Monitoring Autophagy Immunohistochemically and Ultrastructurally during Human Head and Neck Carcinogenesis. Relationship with the DNA Damage Response Pathway †

 ,
,
Abstract
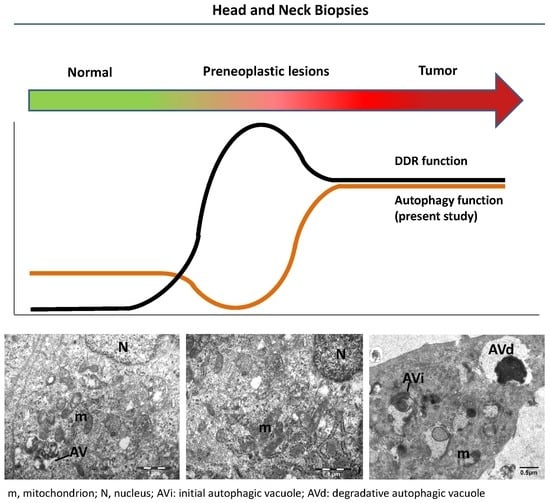
:
1. Introduction
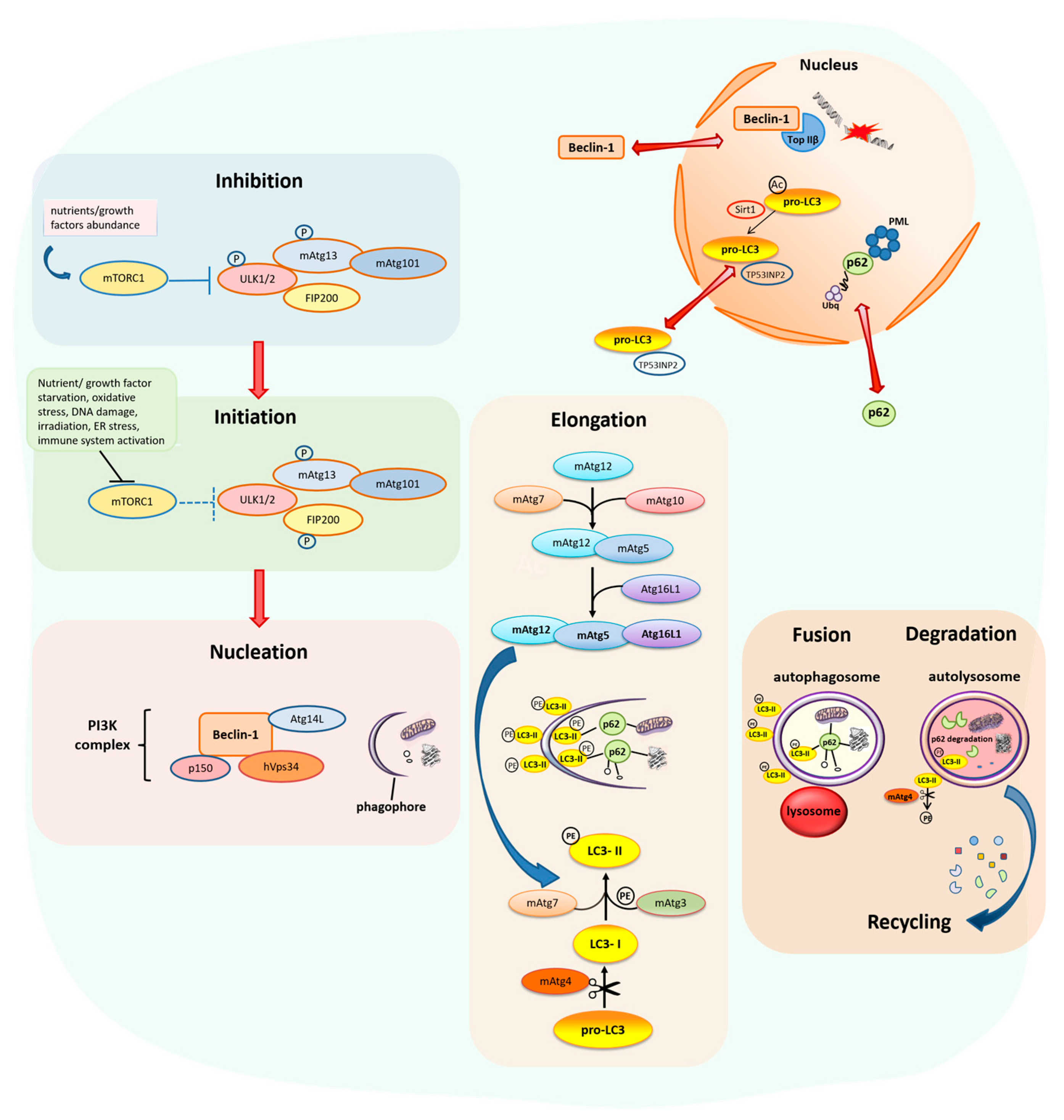
2. Results and Discussion
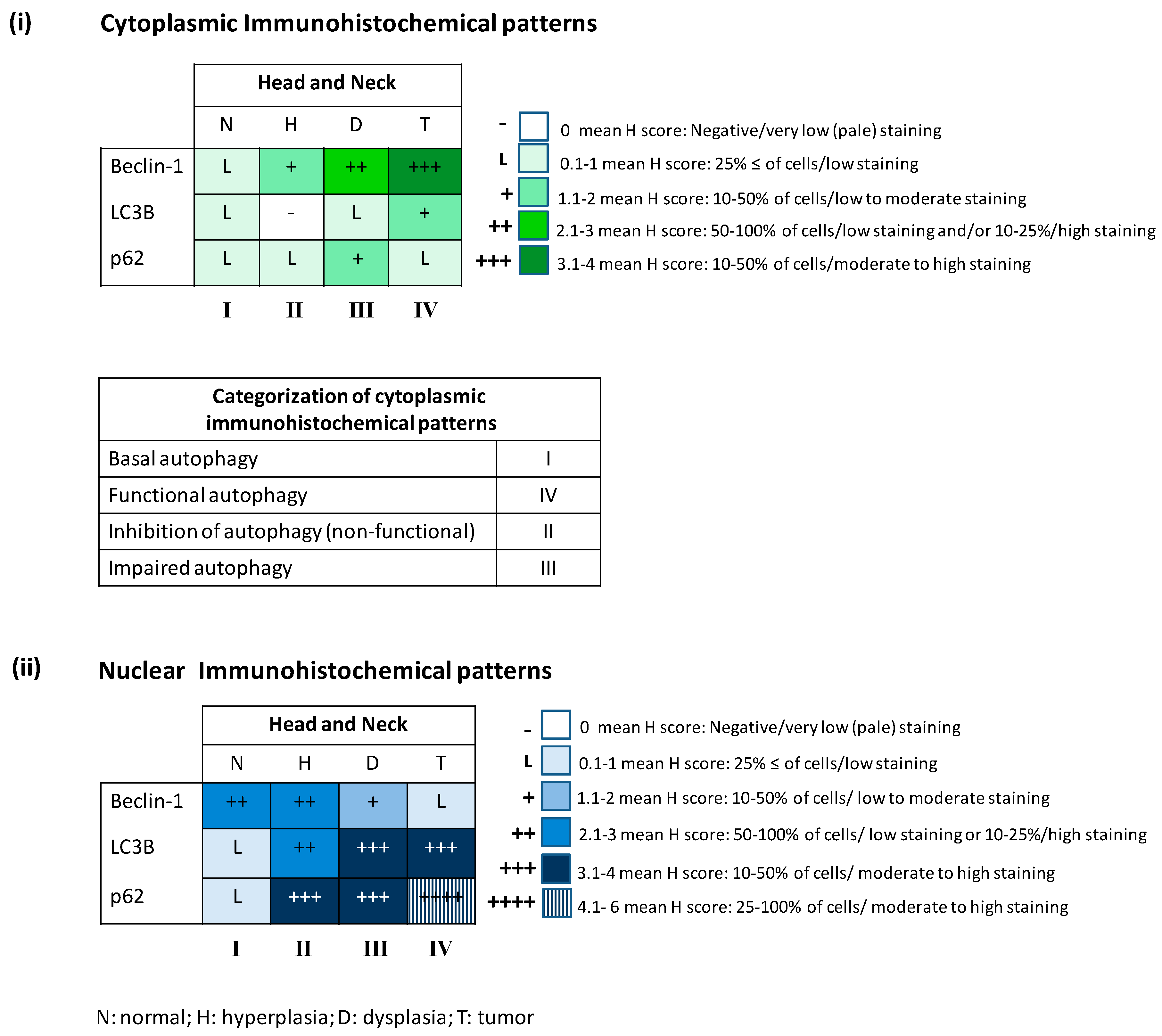
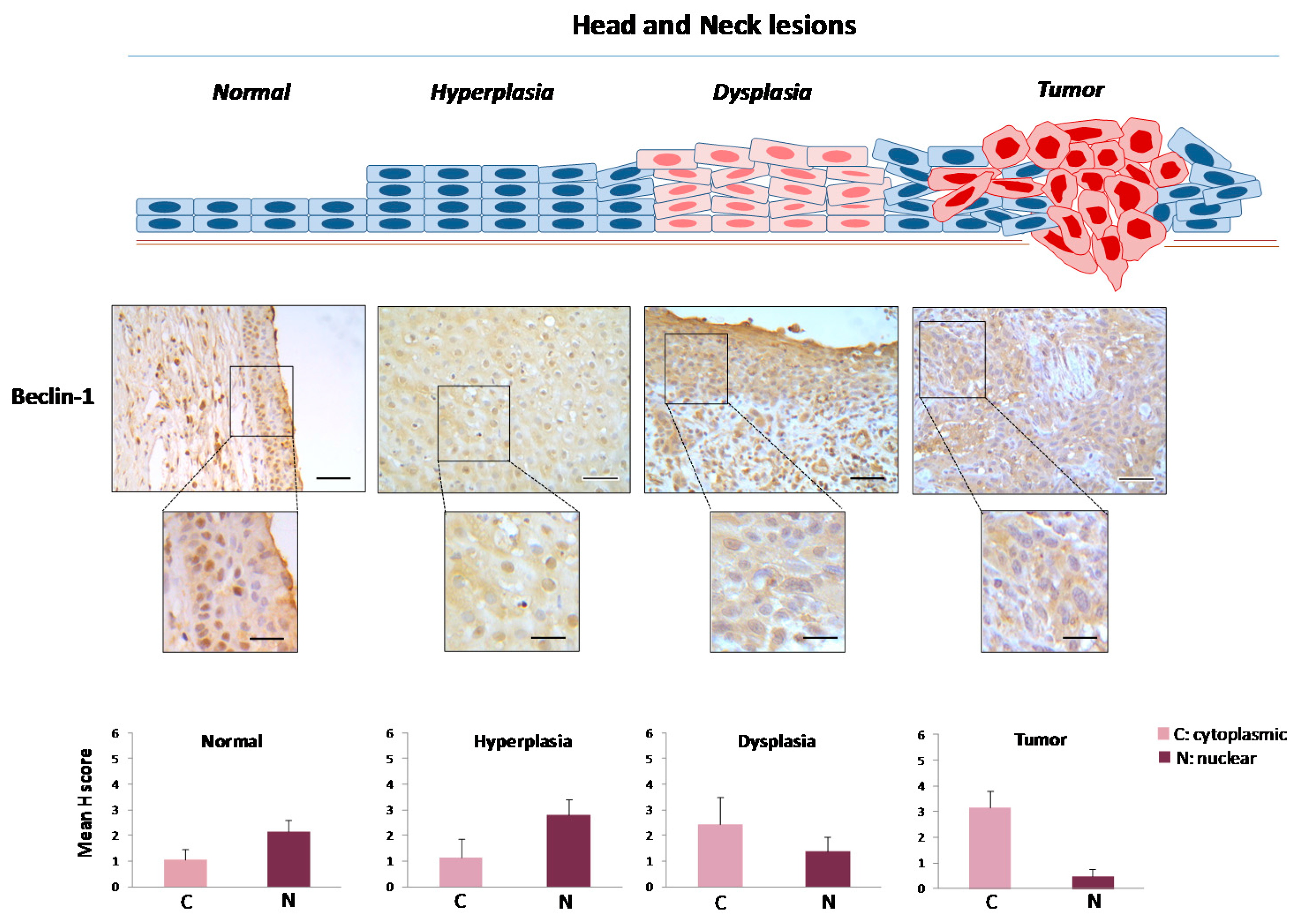
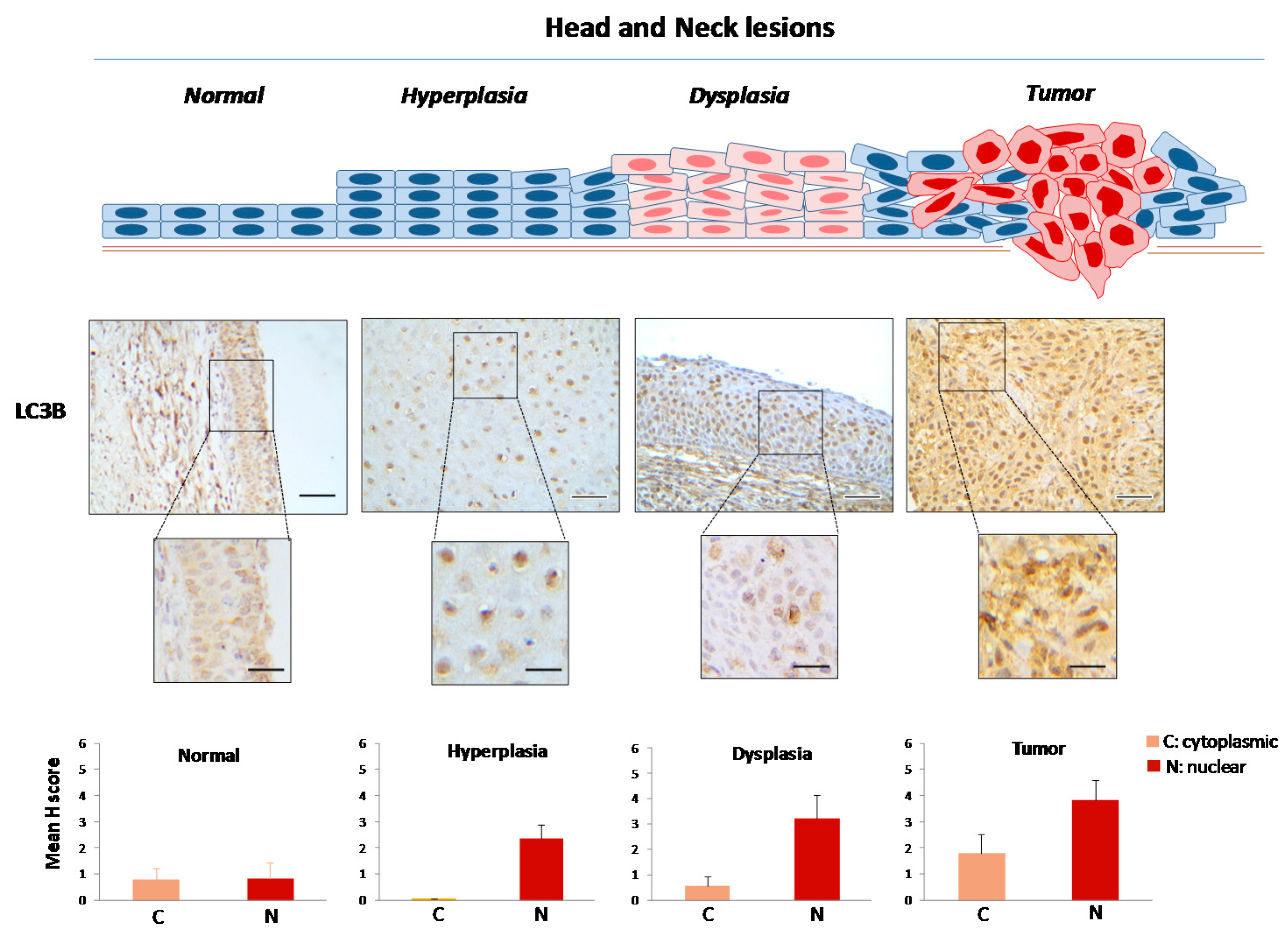
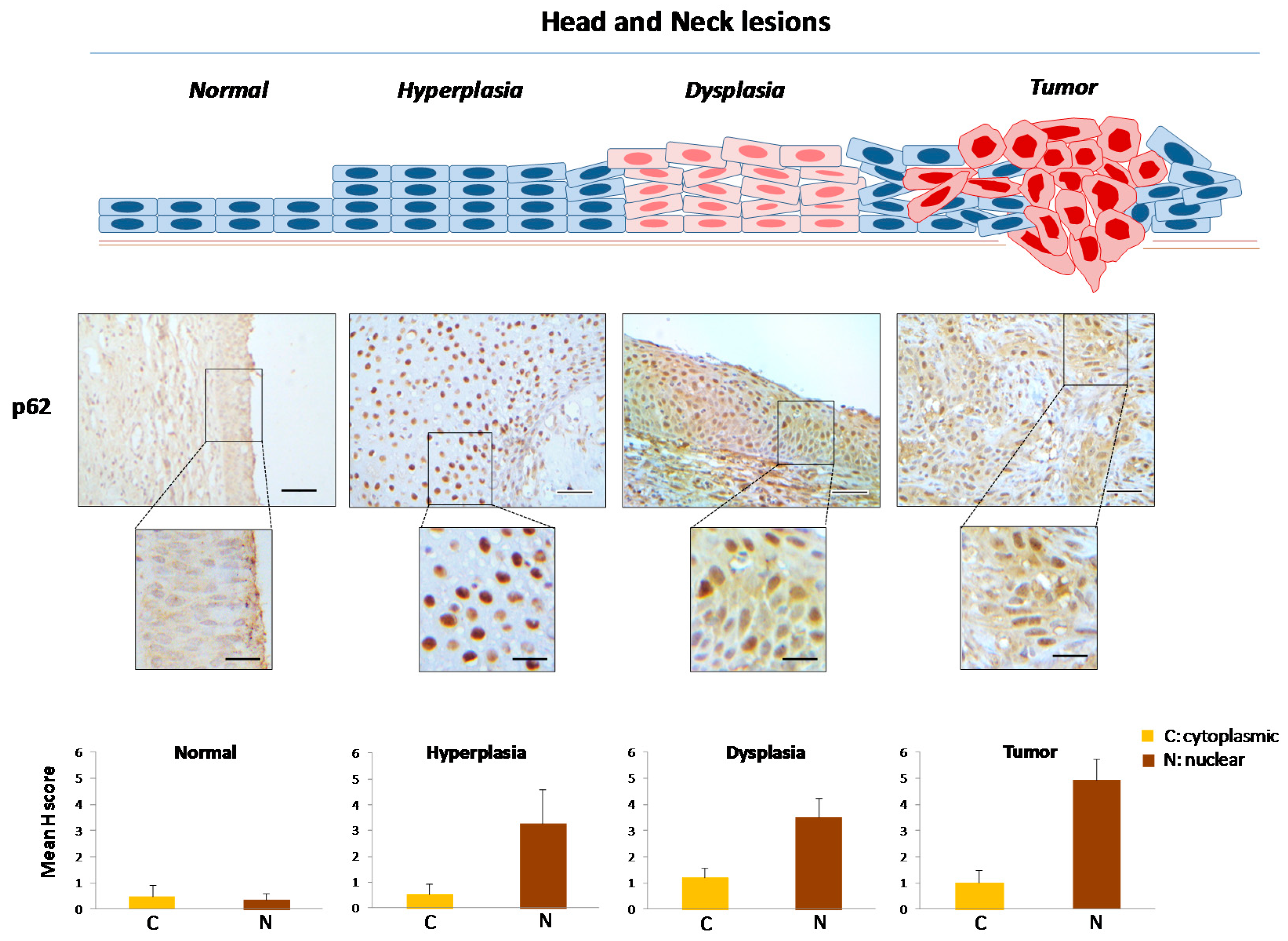
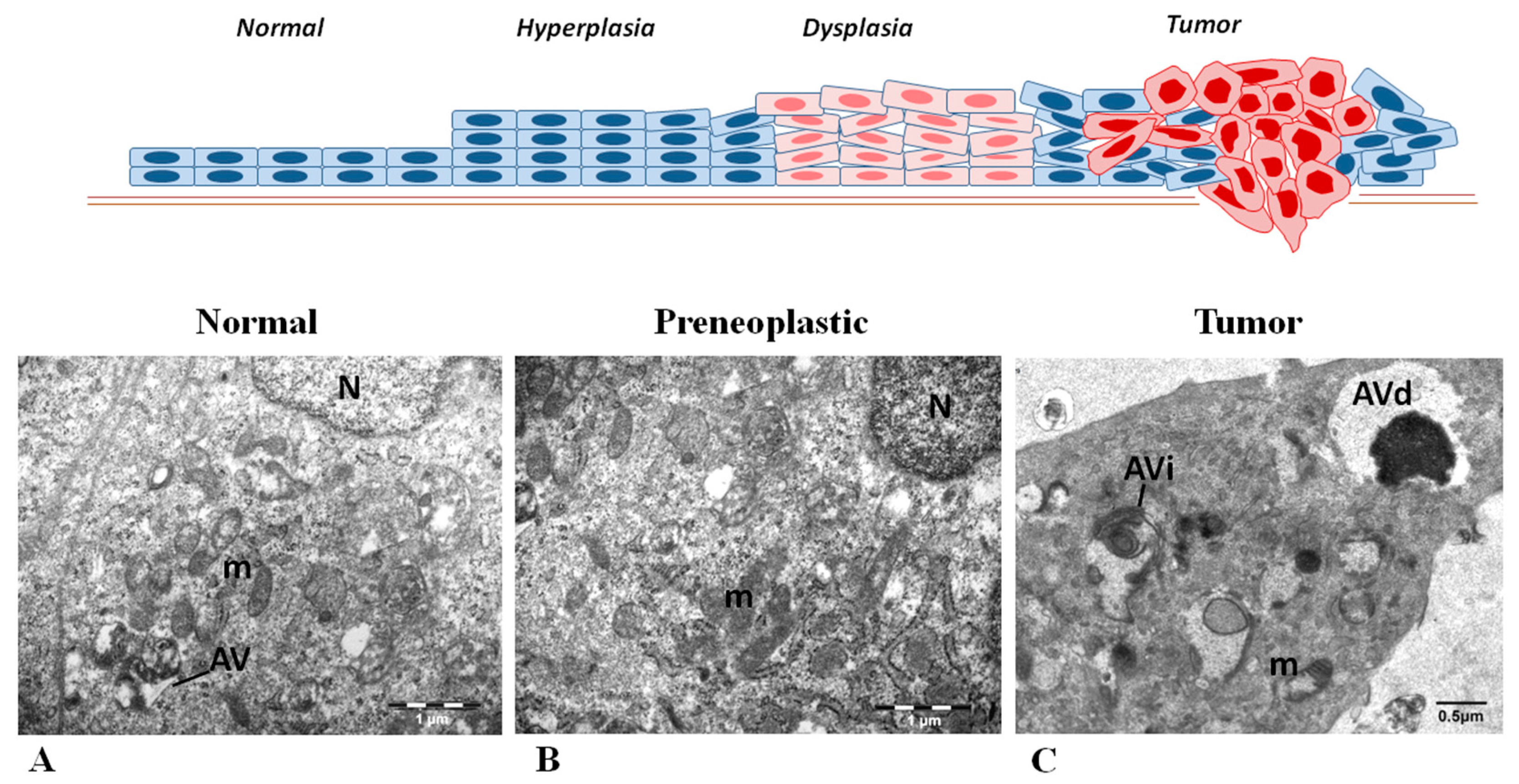
2.1. Defining the Immunohistochemical Profiles of Beclin-1, LC3B and p62 in Human Head and Neck Prencancerous and Cancerous Lesions
2.2. Evaluating Immunohistochemically and Ultrastructurally Autophagy in Human Head and Neck Carcinogenesis
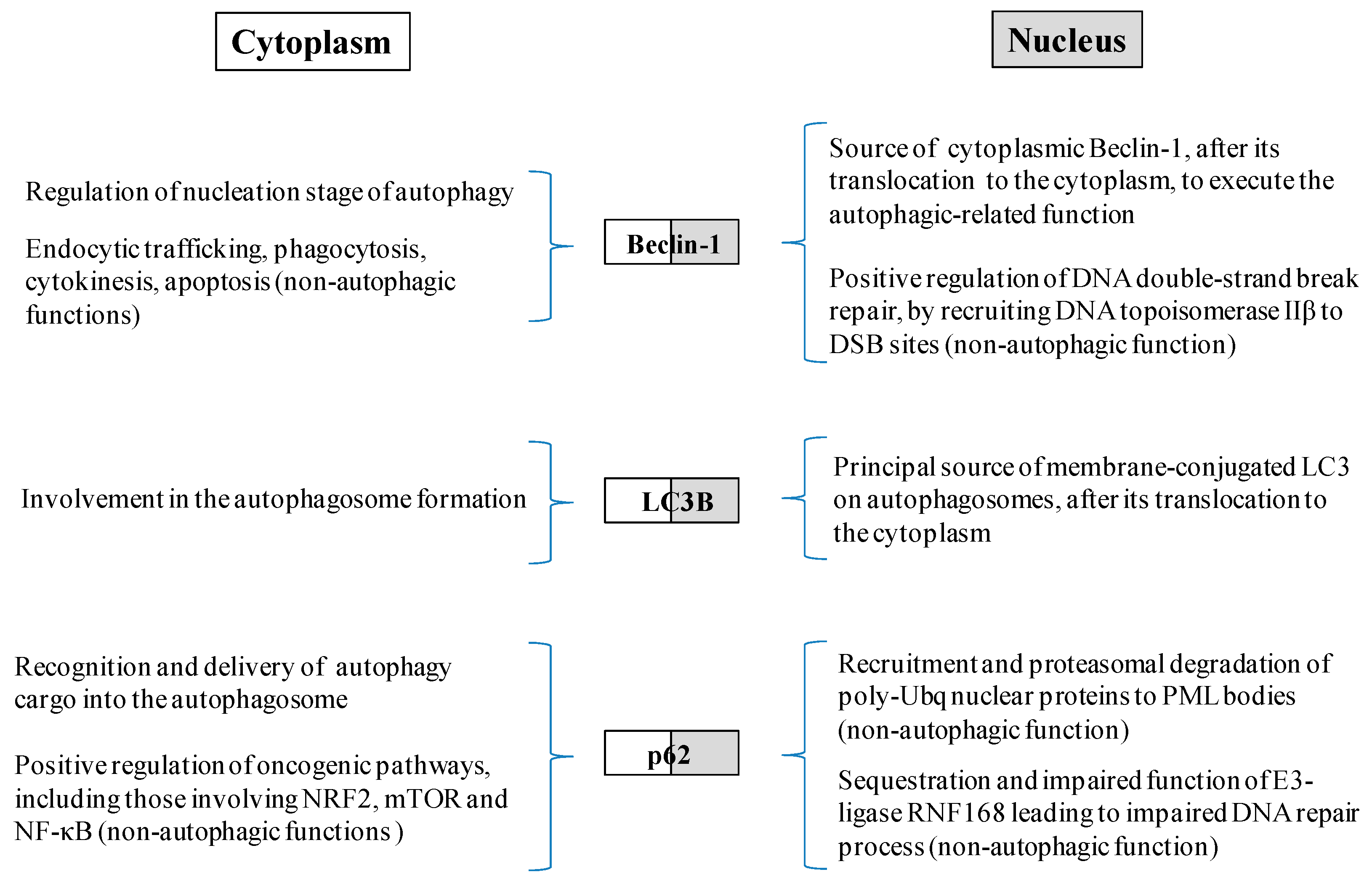
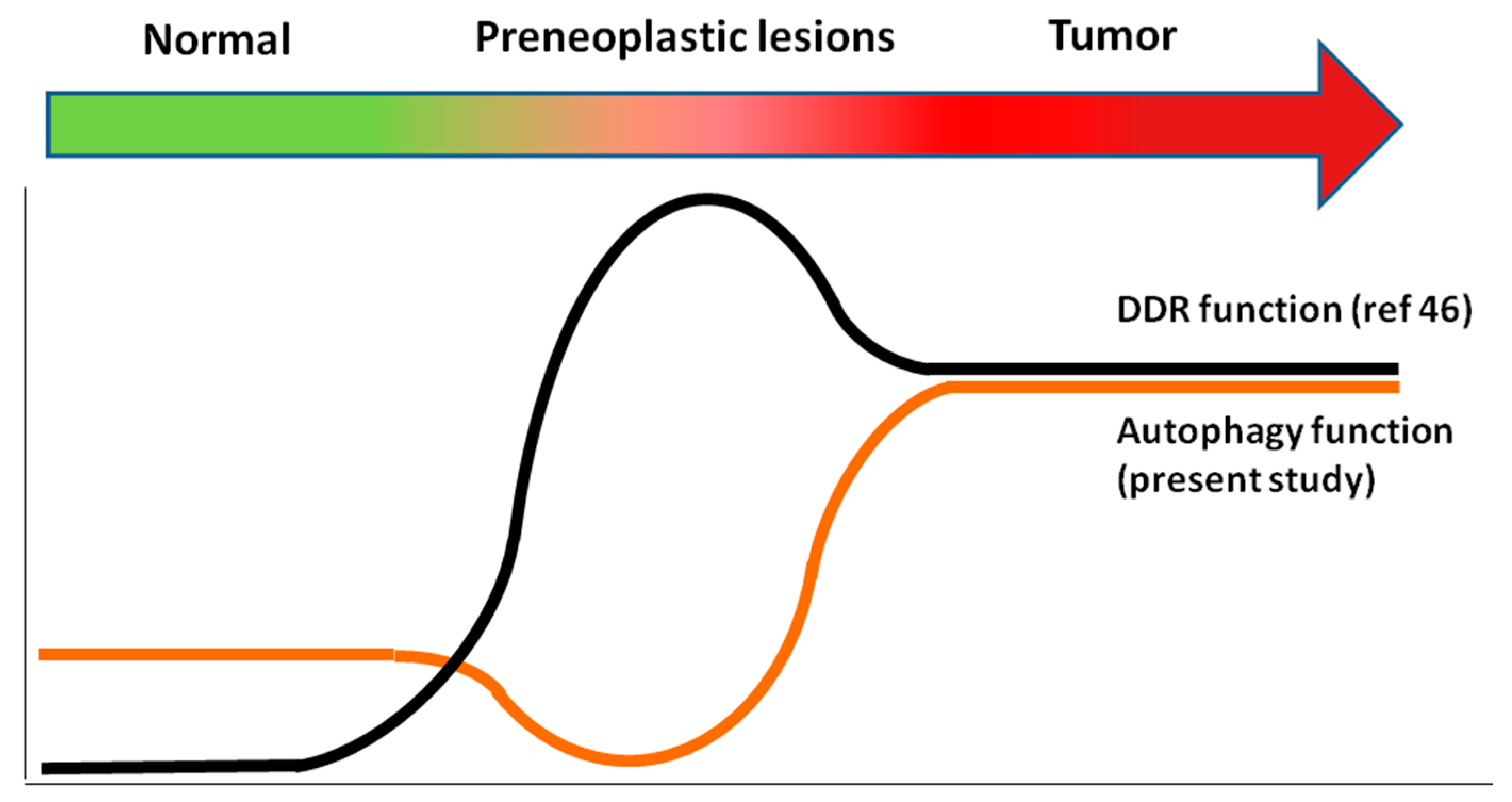
2.3. Relationship between Autophagy and the DDR Pathway during Human Head and Neck Cancer Development
3. Material and Methods
3.1. Human Samples
3.2. Immunohistochemistry (IHC)
Selected Fields Were Identical in all Serial Sections to Enable Optimal Comparison
3.3. Transmission Electron Microscopy (TEM)
4. Conclusions and Further Questions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| ATG | Autophagy-related gene |
| Atg16L1 | Atg16-like 1 protein |
| Atg14L | Atg14-like protein |
| CDK | Cyclin–dependent kinase |
| DDR | DNA damage response |
| DSB | DNA double-strand break |
| FIP200 | Focal adhesion kinase family-interacting protein of 200 kDa |
| hVps34 | Human vacuolar protein sorting-associated protein 34 |
| IHC | Immunohistochemistry |
| LC3 | Microtubule-associated protein 1 light chain 3 |
| mTOR | Mammalian target of rapamycin |
| mTORC1 | Mammalian target of rapamycin complex 1 |
| NES | Nuclear export signal |
| NRF2 | Nuclear factor erythroid 2-related factor |
| NF-κΒ | Nuclear factor-κB |
| PE | Phosphatidylethanolamine |
| PI3K | Phosphatidylinositol 3-kinase |
| PML | Promyelocytic leukemia bodies |
| RNF168 | Ring-finger protein 168 |
| SIRT1 | Sirtuin 1 |
| SQSTMQ/p62 | Sequestosome 1/p62 |
| TP53INP2 | Tumor protein 53 inducible nuclear protein 2 |
| ULK1/2 | Unc-51-like kinase 1/2 |
References
- Klionsky, D.J. Autophagy: From phenomenology to molecular understanding in less than a decade. Nat. Rev. Mol. Cell Biol. 2007, 8, 931–937. [Google Scholar] [CrossRef] [PubMed]
- Rabinowitz, J.D.; White, E. Autophagy and metabolism. Science 2010, 330, 1344–1348. [Google Scholar] [CrossRef] [PubMed]
- Kaur, J.; Debnath, J. Autophagy at the crossroads of catabolism and anabolism. Nat. Rev. Mol. Cell. Biol. 2015, 16, 461–472. [Google Scholar] [CrossRef] [PubMed]
- Levine, B.; Kroemer, G. Autophagy in the pathogenesis of disease. Cell 2008, 132, 27–42. [Google Scholar] [CrossRef] [PubMed]
- Choi, A.M.K.; Ryter, S.W.; Levine, B. Autophagy in human health and disease. N. Engl. J. Med. 2013, 368, 1845–1846. [Google Scholar] [CrossRef] [PubMed]
- Li, W.W.; Li, J.; Bao, J.K. Microautophagy: Lesser-known self-eating. Cell. Mol. Life Sci. 2012, 69, 1125–1136. [Google Scholar] [CrossRef] [PubMed]
- Parzych, K.R.; Klionsky, D.J. An overview of autophagy: Morphology, mechanism, and regulation. Antioxid. Redox Signal. 2014, 20, 460–473. [Google Scholar] [CrossRef] [PubMed]
- Massey, A.; Kiffin, R.; Cuervo, A.M. Pathophysiology of chaperone-mediated autophagy. Int. J. Biochem. Cell. Biol. 2004, 36, 2420–2434. [Google Scholar] [CrossRef] [PubMed]
- Cuervo, A.M.; Wong, E. Chaperone-mediated autophagy: Roles in disease and aging. Cell Res. 2014, 24, 92–104. [Google Scholar] [CrossRef] [PubMed]
- He, C.; Klionsky, D.J. Regulation mechanisms and signalling pathways of autophagy. Annu. Rev. Genet. 2009, 43, 67–93. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N.; Komatsu, M. Autophagy: Renovation of cells and tissues. Cell 2011, 147, 728–741. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Debnath, J. The evolving, multifaceted roles of autophagy in cancer. Adv. Cancer Res. 2016, 130, 1–53. [Google Scholar] [PubMed]
- Pattingre, S.; Espert, L.; Biard-Piechaczyk, M.; Codogno, P. Regulation of macroautophagy by mTOR and Beclin 1 complexes. Biochimie 2008, 90, 313–323. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Klionsky, D.J. Mammalian autophagy: Core molecular machinery and signaling regulation. Curr. Opin. Cell Biol. 2010, 22, 124–131. [Google Scholar] [CrossRef] [PubMed]
- Kabeya, Y.; Mizushima, N.; Yamamoto, A.; Oshitani-Okamoto, S.; Ohsumi, Y.; Yoshimori, T. LC3, GABARAP and GATE16 localize to autophagosomal membrane depending on form-II formation. J. Cell Sci. 2004, 117, 2805–2812. [Google Scholar] [CrossRef] [PubMed]
- Koukourakis, M.I.; Kalamida, D.; Giatromanolaki, A.; Zois, C.E.; Sivridis, E.; Pouliliou, S.; Mitrakas, A.; Gatter, K.C.; Harris, A.L. Autophagosome proteins LC3A, LC3B and LC3C have distinct subcellular distribution kinetics and expression in cancer cell lines. PLoS ONE 2015, 10, e0137675. [Google Scholar] [CrossRef] [PubMed]
- Pankiv, S.; Clausen, T.H.; Lamark, T.; Brech, A.; Bruun, J.A.; Outzen, H.; Øvervatn, A.; Bjørkøy, G.; Johansen, T. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J. Biol. Chem. 2007, 282, 24131–24145. [Google Scholar] [CrossRef] [PubMed]
- Ichimura, Y.; Kumanomidou, T.; Sou, Y.S.; Mizushima, T.; Ezaki, J.; Ueno, T.; Kominami, E.; Yamane, T.; Tanaka, K.; Komatsu, M. Structural basis for sorting mechanism of p62 in selective autophagy. J. Biol. Chem. 2008, 283, 22847–22857. [Google Scholar] [CrossRef] [PubMed]
- Tanida, I.; Waguri, S. Measurement of autophagy in cells and tissues. Methods Mol. Biol. 2010, 648, 193–214. [Google Scholar] [PubMed]
- Drake, K.R.; Kang, M.; Kenworthy, A.K. Nucleocytoplasmic distribution and dynamics of the autophagosome marker EGFP-LC3. PLoS ONE 2010, 5, e9806. [Google Scholar] [CrossRef] [PubMed]
- Pankiv, S.; Lamark, T.; Bruun, J.A.; Øvervatn, A.; Bjørkøy, G.; Johansen, T. Nucleocytoplasmic shuttling of p62/SQSTM1 and its role in recruitment of nuclear polyubiquitinated proteins to promyelocytic leukemia bodies. J. Biol. Chem. 2010, 285, 5941–5953. [Google Scholar] [CrossRef] [PubMed]
- Liang, X.H.; Yu, J.; Brown, K.; Levine, B. Beclin 1 contains a leucine-rich nuclear export signal that is required for its autophagy and tumor suppressor function. Cancer Res. 2001, 61, 3443–3449. [Google Scholar] [PubMed]
- Wirawan, E.; Lippens, S.; Vanden Berghe, T.; Romagnoli, A.; Fimia, G.M.; Piacentini, M.; Vandenabeele, P. Beclin1: A role in membrane dynamics and beyond. Autophagy 2012, 8, 6–17. [Google Scholar] [CrossRef] [PubMed]
- Sahni, S.; Merlot, A.M.; Krishan, S.; Jansson, P.J.; Richardson, D.R. Gene of the month: BECN1. J. Clin. Pathol. 2014, 67, 656–660. [Google Scholar] [CrossRef] [PubMed]
- Taniguchi, K.; Yamachika, S.; He, F.; Karin, M. p62/SQSTM1—Dr. Jekyll and Mr. Hyde that prevents oxidative stress but promotes liver cancer. FEBS Lett. 2016, 590, 2375–2397. [Google Scholar] [CrossRef] [PubMed]
- Moscat, J.; Karin, M.; Diaz-Meco, M.T. p62 in cancer: Signaling adaptor beyond autophagy. Cell 2016, 167, 606–609. [Google Scholar] [CrossRef] [PubMed]
- Xu, F.; Fang, Y.; Yan, L.; Xu, L.; Zhang, S.; Cao, Y.; Xu, L.; Zhang, X.; Xie, J.; Jiang, G.; et al. Nuclear localization of Beclin 1 promotes radiation-induced DNA damage repair independent of autophagy. Sci. Rep. 2017, 7, 45385–45395. [Google Scholar] [CrossRef] [PubMed]
- Huang, R.; Xu, Y.; Wan, W.; Shou, X.; Qian, J.; You, Z.; Liu, B.; Chang, C.; Zhou, T.; Lippincott-Schwartz, J.; et al. Deacetylation of nuclear LC3 drives autophagy initiation under starvation. Mol. Cell 2015, 57, 456–467. [Google Scholar] [CrossRef] [PubMed]
- Huang, R.; Liu, W. Identifying an essential role of nuclear LC3 for autophagy. Autophagy 2015, 11, 852–853. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhang, N.; Zhang, L.; Li, R.; Fu, W.; Ma, K.; Li, X.; Wang, L.; Wang, J.; Zhang, H.; et al. Autophagy regulates chromatin ubiquitination in DNA damage response through elimination of SQSTM1/p62. Mol. Cell 2016, 63, 34–48. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhu, W.-G.; Zhao, Y. Autophagy substrate SQSTM1/p62 regulates chromatin ubiquitination during the DNA damage response. Autophagy 2017, 13, 212–213. [Google Scholar] [CrossRef] [PubMed]
- White, E. Deconvoluting the context-dependent role for autophagy in cancer. Nat. Rev. Cancer 2012, 12, 401–410. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Pietrocola, F.; Bravo-San Pedro, J.M.; Amaravadi, R.K.; Baehrecke, E.H.; Cecconi, F.; Codogno, P.; Debnath, J.; Gewirtz, D.A.; Karantza, V.; et al. Autophagy in malignant transformation and cancer progression. EMBO J. 2015, 34, 856–880. [Google Scholar] [CrossRef] [PubMed]
- White, E. The role for autophagy in cancer. J. Clin. Investig. 2015, 125, 42–46. [Google Scholar] [CrossRef] [PubMed]
- Shintani, T.; Klionsky, D.J. Autophagy in health and disease: A double-edged sword. Science 2004, 306, 990–995. [Google Scholar] [CrossRef] [PubMed]
- Roy, S.; Debnath, J. Autophagy and tumorigenesis. Semin. Immunopathol. 2010, 32, 383–396. [Google Scholar] [CrossRef] [PubMed]
- Adhauliya, N.; Kalappanavar, A.N.; Ali, I.M.; Annigeri, R.G. Autophagy: A boon or bane in oral cancer. Oral Oncol. 2016, 61, 120–126. [Google Scholar] [CrossRef] [PubMed]
- Cosway, B.; Lovat, P. The role of autophagy in squamous cell carcinoma of the head and neck. Oral Oncol. 2016, 54, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Gorgoulis, V.G.; Vassiliou, L.-V.F.; Karakaidos, P.; Zacharatos, P.; Kotsinas, A.; Liloglou, T.; Venere, M.; Ditullio, R.A.; Kastrinakis, N.G.; Levy, B.; et al. Activation of the DNA damage checkpoint and genomic instability in human precancerous lesions. Nature 2005, 434, 907–913. [Google Scholar] [CrossRef] [PubMed]
- Bartkova, J.; Rezaei, N.; Liontos, M.; Karakaidos, P.; Kletsas, D.; Issaeva, N.; Vassiliou, L.V.; Kolettas, E.; Niforou, K.; Zoumpourlis, V.C.; et al. Oncogene-induced senescence is part of the tumorigenesis barrier imposed by DNA damage checkpoints. Nature 2006, 444, 633–637. [Google Scholar] [CrossRef] [PubMed]
- Liontos, M.; Koutsami, M.; Sideridou, M.; Evangelou, K.; Kletsas, D.; Levy, B.; Kotsinas, A.; Nahum, O.; Zoumpourlis, V.; Kouloukoussa, M.; et al. Deregulated overexpression of hCdt1 and hCdc6 promotes malignant behavior. Cancer Res. 2007, 67, 10899–10909. [Google Scholar] [CrossRef] [PubMed]
- Halazonetis, T.D.; Gorgoulis, V.G.; Bartek, J. An oncogene-induced DNA damage model for cancer development. Science 2008, 319, 1352–1355. [Google Scholar] [CrossRef] [PubMed]
- Tsantoulis, P.K.; Kotsinas, A.; Sfikakis, P.P.; Evangelou, K.; Sideridou, M.; Levy, B.; Mo, L.; Kittas, C.; Wu, X.R.; Papavassiliou, A.G.; et al. Oncogene-induced replication stress preferentially targets common fragile sites in preneoplastic lesions. A genome-wide study. Oncogene 2008, 27, 3256–3264. [Google Scholar] [CrossRef] [PubMed]
- Pateras, I.S.; Havaki, S.; Nikitopoulou, X.; Vougas, K.; Townsend, P.A.; Panayiotidis, M.I.; Georgakilas, A.G.; Gorgoulis, V.G. The DNA damage response and immune signaling alliance: Is it good or bad? Nature decides when and where. Pharmacol. Ther. 2015, 154, 36–56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Polo, S.E.; Jackson, S.P. Dynamics of DNA damage response proteins at DNA breaks: A focus on protein modifications. Genes Dev. 2011, 25, 409–433. [Google Scholar] [CrossRef] [PubMed]
- Jackson, S.P.; Bartek, J. The DNA-damage response in human biology and disease. Nature 2009, 461, 1071–1078. [Google Scholar] [CrossRef] [PubMed]
- Gorgoulis, V.G.; Halazonetis, T.D. Oncogene-induced senescence: The bright and dark side of the response. Curr. Opin. Cell Biol. 2010, 22, 816–827. [Google Scholar] [CrossRef] [PubMed]
- Evangelou, K.; Bartkova, J.; Kotsinas, A.; Pateras, I.S.; Liontos, M.; Velimezi, G.; Kosar, M.; Liloglou, T.; Trougakos, I.P.; Dyrskjot, L.; et al. The DNA damage checkpoint precedes activation of ARF in response to escalating oncogenic stress during tumorigenesis. Cell Death Differ. 2013, 20, 1485–1497. [Google Scholar] [CrossRef] [PubMed]
- Mathew, R.; Karantza-Wadsworth, V.; White, E. Role of autophagy in cancer. Nat. Rev. Cancer 2007, 7, 961–967. [Google Scholar] [CrossRef] [PubMed]
- Klionsky, D.J.; Abdelmohsen, K.; Abe, A.; Abedin, M.J.; Abeliovich, H.; Acevedo Arozena, A.; Adachi, H.; Adams, C.M.; Adams, P.D.; Adeli, K.; et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy 2016, 12, 1–222. [Google Scholar] [PubMed]
- Moscat, J.; Diaz-Meco, M.T. p62: A versatile multitasker takes on cancer. Trends Biochem Sci. 2012, 37, 230–236. [Google Scholar] [CrossRef] [PubMed]
- Papandreou, I.; Lim, A.L.; Laderoute, K.; Denko, N.C. Hypoxia signals autophagy in tumor cells via AMPK activity, independent of HIF-1, BNIP3, and BNIP3L. Cell Death Differ. 2008, 15, 1572–1581. [Google Scholar] [CrossRef] [PubMed]
- Kroemer, G.; Mariño, G.; Levine, B. Autophagy and the integrated stress response. Mol. Cell 2010, 40, 280–293. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Vitale, I.; Michels, J.; Brenner, C.; Szabadkai, G.; Harel-Bellan, A.; Castedo, M.; Kroemer, G. Systems biology of cisplatin resistance: Past, present and future. Cell Death Dis. 2014, 5, e1257. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.-L.; Chen, F.-F.; Chang, S.-F.; Chen, C.-N.; Lung, J.; Lo, C.-H.; Lee, F.-H.; Lu, Y.-C.; Hung, C.-H. Expression of Beclin family proteins is associated with tumor progression in oral cancer. PLoS ONE 2015, 10, e0141308. [Google Scholar] [CrossRef] [PubMed]
- Wan, X.B.; Fan, X.J.; Chen, M.Y.; Xiang, J.; Huang, P.Y.; Guo, L.; Wu, X.Y.; Xu, J.; Long, Z.J.; Zhao, Y.; et al. Elevated Beclin 1 expression is correlated with HIF-1 α in predicting poor prognosis of nasopharyngeal carcinoma. Autophagy 2010, 6, 395–404. [Google Scholar] [CrossRef] [PubMed]
- Tang, J.-Y.; Fang, Y.-Y.; Hsi, E.; Huang, Y.-C.; Hsu, N.C.-H.; Yang, W.-C.; Chang, H.-W.; Chai, C.-Y.; Chu, P.-Y. Immunopositivity of Beclin-1 and ATG5 as indicators of survival and disease recurrence in oral squamous cell carcinoma. Anticancer Res. 2013, 33, 5611–5616. [Google Scholar] [PubMed]
- Sakakura, K.; Takahashi, H.; Kaira, K.; Toyoda, M.; Oyama, T.; Chikamatsu, K. Immunological significance of the accumulation of autophagy components in oral squamous cell carcinoma. Cancer Sci. 2015, 106, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Wang, S.; Li, S.-S.; Yang, X.-M. Prognostic significance of Beclin-1 expression in laryngeal squamous cell carcinoma. Pathol. Oncol. Res. 2013, 19, 771–777. [Google Scholar] [CrossRef] [PubMed]
- Hewitt, G.; Carroll, B.; Sarallah, R.; Correia-Melo, C.; Ogrodnik, M.; Nelson, G.; Otten, E.G.; Manni, D.; Antrobus, R.; Morgan, B.A.; et al. SQSTM1/p62 mediates crosstalk between autophagy and the UPS in DNA repair. Autophagy 2016, 12, 1917–1930. [Google Scholar] [CrossRef] [PubMed]
- Galanos, P.; Vougas, K.; Walter, D.; Polyzos, A.; Maya-Mendoza, A.; Haagensen, E.J.; Kokkalis, A.; Roumelioti, F.M.; Gagos, S.; Tzetis, M.; et al. Chronic p53-independent p21 expression causes genomic instability by deregulating replication licensing. Nat. Cell Biol. 2016, 18, 777–789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, H.; He, Z.; Simon, H.U. Autophagy suppresses melanoma tumorigenesis by inducing senescence. Autophagy 2014, 10, 372–373. [Google Scholar] [CrossRef] [PubMed]
- Guo, G.F.; Jiang, W.Q.; Zhang, B.; Cai, Y.C.; Xu, R.H.; Chen, X.X.; Wang, F.; Xia, L.P. Autophagy-related proteins Beclin 1 and LC3 predict cetuximab efficacy in advanced colorectal cancer. World J. Gastroenterol. 2011, 17, 4779–4786. [Google Scholar] [CrossRef] [PubMed]
- Rikiishi, H. Autophagic action of new targeting agents in head and neck oncology. Cancer Biol. Ther. 2012, 13, 978–991. [Google Scholar] [CrossRef] [PubMed]
- Kang, R.; Wang, Z.H.; Wang, B.Q.; Zhang, C.M.; Gao, W.; Feng, Y.; Bai, T.; Zhang, H.L.; Huang-Pu, H.; Wen, S.X. Inhibition of autophagy-potentiated chemosensitivity to cisplatin in laryngeal cancer Hep-2 cells. Am. J. Otolaryngol. 2012, 33, 678–684. [Google Scholar] [CrossRef] [PubMed]
- Chi, K.H.; Wang, Y.S.; Huang, Y.C.; Chiang, H.C.; Chi, M.S.; Chi, C.H.; Wang, H.E.; Kao, S.J. Simultaneous activation and inhibition of autophagy sensitizes cancer cells to chemotherapy. Oncotarget 2016, 7, 58075–58088. [Google Scholar] [CrossRef] [PubMed]
- Carden, D.L.; Granger, D.N. Pathophysiology of ischaemia-reperfusion injury. J. Pathol. 2000, 190, 255–266. [Google Scholar] [CrossRef]
- Evangelou, K.; Lougiakis, N.; Rizou, S.V.; Kotsinas, A.; Kletsas, D.; Muñoz-Espín, D.; Kastrinakis, N.G.; Pouli, N.; Marakos, P.; Townsend, P.; et al. Robust, universal biomarker assay to detect senescent cells in biological specimens. Aging Cell 2016, 16, 192–197. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| (i) Cytoplasmic Immunohistochemical Patterns | (ii) Nuclear Immunohistochemical Patterns | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Autopahgy-Related Factors | Head and Neck | Autopahgy-Related Factors | Head and Neck | ||||||
| N | H | D | T | N | H | D | T | ||
| Beclin-1 | L | + | ++ | +++ | Beclin-1 | ++ | ++ | + | L |
| LC3B | L | − | L | + | LC3B | L | ++ | +++ | +++ |
| p62 | L | L | + | L | p62 | L | +++ | +++ | ++++ |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Havaki, S.; Vlachou, V.; Zampetidis, C.P.; Selemenakis, P.; Kotsinas, A.; Mavrogonatou, E.; Rizou, S.V.; Kyrodimos, E.; Evangelou, K.; Kletsas, D.; et al. Monitoring Autophagy Immunohistochemically and Ultrastructurally during Human Head and Neck Carcinogenesis. Relationship with the DNA Damage Response Pathway. Int. J. Mol. Sci. 2017, 18, 1920. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms18091920
Havaki S, Vlachou V, Zampetidis CP, Selemenakis P, Kotsinas A, Mavrogonatou E, Rizou SV, Kyrodimos E, Evangelou K, Kletsas D, et al. Monitoring Autophagy Immunohistochemically and Ultrastructurally during Human Head and Neck Carcinogenesis. Relationship with the DNA Damage Response Pathway. International Journal of Molecular Sciences. 2017; 18(9):1920. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms18091920
Chicago/Turabian StyleHavaki, Sophia, Vassiliki Vlachou, Christos P. Zampetidis, Platonas Selemenakis, Athanassios Kotsinas, Eleni Mavrogonatou, Sophia V. Rizou, Euthymios Kyrodimos, Konstantinos Evangelou, Dimitris Kletsas, and et al. 2017. "Monitoring Autophagy Immunohistochemically and Ultrastructurally during Human Head and Neck Carcinogenesis. Relationship with the DNA Damage Response Pathway" International Journal of Molecular Sciences 18, no. 9: 1920. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms18091920