Crosstalk between DNA Damage and Inflammation in the Multiple Steps of Carcinogenesis


Abstract
:1. Introduction
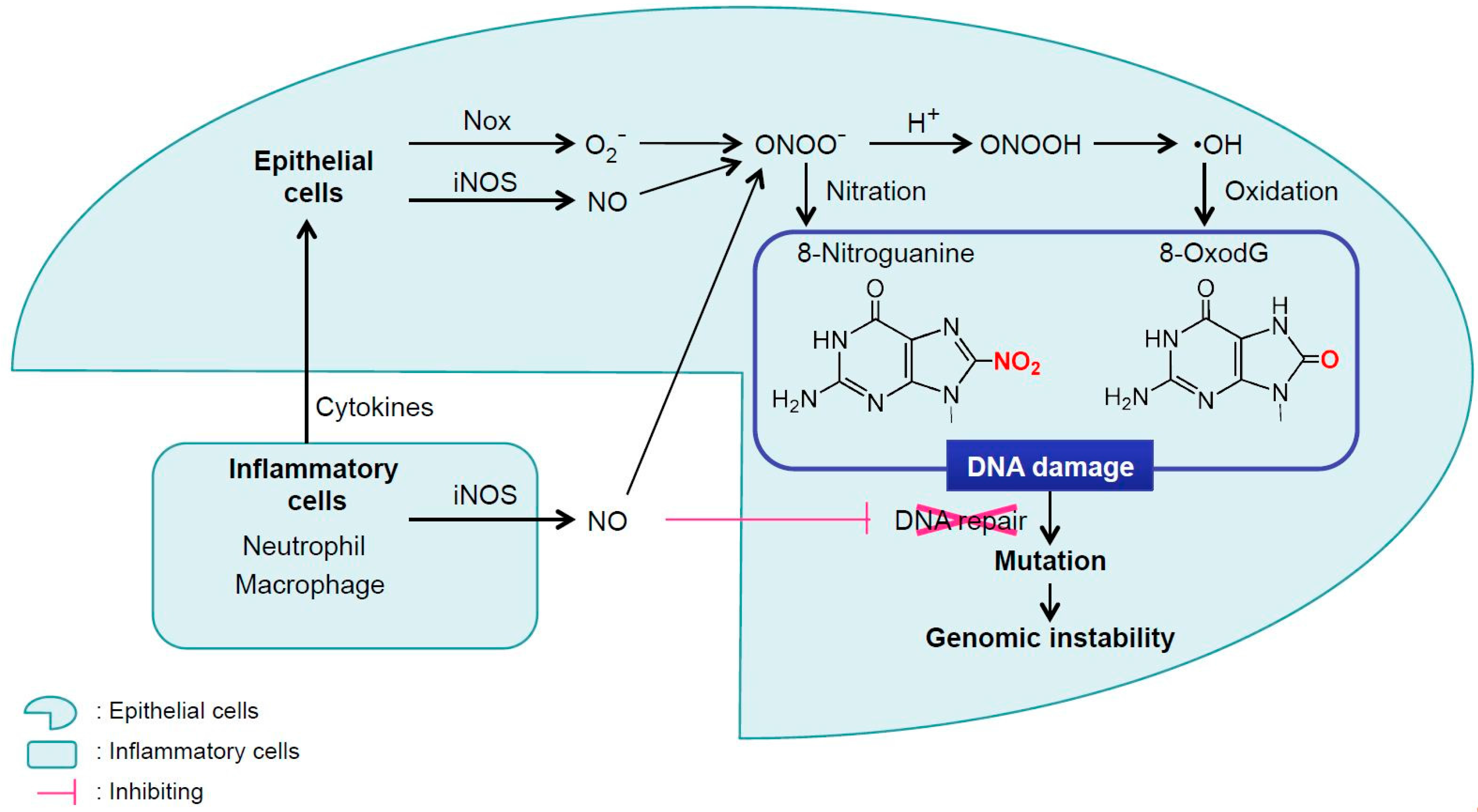
2. Inflammation-Mediated DNA Damage
2.1 Inflammation-Mediated DNA Damage by Infectious Agents
2.2 Inflammation-Mediated DNA Damage in Inflammatory Diseases
2.3 Inflammation-Mediated DNA Damage by Particulate Matters
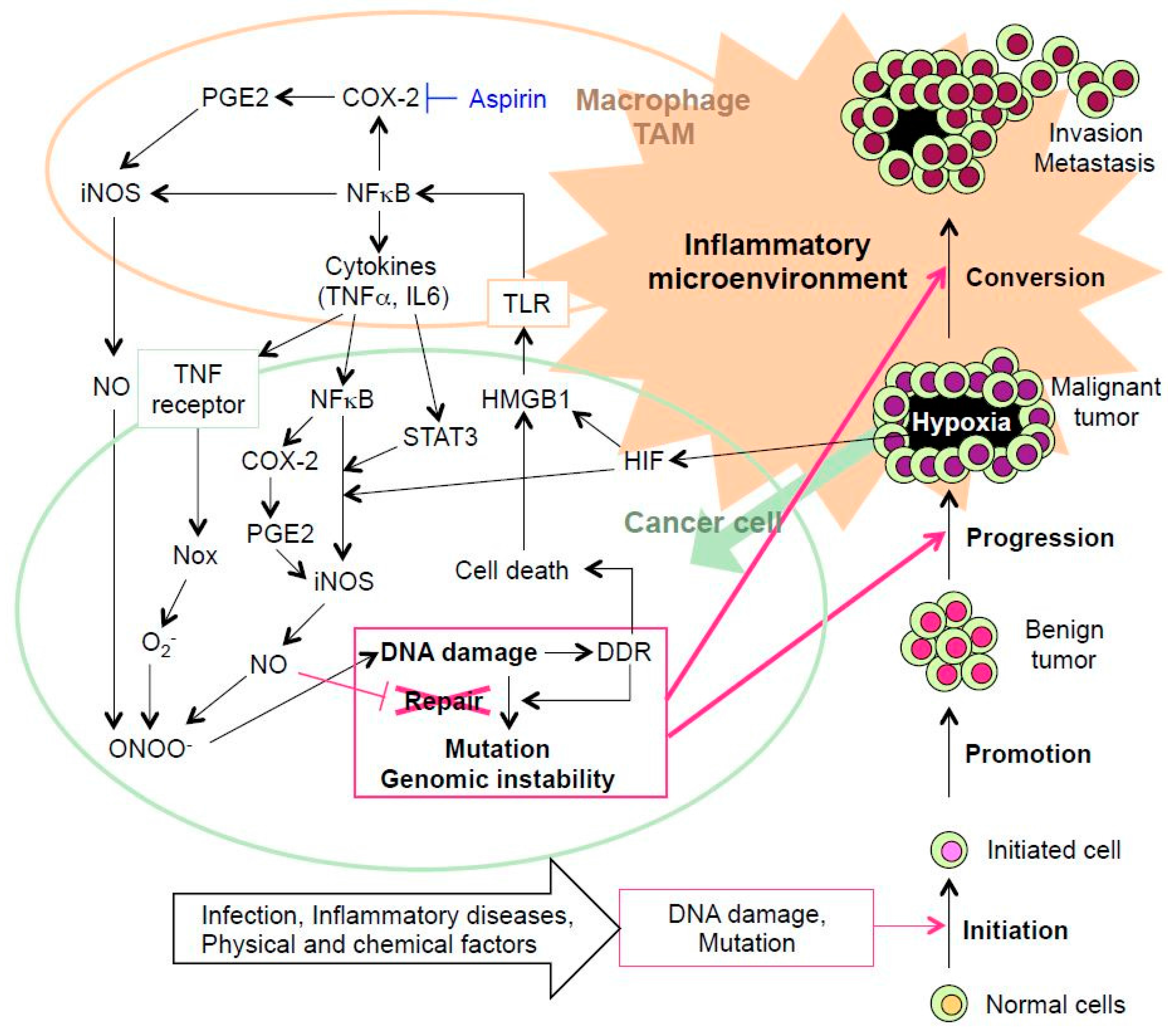
3. DNA Damage and Inflammation Interplay
3.1 Hypoxia-Related DNA Damage and Prognosis
3.2 Tumor Microenvironment-Induced Inflammation Followed by DNA Damage
4. Expectation of Non-Steroidal Anti-Inflammatory Drugs (NSAIDs) as Chemical Cancer Prevention
5. Conclusions
Abbreviations
| 8-oxodG | 8-oxo-7,8-dihydro-2’-deoxyguanosine |
| BE | Barrett’s esophagus |
| BEA | Barrett’s esophageal adenocarcinoma |
| CB | Carbon black |
| CNT | Carbon nanotube |
| COX-2 | Cyclooxygenase 2 |
| DDR | DNA damage response |
| EBV | Epstein-Barr virus |
| H. pylori | Helicobacter pylori |
| HBV | Hepatitis B virus |
| HCV | Hepatitis C virus |
| HMGB1 | High mobility group box 1 |
| HPV | Human papillomavirus |
| HIF | Hypoxia-inducible factor |
| iNOS | Inducible nitric oxide synthase |
| IBDs | Inflammatory bowel diseases |
| IL-6 | Interleukin-6 |
| IARC | International Agency for Research on Cancer |
| MFH | Malignant fibrous histiocytoma |
| Nox | NAD(P)H oxidase |
| NO | Nitric oxide |
| NOS | NO synthase |
| NSAIDs | Non-steroidal anti-inflammatory drugs |
| NF-κB | Nuclear factor-κB |
| OLP | Oral lichen planus |
| OSCC | Oral squamous cell carcinoma |
| OV | Opisthorchis viverrini |
| ONOO− | Peroxynitrite |
| RNS | Reactive nitrogen species |
| PGH2 | Prostaglandin H2 |
| PGE2 | Prostaglandin E2 |
| PPIs | Proton pump inhibitors |
| ROS | Reactive oxygen species |
| SH | Schistosoma haematobium |
| STAT | Signal transducer and activator of transcription |
| O2− | Superoxide |
| TNF-α | Tumor necrosis factor alpha |
| TAM | Tumor-associated macrophage |
| TLR | Toll-like receptor |
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Coussens, L.M.; Werb, Z. Inflammation and cancer. Nature 2002, 420, 860–867. [Google Scholar] [CrossRef] [PubMed]
- Hussain, S.P.; Harris, C.C. Inflammation and cancer: An ancient link with novel potentials. Int. J. Cancer 2007, 121, 2373–2380. [Google Scholar] [CrossRef] [PubMed]
- Hussain, S.P.; Hofseth, L.J.; Harris, C.C. Radical causes of cancer. Nat. Rev. Cancer 2003, 3, 276–285. [Google Scholar] [CrossRef] [PubMed]
- Wiseman, H.; Halliwell, B. Damage to DNA by reactive oxygen and nitrogen species: Role in inflammatory disease and progression to cancer. Biochem. J. 1996, 313, 17–29. [Google Scholar] [CrossRef] [PubMed]
- Kawanishi, S.; Hiraku, Y. Oxidative and nitrative DNA damage as biomarker for carcinogenesis with special reference to inflammation. Antioxid. Redox Signal. 2006, 8, 1047–1058. [Google Scholar] [CrossRef] [PubMed]
- Murata, M.; Thanan, R.; Ma, N.; Kawanishi, S. Role of nitrative and oxidative DNA damage in inflammation-related carcinogenesis. J. Biomed. Biotechnol. 2012, 623019. [Google Scholar] [CrossRef] [PubMed]
- Kawanishi, S.; Ohnishi, S.; Ma, N.; Hiraku, Y.; Oikawa, S.; Murata, M. Nitrative and oxidative DNA damage in infection-related carcinogenesis in relation to cancer stem cells. Genes Environ. 2017, 38, 55. [Google Scholar] [CrossRef] [PubMed]
- Fukumura, D.; Kashiwagi, S.; Jain, R.K. The role of nitric oxide in tumour progression. Nat. Rev. Cancer 2006, 6, 521–534. [Google Scholar] [CrossRef] [PubMed]
- Lala, P.K.; Chakraborty, C. Role of nitric oxide in carcinogenesis and tumour progression. Lancet Oncol. 2001, 2, 149–156. [Google Scholar] [CrossRef]
- Halliwell, B. Oxygen and nitrogen are pro-carcinogens. Damage to DNA by reactive oxygen, chlorine and nitrogen species: Measurement, mechanism and the effects of nutrition. Mutat. Res. 1999, 443, 37–52. [Google Scholar] [CrossRef]
- Sodum, R.S.; Fiala, E.S. Analysis of peroxynitrite reactions with guanine, xanthine, and adenine nucleosides by high-pressure liquid chromatography with electrochemical detection: C8-nitration and -oxidation. Chem. Res. Toxicol. 2001, 14, 438–450. [Google Scholar] [CrossRef] [PubMed]
- Yermilov, V.; Rubio, J.; Ohshima, H. Formation of 8-nitroguanine in DNA treated with peroxynitrite in vitro and its rapid removal from DNA by depurination. FEBS Lett. 1995, 376, 207–210. [Google Scholar] [CrossRef]
- Wu, X.; Takenaka, K.; Sonoda, E.; Hochegger, H.; Kawanishi, S.; Kawamoto, T.; Takeda, S.; Yamazoe, M. Critical roles for polymerase ζ in cellular tolerance to nitric oxide-induced DNA damage. Cancer Res. 2006, 66, 748–754. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, N.; Yasui, M.; Geacintov, N.E.; Shafirovich, V.; Shibutani, S. Miscoding events during DNA synthesis past the nitration-damaged base 8-nitroguanine. Biochemistry 2005, 44, 9238–9245. [Google Scholar] [CrossRef] [PubMed]
- Loeb, L.A.; Preston, B.D. Mutagenesis by apurinic/apyrimidinic sites. Annu. Rev. Genet. 1986, 20, 201–230. [Google Scholar] [CrossRef] [PubMed]
- Carballal, S.; Bartesaghi, S.; Radi, R. Kinetic and mechanistic considerations to assess the biological fate of peroxynitrite. Biochim. Biophys. Acta 2014, 1840, 768–780. [Google Scholar] [CrossRef] [PubMed]
- Grivennikov, S.I.; Greten, F.R.; Karin, M. Immunity, inflammation, and cancer. Cell 2010, 140, 883–899. [Google Scholar] [CrossRef] [PubMed]
- Oshima, H.; Ishikawa, T.; Yoshida, G.J.; Naoi, K.; Maeda, Y.; Naka, K.; Ju, X.; Yamada, Y.; Minamoto, T.; Mukaida, N.; et al. TNF-α/TNFR1 signaling promotes gastric tumorigenesis through induction of Noxo1 and Gna14 in tumor cells. Oncogene 2014, 33, 3820–3829. [Google Scholar] [CrossRef] [PubMed]
- Mikhed, Y.; Görlach, A.; Knaus, U.G.; Daiber, A. Redox regulation of genome stability by effects on gene expression, epigenetic pathways and DNA damage/repair. Redox Biol. 2015, 5, 275–289. [Google Scholar] [CrossRef] [PubMed]
- Moritz, E.; Pauly, K.; Bravard, A.; Hall, J.; Radicella, J.P.; Epe, B. hOGG1-Cys326 variant cells are hypersensitive to DNA repair inhibition by nitric oxide. Carcinogenesis 2014, 35, 1426–1433. [Google Scholar] [CrossRef] [PubMed]
- Tubbs, A.; Nussenzweig, A. Endogenous DNA damage as a source of genomic instability in cancer. Cell 2017, 168, 644–656. [Google Scholar] [CrossRef] [PubMed]
- IARC. Chronic infections. In World Cancer Report; Boyle, P., Levin, B., Eds.; IARC Press: Lyon, France, 2008; pp. 128–135. ISBN 978-92-832-0423-7. [Google Scholar]
- IARC. Opisthorchis viverrini and clonorchis sinensis. In IARC Monographs on the Evaluation of Carcinogenic Risks to Humans; IARC Press: Lyon, France, 2009; pp. 347–376. ISBN 978-92-832-1319-2. [Google Scholar]
- Podolsky, D.K. Inflammatory bowel disease. N. Engl. J. Med. 2002, 347, 417–429. [Google Scholar] [CrossRef] [PubMed]
- Ekbom, A.; Helmick, C.; Zack, M.; Adami, H.O. Increased risk of large-bowel cancer in Crohn’s disease with colonic involvement. Lancet 1990, 336, 357–359. [Google Scholar] [CrossRef]
- Langholz, E.; Munkholm, P.; Davidsen, M.; Binder, V. Colorectal cancer risk and mortality in patients with ulcerative colitis. Gastroenterology 1992, 103, 1444–1451. [Google Scholar] [CrossRef]
- Choi, P.M.; Zelig, M.P. Similarity of colorectal cancer in Crohn’s disease and ulcerative colitis: implications for carcinogenesis and prevention. Gut 1994, 35, 950–954. [Google Scholar] [CrossRef] [PubMed]
- Wild, C.P.; Hardie, L.J. Reflux, Barrett’s oesophagus and adenocarcinoma: Burning questions. Nat. Rev. Cancer 2003, 3, 676–684. [Google Scholar] [CrossRef] [PubMed]
- Rajentheran, R.; McLean, N.R.; Kelly, C.G.; Reed, M.F.; Nolan, A. Malignant transformation of oral lichen planus. Eur. J. Surg. Oncol. 1999, 25, 520–523. [Google Scholar] [CrossRef] [PubMed]
- Mignogna, M.D.; Fedele, S.; lo Russo, L.; lo Muzio, L.; Bucci, E. Immune activation and chronic inflammation as the cause of malignancy in oral lichen planus: Is there any evidence? Oral Oncol. 2004, 40, 120–130. [Google Scholar] [CrossRef] [PubMed]
- Chaiyarit, P.; Ma, N.; Hiraku, Y.; Pinlaor, S.; Yongvanit, P.; Jintakanon, D.; Murata, M.; Oikawa, S.; Kawanishi, S. Nitrative and oxidative DNA damage in oral lichen planus in relation to human oral carcinogenesis. Cancer Sci. 2005, 96, 553–559. [Google Scholar] [CrossRef] [PubMed]
- Ding, X.; Hiraku, Y.; Ma, N.; Kato, T.; Saito, K.; Nagahama, M.; Semba, R.; Kuribayashi, K.; Kawanishi, S. Inducible nitric oxide synthase-dependent DNA damage in mouse model of inflammatory bowel disease. Cancer Sci. 2005, 96, 157–163. [Google Scholar] [CrossRef] [PubMed]
- Thanan, R.; Ma, N.; Hiraku, Y.; Iijima, K.; Koike, T.; Shimosegawa, T.; Murata, M.; Kawanishi, S. DNA damage in CD133-positive cells in Barrett’s esophagus and esophageal adenocarcinoma. Mediat. Inflamm. 2016, 7937814. [Google Scholar] [CrossRef] [PubMed]
- Thanan, R.; Ma, N.; Iijima, K.; Abe, Y.; Koike, T.; Shimosegawa, T.; Pinlaor, S.; Hiraku, Y.; Oikawa, S.; Murata, M.; Kawanishi, S. Proton pump inhibitors suppress iNOS-dependent DNA damage in Barrett's esophagus by increasing Mn-SOD expression. Biochem. Biophys. Res. Commun. 2012, 421, 280–285. [Google Scholar] [CrossRef] [PubMed]
- Ohshima, H.; Tatemichi, M.; Sawa, T. Chemical basis of inflammation-induced carcinogenesis. Arch. Biochem. Biophys. 2003, 417, 3–11. [Google Scholar] [CrossRef]
- Wong, J.; Magun, B.E.; Wood, L.J. Lung inflammation caused by inhaled toxicants: A review. Int. J. Chron. Obstruct. Pulmon. Dis. 2016, 11, 1391–1401. [Google Scholar] [CrossRef] [PubMed]
- Jacobsen, N.R.; Moller, P.; Jensen, K.A.; Vogel, U.; Ladefoged, O.; Loft, S.; Wallin, H. Lung inflammation and genotoxicity following pulmonary exposure to nanoparticles in ApoE-/- mice. Part. Fibre Toxicol. 2009, 6, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Porter, D.W.; Hubbs, A.F.; Mercer, R.R.; Wu, N.; Wolfarth, M.G.; Sriram, K.; Leonard, S.; Battelli, L.; Schwegler-Berry, D.; Friend, S.; et al. Mouse pulmonary dose- and time course-responses induced by exposure to multi-walled carbon nanotubes. Toxicology 2010, 269, 136–147. [Google Scholar] [CrossRef] [PubMed]
- Shvedova, A.A.; Kisin, E.R.; Porter, D.; Schulte, P.; Kagan, V.E.; Fadeel, B.; Castranova, V. Mechanisms of pulmonary toxicity and medical applications of carbon nanotubes: Two faces of Janus? Pharmacol. Ther. 2009, 121, 192–204. [Google Scholar] [CrossRef] [PubMed]
- Heinrich, U.; Fuhst, R.; Rittinghausen, S.; Creutzenberg, O.; Bellmann, B.; Koch, W.; Levsen, K. Chronic inhalation exposure of Wistar rats and two different strains of mice to diesel engine exhaust carbon black, and titanium dioxide. Inhal. Toxicol. 1995, 7, 533–556. [Google Scholar] [CrossRef]
- Hiraku, Y.; Kawanishi, S.; Ichinose, T.; Murata, M. The role of iNOS-mediated DNA damage in infection- and asbestos-induced carcinogenesis. Ann. N. Y. Acad. Sci. 2010, 1203, 15–22. [Google Scholar] [CrossRef] [PubMed]
- Hiraku, Y.; Sakai, K.; Shibata, E.; Kamijima, M.; Hisanaga, N.; Ma, N.; Kawanishi, S.; Murata, M. Formation of the nitrative DNA lesion 8-nitroguanine is associated with asbestos contents in human lung tissues: A pilot study. J. Occup. Health 2014, 56, 186–196. [Google Scholar] [CrossRef] [PubMed]
- IARC. Asbestos (chrysotile, amosite, crocidolite, tremolite, actinolite, and anthophyllite). In IARC Monographs on the Evaluation of Carcinogenic Risks to Humans; IARC Press: Lyon, France, 2009; pp. 219–309. ISBN 978-92 832-1320-8. [Google Scholar]
- Robinson, B.W.; Lake, R.A. advances in malignant mesothelioma. N. Engl. J. Med. 2005, 353, 1591–1603. [Google Scholar] [CrossRef] [PubMed]
- Medina, C.; Santos-Martinez, M.J.; Radomski, A.; Corrigan, O.I.; Radomski, M.W. Nanoparticles: Pharmacological and toxicological significance. Br. J. Pharmacol. 2007, 150, 552–558. [Google Scholar] [CrossRef] [PubMed]
- Pacurari, M.; Yin, X.J.; Zhao, J.; Ding, M.; Leonard, S.S.; Schwegler-Berry, D.; Ducatman, B.S.; Sbarra, D.; Hoover, M.D.; Castranova, V.; et al. Raw single-wall carbon nanotubes induce oxidative stress and activate MAPKs, AP-1, NF-κB, and Akt in normal and malignant human mesothelial cells. Environ. Health Perspect. 2008, 116, 1211–1217. [Google Scholar] [CrossRef] [PubMed]
- Nagai, H.; Okazaki, Y.; Chew, S.H.; Misawa, N.; Yamashita, Y.; Akatsuka, S.; Ishihara, T.; Yamashita, K.; Yoshikawa, Y.; Yasui, H.; et al. Diameter and rigid- ity of multiwalled carbon nanotubes are critical factors in mesothelial injury and carcinogenesis. Proc. Natl. Acad. Sci. USA 2011, 108, 1330–1338. [Google Scholar] [CrossRef] [PubMed]
- Poland, C.A.; Duffin, R.; Kinloch, I.; Maynard, A.; Wallace, W.A.; Seaton, A.; Stone, V.; Brown, S.; MacNee, W.; Donaldson, K. Carbon nanotubes introduced into the abdominal cavity of mice show asbestos-like pathogenicity in a pilot study. Nat. Nanotechnol. 2008, 3, 423–428. [Google Scholar] [CrossRef] [PubMed]
- Takagi, A.; Hirose, A.; Nishimura, T.; Fukumori, N.; Ogata, A.; Ohashi, N.; Kitajima, S.; Kanno, J. Induction of mesothelioma in p53+/- mouse by intraperitoneal application of multi-wall carbon nanotube. J. Toxicol. Sci. 2008, 33, 105–116. [Google Scholar] [CrossRef] [PubMed]
- Sakamoto, Y.; Nakae, D.; Fukumori, N.; Tayama, K.; Maekawa, A.; Imai, K.; Hirose, A.; Nishimura, T.; Ohashi, N.; Ogata, A. Induction of mesothelioma by a single intrascrotal administration of multi-wall carbon nanotube in intact male Fischer 344 rats. J. Toxicol. Sci. 2009, 34, 65–76. [Google Scholar] [CrossRef] [PubMed]
- Guo, F.; Ma, N.; Horibe, Y.; Kawanishi, S.; Murata, M.; Hiraku, Y. Nitrative DNA damage induced by multi-walled carbon nanotube via endocytosis in human lung epithelial cells. Toxicol. Appl. Pharmacol. 2012, 260, 183–192. [Google Scholar] [CrossRef] [PubMed]
- Hiraku, Y.; Guo, F.; Ma, N.; Yamada, T.; Wang, S.; Kawanishi, S.; Murata, M. Multi-walled carbon nanotube induces nitrative DNA damage in human lung epithelial cells via HMGB1-RAGE interaction and Toll-like receptor 9 activation. Part. Fibre Toxicol. 2016, 13, 16. [Google Scholar] [CrossRef] [PubMed]
- IARC. Carbon black, titanium dioxide, and talc. In IARC Monographs on the Evaluation of Carcinogenic Risks to Humans; IARC Press: Lyon, France, 2010; pp. 43–191. ISBN 978-92 832-1293-5. [Google Scholar]
- Hiraku, Y.; Nishikawa, Y.; Ma, N.; Afroz, T.; Mizobuchi, K.; Ishiyama, R.; Matsunaga, Y.; Ichinose, T.; Kawanishi, S.; Murata, M. Nitrative DNA damage induced by carbon-black nanoparticles in macrophages and lung epithelial cells. Mutat. Res. 2017, 818, 7–16. [Google Scholar] [CrossRef] [PubMed]
- Kandoth, C.; McLellan, M.D.; Vandin, F.; Ye, K.; Niu, B.; Lu, C.; Xie, M.; Zhang, Q.; McMichael, J.F.; Wyczalkowski, M.A.; et al. Mutational landscape and significance across 12 major cancer types. Nature 2013, 502, 333–339. [Google Scholar] [CrossRef] [PubMed]
- Loeb, L.A. Human cancers express mutator phenotypes: Origin, consequences and targeting. Nat. Rev. Cancer 2011, 11, 450–457. [Google Scholar] [CrossRef] [PubMed]
- Hoki, Y.; Murata, M.; Hiraku, Y.; Ma, N.; Matsumine, A.; Uchid, A.; Kawanishi, S. 8-Nitroguanine as a potential biomarker for progression of malignant fibrous histiocytoma, a model of inflammation-related cancer. Oncol. Rep. 2007, 18, 1165–1169. [Google Scholar] [PubMed]
- Hoki, Y.; Hiraku, Y.; Ma, N.; Murata, M.; Matsumine, A.; Nagahama, M.; Shintani, K.; Uchid, A.; Kawanishi, S. iNOS-dependent DNA damage in patients with malignant fibrous histiocytoma in relation to prognosis. Cancer Sci. 2007, 98, 163–168. [Google Scholar] [CrossRef] [PubMed]
- Pinlaor, S.; Sripa, B.; Ma, N.; Hiraku, Y.; Yongvanit, P.; Wongkham, S.; Pairojkul, C.; Bhudhisawasdi, V.; Oikawa, S.; Murata, M.; et al. Nitrative and oxidative DNA damage in intrahepatic cholangiocarcinoma patients in relation to tumor invasion. World J. Gastroenterol. 2005, 11, 4644–4649. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, M.I.; Silva-Carvalho, R.; Pires, I.; Prada, J.; Bianchini, R.; Jensen-Jarolim, E.; Queiroga, F.L. A comparative approach of tumor-associated inflammation in mammary cancer between humans and dogs. BioMed Res. Int. 2016, 4917387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mamlouk, S.; Wielockx, B. Hypoxia-inducible factors as key regulators of tumor inflammation. Int. J. Cancer 2013, 132, 2721–2729. [Google Scholar] [CrossRef] [PubMed]
- Guo, Q.; Jin, Z.; Yuan, Y.; Liu, R.; Xu, T.; Wei, H.; Xu, X.; He, S.; Chen, S.; Shi, Z.; et al. New mechanisms of tumor-associated macrophages on promoting tumor progression: Recent research advances and potential targets for tumor immunotherapy. J. Immunol. Res. 2016, 9720912. [Google Scholar] [CrossRef] [PubMed]
- Triner, D.; Shah, Y.M. Hypoxia-inducible factors: A central link between inflammation and cancer. J. Clin. Investig. 2016, 126, 3689–3698. [Google Scholar] [CrossRef] [PubMed]
- Ma, N.; Kawanishi, M.; Hiraku, Y.; Murata, M.; Huang, G.W.; Huang, Y.; Luo, D.Z.; Mo, W.G.; Fukui, Y.; Kawanishi, S. Reactive nitrogen species-dependent DNA damage in EBV-associated nasopharyngeal carcinoma: The relation to STAT3 activation and EGFR expression. Int. J. Cancer 2008, 122, 2517–2525. [Google Scholar] [CrossRef] [PubMed]
- Pistoia, V.; Pezzolo, A. Involvement of HMGB1 in resistance to tumor vessel-targeted, monoclonal antibody-based immunotherapy. J. Immunol. Res. 2016, 3142365. [Google Scholar] [CrossRef] [PubMed]
- Kang, R.; Zhang, Q.; Zeh, H.J., 3rd; Lotze, M.T.; Tang, D. HMGB1 in cancer: Good, bad, or both? Clin. Cancer Res. 2013, 19, 4046–4057. [Google Scholar] [CrossRef] [PubMed]
- Shalapour, S.; Karin, M. Immunity, inflammation, and cancer: An eternal fight between good and evil. J. Clin. Investig. 2015, 125, 3347–3355. [Google Scholar] [CrossRef] [PubMed]
- He, S.J.; Cheng, J.; Feng, X.; Yu, Y.; Tian, L.; Huang, Q. The dual role and therapeutic potential of high-mobility group box 1 in cancer. Oncotarget 2017. [Google Scholar] [CrossRef]
- Tadie, J.M.; Bae, H.B.; Deshane, J.S.; Bell, C.P.; Lazarowski, E.R.; Chaplin, D.D.; Thannickal, V.J.; Abraham, E.; Zmijewski, J.W. Toll-like receptor 4 engagement inhibits adenosine 5′-monophosphate-activated protein kinase activation through a high mobility group box 1 protein-dependent mechanism. Mol. Med. 2012, 18, 659–668. [Google Scholar] [CrossRef] [PubMed]
- Weng, H.; Deng, Y.; Xie, Y.; Liu, H.; Gong, F. Expression and significance of HMGB1, TLR4 and NF-κB p65 in human epidermal tumors. BMC Cancer 2013, 13, 311. [Google Scholar] [CrossRef] [PubMed]
- Jube, S.; Rivera, Z.S.; Bianchi, M.E.; Powers, A.; Wang, E.; Pagano, I.; Pass, H.I.; Gaudino, G.; Carbone, M.; Yang, H. Cancer cell secretion of the DAMP protein HMGB1 supports progression in malignant mesothelioma. Cancer Res. 2012, 72, 3290–3301. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Tracey, K.J. Targeting HMGB1 in inflammation. Biochim. Biophys. Acta 2010, 1799, 149–156. [Google Scholar] [CrossRef] [PubMed]
- Surh, Y.J.; Chun, K.S.; Cha, H.H.; Han, S.S.; Keum, Y.S.; Park, K.K.; Lee, S.S. Molecular mechanisms underlying chemopreventive activities of anti- inflammatory phytochemicals: Down-regulation of COX-2 and iNOS through suppression of NF-κB activation. Mutat. Res. 2001, 480, 243–268. [Google Scholar] [CrossRef]
- Balkwill, F.; Coussens, L.M. Cancer: An inflammatory link. Nature 2004, 431, 4056. [Google Scholar] [CrossRef] [PubMed]
- Echizen, K.; Hirose, O.; Maeda, Y.; Oshima, M. Inflammation in gastric cancer: Interplay of the COX-2/prostaglandin E2 and Toll-like receptor/MyD88 pathways. Cancer Sci. 2016, 107, 391–397. [Google Scholar] [CrossRef] [PubMed]
- Vannini, F.; Kashfi, K.; Nath, N. The dual role of iNOS in cancer. Redox Biol. 2015, 6, 334–343. [Google Scholar] [CrossRef] [PubMed]
- Nathan, C. Nitric oxide as a secretory product of mammalian cells. FASEB J. 1992, 6, 3051–3064. [Google Scholar] [PubMed]
- Ferreiro, C.R.; Chagas, A.C.; Carvalho, M.H.; Dantas, A.P.; Jatene, M.B.; Bento de Souza, L.C.; Lemos da Luz, P. Influence of hypoxia on nitric oxide synthase activity and gene expression in children with congenital heart disease: A novel pathophysiological adaptive mechanism. Circulation 2001, 103, 2272–2276. [Google Scholar] [CrossRef] [PubMed]
- Gupta, J.; Nebreda, A.R. Roles of p38α mitogen-activated protein kinase in mouse models of inflammatory diseases and cancer. FEBS J. 2015, 282, 1841–1857. [Google Scholar] [CrossRef] [PubMed]
- Eliopoulos, A.G.; Havaki, S.; Gorgoulis, V.G. DNA damage response and autophagy: A meaningful partnership. Front. Genet. 2016, 7, 204. [Google Scholar] [CrossRef] [PubMed]
- Moreno-Villanueva, M.; Bürkle, A. Stress hormone-mediated DNA damage response--Implications for cellular senescence and tumour progression. Curr. Drug Targets 2016, 17, 398–404. [Google Scholar] [CrossRef] [PubMed]
- Thun, M.J.; Jacobs, E.J.; Patrono, C. The role of aspirin in cancer prevention. Nat. Rev. Clin. Oncol. 2012, 9, 259–267. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, E.J.; Newton, C.C.; Gapstur, S.M.; Thun, M.J. Daily aspirin use and cancer mortality in a large US cohort. J. Natl. Cancer Inst. 2012, 104, 1208–1217. [Google Scholar] [CrossRef] [PubMed]
- Algra, A.M.; Rothwell, P.M. Effects of regular aspirin on long-term cancer incidence and metastasis: A systematic comparison of evidence from observational studies versus randomised trials. Lancet Oncol. 2012, 13, 518–527. [Google Scholar] [CrossRef]
- Rothwell, P.M.; Wilson, M.; Price, J.F.; Belch, J.F.; Meade, T.W.; Mehta, Z. Effect of daily aspirin on risk of cancer metastasis: A study of incident cancers during randomised controlled trials. Lancet 2012, 379, 1591–1601. [Google Scholar] [CrossRef]
- Rothwell, P.M.; Price, J.F.; Fowkes, F.G.; Zanchetti, A.; Roncaglioni, M.C.; Tognoni, G.; Lee, R.; Belch, J.F.; Wilson, M.; Mehta, Z.; et al. Short-term effects of daily aspirin on cancer incidence, mortality, and non-vascular death: Analysis of the time course of risks and benefits in 51 randomised controlled trials. Lancet 2012, 379, 1602–1612. [Google Scholar] [CrossRef]
- Elwood, P.C.; Morgan, G.; Pickering, J.E.; Galante, J.; Weightman, A.L.; Morris, D.; Kelson, M.; Dolwani, S. Aspirin in the treatment of cancer: Reductions in metastatic spread and in mortality: A systematic review and meta-analyses of published studies. PLoS ONE 2016, 11, e0152402. [Google Scholar] [CrossRef] [PubMed]
- Rothwell, P.M.; Wilson, M.; Elwin, C.E.; Norrving, B.; Algra, A.; Warlow, C.P.; Meade, T.W. Long-term effect of aspirin on colorectal cancer incidence and mortality: 20-year follow-up of five randomised trials. Lancet 2010, 376, 1741–1750. [Google Scholar] [CrossRef]
- Boutaud, O.; Sosa, I.R.; Amin, T.; Oram, D.; Adler, D.; Hwang, H.S.; Crews, B.C.; Milne, G.; Harris, B.K.; Hoeksema, M.; Knollmann, B.C.; Lammers, P.E.; Marnett, L.J.; Massion, P.P.; Oates, J.A. Inhibition of the biosynthesis of prostaglandin E2 by low-dose aspirin: Implications for adenocarcinoma metastasis. Cancer Prev. Res. 2016, 9, 855–865. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Nakayama, T. Inflammation, a link between obesity and cardiovascular disease. Mediat. Inflamm. 2010, 535918. [Google Scholar] [CrossRef] [PubMed]
- Mikawa, S.; Ohta, Y.; Kaji, N.; Islam, M.S.; Murata, T.; Ozaki, H.; Hori, M. Time-dependent changes in inhibitory action of lipopolysaccharide on intestinal motility in rat. J. Vet. Med. Sci. 2015, 77, 1443–1449. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Causative Factors | Cancer Sites | ||
|---|---|---|---|
| Infectious agents | Bacteria | H. pylori | Stomach |
| Viruses | HPV | Cervix and other sites | |
| HBV | Liver | ||
| HCV | |||
| EBV | Lymph node, nasopharynx and other sites | ||
| Parasites | SH | Bladder | |
| OV | Bile duct | ||
| Inflammatory diseases | OLP | Oral cavity | |
| BE | Esophagus | ||
| IBDs | Colon | ||
| MFH | Soft tissue | ||
| Particulate matters | Asbestos | Mesothelium, lung | |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kawanishi, S.; Ohnishi, S.; Ma, N.; Hiraku, Y.; Murata, M. Crosstalk between DNA Damage and Inflammation in the Multiple Steps of Carcinogenesis. Int. J. Mol. Sci. 2017, 18, 1808. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms18081808
Kawanishi S, Ohnishi S, Ma N, Hiraku Y, Murata M. Crosstalk between DNA Damage and Inflammation in the Multiple Steps of Carcinogenesis. International Journal of Molecular Sciences. 2017; 18(8):1808. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms18081808
Chicago/Turabian StyleKawanishi, Shosuke, Shiho Ohnishi, Ning Ma, Yusuke Hiraku, and Mariko Murata. 2017. "Crosstalk between DNA Damage and Inflammation in the Multiple Steps of Carcinogenesis" International Journal of Molecular Sciences 18, no. 8: 1808. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms18081808