FANCD2 and DNA Damage


1
Cancer Biology Program, University of Hawaii Cancer Center, Honolulu, HI 96813, USA
2
Graduate Program of Molecular Biosciences and Bioengineering, University of Hawaii, Honolulu, HI 96813, USA
3
Department of Laboratory Medicine and Pathology, Mayo Clinic Foundation, Rochester, MN 55905, USA
*
Author to whom correspondence should be addressed.
Int. J. Mol. Sci. 2017, 18(8), 1804; https://0-doi-org.brum.beds.ac.uk/10.3390/ijms18081804
Submission received: 31 July 2017
/
Revised: 8 August 2017
/
Accepted: 12 August 2017
/
Published: 19 August 2017
(This article belongs to the Special Issue DNA Injury and Repair Systems)
{kind=link}
Abstract
:Investigators have dedicated considerable effort to understanding the molecular basis underlying Fanconi Anemia (FA), a rare human genetic disease featuring an extremely high incidence of cancer and many congenital defects. Among those studies, FA group D2 protein (FANCD2) has emerged as the focal point of FA signaling and plays crucial roles in multiple aspects of cellular life, especially in the cellular responses to DNA damage. Here, we discuss the recent and relevant studies to provide an updated review on the roles of FANCD2 in the DNA damage response.
1. Introduction of the Fanconi Anemia Pathway
Fanconi anemia (FA) is a rare human genetic disease displaying various clinical symptoms such as severe bone marrow failure, an extremely high incidence of cancer, and many other congenital defects. The incidence of FA is relatively low, manifesting in less than 1 per 136,000 live births. However, the frequency of normal individuals carrying mutations in any of the FA genes is considerably higher, which is estimated at about 1 in 300. Whereby, the incidence of cancer in FA patients is thought to occur at a rate approximately fifty fold higher than the general population and even several hundred-fold higher for particular malignancies [1]. At the cellular level, FA is characterized by chromosomal abnormalities and hypersensitivity to DNA crosslinking agents, such as mitomycin C (MMC), diepoxybutane (DEB), and Cisplatin [2,3,4]. These common features are displayed in at least 22 known complementation groups FANC-A, B, C, D1, D2, E, F, G, I, J, L, M, N, O, P, Q, R, S, T, U, V and W [5,6,7,8,9,10,11,12,13,14,15,16,17], which suggests that the FA proteins all function in a shared signaling transduction pathway, namely, the FA pathway. This pathway is activated either during DNA replication or upon DNA damage [18,19]. Upon activation, Fanconi anemia group D2 protein (FANCD2) is monoubiquitinated at K561 and works in cooperation with other FA and non-FA proteins [9,11,20,21,22,23,24] to repair DNA damage [5]. In regards to the FA pathway, monoubiquitinated or activated FANCD2 is often portrayed as the functional representative for the functions of activated FA signaling. Within FA signaling or any signaling networks, protein complex formation is essential in regulating signal transduction and FA function. This includes the FA core complex, which is comprised of eight FA proteins and contains the E3 function [6,9,11,21], of which FANCL is the catalytic subunit [25]. Silencing FANCL or any other component of the FA E3 complex is a routine tool used in the laboratory to study the functions of activated versus inactivated FANCD2.
In the past decade, we have been greatly interested in studying the role of the FA signaling pathway in human cancer [26,27]. This pathway appears to be an essential part of the DNA-damage repair-signaling network. Interestingly, four FA-gene products (FANCD1/J/N/S) are previously-known DNA-damage repair proteins (BRCA1/2, BRIP1, and PALB2), which are mainly related to breast cancer susceptibility; and many others (FANCG/O/Q/R/U/V/W) are also previously known proteins involved in DNA damage repair (Rad51, Rad51C, XRCC2/4/9, REV7, and RFWD3) [17,28]. With a considerable number of these functionally-significant players in the FA signaling pathway, it is clear that the FA pathway is greatly important in maintaining genome stability in response to a variety of genotoxic stresses in the general population. Therefore, we believe it is crucial to first investigate the characteristics of FANCD2, the representative of the FA pathway, in order to advance our understanding of how the FA signaling pathway protects human cells from genome instability and thus neoplastic transformation.
2. The Importance of Fanconi Anemia Group D2 Protein (FANCD2)
Amongst the FA genes, FANCD2 is the most evolutionarily conserved gene, demonstrating a high sequence similarity from lower eukaryotes to humans [29]. This differs from the other FA genes (with the exception of FANCM [7]), as most of the FA homologs only exist in mammals, acting in concert to respond to DNA damage [30]. DNA crosslinks are one of the few major forms of DNA damage, which presents a major challenge to genomic stability. In eukaryotes, DNA crosslink repair is a complex process; yeast cells require a combination of nucleotide excision repair (NER), homologous recombination (HR) repair, and post replication repair/translesion DNA synthesis (TLS) to remove DNA crosslinks. However, in mammalian cells, the cellular response to DNA crosslinks requires the coordination of complex signaling networks, which includes the FA proteins/pathway [29]. Typically in FA cells, the inability of FANCD2 to be monoubiquitinated appears to be a common molecular defect in response to a variety of genotoxic stresses [12,15,27,31]. Many studies indicate that FANCD2 acts in coordination with many known repair proteins and those yet to be identified, and in nearly all phases of the DNA damage response, damage-sensing, signal transduction, and execution of repair. As such, FANCD2 can perform the signaling transduction role in ATM signaling; specifically, the phosphorylation of FANCD2 at Ser222, initiated by ATM, contributes to arresting cells in the S phase of the cell cycle [32]. Considering checkpoint mechanisms centering on the coordinated events [33], the DNA damage repair function of FANCD2 is equally crucial in arresting or resuming cell proliferation, or in helping eliminate over-damaged cells via apoptosis; however, the latter demands additional studies.
To date, 22 corresponding FA genes have been identified, and their biallelic germline mutations account for the occurrence of FA with the exception of FANCB and FANCR [34,35]. In addition, about 80% of the biallelic germline mutations are attributed to compromised FANCD2 monoubiquitination [36]. As monoubiquitination occurs on the evolutionarily conserved lysine residues, mutations affecting either FANCD2 at K561 or its paralog FANCI at K523 appear to be responsible for major molecular defects, as displayed by the hypersensitivity to DNA damage agents in the FA cells [37,38]. Following monoubiquitination, FANCD2/I is required to work in concert with FANCD1/breast cancer type 2 susceptibility protein (BRCA2), FANCR/RAD51, FANCS/BRCA1, FANCN, FANCJ, FANCO, FANCP, and FANCQ for DNA damage repair [39] in nearly all known repair mechanisms, such as HR, TLS, or postreplication repair, and others [40,41]. To date, accumulated FA studies have indicated that many of the FA and FA-associated proteins not only perform their common role in the FA pathway, but also conduct tasks in an FA pathway-independent manner [42,43,44,45]. Among them, FANCD2 is highly prominent and associated with numerous functions performed independent of the FA pathway. This is further supported by its conservation in regards to its homologous presence in the species, wherein many FA gene-related homologs are absent [27].
3. FANCD2 under Stressed Conditions
The cellular stress response is comprised of a network of signaling transduction pathways; when DNA damage occurs, the signaling transduction pathways in this network are well coordinated and divided into phases of sensing, signal transduction, and DNA-lesion repair. Specifically, surveillance proteins can sense DNA damage, initiate cell growth arrest, perform DNA-damage repair, and/or execute cell death programs. These responses inhibit the generation of potentially deleterious cancerous mutations to guard genome stability [46].
3.1. Cross Talking with Human Homologs of Yeast Rad6 (HHR6) Signaling
A similar sensitivity to crosslinking agents is displayed in FA cells as well as in the Rad6−/− yeast-prompted study on the potential link between FANCD2 monoubiquitination and human homologs of yeast Rad6 (HHR6) [47]. Subsequently, HHR6 was found to be capable of regulating FANCD2 monoubiquitination in a distinct manner from FANCT (the ubiquitin conjugating enzyme E2-UBE2T) [47], which cooperates with the FA complex E3 to monoubiquitinate FANCD2 [12]. This observation questioned whether or not hRad18, a functional partner of HHR6, also participated in the regulation of FANCD2 monoubiquitination. To answer this, several studies showed that hRad18 could also regulate FANCD2 monoubiquitination [48,49,50]. Moreover, monoubiquitinated FANCD2 was found to modulate the activity of translesion DNA synthesis [26], at least partly through interacting with pol eta at known regions previously characterized to interact with proliferating cell nuclear antigen (PCNA) [51,52]. Importantly, upon DNA damage, the interaction between pol eta and FANCD2 occurs earlier than that with PCNA [26]. Furthermore, it has been indicated that FANCD2 monoubiquitination performs an anchoring role, similar to the histone proteins to bind DNA in general, but more specific in regards to FANCD2 regulation of pol eta relocation at the site of damaged DNA [26]. Additionally, FANCD2 monoubiquitination can also occur in vitro in the absence of the FA core complex E3 [53], evidently showing it can act in an FA pathway-independent manner. Together, these studies indicate that in the early phases of the DNA damage response, FANCD2 plays a crucial role as a sensor as well as a messenger for the timely repair of damaged DNA (Figure 1).
3.2. Coupling with Ataxia Telangiectasia and Rad3-Related Protein (ATR)/Ataxia Telangiectasia Mutated (ATM) Signaling
In response to genotoxic stresses, the FA pathway activates the FA core complex harboring the activity of E3 ubiquitin ligase, which in turn leads to the monoubiquitination of FANCI and FANCD2 (Figure 1). The monoubiquitinated FANCI-FANCD2 complex is recruited to DNA damage sites and promotes TLS, NER, and Rad51-medated HR [54,55]. Ataxia telangiectasia mutated (ATM) along with its regulator, the MRN (Mre1 1-Rad50-NBS1) complex, sense double strand breaks (DSBs) [56]. Whereas, ATR with its regulator ATRIP (ATR-interacting protein) sense single strand DNA (ssDNA) that was generated by processing DSBs, as well as the ssDNA present at the stalled replication forks. Both kinases then phosphorylate proteins to initiate signaling cascades, which includes checkpoint kinases (CHK1) and (CHK2), both of which can initiate a secondary wave of phosphorylation events to extend signaling and promote DNA-damage repair signaling [57]. Typically, ionizing radiation can lead to ATM phosphorylation of FANCD2 and Nijmegen breakage syndrome protein 1 (NBS1), and an S-phase arrest [58]. Thus, FANCD2 not only performs a critical role in orchestrating the FA proteins in the FA signaling pathway but also closely cooperates with ATM to issue an S phase arrest to modulate cell proliferation and eventually prevent cells from genome instability.
In the FA pathway, the FANCD2/I complex has been known for its close partnership between FANCD2 and its paralog FANCI as early as 2007 when FANCI was identified [59]. Studies have shown that many functions of FANCD2 in the FA signaling pathway are largely dependent on the phosphorylation of FANCI that is performed by ATR [60]. This is consistent with the non-ubiquitinated state of the FANCD2-FANCI complex when recruited to DNA interstrand crosslinks [61]. BLM was also found to be involved in the activation of FANCD2 in stressed cells [62]. Certainly, this discovery promotes its function in maintaining genome stability, which includes a newly identified role in governing the stability of replication forks [63]. FANCD2 was also found to be required for proper phosphorylation of H2AX (a variant of the H2A protein family) and hence activation of ATM in rhabdomyosarcoma Rh30 cells, but not essential for ATR-Chk1 activation. This observation spans beyond the roles that FANCD2 plays in response to ICL damage [54,55] (as the focal point in the FA signaling pathway). Here, FANCD2 acts more like a sensor, similar to those sensors passing signaling in the early phase of the DNA damage response (DDR). This substantially contributes to ATM functions in maintaining genome integrity in response to DNA DSB [64]. Furthermore, FANCD2 was also found to be capable of modulating the enzymatic activities of FAN1 and pol eta [26,65,66,67,68], needed in the later phase of the DDR for DNA damage repair. However, whether FANCD2 directly plays some enzymatic roles in the DNA damage responses waits to be further investigated, although its nuclease activity was proposed [69].
3.3. Cooperating with Other Signaling Pathways
Previously, studies have suggested that signaling pathways such as PI3K and Ras are deregulated during the progression of human malignancy. Additionally, the defective FA pathway function is also a likely contributor during the course of the neoplastic transformation, which originates from these defective signaling pathways. Moreover, in yeast, mechanistic target of rapamycin (mTOR) promotes cell survival but at the cost of an increase in the alkylation agent melphalan, accompanied with significant down-regulation of FANCD2 [70]. Experiments have demonstrated that mTOR signaling controls FANCD2 gene transcription via cyclin-dependent kinase 4 (CDK4), supporting the observation that FANCD2 is regulated by Rb-E2F1(retinoblastoma protein-E2 transcription factor 1) [71]. Furthermore, Ras, p53, and Rb signaling was also found to be able to circuit the signaling control of FANCD2 by CDK4 [72]. This suggests that cancer cells with self-sufficiency in growth signaling and resistance to anti-proliferation signaling may depend on the functional status of FANCD2 for survival and the activation of the DNA damage checkpoint mechanisms discussed above. However, this field of studies regarding the FA proteins is relatively under-investigated, requiring more studies to continue.
4. FANCD2 in Non-Stressed Condition
As viewed above, huge attention has been given to the stress condition of cells where monoubiquitination/activation of FANCD2 takes place to eventually repair damaged DNA. In contrast, little attention has been given to FANCD2 under normal (non-stressed) conditions. In non-stressed cells, the FA pathway is not constitutively active; rather, it is activated in the S phase of the cell cycle [31]. This basal level of FANCD2 monoubiquitination occurring in normally growing cells has been demonstrated to be essential for replication origins to fire at a normal rate [73]. Conversely, the loss of the basal level of FANCD2 monoubiquitination leads to a slow rate of replication origin firing. For the first time, this observation was able to give a rational explanation of the aging phenotype displayed in many FA patients. With the studies focused on the FA proteins for their roles in resolving stalled replication forks [62,74], whether FANCD2 also plays roles in replication elongation and/or termination appears to be another important aspect for future research on FANCD2 under non-stressed conditions. Recently, with the progress of many modern research technologies, metabolomics has attracted many cancer researchers to study the cancer mechanism at the metabolic level. Accordingly, the first metabolomics study for the tumor promotion role played by a compromised FA signaling pathway filled the bank for this related field of studies in metabolism, consistent with the well-accepted concept that cancer is one of four major metabolic disorders, which also include aging, diabetes, and stroke [75]. These recent studies on the involvement of FANCD2 in metabolism via the mitochondria [36,76] further indicate the importance of FANCD2 functions in non-stressed cells, which unveil crucial biological functions that were not previously considered.
5. Conclusive Remarks
FANCD2 critically governs the FA signaling pathway by interacting with FA and non-FA protein partners for DNA damage repair to guard genome stability. On the other hand, FANCD2 can also function as a veteran checkpoint-player (Figure 1), coupling with a variety of cellular processes outside the FA signaling pathway. As DNA damage causes cancer, it is also acknowledged that, conversely, DNA damage is also beneficial to the efficacy of cancer treatments. Recently, the development of molecular inhibitors has become a promising therapeutic strategy to target DNA-damage repair by inhibiting the DNA repair process. From viewing functions focused on FANCD2, the inhibition of a DNA-damage repair player or even a whole process is harder to achieve than expected, because the redundancy, overlapping, and multifaceted natures are rooted in the human DNA repair signaling network, for which we shall need to have more in-depth studies to get into the bottom of these properties.
Acknowledgments
The referenced work from our own laboratory was supported in part by NIH grants (R01CA136532 & R01CA188251 to PF) and institutional supports from the University of Hawaii Cancer Center and Mayo Clinic Foundation. We apologize for the absence of citations supporting the similar findings.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Alter, B.P. Cancer in Fanconi anemia, 1927–2001. Cancer 2003, 97, 425–440. [Google Scholar] [CrossRef] [PubMed]
- D’Andrea, A.D. The Fanconi road to cancer. Genes Dev. 2003, 17, 1933–1936. [Google Scholar] [CrossRef] [PubMed]
- Bagby, G.C., Jr. Genetic basis of Fanconi anemia. Curr. Opin. Hematol. 2003, 10, 68–76. [Google Scholar] [CrossRef] [PubMed]
- D’Andrea, A.D.; Grompe, M. The Fanconi anaemia/BRCA pathway. Nat. Rev. Cancer 2003, 3, 23–34. [Google Scholar] [CrossRef] [PubMed]
- Hahn, S.A.; Greenhalf, B.; Ellis, I.; Sina-Frey, M.; Rieder, H.; Korte, B.; Gerdes, B.; Kress, R.; Ziegler, A.; Raeburn, J.A.; et al. BRCA2 germline mutations in familial pancreatic carcinoma. J. Natl. Cancer Inst. 2003, 95, 214–221. [Google Scholar] [CrossRef] [PubMed]
- Taniguchi, T.; D’Andrea, A.D. Molecular pathogenesis of Fanconi anemia: Recent progress. Blood 2006, 107, 4223–4233. [Google Scholar] [CrossRef] [PubMed]
- Meetei, A.R.; Medhurst, A.L.; Ling, C.; Xue, Y.; Singh, T.R.; Bier, P.; Steltenpool, J.; Stone, S.; Dokal, I.; Mathew, C.G.; et al. A human ortholog of archaeal DNA repair protein Hef is defective in Fanconi anemia complementation group M. Nat. Genet. 2005, 37, 958–963. [Google Scholar] [CrossRef] [PubMed]
- Huang, T.T.; Nijman, S.M.; Mirchandani, K.D.; Galardy, P.J.; Cohn, M.A.; Haas, W.; Gygi, S.P.; Ploegh, H.L.; Bernards, R.; D’Andrea, A.D. Regulation of monoubiquitinated PCNA by DUB autocleavage. Nat. Cell Biol. 2006, 8, 339–347. [Google Scholar] [CrossRef] [PubMed]
- Reid, S.S.D.; Hanenberg, H.; Barker, K.; Hanks, S.; Kalb, R.; Neveling, K.; Kelly, P.; Seal, S.; Reund, M.; Wurm, M.; et al. Biallelic mutations in PALB2 cause Fanconi anemia subtype FA-N and predispose to childhood cancer. Nat. Genet. 2006, 39, 162–164. [Google Scholar] [CrossRef] [PubMed]
- Roest, H.P.; Baarends, W.M.; de Wit, J.; van Klaveren, J.W.; Wassenaar, E.; Hoogerbrugge, J.W.; van Cappellen, W.A.; Hoeijmakers, J.H.; Grootegoed, J.A. The ubiquitin-conjugating DNA repair enzyme HR6A is a maternal factor essential for early embryonic development in mice. Mol. Cell. Biol. 2004, 24, 5485–5495. [Google Scholar] [CrossRef] [PubMed]
- Xia, B.; Dorsman, J.C.; Ameziane, N.; de Vries, Y.; Rooimans, M.A.; Sheng, Q.; Pals, G.; Errami, A.; Gluckman, E.; Llera, J.; et al. Fanconi anemia is associated with a defect in the BRCA2 partner PALB2. Nat. Genet. 2006, 39, 159–161. [Google Scholar] [CrossRef] [PubMed]
- Wang, W. Emergence of a DNA-damage response network consisting of Fanconi aneamia and BRCA proteins. Nat. Rev. Genet. 2007, 8, 735–748. [Google Scholar] [CrossRef] [PubMed]
- Deans, A.J.; West, S.C. DNA interstrand crosslink repair and cancer. Nat. Rev. Genet. 2011, 11, 467–480. [Google Scholar] [CrossRef] [PubMed]
- Bogliolo, M.; Schuster, B.; Stoepker, C.; Derkunt, B.; Su, Y.; Raams, A.; Trujillo, J.P.; Minguillon, J.; Ramirez, M.J.; Pujol, R.; et al. Mutations in ERCC4, encoding the DNA-repair endonuclease XPF, cause Fanconi anemia. Am. J. Hum. Genet. 2013, 92, 800–806. [Google Scholar] [CrossRef] [PubMed]
- Pickering, A.; Zhang, J.; Panneerselvam, J.; Fei, P. Advances in the understanding of Fanconi anemia tumor suppressor pathway. Cancer Biol. Ther. 2013, 14, 1089–1091. [Google Scholar] [CrossRef] [PubMed]
- Bogliolo, M.; Surralles, J. Fanconi anemia: A model disease for studies on human genetics and advanced therapeutics. Curr. Opin. Genet. Dev. 2015, 33, 32–40. [Google Scholar] [CrossRef] [PubMed]
- Knies, K.; Inano, S.; Ramirez, M.J.; Ishiai, M.; Surralles, J.; Takata, M.; Schindler, D. Biallelic mutations in the ubiquitin ligase RFWD3 cause Fanconi anemia. J. Clin. Investig. 2017, 127, 3013–3027. [Google Scholar] [CrossRef] [PubMed]
- Sala-Trepat, M.; Rouillard, D.; Escarceller, M.; Laquerbe, A.; Moustacchi, E.; Papadopoulo, D. Arrest of S-phase progression is impaired in Fanconi anemia cells. Exp. Cell Res. 2000, 260, 208–215. [Google Scholar] [CrossRef] [PubMed]
- Grompe, M. FANCL, as in ligase. Nat. Genet. 2003, 35, 113–114. [Google Scholar] [CrossRef]
- Howlett, N.G.; Taniguchi, T.; Olson, S.; Cox, B.; Waisfisz, Q.; de Die-Smulders, C.; Persky, N.; Grompe, M.; Joenje, H.; Pals, G.; et al. Biallelic inactivation of BRCA2 in Fanconi anemia. Science 2002, 297, 606–609. [Google Scholar] [CrossRef] [PubMed]
- Rahman, N.S.S.; Thompson, D.; Kelly, P.; Renwick, A.; Elliott, A.; Reid, S.; Spanova, K.; Barfoot, R.; Chagtai, T.; Jayatilake, H.; et al. PALB2, which encodes a BRCA2-interacting protein, is a breast cancer susceptibility gene. Nat. Genet. 2006, 39, 165–167. [Google Scholar] [CrossRef] [PubMed]
- Xia, B.; Sheng, Q.; Nakanishi, K.; Ohashi, A.; Wu, J.; Christ, N.; Liu, X.; Jasin, M.; Couch, F.J.; Livingston, D.M. Control of BRCA2 cellular and clinical functions by a nuclear partner, PALB2. Mol. Cell 2006, 22, 719–729. [Google Scholar] [CrossRef] [PubMed]
- Levitus, M.; Waisfisz, Q.; Godthelp, B.C.; de Vries, Y.; Hussain, S.; Wiegant, W.W.; Elghalbzouri-Maghrani, E.; Steltenpool, J.; Rooimans, M.A.; Pals, G.; et al. The DNA helicase BRIP1 is defective in Fanconi anemia complementation group J. Nat. Genet. 2005, 37, 934–935. [Google Scholar] [CrossRef] [PubMed]
- Litman, R.; Peng, M.; Jin, Z.; Zhang, F.; Zhang, J.; Powell, S.; Andreassen, P.R.; Cantor, S.B. BACH1 is critical for homologous recombination and appears to be the Fanconi anemia gene product FANCJ. Cancer Cell 2005, 8, 255–265. [Google Scholar] [CrossRef] [PubMed]
- Meetei, A.R.; de Winter, J.P.; Medhurst, A.L.; Wallisch, M.; Waisfisz, Q.; van de Vrugt, H.J.; Oostra, A.B.; Yan, Z.; Ling, C.; Bishop, C.E.; et al. A novel ubiquitin ligase is deficient in Fanconi anemia. Nat. Genet. 2003, 35, 165–170. [Google Scholar] [CrossRef] [PubMed]
- Fu, D.; Dudimah, F.D.; Zhang, J.; Pickering, A.; Paneerselvam, J.; Palrasu, M.; Wang, H.; Fei, P. Recruitment of DNA polymerase eta by FANCD2 in the early response to DNA damage. Cell Cycle 2013, 12, 803–809. [Google Scholar] [CrossRef] [PubMed]
- Fei, P.; Yin, J.; Wang, W. New advances in the DNA damage response network of Fanconi anemia and BRCA proteins. FAAP95 replaces BRCA2 as the true FANCB protein. Cell Cycle 2005, 4, 80–86. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Masson, J.Y.; Shah, R.; O’Regan, P.; West, S.C. RAD51C is required for Holliday junction processing in mammalian cells. Science 2004, 303, 243–246. [Google Scholar] [CrossRef] [PubMed]
- McHugh, P.J.; Ward, T.A.; Chovanec, M. A prototypical Fanconi anemia pathway in lower eukaryotes? Cell Cycle 2012, 11, 3739–3744. [Google Scholar] [CrossRef] [PubMed]
- Alpi, A.F.; Patel, K.J. Monoubiquitylation in the Fanconi anemia DNA damage response pathway. DNA Repair (Amst.) 2009, 8, 430–435. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; D’Andrea, A.D. Regulation of DNA cross-link repair by the Fanconi anemia/BRCA pathway. Genes Dev. 2012, 26, 1393–1408. [Google Scholar] [CrossRef] [PubMed]
- Bartek, J.; Lukas, C.; Lukas, J. Checking on DNA damage in S phase. Nat. Rev. Mol. Cell Biol. 2004, 5, 792–804. [Google Scholar] [CrossRef] [PubMed]
- Shaltiel, I.A.; Krenning, L.; Bruinsma, W.; Medema, R.H. The same, only different—DNA damage checkpoints and their reversal throughout the cell cycle. J. Cell Sci. 2015, 128, 607–620. [Google Scholar] [CrossRef] [PubMed]
- Meetei, A.R.; Levitus, M.; Xue, Y.; Medhurst, A.L.; Zwaan, M.; Ling, C.; Rooimans, M.A.; Bier, P.; Hoatlin, M.; Pals, G.; et al. X-linked inheritance of Fanconi anemia complementation group B. Nat. Genet. 2004, 36, 1219–1224. [Google Scholar] [CrossRef] [PubMed]
- Ameziane, N.; May, P.; Haitjema, A.; van de Vrugt, H.J.; van Rossum-Fikkert, S.E.; Ristic, D.; Williams, G.J.; Balk, J.; Rockx, D.; Li, H.; et al. A novel Fanconi anaemia subtype associated with a dominant-negative mutation in RAD51. Nat. Commun. 2015, 6, 8829. [Google Scholar] [CrossRef] [PubMed]
- Jayabal, P.; Ma, C.; Nepal, M.; Shen, Y.; Che, R.; Turkson, J.; Fei, P. Involvement of FANCD2 in Energy Metabolism via ATP5alpha. Sci. Rep. 2017, 7, 4921. [Google Scholar] [CrossRef] [PubMed]
- Romick-Rosendale, L.E.; Lui, V.W.; Grandis, J.R.; Wells, S.I. The Fanconi anemia pathway: Repairing the link between DNA damage and squamous cell carcinoma. Mutat. Res. 2013, 743, 78–88. [Google Scholar] [CrossRef] [PubMed]
- Alpi, A.F.; Pace, P.E.; Babu, M.M.; Patel, K.J. Mechanistic insight into site-restricted monoubiquitination of FANCD2 by Ube2t, FANCL, and FANCI. Mol. Cell 2008, 32, 767–777. [Google Scholar] [CrossRef] [PubMed]
- Grompe, M.; D’Andrea, A. Fanconi anemia and DNA repair. Hum. Mol. Genet. 2001, 10, 2253–2259. [Google Scholar] [CrossRef] [PubMed]
- Murina, O.; Von Aesch, C.; Karakus, U.; Ferretti, L.P.; Bolck, H.A.; Hanggi, K.; Sartori, A.A. FANCD2 and CtIP cooperate to repair DNA interstrand crosslinks. Cell Rep. 2014, 7, 1030–1038. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lachaud, C.; Moreno, A.; Marchesi, F.; Toth, R.; Blow, J.J.; Rouse, J. Ubiquitinated FANCD2 recruits Fan1 to stalled replication forks to prevent genome instability. Science 2016, 351, 846–849. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Sipple, J.; Maynard, S.; Mehta, P.A.; Rose, S.R.; Davies, S.M.; Pang, Q. Fanconi anemia links reactive oxygen species to insulin resistance and obesity. Antioxid. Redox Signal. 2012, 17, 1083–1098. [Google Scholar] [CrossRef] [PubMed]
- Matsuzaki, K.; Borel, V.; Adelman, C.A.; Schindler, D.; Boulton, S.J. FANCJ suppresses microsatellite instability and lymphomagenesis independent of the Fanconi anemia pathway. Genes Dev. 2015, 29, 2532–2546. [Google Scholar] [CrossRef] [PubMed]
- Daschkey, S.; Bienemann, K.; Schuster, V.; Kreth, H.W.; Linka, R.M.; Honscheid, A.; Fritz, G.; Johannes, C.; Fleckenstein, B.; Kempkes, B.; et al. Fatal Lymphoproliferative Disease in Two Siblings Lacking Functional FAAP24. J. Clin. Immunol. 2016, 36, 684–692. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Lu, X.; Akhter, S.; Georgescu, M.M.; Legerski, R.J. FANCI is a negative regulator of Akt activation. Cell Cycle 2016, 15, 1134–1143. [Google Scholar] [CrossRef] [PubMed]
- Jackson, S.P.; Bartek, J. The DNA-damage response in human biology and disease. Nature 2009, 461, 1071–1078. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Zhao, D.; Wang, H.; Lin, C.J.; Fei, P. FANCD2 monoubiquitination provides a link between the HHR6 and FA-BRCA pathways. Cell Cycle 2008, 7, 407–413. [Google Scholar] [CrossRef] [PubMed]
- Park, H.K.; Wang, H.; Zhang, J.; Datta, S.; Fei, P. Convergence of Rad6/Rad18 and Fanconi anemia tumor suppressor pathways upon DNA damage. PLoS ONE 2010, 5, e13313. [Google Scholar] [CrossRef] [PubMed]
- Song, I.Y.; Palle, K.; Gurkar, A.; Tateishi, S.; Kupfer, G.M.; Vaziri, C. Rad18-mediated trans-lesion synthesis of bulky DNA adducts is coupled to activation of the fanconi anemia DNA repair pathway. J. Biol. Chem. 2010, 285, 1525–1536. [Google Scholar] [CrossRef] [PubMed]
- Geng, L.; Huntoon, C.J.; Karnitz, L.M. RAD18-mediated ubiquitination of PCNA activates the Fanconi anemia DNA repair network. J. Cell Biol. 2010, 191, 249–257. [Google Scholar] [CrossRef] [PubMed]
- Bienko, M.; Green, C.M.; Crosetto, N.; Rudolf, F.; Zapart, G.; Coull, B.; Kannouche, P.; Wider, G.; Peter, M.; Lehmann, A.R.; et al. Ubiquitin-binding domains in Y-family polymerases regulate translesion synthesis. Science 2005, 310, 1821–1824. [Google Scholar] [CrossRef] [PubMed]
- Kannouche, P.L.; Wing, J.; Lehmann, A.R. Interaction of human DNA polymerase eta with monoubiquitinated PCNA: A possible mechanism for the polymerase switch in response to DNA damage. Mol. Cell 2004, 14, 491–500. [Google Scholar] [CrossRef]
- Pickering, A.; Panneerselvam, J.; Zhang, J.; Zheng, J.; Zhang, Y.; Fei, P. In vitro FANCD2 monoubiquitination by HHR6 and hRad18. Cell Cycle 2013, 12, 3448–3449. [Google Scholar] [CrossRef] [PubMed]
- Joo, W.; Xu, G.; Persky, N.S.; Smogorzewska, A.; Rudge, D.G.; Buzovetsky, O.; Elledge, S.J.; Pavletich, N.P. Structure of the FANCI-FANCD2 complex: Insights into the Fanconi anemia DNA repair pathway. Science 2011, 333, 312–316. [Google Scholar] [CrossRef] [PubMed]
- Knipscheer, P.; Raschle, M.; Smogorzewska, A.; Enoiu, M.; Ho, T.V.; Scharer, O.D.; Elledge, S.J.; Walter, J.C. The Fanconi anemia pathway promotes replication-dependent DNA interstrand cross-link repair. Science 2009, 326, 1698–1701. [Google Scholar] [CrossRef] [PubMed]
- Kastan, M.B.; Bartek, J. Cell-cycle checkpoints and cancer. Nature 2004, 432, 316–323. [Google Scholar] [CrossRef] [PubMed]
- Shiloh, Y. The ATM-mediated DNA-damage response: Taking shape. Trends Biochem. Sci. 2006, 31, 402–410. [Google Scholar] [CrossRef] [PubMed]
- Nakanishi, K.; Taniguchi, T.; Ranganathan, V.; New, H.V.; Moreau, L.A.; Stotsky, M.; Mathew, C.G.; Kastan, M.B.; Weaver, D.T.; D’Andrea, A.D. Interaction of FANCD2 and NBS1 in the DNA damage response. Nat. Cell Biol. 2002, 4, 913–920. [Google Scholar] [CrossRef] [PubMed]
- Smogorzewska, A.; Matsuoka, S.; Vinciguerra, P.; McDonald, E.R., 3rd; Hurov, K.E.; Luo, J.; Ballif, B.A.; Gygi, S.P.; Hofmann, K.; D’Andrea, A.D.; et al. Identification of the FANCI protein, a monoubiquitinated FANCD2 paralog required for DNA repair. Cell 2007, 129, 289–301. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.H.; Jones, M.J.; Yin, Y.; Crist, S.B.; Colnaghi, L.; Sims, R.J., 3rd; Rothenberg, E.; Jallepalli, P.V.; Huang, T.T. ATR-mediated phosphorylation of FANCI regulates dormant origin firing in response to replication stress. Mol. Cell 2015, 58, 323–338. [Google Scholar] [CrossRef] [PubMed]
- Liang, C.C.; Li, Z.; Lopez-Martinez, D.; Nicholson, W.V.; Venien-Bryan, C.; Cohn, M.A. The FANCD2-FANCI complex is recruited to DNA interstrand crosslinks before monoubiquitination of FANCD2. Nat. Commun. 2016, 7, 12124. [Google Scholar] [CrossRef] [PubMed]
- Panneerselvam, J.; Wang, H.; Zhang, J.; Che, R.; Yu, H.; Fei, P. BLM promotes the activation of Fanconi Anemia signaling pathway. Oncotarget 2016, 7, 32351–32361. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Su, F.; Mukherjee, S.; Mori, E.; Hu, B.; Asaithamby, A. FANCD2 influences replication fork processes and genome stability in response to clustered DSBs. Cell Cycle 2015, 14, 1809–1822. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Cheng, H.; Roberts, T.M.; Zhao, J.J. Targeting the phosphoinositide 3-kinase pathway in cancer. Nat. Rev. Drug Discov. 2009, 8, 627–644. [Google Scholar] [CrossRef] [PubMed]
- Smogorzewska, A.; Desetty, R.; Saito, T.T.; Schlabach, M.; Lach, F.P.; Sowa, M.E.; Clark, A.B.; Kunkel, T.A.; Harper, J.W.; Colaiacovo, M.P.; et al. A genetic screen identifies FAN1, a Fanconi anemia-associated nuclease necessary for DNA interstrand crosslink repair. Mol. Cell 2010, 39, 36–47. [Google Scholar] [CrossRef] [PubMed]
- Lachaud, C.; Slean, M.; Marchesi, F.; Lock, C.; Odell, E.; Castor, D.; Toth, R.; Rouse, J. Karyomegalic interstitial nephritis and DNA damage-induced polyploidy in Fan1 nuclease-defective knock-in mice. Genes Dev. 2016, 30, 639–644. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pizzolato, J.; Mukherjee, S.; Scharer, O.D.; Jiricny, J. FANCD2-associated nuclease 1, but not exonuclease 1 or flap endonuclease 1, is able to unhook DNA interstrand cross-links in vitro. J. Biol. Chem. 2015, 290, 22602–22611. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Ghosal, G.; Yuan, J.; Chen, J.; Huang, J. FAN1 acts with FANCI-FANCD2 to promote DNA interstrand cross-link repair. Science 2010, 329, 693–696. [Google Scholar] [CrossRef] [PubMed]
- Pace, P.; Mosedale, G.; Hodskinson, M.R.; Rosado, I.V.; Sivasubramaniam, M.; Patel, K.J. Ku70 corrupts DNA repair in the absence of the Fanconi anemia pathway. Science 2010, 329, 219–223. [Google Scholar] [CrossRef] [PubMed]
- Shen, C.; Oswald, D.; Phelps, D.; Cam, H.; Pelloski, C.E.; Pang, Q.; Houghton, P.J. Regulation of FANCD2 by the mTOR pathway contributes to the resistance of cancer cells to DNA double-strand breaks. Cancer Res. 2013, 73, 3393–3401. [Google Scholar] [CrossRef] [PubMed]
- Hoskins, E.E.; Gunawardena, R.W.; Habash, K.B.; Wise-Draper, T.M.; Jansen, M.; Knudsen, E.S.; Wells, S.I. Coordinate regulation of Fanconi anemia gene expression occurs through the Rb/E2F pathway. Oncogene 2008, 27, 4798–4808. [Google Scholar] [CrossRef] [PubMed]
- Shen, C.; Houghton, P.J. Targeting FANCD2 for therapy sensitization. Oncotarget 2014, 5, 3426–3427. [Google Scholar] [CrossRef] [PubMed]
- Panneerselvam, J.; Pickering, A.; Han, B.; Li, L.; Zheng, J.; Zhang, J.; Zhang, Y.; Fei, P. Basal level of FANCD2 monoubiquitination is required for the maintenance of a sufficient number of licensed-replication origins to fire at a normal rate. Oncotarget 2014, 5, 1326–1337. [Google Scholar] [CrossRef] [PubMed]
- Ling, C.; Huang, J.; Yan, Z.; Li, Y.; Ohzeki, M.; Ishiai, M.; Xu, D.; Takata, M.; Seidman, M.; Wang, W. Bloom syndrome complex promotes FANCM recruitment to stalled replication forks and facilitates both repair and traverse of DNA interstrand crosslinks. Cell Discov. 2016, 2, 16047. [Google Scholar] [CrossRef] [PubMed]
- Panneerselvam, J.; Xie, G.; Che, R.; Su, M.; Zhang, J.; Jia, W.; Fei, P. Distinct Metabolic Signature of Human Bladder Cancer Cells Carrying an Impaired Fanconi Anemia Tumor-Suppressor Signaling Pathway. J. Proteom. Res. 2016, 15, 1333–1341. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Du, W.; Wilson, A.F.; Namekawa, S.H.; Andreassen, P.R.; Meetei, A.R.; Pang, Q. Fancd2 in vivo interaction network reveals a non-canonical role in mitochondrial function. Sci. Rep. 2017, 7, 45626. [Google Scholar] [CrossRef] [PubMed]
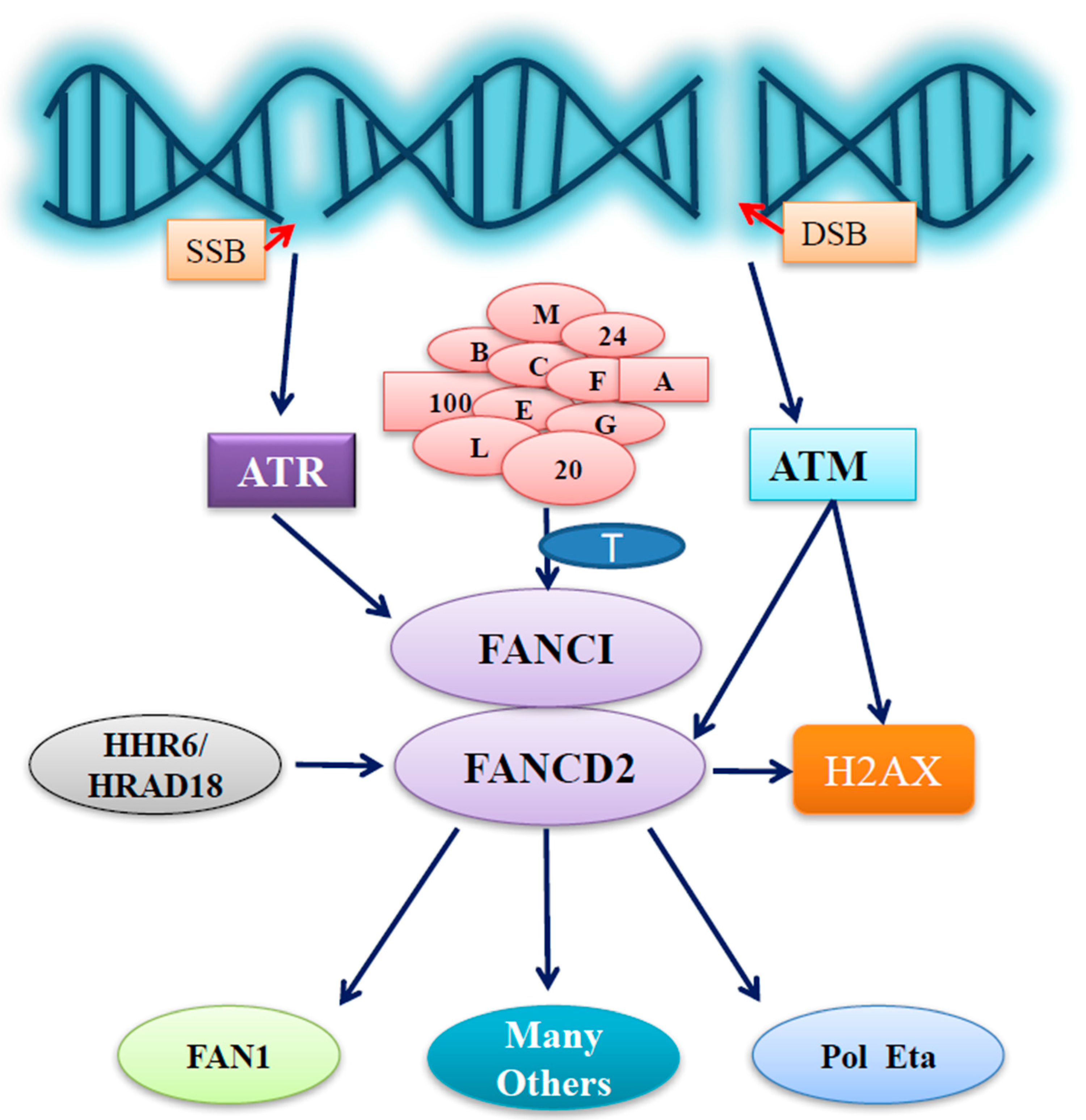
Figure 1.
Outline of FA group D2 protein (FANCD2) functions under stressed conditions. In stressed cells, ataxia telangiectasia and Rad3-related protein (ATR) or ataxia telangiectasia mutated (ATM) is activated upon the generation of DNA single or double strand breaks (SSB or DSB), respectively. FANCD2 activation/monoubiquitination issued from the Fanconi anemia (FA) core complex E3 and an E2 (FANCT) can be promoted by the phosphorylation of FANCI triggered by activated ATR, thus conducting important roles that have originated from the activation of ATR. FANCD2 can also play roles not only in aiding ATM signaling for S phase arrest through ATM-dependent phosphorylation at S222 but also possibly in facilitating the initiation of ATM signaling far more upstream via its involvement in the phosphorylation of H2AX. Furthermore, human homologs of yeast Rad6 (HHR6) & hRad18 are also capable of regulating the functions of FANCD2, together influencing the functions of the downstream partners of FANCD2, including Fanconi-associated nuclease 1 (FAN1), DNA polymerase eta (Pol eta), and many others known or yet to be identified.
Figure 1.
Outline of FA group D2 protein (FANCD2) functions under stressed conditions. In stressed cells, ataxia telangiectasia and Rad3-related protein (ATR) or ataxia telangiectasia mutated (ATM) is activated upon the generation of DNA single or double strand breaks (SSB or DSB), respectively. FANCD2 activation/monoubiquitination issued from the Fanconi anemia (FA) core complex E3 and an E2 (FANCT) can be promoted by the phosphorylation of FANCI triggered by activated ATR, thus conducting important roles that have originated from the activation of ATR. FANCD2 can also play roles not only in aiding ATM signaling for S phase arrest through ATM-dependent phosphorylation at S222 but also possibly in facilitating the initiation of ATM signaling far more upstream via its involvement in the phosphorylation of H2AX. Furthermore, human homologs of yeast Rad6 (HHR6) & hRad18 are also capable of regulating the functions of FANCD2, together influencing the functions of the downstream partners of FANCD2, including Fanconi-associated nuclease 1 (FAN1), DNA polymerase eta (Pol eta), and many others known or yet to be identified.

© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Nepal, M.; Che, R.; Ma, C.; Zhang, J.; Fei, P. FANCD2 and DNA Damage. Int. J. Mol. Sci. 2017, 18, 1804. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms18081804
AMA Style
Nepal M, Che R, Ma C, Zhang J, Fei P. FANCD2 and DNA Damage. International Journal of Molecular Sciences. 2017; 18(8):1804. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms18081804
Chicago/Turabian StyleNepal, Manoj, Raymond Che, Chi Ma, Jun Zhang, and Peiwen Fei. 2017. "FANCD2 and DNA Damage" International Journal of Molecular Sciences 18, no. 8: 1804. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms18081804
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.