The Role of PALB2 in the DNA Damage Response and Cancer Predisposition

 and
and
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
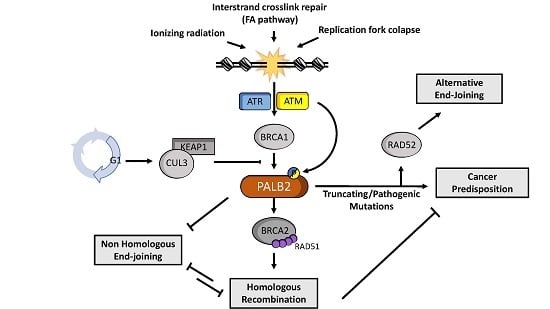
2. PALB2 in Cancer Predisposition and Clinical Management
2.1. Cancer Predisposition
2.2. Clinical Management
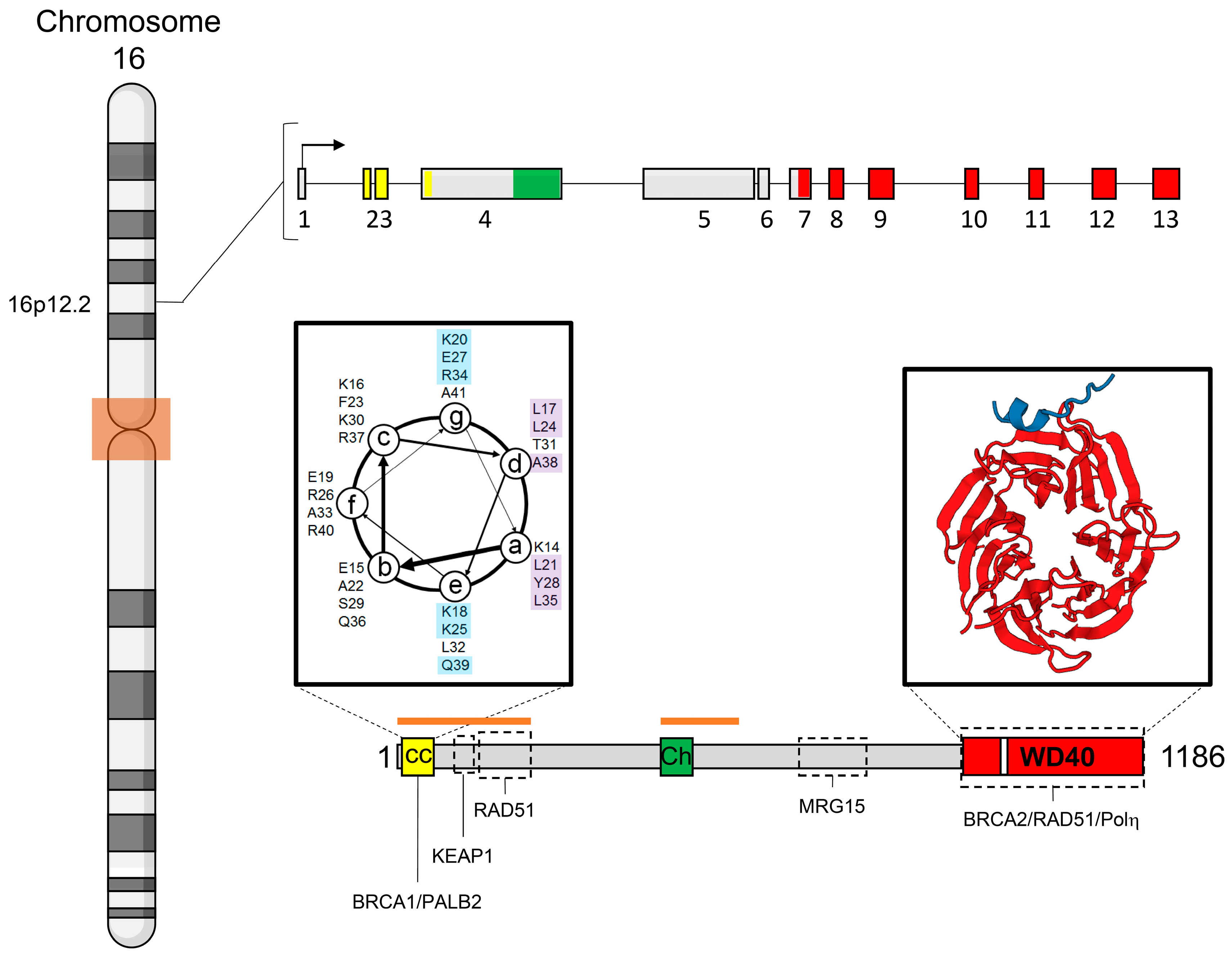
3. Gene and Protein Structures
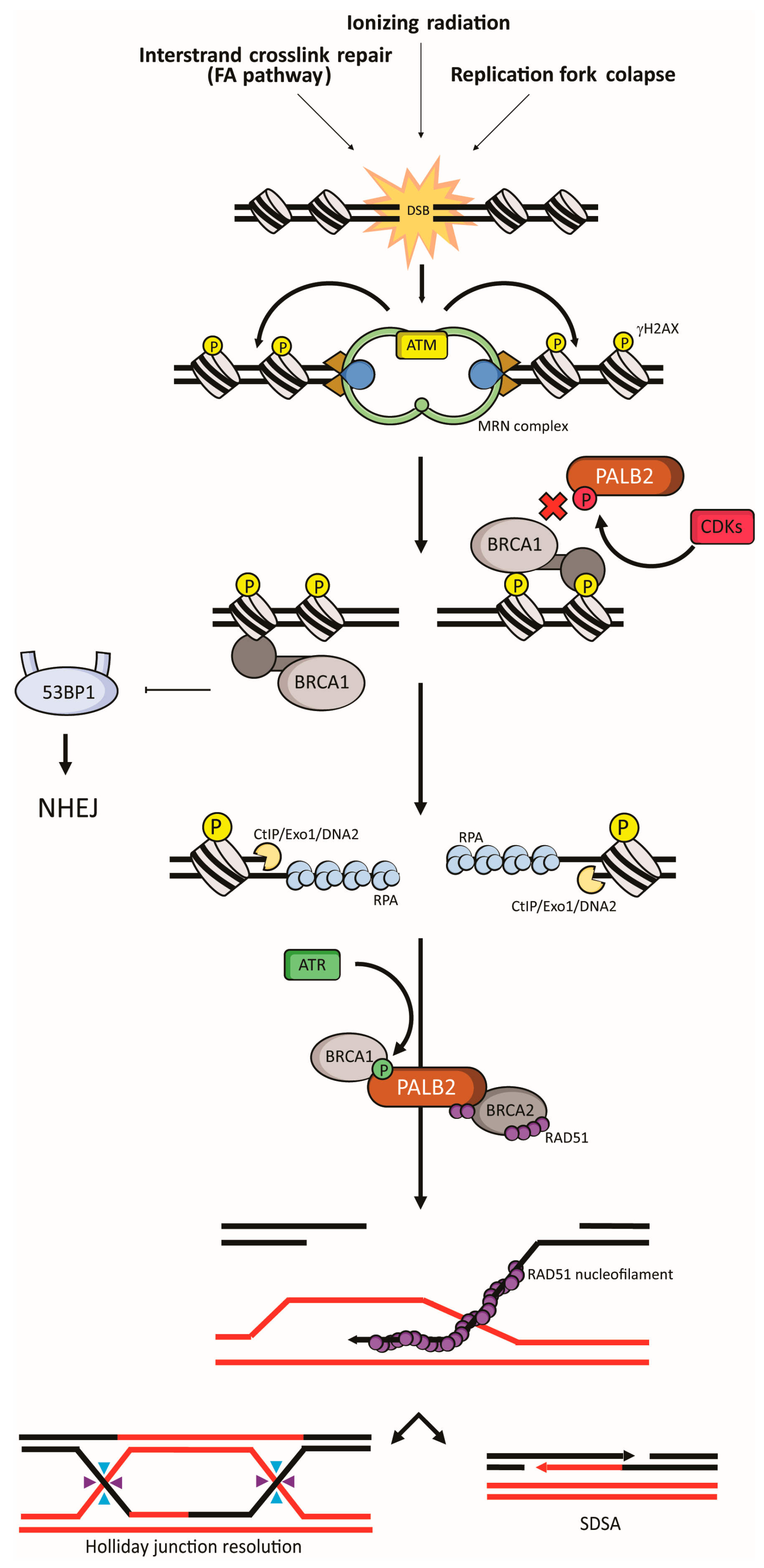
4. PALB2 in Fanconi Anemia and Homologous Recombination
4.1. Fanconi Anemia
4.2. Homologous Recombination
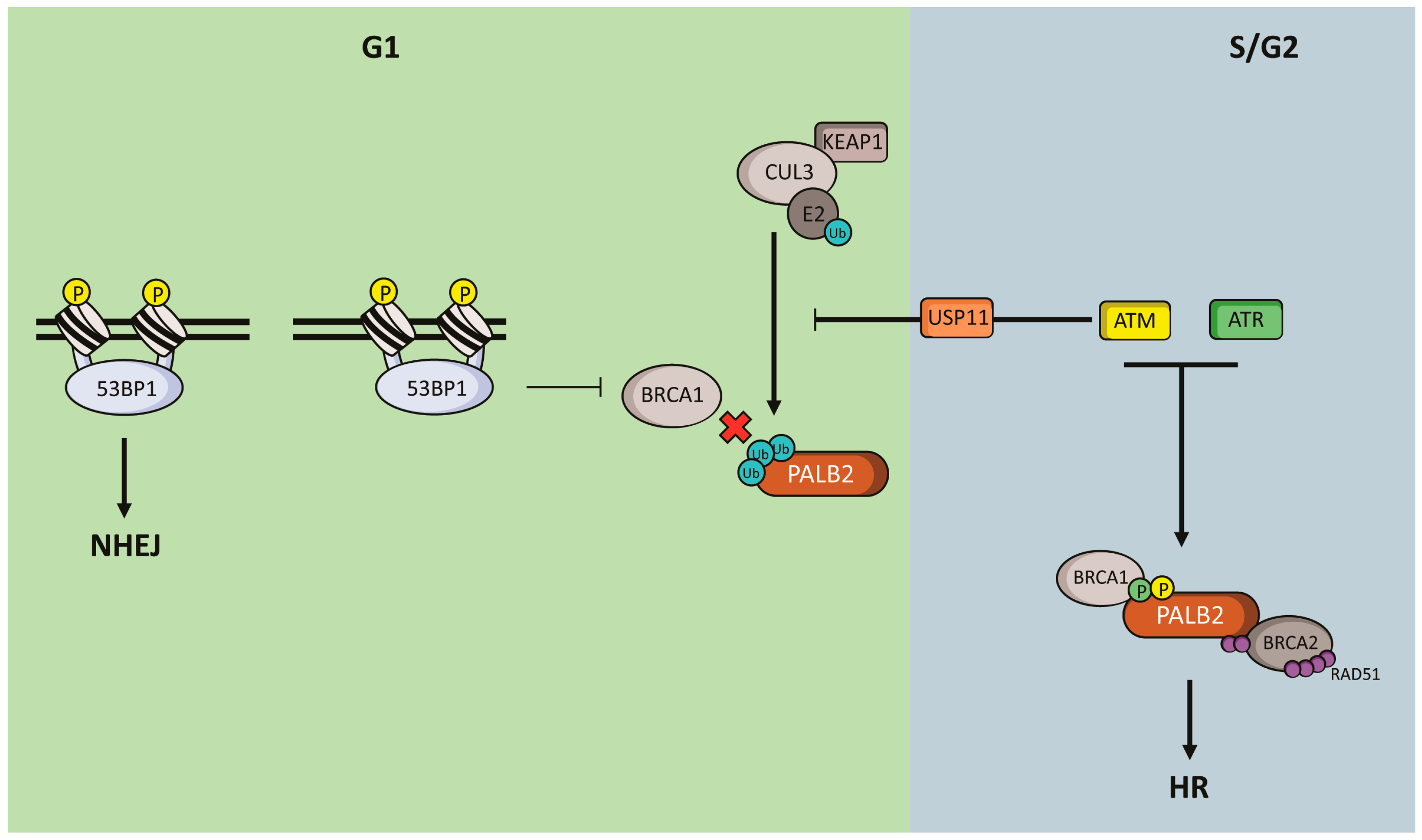
5. Regulation of PALB2 Functions
6. PALB2 and RAD52
7. Concluding Remarks
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| ATM | Ataxia Telangiectasia Mutated |
| ATR | ATM- and Rad3-Related |
| BC | Breast cancer |
| BRCA1 | Breast Cancer 1 |
| BRCA2 | Breast Cancer 2 |
| CDK | Cyclin-Dependent Kinase |
| ChAM | Chromatin-associated motif |
| CtBP | C-terminal binding protein |
| CtIP | CtBP-Interacting protein |
| DDR | DNA damage response |
| DNA | Deoxyribonucleic Acid |
| DSB | Double strand break |
| FA | Fanconi anemia |
| FANCN | Fanconi Anemia Group N protein |
| HR | Homologous recombination |
| ICL | Interstrand crosslink |
| MMC | Mitomycin C |
| NES | Nuclear export sequence |
| OC | Ovarian cancer |
| OR | Odds ratio |
| PALB2 | Partner and Localizer of BRCA2 |
| PC | Pancreatic cancer |
| RCSB | Research Collaboratory for Structural Bioinformatics |
| RPA | Replication Protein A |
| SDSA | Synthesis dependent strand annealing |
| ssDNA | Single strand DNA |
| TAP-MS | Tandem affinity purification followed by mass spectrometry |
| TLS | Translesion synthesis |
| VUS | Variant of uncertain significance |
References
- Hall, J.M.; Lee, M.K.; Newman, B.; Morrow, J.E.; Anderson, L.A.; Huey, B.; King, M.-C. Linkage of Early-Onset Familial Breast Cancer to Chromosome 17q21. Science 1990, 250, 1684–1689. [Google Scholar] [CrossRef] [PubMed]
- Miki, Y.; Swensen, J.; Shattuck-Eidens, D.; Futreal, P.A.; Harshman, K.; Tavtigian, S.; Liu, Q.; Cochran, C.; Bennett, L.M.; Ding, W. A Strong Candidate for the Breast and Ovarian Cancer Susceptibility Gene BRCA1. Science 1994, 266, 66–71. [Google Scholar] [CrossRef] [PubMed]
- Wooster, R.; Neuhausen, S.L.; Mangion, J.; Quirk, Y.; Ford, D.; Collins, N.; Nguyen, K.; Seal, S.; Tran, T.; Averill, D.; et al. Localization of a breast cancer susceptibility gene, BRCA2, to chromosome 13q12–13. Science 1994, 265, 2088–2090. [Google Scholar] [CrossRef] [PubMed]
- Wooster, R.; Bignell, G.; Lancaster, J.; Swift, S.; Seal, S.; Mangion, J.; Collins, N.; Gregory, S.; Gumbs, C.; Micklem, G.; et al. Identification of the breast cancer susceptibility gene BRCA2. Nature 1995. [Google Scholar] [CrossRef] [PubMed]
- Scully, R.; Chen, J.; Plug, A.; Xiao, Y.; Weaver, D.; Feunteun, J.; Ashley, T.; Livingston, D.M. Association of BRCA1 with Rad51 in mitotic and meiotic cells. Cell 1997, 88, 265–275. [Google Scholar] [CrossRef]
- Sharan, S.K.; Morimatsu, M.; Albrecht, U.; Lim, D.S.; Regel, E.; Dinh, C.; Sands, A.; Eichele, G.; Hasty, P.; Bradley, A. Embryonic lethality and radiation hypersensitivity mediated by Rad51 in mice lacking Brca2. Nature 1997, 386, 804–810. [Google Scholar] [CrossRef] [PubMed]
- Gowen, L.C.; Johnson, B.L.; Latour, A.M.; Sulik, K.K.; Koller, B.H. Brca1 deficiency results in early embryonic lethality characterized by neuroepithelial abnormalities. Nat. Genet. 1996, 12, 191–194. [Google Scholar] [CrossRef] [PubMed]
- Ludwig, T.; Chapman, D.L.; Papaioannou, V.E. Targeted mutations of breast cancer susceptibility gene homologs in mice: Lethal phenotypes of Brca1, Brca2, Brca1/Brca2, Brca1/p53, and Brca2/p53 nullizygous embryos. Genes Dev. 1997. [Google Scholar] [CrossRef]
- Chen, J.; Silver, D.P.; Walpita, D.; Cantor, S.B.; Gazdar, A.F.; Tomlinson, G.; Couch, F.J.; Weber, B.L.; Ashley, T.; Livingston, D.M.; et al. Stable Interaction between the Products of the BRCA1 and BRCA2 Tumor Suppressor Genes in Mitotic and Meiotic Cells. Mol. Cell 1998, 2, 317–328. [Google Scholar] [CrossRef]
- Xia, B.; Sheng, Q.; Nakanishi, K.; Ohashi, A.; Wu, J.; Christ, N.; Liu, X.; Jasin, M.; Couch, F.J.; Livingston, D.M. Control of BRCA2 Cellular and Clinical Functions by a Nuclear Partner, PALB2. Mol. Cell 2006, 22, 719–729. [Google Scholar] [CrossRef] [PubMed]
- Erkko, H.; Xia, B.; Nikkilä, J.; Schleutker, J.; Syrjäkoski, K.; Mannermaa, A.; Kallioniemi, A.; Pylkäs, K.; Karppinen, S.-M.; Rapakko, K.; et al. A recurrent mutation in PALB2 in Finnish cancer families. Nature 2007, 446, 316–319. [Google Scholar] [CrossRef] [PubMed]
- Tischkowitz, M.; Xia, B.; Sabbaghian, N.; Reis-Filho, J.S.; Hamel, N.; Li, G.; van Beers, E.H.; Li, L.; Khalil, T.; Quenneville, L.A.; et al. Analysis of PALB2/FANCN-associated breast cancer families. Proc. Natl. Acad. Sci. USA 2007, 104, 6788–6793. [Google Scholar] [CrossRef] [PubMed]
- Dansonka-Mieszkowska, A.; Kluska, A.; Moes, J.; Dabrowska, M.; Nowakowska, D.; Niwinska, A.; Derlatka, P.; Cendrowski, K.; Kupryjanczyk, J. A novel germline PALB2 deletion in Polish breast and ovarian cancer patients. BMC Med. Genet. 2010. [Google Scholar] [CrossRef] [PubMed]
- Stadler, Z.K.; Salo-Mullen, E.; Sabbaghian, N.; Simon, J.A.; Zhang, L.; Olson, S.H.; Kurtz, R.; Offit, K.; Foulkes, W.D.; Robson, M.E.; et al. Germline PALB2 mutation analysis in breast-pancreas cancer families. J. Med. Genet. 2011, 48, 523–525. [Google Scholar] [CrossRef] [PubMed]
- Southey, M.C.; Goldgar, D.E.; Winqvist, R.; Pylkäs, K.; Couch, F.; Tischkowitz, M.; Foulkes, W.D.; Dennis, J.; Michailidou, K.; van Rensburg, E.J.; et al. PALB2, CHEK2 and ATM rare variants and cancer risk: Data from COGS. J. Med. Genet. 2016. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Ma, J.; Wu, J.; Ye, L.; Cai, H.; Xia, B.; Yu, X. PALB2 Links BRCA1 and BRCA2 in the DNA-Damage Response. Curr. Biol. 2009, 19, 524–529. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Fan, Q.; Ren, K.; Andreassen, P.R. PALB2 functionally connects the breast cancer susceptibility proteins BRCA1 and BRCA2. Mol. Cancer Res. 2009, 7, 1110–1118. [Google Scholar] [CrossRef] [PubMed]
- Sy, S.M.H.; Huen, M.S.Y.; Chen, J. PALB2 is an integral component of the BRCA complex required for homologous recombination repair. Proc. Natl. Acad. Sci. USA 2009, 106, 7155–7160. [Google Scholar] [CrossRef] [PubMed]
- Reid, S.; Schindler, D.; Hanenberg, H.; Barker, K.; Hanks, S.; Kalb, R.; Neveling, K.; Kelly, P.; Seal, S.; Freund, M.; et al. Biallelic mutations in PALB2 cause Fanconi anemia subtype FA-N and predispose to childhood cancer. Nat. Genet. 2007, 39, 162–164. [Google Scholar] [CrossRef] [PubMed]
- Xia, B.; Dorsman, J.C.; Ameziane, N.; de Vries, Y.; Rooimans, M.A.; Sheng, Q.; Pals, G.; Errami, A.; Gluckman, E.; Llera, J.; et al. Fanconi anemia is associated with a defect in the BRCA2 partner PALB2. Nat. Genet. 2007, 39, 159–161. [Google Scholar] [CrossRef] [PubMed]
- Rahman, N.; Seal, S.; Thompson, D.; Kelly, P.; Renwick, A.; Elliott, A.; Reid, S.; Spanova, K.; Barfoot, R.; Chagtai, T.; et al. PALB2, which encodes a BRCA2-interacting protein, is a breast cancer susceptibility gene. Cancer 2007, 39, 165–167. [Google Scholar] [CrossRef] [PubMed]
- Southey, M.C.; Teo, Z.L.; Dowty, J.G.; Odefrey, F.A.; Park, D.J.; Tischkowitz, M.; Sabbaghian, N.; Apicella, C.; Byrnes, G.B.; Winship, I.; et al. A PALB2 mutation associated with high risk of breast cancer. Breast Cancer Res. 2010, 12, R109. [Google Scholar] [CrossRef] [PubMed]
- Antoniou, A.C.; Casadei, S.; Heikkinen, T.; Barrowdale, D.; Pylkäs, K.; Roberts, J.; Lee, A.; Subramanian, D.; De Leeneer, K.; Fostira, F.; et al. Breast-cancer risk in families with mutations in PALB2. N. Engl. J. Med. 2014, 371, 497–506. [Google Scholar] [CrossRef] [PubMed]
- Slavin, T.P.; Maxwell, K.N.; Lilyquist, J.; Vijai, J.; Neuhausen, S.L.; Hart, S.N.; Ravichandran, V.; Thomas, T.; Maria, A.; Villano, D.; et al. The contribution of pathogenic variants in breast cancer susceptibility genes to familial breast cancer risk. NPJ Breast Cancer 2017. [Google Scholar] [CrossRef] [PubMed]
- Susswein, L.R.; Marshall, M.L.; Nusbaum, R.; Vogel Postula, K.J.; Weissman, S.M.; Yackowski, L.; Vaccari, E.M.; Bissonnette, J.; Booker, J.K.; Cremona, M.L.; et al. Pathogenic and likely pathogenic variant prevalence among the first 10,000 patients referred for next-generation cancer panel testing. Genet. Med. 2015, 18, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Silvestri, V.; Zelli, V.; Valentini, V.; Rizzolo, P.; Navazio, A.S.; Coppa, A.; Agata, S.; Oliani, C.; Barana, D.; Castrignan, T.; et al. Whole-exome sequencing and targeted gene sequencing provide insights into the role of PALB2 as a male breast cancer susceptibility gene. Cancer 2017, 123, 210–218. [Google Scholar] [CrossRef] [PubMed]
- Reddy, N.; Malipatil, B.; Kumar, S. A rare case of familial multiple subcutaneous lipomatosis with novel PALB2 mutation and increased predilection to cancers. Hematol. Oncol. Stem Cell Ther. 2016, 9, 154–156. [Google Scholar] [CrossRef] [PubMed]
- Pritzlaff, M.; Summerour, P.; McFarland, R.; Li, S.; Reineke, P.; Dolinsky, J.S.; Goldgar, D.E.; Shimelis, H.; Couch, F.J.; Chao, E.C.; et al. Male breast cancer in a multi-gene panel testing cohort: Insights and unexpected results. Breast Cancer Res. Treat. 2017, 161, 575–586. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.R.; Devi, B.C.R.; Sung, H.; Guida, J.; Mucaki, E.J.; Xiao, Y.; Best, A.; Garland, L.; Xie, Y.; Hu, N.; et al. Prevalence and spectrum of germline rare variants in BRCA1/2 and PALB2 among breast cancer cases in Sarawak, Malaysia. Breast Cancer Res. Treat. 2017. [Google Scholar] [CrossRef] [PubMed]
- Cybulski, C.; Kluźniak, W.; Huzarski, T.; Wokołorczyk, D.; Kashyap, A.; Jakubowska, A.; Szwiec, M.; Byrski, T.; Dębniak, T.; Górski, B.; et al. Clinical outcomes in women with breast cancer and a PALB2 mutation: A prospective cohort analysis. Lancet Oncol. 2015, 16, 638–644. [Google Scholar] [CrossRef]
- Damiola, F.; Schultz, I.; Barjhoux, L.; Sornin, V.; Dondon, M.G.; Eon-Marchais, S.; Marcou, M.; Caron, O.; Gauthier-Villars, M.; de Pauw, A.; et al. Mutation analysis of PALB2 gene in French breast cancer families. Breast Cancer Res. Treat. 2015, 154, 463–471. [Google Scholar] [CrossRef] [PubMed]
- Haanpää, M.; Pylkäs, K.; Moilanen, J.S.; Winqvist, R. Evaluation of the need for routine clinical testing of PALB2 c.1592delT mutation in BRCA negative Northern Finnish breast cancer families. BMC Med. Genet. 2013. [Google Scholar] [CrossRef] [PubMed]
- Casadei, S.; Norquist, B.M.; Walsh, T.; Stray, S.; Mandell, J.B.; Lee, M.K.; Stamatoyannopoulos, J.A.; King, M.C. Contribution of inherited mutations in the BRCA2-interacting protein PALB2 to familial breast cancer. Cancer Res. 2011, 71, 2222–2229. [Google Scholar] [CrossRef] [PubMed]
- Catucci, I.; Milgrom, R.; Kushnir, A.; Laitman, Y.; Paluch-Shimon, S.; Volorio, S.; Ficarazzi, F.; Bernard, L.; Radice, P.; Friedman, E.; et al. Germline mutations in BRIP1 and PALB2 in Jewish high cancer risk families. Fam. Cancer 2012, 11, 483–491. [Google Scholar] [CrossRef] [PubMed]
- McInerney, N.M.; Miller, N.; Rowan, A.; Colleran, G.; Barclay, E.; Curran, C.; Kerin, M.J.; Tomlinson, I.P.; Sawyer, E. Evaluation of variants in the CHEK2, BRIP1 and PALB2 genes in an Irish breast cancer cohort. Breast Cancer Res. Treat. 2010, 121, 203–210. [Google Scholar] [CrossRef] [PubMed]
- Nakagomi, H.; Sakamoto, I.; Hirotsu, Y.; Amemiya, K.; Mochiduki, H.; Omata, M. Analysis of PALB2 mutations in 155 Japanese patients with breast and/or ovarian cancer. Int. J. Clin. Oncol. 2016, 21, 270–275. [Google Scholar] [CrossRef] [PubMed]
- Harinck, F.; Kluijt, I.; van Mil, S.E.; Waisfisz, Q.; van Os, T.A.M.; Aalfs, C.M.; Wagner, A.; Olderode-Berends, M.; Sijmons, R.H.; Kuipers, E.J.; et al. Routine testing for PALB2 mutations in familial pancreatic cancer families and breast cancer families with pancreatic cancer is not indicated. Eur. J. Hum. Genet. 2012, 20, 577–579. [Google Scholar] [CrossRef] [PubMed]
- Norquist, B.M.; Harrell, M.I.; Brady, M.F.; Walsh, T.; Lee, M.K.; Gulsuner, S.; Bernards, S.S.; Casadei, S.; Yi, Q.; Burger, R.A.; et al. Inherited Mutations in Women With Ovarian Carcinoma. JAMA Oncol. 2015, 6460, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Walsh, T.; Casadei, S.; Lee, M.K.; Pennil, C.C.; Nord, A.S.; Thornton, A.M.; Roeb, W.; Agnew, K.J.; Stray, S.M.; Wickramanayake, A.; et al. Mutations in 12 genes for inherited ovarian, fallopian tube, and peritoneal carcinoma identified by massively parallel sequencing. Proc. Natl. Acad. Sci. USA 2011, 108, 18032–18037. [Google Scholar] [CrossRef] [PubMed]
- Pennington, K.P.; Walsh, T.; Harrell, M.I.; Lee, M.K.; Pennil, C.C.; Rendi, M.H.; Thornton, A.; Norquist, B.M.; Casadei, S.; Nord, A.S.; et al. Germline and somatic mutations in homologous recombination genes predict platinum response and survival in ovarian, fallopian tube, and peritoneal carcinomas. Clin. Cancer Res. 2014, 20, 764–775. [Google Scholar] [CrossRef] [PubMed]
- Kotsopoulos, J.; Sopik, V.; Rosen, B.; Fan, I.; McLaughlin, J.R.; Risch, H.; Sun, P.; Narod, S.A.; Akbari, M.R. Frequency of germline PALB2 mutations among women with epithelial ovarian cancer. Fam. Cancer 2017, 16, 29–34. [Google Scholar] [CrossRef] [PubMed]
- Salo-Mullen, E.E.; O’Reilly, E.; Kelsen, D.; Ashraf, A.M.; Lowery, M.; Yu, K.; Reidy, D.; Epstein, A.S.; Lincoln, A.; Saldia, A.; et al. Identification of Germline Genetic Mutations in Pancreatic with pancreatic cancer. Cancer 2015, 121, 4382–4388. [Google Scholar] [CrossRef] [PubMed]
- Waddell, N.; Pajic, M.; Patch, A.; Chang, D.; Kassahn, K.; Bailey, P.; Johns, A.; Miller, D.; Nones, K.; Quek, K.; et al. Whole genomes redefine the mutational landscape of pancreatic cancer. Obs. Gynecol. 2015, 125, 628–635. [Google Scholar] [CrossRef]
- Slater, E.P.; Langer, P.; Niemczyk, E.; Strauch, K.; Butler, J.; Habbe, N.; Neoptolemos, J.P.; Greenhalf, W.; Bartsch, D.K. PALB2 mutations in European familial pancreatic cancer families. Clin. Genet. 2010, 78, 490–494. [Google Scholar] [CrossRef] [PubMed]
- Jones, S.; Hruban, R.H.; Kamiyama, M.; Borges, M.; Zhang, X.; Parsons, D.W.; Lin, J.C.H.; Palmisano, E.; Brune, K.; Jaffee, E.M.; et al. Exomic sequencing identifies PALB2 as a pancreatic cancer susceptibility gene. Science 2009. [Google Scholar] [CrossRef] [PubMed]
- Hofstatter, E.W. PALB2 mutations in familial breast and pancreatic cancer. Fam. Cancer 2013, 10, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Takai, E.; Yachida, S.; Shimizu, K.; Furuse, J.; Kubo, E.; Ohmoto, A.; Suzuki, M.; Hruban, R.H.; Okusaka, T.; Morizane, C.; et al. Germline mutations in Japanese familial pancreatic cancer patients. Oncotarget 2016. [Google Scholar] [CrossRef] [PubMed]
- Schneider, R.; Slater, E.P.; Sina, M.; Habbe, N.; Fendrich, V.; Matthäi, E.; Langer, P.; Bartsch, D.K. German national case collection for familial pancreatic cancer (FaPaCa): Ten years experience. Fam. Cancer 2011, 10, 323–330. [Google Scholar] [CrossRef] [PubMed]
- Ghiorzo, P.; Pensotti, V.; Fornarini, G.; Sciallero, S.; Battistuzzi, L.; Belli, F.; Bonelli, L.; Borgonovo, G.; Bruno, W.; Gozza, A.; et al. Contribution of germline mutations in the BRCA and PALB2 genes to pancreatic cancer in Italy. Fam. Cancer 2012, 11, 41–47. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.; Xie, M.; Wendl, M.C.; Wang, J.; McLellan, M.D.; Leiserson, M.D.M.; Huang, K.-L.; Wyczalkowski, M.A.; Jayasinghe, R.; Banerjee, T.; et al. Patterns and functional implications of rare germline variants across 12 cancer types. Nat. Commun. 2015. [Google Scholar] [CrossRef] [PubMed]
- Pritchard, C.C.; Mateo, J.; Walsh, M.F.; De Sarkar, N.; Abida, W.; Beltran, H.; Garofalo, A.; Gulati, R.; Carreira, S.; Eeles, R.; et al. Inherited DNA-Repair Gene Mutations in Men with Metastatic Prostate Cancer. N. Engl. J. Med. 2016, 375, 443–453. [Google Scholar] [CrossRef] [PubMed]
- Pearlman, R.; Frankel, W.L.; Swanson, B.; Zhao, W.; Yilmaz, A.; Miller, K.; Bacher, J.; Bigley, C.; Nelsen, L.; Goodfellow, P.J.; et al. Prevention Initiative Study Group Prevalence and Spectrum of Germline Cancer Susceptibility Gene Mutations Among Patients With Early-Onset Colorectal Cancer. JAMA Oncol. 2016, 354, 2751–2763. [Google Scholar] [CrossRef]
- Sahasrabudhe, R.; Lott, P.; Bohorquez, M.; Toal, T.; Estrada, A.P.; Suarez, J.J.; Brea-Fernández, A.; Cameselle-Teijeiro, J.; Pinto, C.; Ramos, I.; et al. Germline Mutations in PALB2, BRCA1, and RAD51C, Which Regulate DNA Recombination Repair, in Patients with Gastric Cancer. Gastroenterology 2017, 152, 983–986. [Google Scholar] [CrossRef] [PubMed]
- Pilié, P.G.; Johnson, A.M.; Hanson, K.L.; Dayno, M.E.; Kapron, A.L.; Stoffel, E.M.; Cooney, K.A. Germline genetic variants in men with prostate cancer and one or more additional cancers. Cancer 2017. [Google Scholar] [CrossRef] [PubMed]
- Blanco, A.; de la Hoya, M.; Osorio, A.; Diez, O.; Miramar, M.D.; Infante, M.; Martinez-Bouzas, C.; Torres, A.; Lasa, A.; Llort, G.; et al. Analysis of PALB2 Gene in BRCA1/BRCA2 Negative Spanish Hereditary Breast/Ovarian Cancer Families with Pancreatic Cancer Cases. PLoS ONE 2013. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.T.; Jiang, W.H.; Wang, X.W.; Zhang, M.S.; Zhang, C.G.; Yi, L.N.; WuwaliKhan, F.; Ayoufu, A.; Ou, J.H. PALB2 mutations in breast cancer patients from a multi-ethnic region in northwest China. Eur. J. Med. Res. 2015, 20, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Foo, T.K.; Tischkowitz, M.; Simhadri, S.; Boshari, T.; Zayed, N.; Burke, K.A.; Berman, S.H.; Blecua, P.; Riaz, N.; Huo, Y.; et al. Compromised BRCA1-PALB2 interaction is associated with breast cancer risk. Oncogene 2017. [Google Scholar] [CrossRef] [PubMed]
- Heikkinen, T.; Kärkkäinen, H.; Aaltonen, K.; Milne, R.L.; Heikkilä, P.; Aittomäki, K.; Blomqvist, C.; Nevanlinna, H. The breast cancer susceptibility mutation PALB2 1592delT is associated with an aggressive tumor phenotype. Clin. Cancer Res. 2009, 15, 3214–3222. [Google Scholar] [CrossRef] [PubMed]
- Tischkowitz, M.; Xia, B. PALB2/FANCN: Recombining cancer and fanconi anemia. Cancer Res. 2010, 70, 7353–7359. [Google Scholar] [CrossRef] [PubMed]
- Lefebvre, C.; Bachelot, T.; Filleron, T.; Pedrero, M.; Campone, M.; Soria, J.C.; Massard, C.; Lévy, C.; Arnedos, M.; Lacroix-Triki, M.; et al. Mutational Profile of Metastatic Breast Cancers: A Retrospective Analysis. PLoS Med. 2016, 13, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Villarroel, M.C.; Rajeshkumar, N.V.; Garrido-Laguna, I.; De Jesus-Acosta, A.; Jones, S.; Maitra, A.; Hruban, R.H.; Eshleman, J.R.; Klein, A.; Laheru, D.; et al. Personalizing cancer treatment in the age of global genomic analyses: PALB2 gene mutations and the response to DNA damaging agents in pancreatic cancer. Mol. Cancer Ther. 2011, 10, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Xiao, H.; Zhang, K.J.; Xia, B. Defects of FA/BRCA pathway in lymphoma cell lines. Int. J. Hematol. 2009, 88, 543–550. [Google Scholar] [CrossRef] [PubMed]
- Spugnesi, L.; Gabriele, M.; Scarpitta, R.; Tancredi, M.; Maresca, L.; Gambino, G.; Collavoli, A.; Aretini, P.; Bertolini, I.; Salvadori, B.; et al. Germline Mutations in DNA Repair Genes May Predict Neoadjuvant Therapy Response in Triple Negative Breast Patients. Genes Chromosomes Cancer 2016, 55, 915–924. [Google Scholar] [CrossRef] [PubMed]
- Goodall, J.; Mateo, J.; Yuan, W.; Mossop, H.; Porta, N.; Miranda, S.; Perez-lopez, R.; Dolling, D.; Robinson, D.R.; Sandhu, S.; et al. Circulating Free DNA to Guide Prostate Cancer Treatment with PARP Inhibition. Cancer Discov. 2017. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Zhou, J.; Xu, M.; Yuan, G. Investigation on the formation, conversion and bioactivity of a G-quadruplex structure in the PALB2 gene. Int. J. Biol. Macromol. 2016, 83, 242–248. [Google Scholar] [CrossRef] [PubMed]
- Rhodes, D.; Lipps, H. G-quadruplex and their regulatory roles in biology. Nucleic Acids Res. 2015, 43, 8627–8637. [Google Scholar] [CrossRef] [PubMed]
- Simonsson, T.; Pecinka, P.; Kubista, M. DNA tetraplex formation in the control region of c-myc. Nucleic Acids Res. 1998, 26, 1167–1172. [Google Scholar] [CrossRef] [PubMed]
- De Armond, R.; Wood, S.; Sun, D.; Hurley, L.H.; Ebbinghaus, S.W. Evidence for the presence of a guanine quadruplex forming region within a polypurine tract of the hypoxia inducible factor 1α promoter. Biochemistry 2005, 44, 16341–16350. [Google Scholar] [CrossRef] [PubMed]
- Scott, C.M.; Joo, J.H.E.; O’Callaghan, N.; Buchanan, D.D.; Clendenning, M.; Giles, G.G.; Hopper, J.L.; Wong, E.M.; Southey, M.C. Methylation of breast cancer predisposition genes in early-onset breast cancer: Australian breast cancer family registry. PLoS ONE 2016, 11, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Potapova, A.; Hoffman, A.M.; Godwin, A.K.; Al-Saleem, T.; Cairns, P. Promoter hypermethylation of the PALB2 susceptibility gene in inherited and sporadic breast and ovarian cancer. Cancer Res. 2008, 68, 998–1002. [Google Scholar] [CrossRef] [PubMed]
- Rose, P.W.; Prlić, A.; Bi, C.; Bluhm, W.F.; Christie, C.H.; Dutta, S.; Green, R.K.; Goodsell, D.S.; Westbrook, J.D.; Woo, J.; et al. The RCSB Protein Data Bank: Views of structural biology for basic and applied research and education. Nucleic Acids Res. 2015, 43, D345–D356. [Google Scholar] [CrossRef] [PubMed]
- Rose, P.W.; Prli, A.; Altunkaya, A.; Bi, C.; Bradley, A.R.; Christie, C.H.; Di Costanzo, L.; Duarte, J.M.; Dutta, S.; Feng, Z.; et al. The RCSB protein data bank: Integrative view of protein, gene and 3D structural information. Nucleic Acids Res. 2017, 45, D271–D281. [Google Scholar] [CrossRef] [PubMed]
- Oliver, A.W.; Swift, S.; Lord, C.J.; Ashworth, A.; Pearl, L.H. Structural basis for recruitment of BRCA2 by PALB2. EMBO Rep. 2009, 10, 990–996. [Google Scholar] [CrossRef] [PubMed]
- Landschulz, W.; Johnson, P.; McKnight, S. The leucine zipper: A hypothetical structure common to a new class of DNA binding proteins. Science 1988, 240, 1759–1764. [Google Scholar] [CrossRef] [PubMed]
- Lupas, A. Coiled coils: New structures and new functions. Trends Biochem. Sci. 1996, 21, 375–382. [Google Scholar] [CrossRef]
- Lupas, A.; Van Dyke, M.; Stock, J. Predicting coiled coils from protein sequences. Science 1991, 252, 1162–1164. [Google Scholar] [CrossRef]
- O’Shea, E.K.; Lumb, K.J.; Kim, P.S. Peptide “Velcro”: Design of a heterodimeric coiled coil. Curr. Biol. 1993, 3, 658–667. [Google Scholar] [CrossRef]
- Buisson, R.; Masson, J.Y. PALB2 self-interaction controls homologous recombination. Nucleic Acids Res. 2012, 40, 10312–10323. [Google Scholar] [CrossRef] [PubMed]
- Sy, S.M.H.; Huen, M.S.Y.; Zhu, Y.; Chen, J. PALB2 regulates recombinational repair through chromatin association and oligomerization. J. Biol. Chem. 2009, 284, 18302–18310. [Google Scholar] [CrossRef] [PubMed]
- Dray, E.; Etchin, J.; Wiese, C.; Saro, D.; Williams, G.J.; Yu, X.; Galkin, V.E.; Liu, D.; Tsai, M.; Sy, S.M.-H.; et al. Enhancement of the RAD51 Recombinase Activity by the Tumor Suppressor PALB2. Nat. Struct. Mol. Biol. 2011, 17, 1255–1259. [Google Scholar] [CrossRef] [PubMed]
- Buisson, R.; Dion-Côté, A.-M.; Coulombe, Y.; Launay, H.; Cai, H.; Stasiak, A.Z.; Stasiak, A.; Xia, B.; Masson, J.-Y. Cooperation of breast cancer proteins PALB2 and piccolo BRCA2 in stimulating homologous recombination. Nat. Struct. Mol. Biol. 2010, 17, 1247–1254. [Google Scholar] [CrossRef] [PubMed]
- Bleuyard, J.-Y.; Buisson, R.; Masson, J.-Y.; Esashi, F. ChAM, a novel motif that mediates PALB2 intrinsic chromatin binding and facilitates DNA repair. EMBO Rep. 2012, 13, 135–141. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.H.; Chen, R.C.; Gao, Y.; Wu, Y.D. The effect of Asp-His-Ser/Thr-Trp tetrad on the thermostability of WD40-repeat proteins. Biochemistry 2010, 49, 10237–10245. [Google Scholar] [CrossRef] [PubMed]
- Smith, T.F.; Gaitatzes, C.; Saxena, K.; Neer, E.J. The WD repeat: A common architecture for diverse functions. Trends Biochem. Sci. 1999, 24, 181–185. [Google Scholar] [CrossRef]
- Pauty, J.; Couturier, A.M.; Rodrigue, A.; Caron, M.C.; Coulombe, Y.; Dellaire, G.; Masson, J.Y. Cancer-causing mutations in the tumor suppressor PALB2 reveal a novel cancer mechanism using a hidden nuclear export signal in the WD40 repeat motif. Nucleic Acids Res. 2017, 45, 2644–2657. [Google Scholar] [CrossRef] [PubMed]
- Taniguchi, T.; Andrea, A.D.D. Molecular pathogenesis of Fanconi anemia: Recent progress. Blood 2006, 107, 4223–4234. [Google Scholar] [CrossRef] [PubMed]
- Wang, W. Emergence of a DNA-damage response network consisting of Fanconi anaemia and BRCA proteins. Nat. Rev. Genet 2007, 8, 735–748. [Google Scholar] [CrossRef] [PubMed]
- Dong, H.; Nebert, D.W.; Bruford, E.A.; Thompson, D.C.; Joenje, H.; Vasiliou, V. Update of the human and mouse Fanconi anemia genes. Hum. Genom. 2015. [Google Scholar] [CrossRef] [PubMed]
- Castella, M.; Jacquemont, C.; Thompson, E.L.; Yeo, J.E.; Cheung, R.S.; Huang, J.W.; Sobeck, A.; Hendrickson, E.A.; Taniguchi, T. FANCI Regulates Recruitment of the FA Core Complex at Sites of DNA Damage Independently of FANCD2. PLoS Genet. 2015, 11, 1–27. [Google Scholar] [CrossRef] [PubMed]
- Hira, A.; Yoshida, K.; Sato, K.; Okuno, Y.; Shiraishi, Y.; Chiba, K.; Tanaka, H.; Miyano, S.; Shimamoto, A.; Tahara, H.; et al. Mutations in the gene encoding the E2 conjugating enzyme UBE2T cause fanconi anemia. Am. J. Hum. Genet. 2015, 96, 1001–1007. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miles, J.A.; Frost, M.G.; Carroll, E.; Rowe, M.L.; Howard, M.J.; Sidhu, A.; Chaugule, V.K.; Alpi, A.F.; Walden, H. The Fanconi Anemia DNA repair pathway is regulated by an interaction between ubiquitin and the E2-like fold domain of FANCL. J. Biol. Chem. 2015, 290, 20995–21006. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knipscheer, P.; Räschle, M.; Smogorzewska, A.; Enoiu, M.; Ho, T.; Schärer, O.; Elledge, S.; Walter, J. The Fanconi anemia pathway promotes replication-dependent DNA interstrand crosslink repair. Science 2010, 23, 83–88. [Google Scholar] [CrossRef]
- Hodskinson, M.R.G.; Silhan, J.; Crossan, G.P.; Garaycoechea, J.I.; Mukherjee, S.; Johnson, C.M.; Schärer, O.D.; Patel, K.J. Mouse SLX4 Is a Tumor Suppressor that Stimulates the Activity of the Nuclease XPF-ERCC1 in DNA Crosslink Repair. Mol. Cell 2014, 54, 472–484. [Google Scholar] [CrossRef] [PubMed]
- Douwel, D.; Boonen, R.; Long, D.; Szypowska, A.; Räschle, M.; Walter, J.; Knipsheer, P. XPF-ERCC1 acts in unhooking DNA interstrand crosslinks in cooperation with FANCD2 and FANCP/SLX4. Mol. cell 2015, 2, 460–471. [Google Scholar] [CrossRef]
- Budzowska, M.; Graham, T.G.; Sobeck, A.; Waga, S.; Walter, J.C. Regulation of the Rev1–pol f complex during bypass of a DNA interstrand cross-link. EMBO J. 2015, 34, 1971–1985. [Google Scholar] [CrossRef] [PubMed]
- Matsuoka, S.; Ballif, B.A.; Smogorzewska, A.; McDonald, E.R.; Hurov, K.E.; Luo, J.; Bakalarski, C.E.; Zhao, Z.; Solimini, N.; Lerenthal, Y.; et al. ATM and ATR substrate analysis reveals extensive protein networks responsive to DNA damage. Science 2007, 316, 1160–1166. [Google Scholar] [CrossRef] [PubMed]
- Uziel, T.; Lerenthal, Y.; Moyal, L.; Andegeko, Y.; Mittelman, L.; Shiloh, Y. Requirement of the MRN complex for ATP activation by DNA damage. EMBO J. 2003, 22, 5612–5621. [Google Scholar] [CrossRef] [PubMed]
- Kumagai, A.; Lee, J.; Yoo, H.Y.; Dunphy, W.G. TopBP1 activates the ATR-ATRIP complex. Cell 2006, 124, 943–955. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Huang, J. DNA End Resection: Facts and Mechanisms. Genom. Proteom. Biol. 2016, 14, 126–130. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Holzschu, D.L.; Sugiyama, T. PCNA is efficiently loaded on the DNA recombination intermediate to modulate polymerase δ, η, and ζ activities. Proc. Natl. Acad. Sci. USA 2013, 110, 7672–7677. [Google Scholar] [CrossRef] [PubMed]
- Mcllwraith, M.J.; Vaisman, A.; Liu, Y.; Fanning, E.; Woodgate, R.; West, S.C. Human DNA polymerase η promotes DNA synthesis from strand invasion intermediates of homologous recombination. Mol. Cell 2005, 20, 783–792. [Google Scholar] [CrossRef] [PubMed]
- Sneeden, J.L.; Grossi, S.M.; Tappin, I.; Hurwitz, J.; Heyer, W.D. Reconstitution of recombination-associated DNA synthesis with human proteins. Nucleic Acids Res. 2013, 41, 4913–4925. [Google Scholar] [CrossRef] [PubMed]
- Buisson, R.; Niraj, J.; Pauty, J.; Maity, R.; Zhao, W.X.; Coulombe, Y.; Sung, P.; Masson, J.-Y. Breast cancer proteins PALB2 and BRCA2 stimulate polymerase η in recombination-associated DNA synthesis at blocked replication forks. Cell Rep. 2014, 76, 211–220. [Google Scholar] [CrossRef] [PubMed]
- Paliwal, S.; Kanagaraj, R.; Sturzenegger, A.; Burdova, K.; Janscak, P. Human RECQ5 helicase promotes repair of DNA double-strand breaks by synthesis-dependent strand annealing. Nucleic Acids Res. 2014, 42, 2380–2390. [Google Scholar] [CrossRef] [PubMed]
- Fekairi, S.; Scaglione, S.; Chahwan, C.; Taylor, E.R.; Tissier, A.; Coulon, S.; Dong, M.Q.; Ruse, C.; Yates, J.R.; Russell, P.; et al. Human SLX4 Is a Holliday Junction Resolvase Subunit that Binds Multiple DNA Repair/Recombination Endonucleases. Cell 2009, 138, 78–89. [Google Scholar] [CrossRef] [PubMed]
- You, Z.; Bailis, J.M. DNA damage and decisions: CtIP coordinates DNA repair and cell cycle checkpoints. Trends Cell Biol. 2010, 20, 402–409. [Google Scholar] [CrossRef] [PubMed]
- Buisson, R.; Niraj, J.; Rodrigue, A.; Ho, C.K.; Kreuzer, J.; Foo, T.K.; Hardy, E.J.L.; Dellaire, G.; Haas, W.; Xia, B.; et al. Coupling of Homologous Recombination and the Checkpoint by ATR. Mol. Cell 2017, 65, 336–346. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Feng, W.; Sy, S.M.H.; Huen, M.S.Y. ATM-dependent phosphorylation of the Fanconi anemia protein PALB2 promotes the DNA damage response. J. Biol. Chem. 2015, 290, 27545–27556. [Google Scholar] [CrossRef] [PubMed]
- Ahlskog, J.K.; Larsen, B.D.; Achanta, K.; Sørensen, C.S. ATM/ATR-mediated phosphorylation of PALB 2 promotes RAD51 function. EMBO Rep. 2016, 17, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Escribano-Díaz, C.; Orthwein, A.; Fradet-Turcotte, A.; Xing, M.; Young, J.T.F.; Tkáč, J.; Cook, M.A.; Rosebrock, A.P.; Munro, M.; Canny, M.D.; et al. A Cell Cycle-Dependent Regulatory Circuit Composed of 53BP1-RIF1 and BRCA1-CtIP Controls DNA Repair Pathway Choice. Mol. Cell 2013, 49, 872–883. [Google Scholar] [CrossRef] [PubMed]
- Orthwein, A.; Noordermeer, S.M.; Wilson, M.D.; Landry, S.; Enchev, R.I.; Sherker, A.; Munro, M.; Pinder, J.; Salsman, J.; Dellaire, G.; et al. A mechanism for the suppression of homologous recombination in G1 cells. Nature 2015. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Cai, H.; Wu, T.; Sobhian, B.; Huo, Y.; Alcivar, A.; Mehta, M.; Cheung, K.L.; Ganesan, S.; Kong, A.-N.T.; et al. PALB2 interacts with KEAP1 to promote NRF2 nuclear accumulation and function. Mol. Cell. Biol. 2012, 32, 1506–1517. [Google Scholar] [CrossRef] [PubMed]
- Hayakawa, T.; Zhang, F.; Hayakawa, N.; Ohtani, Y.; Shinmyozu, K.; Nakayama, J.; Andreassen, P.R. MRG15 binds directly to PALB2 and stimulates homology-directed repair of chromosomal breaks. J. Cell Sci. 2010, 123, 1124–1130. [Google Scholar] [CrossRef] [PubMed]
- Sy, S.M.H.; Huen, M.S.Y.; Chen, J. MRG15 is a novel PALB2-interacting factor involved in homologous recombination. J. Biol. Chem. 2009, 284, 21127–21131. [Google Scholar] [CrossRef] [PubMed]
- Anantha, R.W.; Alcivar, A.L.; Ma, J.; Cai, H.; Simhadri, S.; Ule, J.; König, J.; Xia, B. Requirement of Heterogeneous Nuclear Ribonucleoprotein C for BRCA Gene Expression and Homologous Recombination. PLoS ONE 2013. [Google Scholar] [CrossRef] [PubMed]
- Doyon, Y.; Selleck, W.; Lane, W.S.; Tan, S.; Côté, J. Structural and functional conservation of the NuA4 histone acetyltransferase complex from yeast to humans. Mol. Cell. Biol. 2004, 24, 1884–1896. [Google Scholar] [CrossRef] [PubMed]
- Sivanand, S.; Rhoades, S.; Jiang, Q.; Lee, J.V.; Benci, J.; Zhang, J.; Yuan, S.; Viney, I.; Zhao, S.; Carrer, A.; et al. Nuclear Acetyl-CoA Production by ACLY Promotes Homologous Recombination. Mol. Cell 2017. [Google Scholar] [CrossRef] [PubMed]
- Tominaga, K.; Kirtane, B. MRG15 regulates embryonic development and cell proliferation. Mol. Cell. Biol. 2005, 25, 2924–2937. [Google Scholar] [CrossRef] [PubMed]
- Garcia, S.N.; Kirtaneb, B.M.; Podlutskya, A.J.; Pereira-Smitha, O.M.; Tominaga, K. Mrg15 null and heterozygous mouse embryonic fibroblasts exhibit DNA repair defects post exposure to gamma ionizing radiation. FEBS Lett. 2007, 581, 5275–5281. [Google Scholar] [CrossRef] [PubMed]
- Bleuyard, J.-Y.; Fournier, M.; Nakato, R.; Couturier, A.M.; Katou, Y.; Ralf, C.; Hester, S.S.; Dominguez, D.; Rhodes, D.; Humphrey, T.C.; et al. MRG15-mediated tethering of PALB2 to unperturbed chromatin protects active genes from genotoxic stress. Proc. Natl. Acad. Sci. USA 2017. [Google Scholar] [CrossRef] [PubMed]
- Martrat, G.; Maxwell, C.A.; Tominaga, E.; Bonifaci, N.; Gómez-baldó, L.; Bogliolo, M.; Lázaro, C.; Blanco, I.; Brunet, J.; Aguilar, H. Exploring the link between MORF4L1 and risk of breast cancer. Breast Cancer Res. 2011, 13, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rio Frio, T.; Haanpaa, M.; Pouchet, C.; Pylkas, K.; Vuorela, M.; Tischkowitz, M.; Winqvist, R.; Foulkes, W.D. Mutation analysis of the gene encoding the PALB2-binding protein MRG15 in BRCA1/2-negative breast cancer families. J. Hum. Genet. 2010, 55, 842–843. [Google Scholar] [CrossRef] [PubMed]
- Mladenov, E.; Magin, S.; Soni, A.; Iliakis, G. DNA double-strand-break repair in higher eukaryotes and its role in genomic instability and cancer: Cell cycle and proliferation-dependent regulation. Semin. Cancer Biol. 2016, 37, 51–64. [Google Scholar] [CrossRef] [PubMed]
- Arcas, A.; Fernández-Capetillo, O.; Cases, I.; Rojas, A.M. Emergence and evolutionary analysis of the human DDR network: Implications in comparative genomics and downstream analyses. Mol. Biol. Evol. 2014, 31, 940–961. [Google Scholar] [CrossRef] [PubMed]
- Prakash, L.; Taillon-Miller, P. Effects of the rad52 gene on sister chromatid recombination in Saccharomyces cerevisiae. Curr. Genet. 1981, 3, 247–250. [Google Scholar] [CrossRef] [PubMed]
- Haber, J.E.; Hearn, M. Rad52-independent mitotic gene conversion in Saccharomyces cerevisiae frequently results in chromosomal loss. Genetics 1985, 111, 7–22. [Google Scholar] [PubMed]
- Shinohara, A.; Shinohara, M.; Ohta, T.; Matsuda, S.; Ogawa, T. Rad52 forms ring structures and co-operates with RPA in single-strand DNA annealing. Genes Cells 1998, 3, 145–156. [Google Scholar] [CrossRef] [PubMed]
- New, J.H.; Sugiyama, T.; Zaitseva, E.; Kowalczykowski, S.C. Rad52 protein stimulates DNA strand exchange by Rad51 and replication protein A. Nature 1998, 391, 407–410. [Google Scholar] [CrossRef] [PubMed]
- Sugiyama, T.; New, J.H.; Kowalczykowski, S.C. DNA annealing by Rad52 Protein is stimulated by specific interaction with the complex of replication protein A and single-stranded DNA. Proc. Natl. Acad. Sci. USA 1998, 95, 6049–6054. [Google Scholar] [CrossRef] [PubMed]
- Game, J.C.; Mortimer, R.K. A genetic study of X-ray sensitive mutants in yeast. Mutat. Res. Fundam. Mol. Mech. Mutagen. 1974, 24, 281–292. [Google Scholar] [CrossRef]
- Pâques, F.; Haber, J.E. Multiple pathways of recombination induced by double-strand breaks in Saccharomyces cerevisiae. Microbiol. Mol. Biol. Rev. 1999, 63, 349–404. [Google Scholar]
- Rijkers, T.; Van Den Ouweland, J.; Morolli, B.; Rolink, A.G.; Baarends, W.M.; Van Sloun, P.P.H.; Lohman, P.H.M.; Pastink, A. Targeted Inactivation of Mouse RAD52 Reduces Homologous Recombination but Not Resistance to Ionizing Radiation. Mol. Cell. Biol. 1998, 18, 6423–6429. [Google Scholar] [CrossRef] [PubMed]
- Stark, J.M.; Pierce, A.J.; Oh, J.; Pastink, A.; Jasin, M. Genetic steps of mammalian homologous repair with distinct mutagenic consequences. Mol. Cell. Biol. 2004, 24, 9305–9316. [Google Scholar] [CrossRef] [PubMed]
- Bennardo, N.; Cheng, A.; Huang, N.; Stark, J.M. Alternative-NHEJ is a mechanistically distinct pathway of mammalian chromosome break repair. PLoS Genet. 2008. [Google Scholar] [CrossRef] [PubMed]
- Ochs, F.; Somyajit, K.; Altmeyer, M.; Rask, M.-B.; Lukas, J.; Lukas, C. 53BP1 fosters fidelity of homology-directed DNA repair. Nat. Struct. Mol. Biol. 2016, 23, 714–721. [Google Scholar] [CrossRef] [PubMed]
- Davis, A.P.; Symington, L.S. The yeast recombinational repair protein Rad59 interacts with Rad52 and stimulates single-strand annealing. Genetics 2001, 159, 515–525. [Google Scholar] [PubMed]
- Jensen, R.B.; Carreira, A.; Kowalczykowski, S.C. Purified human BRCA2 stimulates RAD51-mediated recombination. Nature 2010, 467, 678–683. [Google Scholar] [CrossRef] [PubMed]
- Jensen, R.B. BRCA2: One small step for DNA repair, one giant protein purified. Yale J. Biol. Med. 2013, 86, 479–489. [Google Scholar] [PubMed]
- Liu, J.; Doty, T.; Gibson, B.; Heyer, W.-D. Human BRCA2 protein promotes RAD51 filament formation on RPA-covered single-stranded DNA. Nat. Struct. Mol. Biol. 2010, 17, 1260–1262. [Google Scholar] [CrossRef] [PubMed]
- Moriel-Carretero, M.; Aguilera, A. A Postincision-Deficient TFIIH Causes Replication Fork Breakage and Uncovers Alternative Rad51-or Pol32-Mediated Restart Mechanisms. Mol. Cell 2010, 37, 690–701. [Google Scholar] [CrossRef] [PubMed]
- Murphy, A.K.; Fitzgerald, M.; Ro, T.; Kim, J.H.; Rabinowitsch, A.I.; Chowdhury, D.; Schildkraut, C.L.; Borowiec, J.A. Phosphorylated RPA recruits PALB2 to stalled DNA replication forks to facilitate fork recovery. J. Cell Biol. 2014, 206, 493–507. [Google Scholar] [CrossRef] [PubMed]
- Feng, Z.; Scott, S.P.; Bussen, W.; Sharma, G.G.; Guo, G.; Pandita, T.K.; Powell, S.N. Rad52 inactivation is synthetically lethal with BRCA2 deficiency. Proc. Natl. Acad. Sci. USA 2011, 108, 686–691. [Google Scholar] [CrossRef] [PubMed]
- Lok, B.H.; Carley, a C.; Tchang, B.; Powell, S.N. RAD52 inactivation is synthetically lethal with deficiencies in BRCA1 and PALB2 in addition to BRCA2 through RAD51-mediated homologous recombination. Oncogene 2013, 32, 3552–3558. [Google Scholar] [CrossRef] [PubMed]
- Park, J.-Y.; Singh, T.R.; Nassar, N.; Zhang, F.; Freund, M.; Hanenberg, H.; Meetei, A.R.; Andreassen, P.R. Breast cancer-associated missense mutants of the PALB2 WD40 domain, which directly binds RAD51C, RAD51 and BRCA2, disrupt DNA repair. Oncogene 2014, 33, 4803–4812. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poumpouridou, N.; Kroupis, C. Hereditary breast cancer: Beyond BRCA genetic analysis; PALB2 emerges. Clin. Chem. Lab. Med. 2012, 50, 423–434. [Google Scholar] [CrossRef] [PubMed]
- Shah, S.P.; Morin, R.D.; Khattra, J.; Prentice, L.; Pugh, T.; Burleigh, A.; Delaney, A.; Gelmon, K.; Guliany, R.; Senz, J.; et al. Mutational evolution in a lobular breast tumour profiled at single nucleotide resolution. Nature 2009, 461, 809–813. [Google Scholar] [CrossRef] [PubMed]
- Obermeier, K.; Sachsenweger, J.; Friedl, T.W.P.; Pospiech, H.; Winqvist, R.; Wiesmüller, L. Heterozygous PALB2 c.1592delT mutation channels DNA double-strand break repair into error-prone pathways in breast cancer patients. Oncogene 2015. [Google Scholar] [CrossRef] [PubMed]
- Huang, F.; Goyal, N.; Sullivan, K.; Hanamshet, K.; Patel, M.; Mazina, O.M.; Wang, C.X.; An, W.F.; Spoonamore, J.; Metkar, S.; et al. Targeting BRCA1- and BRCA2-deficient cells with RAD52 small molecule inhibitors. Nucleic Acids Res. 2016, 44, 4189–4199. [Google Scholar] [CrossRef] [PubMed]



© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nepomuceno, T.C.; De Gregoriis, G.; De Oliveira, F.M.B.; Suarez-Kurtz, G.; Monteiro, A.N.; Carvalho, M.A. The Role of PALB2 in the DNA Damage Response and Cancer Predisposition. Int. J. Mol. Sci. 2017, 18, 1886. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms18091886
Nepomuceno TC, De Gregoriis G, De Oliveira FMB, Suarez-Kurtz G, Monteiro AN, Carvalho MA. The Role of PALB2 in the DNA Damage Response and Cancer Predisposition. International Journal of Molecular Sciences. 2017; 18(9):1886. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms18091886
Chicago/Turabian StyleNepomuceno, Thales C., Giuliana De Gregoriis, Francisco M. Bastos De Oliveira, Guilherme Suarez-Kurtz, Alvaro N. Monteiro, and Marcelo A. Carvalho. 2017. "The Role of PALB2 in the DNA Damage Response and Cancer Predisposition" International Journal of Molecular Sciences 18, no. 9: 1886. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms18091886