niche in cancer progression. J. Cell Biol. 2012, 196, 395–406. [Google Scholar] [CrossRef] [PubMed]
- Page-McCaw, A.; Ewald, A.J.; Werb, Z. Matrix metalloproteinases and the regulation of tissue remodelling. Nat. Rev. Mol. Cell Biol. 2007, 8, 221–233. [Google Scholar] [CrossRef] [PubMed]
- Cox, T.R.; Erler, J.T. Remodeling and homeostasis of the extracellular matrix: Implications for fibrotic diseases and cancer. Dis. Models Mech. 2011, 4, 165–178. [Google Scholar] [CrossRef] [PubMed]
- Paszek, M.J.; Weaver, V.M. The tension mounts: Mechanics meets morphogenesis and malignancy. J. Mammary Gland Biol. Neoplasia 2004, 9, 325–342. [Google Scholar] [CrossRef] [PubMed]
- Kass, L.; Erler, J.T.; Dembo, M.; Weaver, V.M. Mammary epithelial cell: Influence of extracellular matrix composition and organization during development and tumorigenesis. Int. J. Biochem. Cell Biol. 2007, 39, 1987–1994. [Google Scholar] [CrossRef] [PubMed]
- Bordeleau, F.; Mason, B.N.; Lollis, E.M.; Mazzola, M.; Zanotelli, M.R.; Somasegar, S.; Califano, J.P.; Montague, C.; LaValley, D.J.; Huynh, J.; et al. Matrix stiffening promotes a tumor vasculature phenotype. Proc. Natl. Acad. Sci. USA 2017, 114, 492–497. [Google Scholar] [CrossRef] [PubMed]
- Hui, L.; Zhang, J.; Ding, X.; Guo, X.; Jiang, X. Matrix stiffness regulates the proliferation, stemness and chemoresistance of laryngeal squamous cancer cells. Int. J. Oncol. 2017, 50, 1439–1447. [Google Scholar] [CrossRef] [PubMed]
- Hoon, J.L.; Tan, M.H.; Koh, C.G. The regulation of cellular responses to mechanical cues by Rho GTPases. Cells 2016, 5, 17. [Google Scholar] [CrossRef] [PubMed]
- Grantab, R.H.; Tannock, I.F. Penetration of anticancer drugs through tumour tissue as a function of cellular packing density and interstitial fluid pressure and its modification by bortezomib. BMC Cancer 2012, 12, 214. [Google Scholar] [CrossRef] [PubMed]
- Harisi, R.; Jeney, A. Extracellular matrix as target for antitumor therapy. OncoTargets Ther. 2015, 8, 1387–1398. [Google Scholar]
- Holle, A.W.; Young, J.L.; Spatz, J.P. In vitro cancer cell-ecm interactions inform in vivo cancer treatment. Adv. Drug Deliv. Rev. 2016, 97, 270–279. [Google Scholar] [CrossRef] [PubMed]
- Mittal, V.; El Rayes, T.; Narula, N.; McGraw, T.E.; Altorki, N.K.; Barcellos-Hoff, M.H. The microenvironment of lung cancer and therapeutic implications. Adv. Exp. Med. Biol. 2016, 890, 75–110. [Google Scholar] [PubMed]
- Nieponice, A.; McGrath, K.; Qureshi, I.; Beckman, E.J.; Luketich, J.D.; Gilbert, T.W.; Badylak, S.F. An extracellular matrix scaffold for esophageal stricture prevention after circumferential EMR. Gastrointest. Endosc. 2009, 69, 289–296. [Google Scholar] [CrossRef] [PubMed]
- Barcus, C.E.; Holt, E.C.; Keely, P.J.; Eliceiri, K.W.; Schuler, L.A. Dense collagen-I matrices enhance pro-tumorigenic estrogen-prolactin crosstalk in MCF-7 and T47D breast cancer cells. PLoS ONE 2015, 10, e0116891. [Google Scholar] [CrossRef] [PubMed]
- Barcus, C.E.; O’Leary, K.A.; Brockman, J.L.; Rugowski, D.E.; Liu, Y.; Garcia, N.; Yu, M.; Keely, P.J.; Eliceiri, K.W.; Schuler, L.A. Elevated collagen-I augments tumor progressive signals, intravasation and metastasis of prolactin-induced estrogen receptor α positive mammary tumor cells. Breast Cancer Res. 2017, 19, 9. [Google Scholar] [CrossRef] [PubMed]
- Brechbuhl, H.M.; Finlay-Schultz, J.; Yamamoto, T.; Gillen, A.; Cittelly, D.M.; Tan, A.C.; Sams, S.B.; Pillai, M.; Elias, A.; Robinson, W.A.; et al. Fibroblast subtypes regulate responsiveness of luminal breast cancer to estrogen. Clin. Cancer Res. 2016, 23, 1710–1721. [Google Scholar] [CrossRef] [PubMed]
- Cun, X.; Ruan, S.; Chen, J.; Zhang, L.; Li, J.; He, Q.; Gao, H. A dual strategy to improve the penetration and treatment of breast cancer by combining shrinking nanoparticles with collagen depletion by losartan. Acta Biomater. 2016, 31, 186–196. [Google Scholar] [CrossRef] [PubMed]
- Mbeunkui, F.; Johann, D.J. Cancer and the tumor microenvironment: A review of an essential relationship. Cancer Chemother. Pharmacol. 2009, 63, 571–582. [Google Scholar] [CrossRef] [PubMed]
- Levental, K.R.; Yu, H.; Kass, L.; Lakins, J.N.; Egeblad, M.; Erler, J.T.; Fong, S.F.; Csiszar, K.; Giaccia, A.; Weninger, W.; et al. Matrix crosslinking forces tumor progression by enhancing integrin signaling. Cell 2009, 139, 891–906. [Google Scholar] [CrossRef] [PubMed]
- Villegas-Pineda, J.C.; Toledo-Leyva, A.; Osorio-Trujillo, J.C.; Hernandez-Ramirez, V.I.; Talamas-Rohana, P. The translational blocking of α5 and α6 integrin subunits affects migration and invasion, and increases sensitivity to carboplatin of SKOV-3 ovarian cancer cell line. Exp. Cell Res. 2017, 351, 127–134. [Google Scholar] [CrossRef] [PubMed]
- Meenakshi Sundaram, D.N.; Kucharski, C.; Parmar, M.B.; Kc, R.B.; Uludag, H. Polymeric delivery of sirna against integrin-β1 (CD29) to reduce attachment and migration of breast cancer cells. Macromol. Biosci. 2017. [Google Scholar] [CrossRef] [PubMed]
- Gopal, S.; Veracini, L.; Grall, D.; Butori, C.; Schaub, S.; Audebert, S.; Camoin, L.; Baudelet, E.; Radwanska, A.; Beghelli-de la Forest Divonne, S.; et al. Fibronectin-guided migration of carcinoma collectives. Nat. Commun. 2017, 8, 14105. [Google Scholar] [CrossRef] [PubMed]
- Gehler, S.; Compere, F.V.; Miller, A.M. Semaphorin 3a increases FAK phosphorylation at focal adhesions to modulate MDA-MB-231 cell migration and spreading on different substratum concentrations. Int. J. Breast Cancer 2017, 2017, 9619734. [Google Scholar] [CrossRef] [PubMed]
- Morin, P.J. Drug resistance and the microenvironment: Nature and nurture. Drug Resist. Updat. 2003, 6, 169–172. [Google Scholar] [CrossRef]
- Sato, N.; Kohi, S.; Hirata, K.; Goggins, M. Role of hyaluronan in pancreatic cancer biology and therapy: Once again in the spotlight. Cancer Sci. 2016, 107, 569–575. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, T.; Packham, G.; Murphy, L.B.; Bateman, A.C.; Conti, J.A.; Fine, D.R.; Johnson, C.D.; Benyon, R.C.; Iredale, J.P. Type I collagen promotes the malignant phenotype of pancreatic ductal adenocarcinoma. Clin. Cancer Res. 2004, 10, 7427–7437. [Google Scholar] [CrossRef] [PubMed]
- Sethi, T.; Rintoul, R.C.; Moore, S.M.; MacKinnon, A.C.; Salter, D.; Choo, C.; Chilvers, E.R.; Dransfield, I.; Donnelly, S.C.; Strieter, R.; et al. Extracellular matrix proteins protect small cell lung cancer cells against apoptosis: A mechanism for small cell lung cancer growth and drug resistance in vivo. Nat. Med. 1999, 5, 662–668. [Google Scholar] [CrossRef] [PubMed]
- Januchowski, R.; Swierczewska, M.; Sterzynska, K.; Wojtowicz, K.; Nowicki, M.; Zabel, M. Increased expression of several collagen genes is associated with drug resistance in ovarian cancer cell lines. J. Cancer 2016, 7, 1295–1310. [Google Scholar] [CrossRef] [PubMed]
- Chauhan, V.P.; Stylianopoulos, T.; Boucher, Y.; Jain, R.K. Delivery of molecular and nanoscale medicine to tumors: Transport barriers and strategies. Annu. Rev. Chem. Biomol. Eng. 2011, 2, 281–298. [Google Scholar] [CrossRef] [PubMed]
- Jain, R.K. Transport of molecules in the tumor interstitium: A review. Cancer Res. 1987, 47, 3039–3051. [Google Scholar] [PubMed]
- St Croix, B.; Kerbel, R.S. Cell adhesion and drug resistance in cancer. Curr. Opin. Oncol. 1997, 9, 549–556. [Google Scholar] [CrossRef] [PubMed]
- Iseri, O.D.; Kars, M.D.; Arpaci, F.; Gunduz, U. Gene expression analysis of drug-resistant MCF-7 cells: Implications for relation to extracellular matrix proteins. Cancer Chemother. Pharmacol. 2010, 65, 447–455. [Google Scholar] [CrossRef] [PubMed]
- Netti, P.A.; Berk, D.A.; Swartz, M.A.; Grodzinsky, A.J.; Jain, R.K. Role of extracellular matrix assembly in interstitial transport in solid tumors. Cancer Res. 2000, 60, 2497–2503. [Google Scholar] [PubMed]
- Berchtold, S.; Grunwald, B.; Kruger, A.; Reithmeier, A.; Hahl, T.; Cheng, T.; Feuchtinger, A.; Born, D.; Erkan, M.; Kleeff, J.; et al. Collagen type V promotes the malignant phenotype of pancreatic ductal adenocarcinoma. Cancer Lett. 2015, 356, 721–732. [Google Scholar] [CrossRef] [PubMed]
- Shields, M.A.; Dangi-Garimella, S.; Redig, A.J.; Munshi, H.G. Biochemical role of the collagen-rich tumour microenvironment in pancreatic cancer progression. Biochem. J. 2012, 441, 541–552. [Google Scholar] [CrossRef] [PubMed]
- Provenzano, P.P.; Eliceiri, K.W.; Campbell, J.M.; Inman, D.R.; White, J.G.; Keely, P.J. Collagen reorganization at the tumor-stromal interface facilitates local invasion. BMC Med. 2006, 4, 38. [Google Scholar] [CrossRef] [PubMed]
- Tavazoie, S.F.; Alarcon, C.; Oskarsson, T.; Padua, D.; Wang, Q.; Bos, P.D.; Gerald, W.L.; Massague, J. Endogenous human micrornas that suppress breast cancer metastasis. Nature 2008, 451, 147–152. [Google Scholar] [CrossRef] [PubMed]
- Sahai, V.; Dangi-Garimella, S.; Ebine, K.; Kumar, K.; Munshi, H.G. Promotion of gemcitabine resistance in pancreatic cancer cells by three-dimensional collagen I through HMGA2-dependent histone acetyltransferase expression. J. Clin. Oncol. 2013, 31, 172. [Google Scholar] [CrossRef]
- Li, J.; Wood, W.H., 3rd; Becker, K.G.; Weeraratna, A.T.; Morin, P.J. Gene expression response to cisplatin treatment in drug-sensitive and drug-resistant ovarian cancer cells. Oncogene 2007, 26, 2860–2872. [Google Scholar] [CrossRef] [PubMed]
- Teng, P.N.; Wang, G.; Hood, B.L.; Conrads, K.A.; Hamilton, C.A.; Maxwell, G.L.; Darcy, K.M.; Conrads, T.P. Identification of candidate circulating cisplatin-resistant biomarkers from epithelial ovarian carcinoma cell secretomes. Br. J. Cancer 2014, 110, 123–132. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.H.; Chang, T.H.; Huang, Y.F.; Chen, C.C.; Chou, C.Y. Col11a1 confers chemoresistance on ovarian cancer cells through the activation of Akt/c/EBPβ pathway and PDK1 stabilization. Oncotarget 2015, 6, 23748–23763. [Google Scholar] [CrossRef] [PubMed]
- Januchowski, R.; Zawierucha, P.; Ruciński, M.; Nowicki, M.; Zabel, M. Extracellular matrix proteins expression profiling in chemoresistant variants of the A2780 ovarian cancer cell line. BioMed Res. Int. 2014, 2014, 365867. [Google Scholar] [CrossRef] [PubMed]
- Timpl, R.; Rohde, H.; Robey, P.G.; Rennard, S.I.; Foidart, J.M.; Martin, G.R. Laminin—A glycoprotein from basement membranes. J. Biol. Chem. 1979, 254, 9933–9937. [Google Scholar] [PubMed]
- Fukazawa, S.; Shinto, E.; Tsuda, H.; Ueno, H.; Shikina, A.; Kajiwara, Y.; Yamamoto, J.; Hase, K. Laminin β3 expression as a prognostic factor and a predictive marker of chemoresistance in colorectal cancer. Jpn. J. Clin. Oncol. 2015, 45, 533–540. [Google Scholar] [PubMed]
- Govaere, O.; Wouters, J.; Petz, M.; Vandewynckel, Y.P.; van den Eynde, K.; van den Broeck, A.; Verhulst, S.; Dolle, L.; Gremeaux, L.; Ceulemans, A.; et al. Laminin-332 sustains chemoresistance and quiescence as part of the human hepatic cancer stem cell niche. J. Hepatol. 2016, 64, 609–617. [Google Scholar] [CrossRef] [PubMed]
- Giannelli, G.; Azzariti, A.; Fransvea, E.; Porcelli, L.; Antonaci, S.; Paradiso, A. Laminin-5 offsets the efficacy of gefitinib (‘iressa’) in hepatocellular carcinoma cells. Br. J. Cancer 2004, 91, 1964–1969. [Google Scholar] [CrossRef] [PubMed]
- Tsurutani, J.; West, K.A.; Sayyah, J.; Gills, J.J.; Dennis, P.A. Inhibition of the phosphatidylinositol 3-kinase/Akt/mammalian target of rapamycin pathway but not the MEK/ERK pathway attenuates laminin-mediated small cell lung cancer cellular survival and resistance to imatinib mesylate or chemotherapy. Cancer Res. 2005, 65, 8423–8432. [Google Scholar] [CrossRef] [PubMed]
- Huanwen, W.; Zhiyong, L.; Xiaohua, S.; Xinyu, R.; Kai, W.; Tonghua, L. Intrinsic chemoresistance to gemcitabine is associated with constitutive and laminin-induced phosphorylation of FAK in pancreatic cancer cell lines. Mol. Cancer 2009, 8, 125. [Google Scholar] [CrossRef] [PubMed]
- Pankov, R.; Yamada, K.M. Fibronectin at a glance. J. Cell Sci. 2002, 115, 3861–3863. [Google Scholar] [CrossRef] [PubMed]
- Rintoul, R.C.; Sethi, T. Extracellular matrix regulation of drug resistance in small-cell lung cancer. Clin. Sci. 2002, 102, 417–424. [Google Scholar] [CrossRef] [PubMed]
- Kosmehl, H.; Berndt, A.; Strassburger, S.; Borsi, L.; Rousselle, P.; Mandel, U.; Hyckel, P.; Zardi, L.; Katenkamp, D. Distribution of laminin and fibronectin isoforms in oral mucosa and oral squamous cell carcinoma. Br. J. Cancer 1999, 81, 1071–1079. [Google Scholar] [CrossRef] [PubMed]
- Han, S.; Sidell, N.; Roser-Page, S.; Roman, J. Fibronectin stimulates human lung carcinoma cell growth by inducing cyclooxygenase-2 (COX-2) expression. Int. J. Cancer 2004, 111, 322–331. [Google Scholar] [CrossRef] [PubMed]
- Han, S.; Sidell, N.; Roman, J. Fibronectin stimulates human lung carcinoma cell proliferation by suppressing p21 gene expression via signals involving Erk and rho kinase. Cancer Lett. 2005, 219, 71–81. [Google Scholar] [CrossRef] [PubMed]
- Xing, H.; Weng, D.; Chen, G.; Tao, W.; Zhu, T.; Yang, X.; Meng, L.; Wang, S.; Lu, Y.; Ma, D. Activation of fibronectin/PI-3K/Akt2 leads to chemoresistance to docetaxel by regulating survivin protein expression in ovarian and breast cancer cells. Cancer Lett. 2008, 261, 108–119. [Google Scholar] [CrossRef] [PubMed]
- Horiuchi, K.; Amizuka, N.; Takeshita, S.; Takamatsu, H.; Katsuura, M.; Ozawa, H.; Toyama, Y.; Bonewald, L.F.; Kudo, A. Identification and characterization of a novel protein, periostin, with restricted expression to periosteum and periodontal ligament and increased expression by transforming growth factor β. J. Bone Miner. Res. 1999, 14, 1239–1249. [Google Scholar] [CrossRef] [PubMed]
- Moniuszko, T.; Wincewicz, A.; Koda, M.; Domysławska, I.; Sulkowski, S. Role of periostin in esophageal, gastric and colon cancer. Oncol. Lett. 2016, 12, 783–787. [Google Scholar] [CrossRef] [PubMed]
- Gillan, L.; Matei, D.; Fishman, D.A.; Gerbin, C.S.; Karlan, B.Y.; Chang, D.D. Periostin secreted by epithelial ovarian carcinoma is a ligand for α(v)β(3) and α(v)β(5) integrins and promotes cell motility. Cancer Res. 2002, 62, 5358–5364. [Google Scholar] [PubMed]
- Zhu, M.; Fejzo, M.S.; Anderson, L.; Dering, J.; Ginther, C.; Ramos, L.; Gasson, J.C.; Karlan, B.Y.; Slamon, D.J. Periostin promotes ovarian cancer angiogenesis and metastasis. Gynecol. Oncol. 2010, 119, 337–344. [Google Scholar] [CrossRef] [PubMed]
- Tumbarello, D.A.; Temple, J.; Brenton, J.D. β3 integrin modulates transforming growth factor β induced (TGFBI) function and paclitaxel response in ovarian cancer cells. Mol. Cancer 2012, 11, 36. [Google Scholar] [CrossRef] [PubMed]
- Sung, P.-L.; Jan, Y.-H.; Lin, S.-C.; Huang, C.-C.; Lin, H.; Wen, K.-C.; Chao, K.-C.; Lai, C.-R.; Wang, P.-H.; Chuang, C.-M.; et al. Periostin in tumor microenvironment is associated with poor prognosis and platinum resistance in epithelial ovarian carcinoma. Oncotarget 2016, 7, 4036–4047. [Google Scholar] [PubMed]
- Abdel-Qadir, H.; Ethier, J.L.; Lee, D.S.; Thavendiranathan, P.; Amir, E. Cardiovascular toxicity of angiogenesis inhibitors in treatment of malignancy: A systematic review and meta-analysis. Cancer Treat. Rev. 2016, 53, 120–127. [Google Scholar] [CrossRef] [PubMed]
- Cantelmo, A.R.; Pircher, A.; Kalucka, J.; Carmeliet, P. Vessel pruning or healing: Endothelial metabolism as a novel target? Expert Opin. Ther. Targets 2017, 21, 239–247. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.T.; Oh, D.Y.; Ryu, M.H.; Yeh, K.H.; Yeo, W.; Carlesi, R.; Cheng, R.; Kim, J.; Orlando, M.; Kang, Y.K. Anti-angiogenic therapy in patients with advanced gastric and gastroesophageal junction cancer: A systematic review. Cancer Res. Treat. 2017. [Google Scholar] [CrossRef] [PubMed]
- Torok, S.; Rezeli, M.; Kelemen, O.; Vegvari, A.; Watanabe, K.; Sugihara, Y.; Tisza, A.; Marton, T.; Kovacs, I.; Tovari, J.; et al. Limited tumor tissue drug penetration contributes to primary resistance against angiogenesis inhibitors. Theranostics 2017, 7, 400–412. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, Y.; Hisano, H.; Nishikawa, Y.; Nagasaki, Y. Targeting and treatment of tumor hypoxia by newly designed prodrug possessing high permeability in solid tumors. Mol. Pharm. 2016, 13, 2283–2289. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.Y.; Lee, G.Y.; Lane, L.A.; Li, B.; Wang, J.; Lu, Q.; Wang, Y.; Nie, S. Functionalized, long-circulating, and ultrasmall gold nanocarriers for overcoming the barriers of low nanoparticle delivery efficiency and poor tumor penetration. Bioconjug. Chem. 2017, 28, 244–252. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Li, P.; He, Y.; Bo, X.; Li, X.; Li, D.; Chen, H.; Xu, H. Synergistic retention strategy of RGD active targeting and radiofrequency-enhanced permeability for intensified RF & chemotherapy synergistic tumor treatment. Biomaterials 2016, 99, 34–46. [Google Scholar] [PubMed]
- Jin, J.; Pastrello, D.; Penning, N.A.; Jones, A.T. Clustering of endocytic organelles in parental and drug-resistant myeloid leukaemia cell lines lacking centrosomally organised microtubule arrays. Int. J. Biochem. Cell Biol. 2008, 40, 2240–2252. [Google Scholar] [CrossRef] [PubMed]
- Kitatani, K.; Idkowiak-Baldys, J.; Hannun, Y.A. Mechanism of inhibition of sequestration of protein kinase C α/βII by ceramide. Roles of ceramide-activated protein phosphatases and phosphorylation/dephosphorylation of protein kinase C α/βII on threonine 638/641. J. Biol. Chem. 2007, 282, 20647–20656. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.M.; Tannock, I.F. Inhibition of endosomal sequestration of basic anticancer drugs: Influence on cytotoxicity and tissue penetration. Br. J. Cancer 2006, 94, 863–869. [Google Scholar] [CrossRef] [PubMed]
- Seebacher, N.A.; Lane, D.J.; Jansson, P.J.; Richardson, D.R. Glucose modulation induces lysosome formation and increases lysosomotropic drug sequestration via the p-glycoprotein drug transporter. J. Biol. Chem. 2016, 291, 3796–3820. [Google Scholar] [CrossRef] [PubMed]
- Williams, M.; Catchpoole, D. Sequestration of AS-DACA into acidic compartments of the membrane trafficking system as a mechanism of drug resistance in rhabdomyosarcoma. Int. J. Mol. Sci. 2013, 14, 13042–13062. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Vaananen, H.K. Pharmacological sequestration of intracellular cholesterol in late endosomes disrupts ruffled border formation in osteoclasts. J. Bone Miner. Res. 2006, 21, 456–465. [Google Scholar] [CrossRef] [PubMed]
- Parodi, A.; Haddix, S.G.; Taghipour, N.; Scaria, S.; Taraballi, F.; Cevenini, A.; Yazdi, I.K.; Corbo, C.; Palomba, R.; Khaled, S.Z.; et al. Bromelain surface modification increases the diffusion of silica nanoparticles in the tumor extracellular matrix. ACS Nano 2014, 8, 9874–9883. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Shen, S.; Liao, Z.; Shi, W.; Wang, Y.; Zhao, J.; Hu, Y.; Yang, J.; Chen, J.; Mei, H.; et al. Targeting fibronectins of glioma extracellular matrix by CLT1 peptide-conjugated nanoparticles. Biomaterials 2014, 35, 4088–4098. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Fan, Z.; Deng, J.; Lemons, P.K.; Arhontoulis, D.C.; Bowne, W.B.; Cheng, H. Hyaluronidase embedded in nanocarrier peg shell for enhanced tumor penetration and highly efficient antitumor efficacy. Nano Lett. 2016, 16, 3268–3277. [Google Scholar] [CrossRef]
- Fink, K.; Boratynski, J. The role of metalloproteinases in modification of extracellular matrix in invasive tumor growth, metastasis and angiogenesis. Postepy Hig. Med. Dosw. 2012, 66, 609–628. [Google Scholar] [CrossRef]
- Kotula, E.; Berthault, N.; Agrario, C.; Lienafa, M.C.; Simon, A.; Dingli, F.; Loew, D.; Sibut, V.; Saule, S.; Dutreix, M. DNA-PKcs plays role in cancer metastasis through regulation of secreted proteins involved in migration and invasion. Cell Cycle 2015, 14, 1961–1972. [Google Scholar] [CrossRef]
- Ortiz, R.; Diaz, J.; Diaz, N.; Lobos-Gonzalez, L.; Cardenas, A.; Contreras, P.; Diaz, M.I.; Otte, E.; Cooper-White, J.; Torres, V.; et al. Extracellular matrix-specific caveolin-1 phosphorylation on tyrosine 14 is linked to augmented melanoma metastasis but not tumorigenesis. Oncotarget 2016, 7, 40571–40593. [Google Scholar] [CrossRef]
- Rucci, N.; Sanita, P.; Angelucci, A. Roles of metalloproteases in metastatic niche. Curr. Mol. Med. 2011, 11, 609–622. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Guo, H.; Terajima, M.; Banerjee, P.; Liu, X.; Yu, J.; Momin, A.A.; Katayama, H.; Hanash, S.M.; Burns, A.R.; et al. Lysyl hydroxylase 2 is secreted by tumor cells and can modify collagen in the extracellular space. J. Biol. Chem. 2016, 291, 25799–25808. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, M.F.; Mortensen, M.B.; Detlefsen, S. Key players in pancreatic cancer-stroma interaction: Cancer-associated fibroblasts, endothelial and inflammatory cells. World J. Gastroenterol. 2016, 22, 2678–2700. [Google Scholar] [CrossRef] [Green Version]
- Spicer, G.L.; Azarin, S.M.; Yi, J.; Young, S.T.; Ellis, R.; Bauer, G.M.; Shea, L.D.; Backman, V. Detection of extracellular matrix modification in cancer models with inverse spectroscopic optical coherence tomography. Phys. Med. Biol. 2016, 61, 6892–6904. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Lu, L.; Feng, Y.; Wang, H.; Dai, L.; Li, Y.; Zhang, P. PKD2 mediates multi-drug resistance in breast cancer cells through modulation of P-glycoprotein expression. Cancer Lett. 2011, 300, 48–56. [Google Scholar] [CrossRef]
- Slamon, D.J.; Leyland-Jones, B.; Shak, S.; Fuchs, H.; Paton, V.; Bajamonde, A.; Fleming, T.; Eiermann, W.; Wolter, J.; Pegram, M.; et al. Use of chemotherapy plus a monoclonal antibody against HER2 for metastatic breast cancer that overexpresses HER2. N. Engl. J. Med. 2001, 344, 783–792. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, D.; Humblet, Y.; Siena, S.; Khayat, D.; Bleiberg, H.; Santoro, A.; Bets, D.; Mueser, M.; Harstrick, A.; Verslype, C.; et al. Cetuximab monotherapy and cetuximab plus irinotecan in irinotecan-refractory metastatic colorectal cancer. N. Engl. J. Med. 2004, 351, 337–345. [Google Scholar] [CrossRef] [PubMed]








© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Senthebane, D.A.; Rowe, A.; Thomford, N.E.; Shipanga, H.; Munro, D.; Mazeedi, M.A.M.A.; Almazyadi, H.A.M.; Kallmeyer, K.; Dandara, C.; Pepper, M.S.; et al. The Role of Tumor Microenvironment in Chemoresistance: To Survive, Keep Your Enemies Closer. Int. J. Mol. Sci. 2017, 18, 1586. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms18071586
Senthebane DA, Rowe A, Thomford NE, Shipanga H, Munro D, Mazeedi MAMA, Almazyadi HAM, Kallmeyer K, Dandara C, Pepper MS, et al. The Role of Tumor Microenvironment in Chemoresistance: To Survive, Keep Your Enemies Closer. International Journal of Molecular Sciences. 2017; 18(7):1586. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms18071586
Chicago/Turabian StyleSenthebane, Dimakatso Alice, Arielle Rowe, Nicholas Ekow Thomford, Hendrina Shipanga, Daniella Munro, Mohammad A. M. Al Mazeedi, Hashim A. M. Almazyadi, Karlien Kallmeyer, Collet Dandara, Michael S. Pepper, and et al. 2017. "The Role of Tumor Microenvironment in Chemoresistance: To Survive, Keep Your Enemies Closer" International Journal of Molecular Sciences 18, no. 7: 1586. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms18071586