Cytokine Disturbances in Coronary Artery Ectasia Do Not Support Atherosclerosis Pathogenesis

 , ,
, ,
Abstract
:1. Introduction
2. Methods
2.1. Patient Selection
2.2. Sample Preparation and Analysis
2.3. Statistical Analysis
3. Results
3.1. Demographics and CV Risk Factors
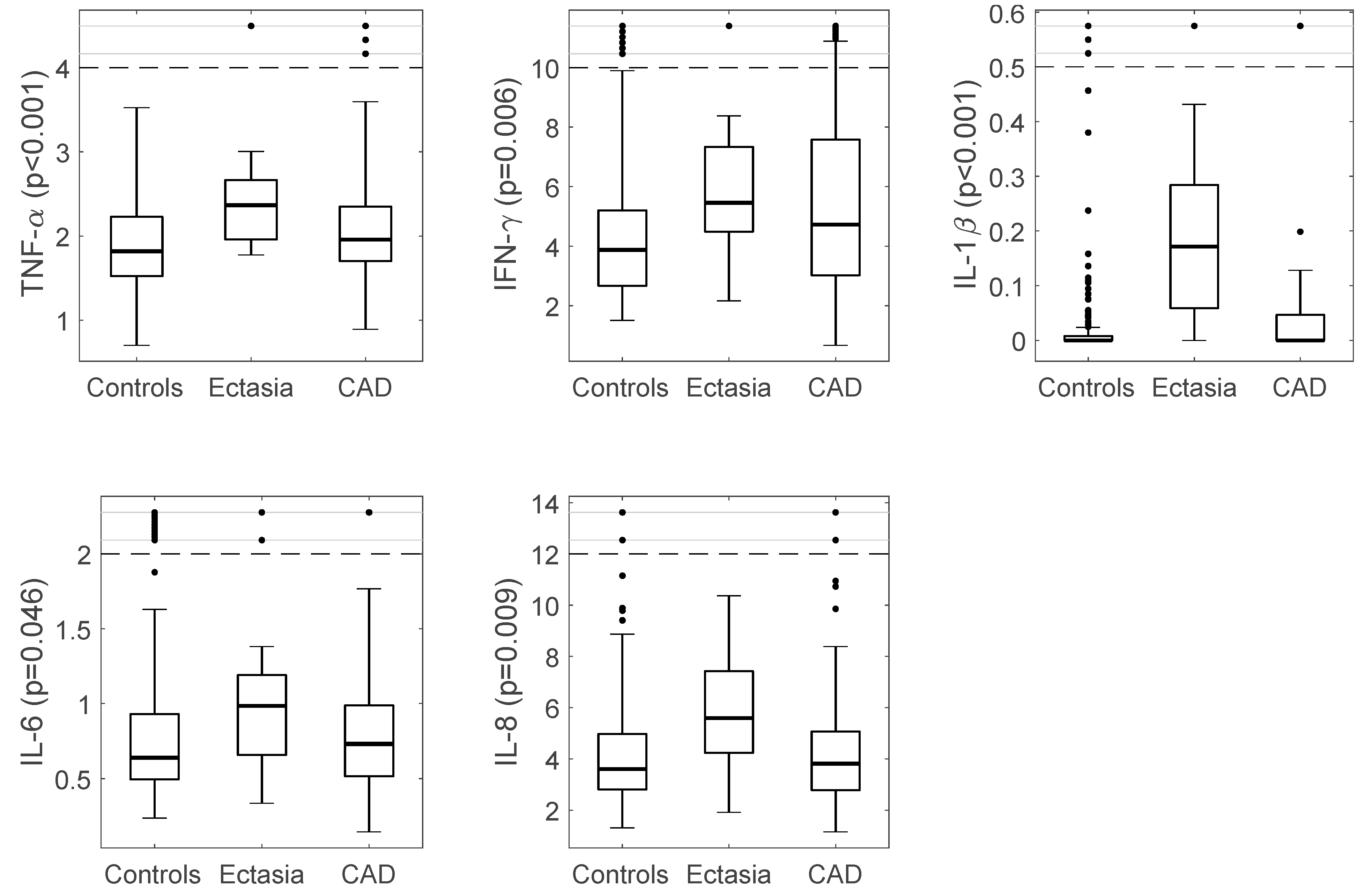
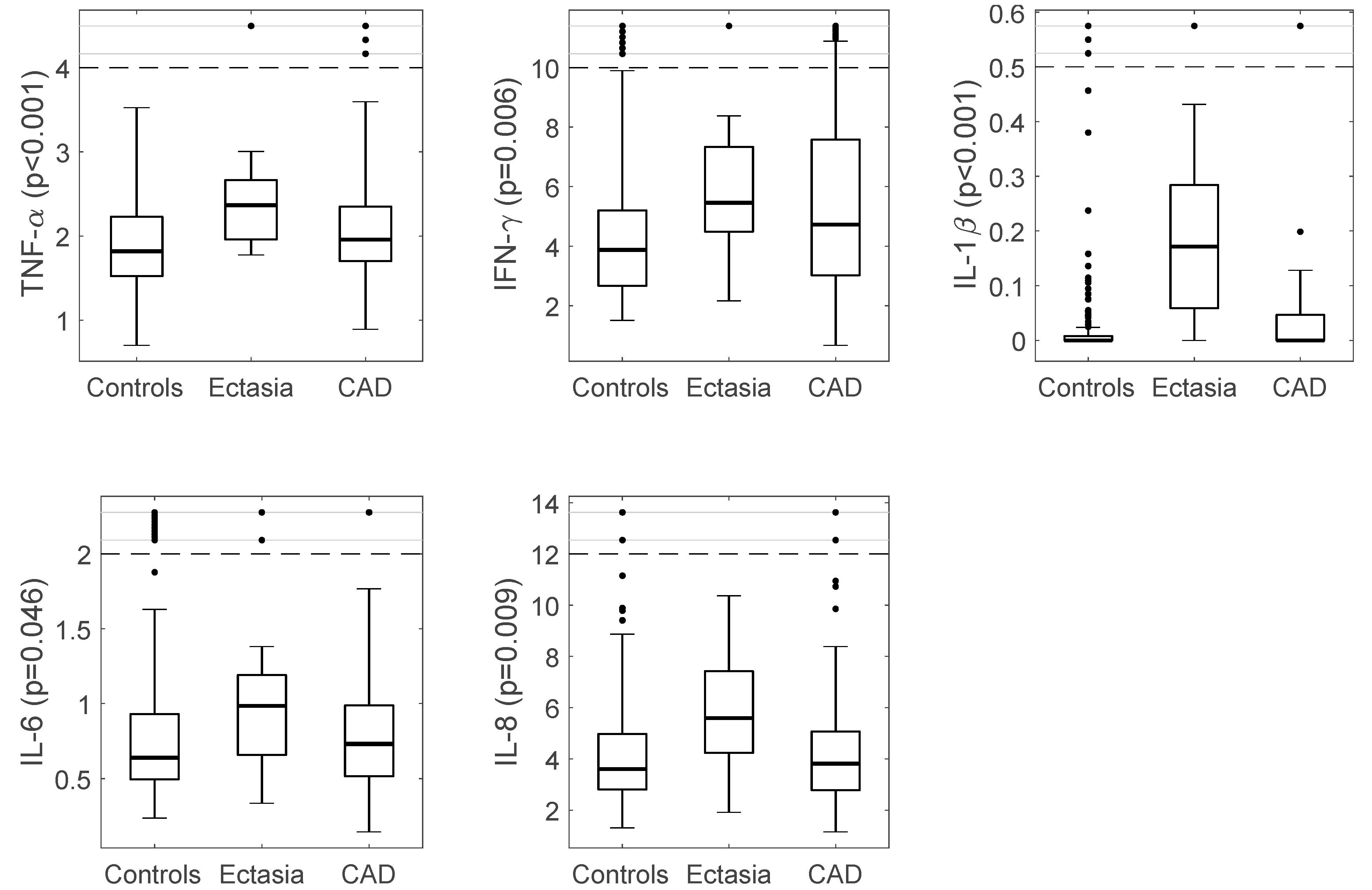
3.2. Immuno-Inflammatory Response in CAE vs. Controls
3.3. Immuno-Inflammatory Response in CAE vs. CAD
3.4. Pure Ectasia vs. Mixed Ectasia
3.5. Data Reproducibility
4. Discussion
4.1. Summary of Findings
4.2. Immuno-Inflammatory Response in CAE vs. Atherosclerosis
4.3. The Role of IL-6 in CAE
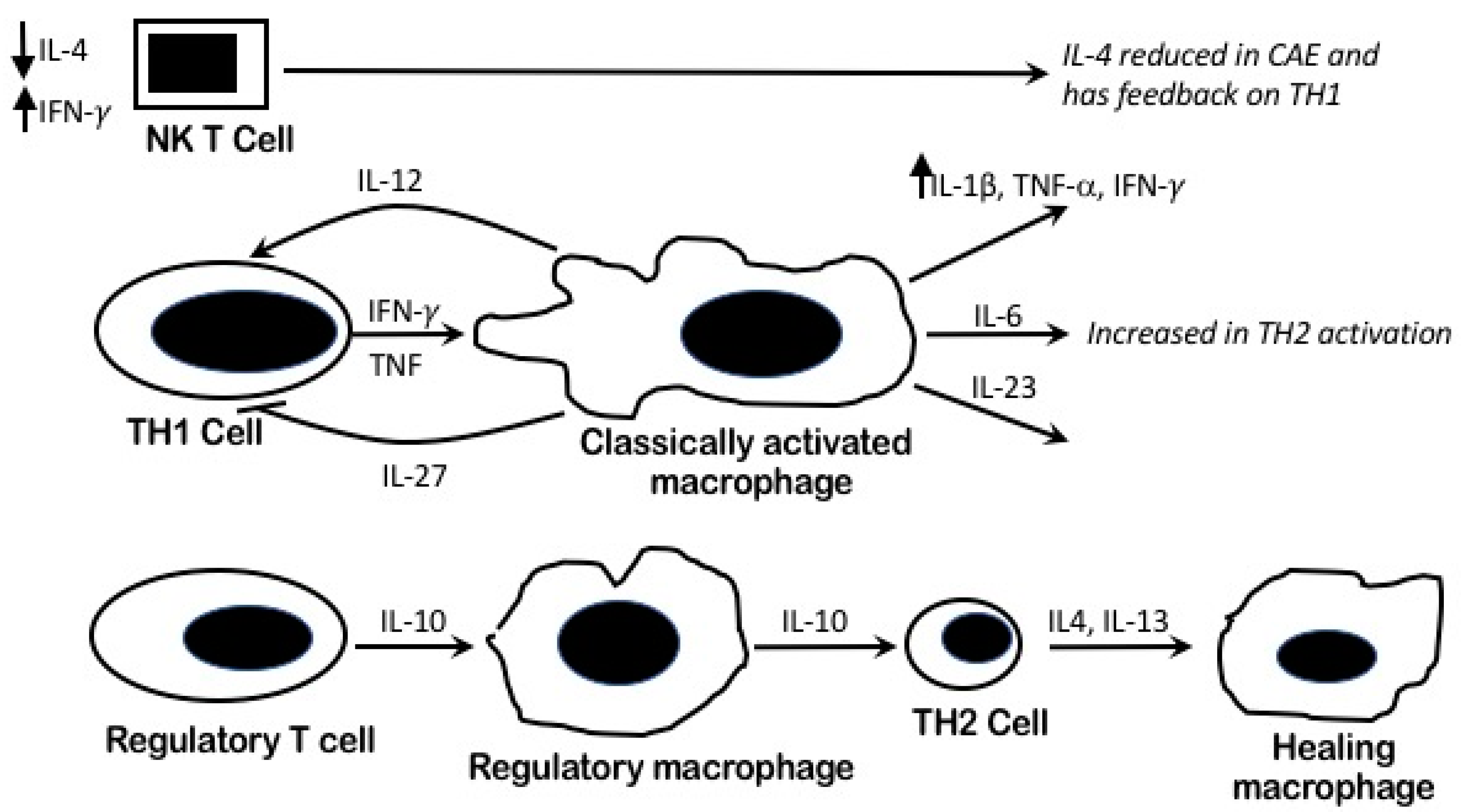
4.4. Pro Inflammatory Pathophysiology and Macrophages Heterogeneity in CAE
4.5. IL-4/TH2 Role in CAE
4.6. Perturbed IL-2 Systemic Levels in CAE
4.7. The Immuno-Inflammatory Role of CAE (TNF-α and IFN-γ) on Vascular Remodeling
4.8. Pure vs. Mixed CAE Cytokines Profiling
4.9. Study Limitations
4.10. Methodological Strength
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Boles, U.; Eriksson, P.; Zhao, Y.; Henein, M.Y. Coronary artery ectasia: Remains a clinical dilemma. Coron. Artery Dis. 2010, 21, 318–320. [Google Scholar] [CrossRef] [PubMed]
- Endoh, S.; Andoh, H.; Sonoyama, K.; Furuse, Y.; Ohtahara, A.; Kasahara, T. Clinical features of coronary artery ectasia. J. Cardiol. 2004, 43, 45–52. [Google Scholar] [PubMed]
- Eitan, A.; Roguin, A. Coronary artery ectasia: New insights into pathophysiology, diagnosis, and treatment. Coron. Artery Dis. 2016, 27, 420–428. [Google Scholar] [CrossRef] [PubMed]
- Yetkin, E.; Waltenberger, J. Novel insights into an old controversy: Is coronary artery ectasia a variant of coronary atherosclerosis? Clin. Res. Cardiol. 2007, 96, 331–339. [Google Scholar] [CrossRef] [PubMed]
- Boles, U.; Pinto, R.C.; David, S.; Abdullah, A.S.; Henein, M.Y. Dysregulated fatty acid metabolism in coronary ectasia: An extended lipidomic analysis. Int. J. Cardiol. 2017, 228, 303–308. [Google Scholar] [CrossRef] [PubMed]
- Hartnell, G.G.; Parnell, B.M.; Pridie, R.B. Coronary artery ectasia. Its prevalence and clinical significance in 4993 patients. Br. Heart J. 1985, 54, 392–395. [Google Scholar] [CrossRef] [PubMed]
- Kornowski, R.; Mintz, G.S.; Lansky, A.J.; Hong, M.K.; Kent, K.M.; Pichard, A.D.; Pichard, A.D.; Satler, L.F.; Popma, J.J.; Bucher, T.A.; et al. Paradoxic decreases in atherosclerotic plaque mass in insulin-treated diabetic patients. Am. J. Cardiol. 1998, 81, 1298–1304. [Google Scholar] [CrossRef]
- Walford, G.D.; Midei, M.G.; Aversano, T.R.; Gottlieb, S.O.; Chew, P.H.; Brinker, J.A. Coronary artery aneurysm formation following percutaneous transluminal coronary angioplasty: Treatment of associated restenosis with repeat percutaneous transluminal coronary angioplasty. Cathether. Cardiovasc. Interv. 1990, 20, 77–83. [Google Scholar] [CrossRef]
- Bal, E.T.; Thijs Plokker, H.W.; van den Berg, E.M.; Ernst, S.M.; Gijs Mast, E.; Gin, R.M.; Ascoop, C.A. Predictability and prognosis of PTCA-induced coronary artery aneurysms. Cathether. Cardiovasc. Interv. 1991, 22, 85–88. [Google Scholar] [CrossRef]
- Malik, I.S.; Harare, O.; AL-Nahhas, A.; Beatt, K.; Mason, J. Takayasu’s arteritis: Management of left main stem stenosis. Heart 2003, 89, e9. [Google Scholar] [CrossRef] [PubMed]
- Ross, R. Atherosclerosis—An inflammatory disease. N. Engl. J. Med. 1999, 340, 115–126. [Google Scholar] [CrossRef] [PubMed]
- Ross, R.; Glomset, J.A. The pathogenesis of atherosclerosis (second of two parts). N. Engl. J. Med. 1976, 295, 420–425. [Google Scholar] [CrossRef] [PubMed]
- Ross, R.; Glomset, J.A. The pathogenesis of atherosclerosis (first of two parts). N. Engl. J. Med. 1976, 295, 369–377. [Google Scholar] [CrossRef] [PubMed]
- Ross, R.; Glomset, J.; Harker, L. Response to injury and atherogenesis. Am. J. Pathol. 1977, 86, 675–684. [Google Scholar] [PubMed]
- Zhou, X.; Nicoletti, A.; Elhage, R.; Hansson, G.K. Transfer of CD4(+) T cells aggravates atherosclerosis in immunodeficient apolipoprotein E knockout mice. Circulation 2000, 102, 2919–2922. [Google Scholar] [CrossRef] [PubMed]
- Stoger, J.L.; Goossens, P.; de Winther, M.P. Macrophage heterogeneity: Relevance and functional implications in atherosclerosis. Curr. Vasc. Pharmacol. 2010, 8, 233–248. [Google Scholar] [CrossRef] [PubMed]
- Berman, R.M.; Suzuki, T.; Tahara, H.; Robbins, P.D.; Narula, S.K.; Lotze, M.T. Systemic administration of cellular IL-10 induces an effective, specific, and long-lived immune response against established tumors in mice. J. Immunol. 1996, 157, 231–238. [Google Scholar] [PubMed]
- Bogdan, C.; Vodovotz, Y.; Nathan, C. Macrophage deactivation by interleukin-10. J. Exp. Med. 1991, 174, 1549–1555. [Google Scholar] [CrossRef] [PubMed]
- Heeschen, C.; Dimmeler, S.; Hamm, C.W.; Fichtlscherer, S.; Boersma, E.; Simoons, M.L.; Zeiher, A.M. Serum level of the antiinflammatory cytokine interleukin-10 is an important prognostic determinant in patients with acute coronary syndromes. Circulation 2003, 107, 2109–2114. [Google Scholar] [CrossRef] [PubMed]
- King, V.L.; Szilvassy, S.J.; Daugherty, A. Interleukin-4 deficiency decreases atherosclerotic lesion formation in a site-specific manner in female LDL receptor−/− mice. Arterioscler. Thromb. Vasc. Biol. 2002, 22, 456–461. [Google Scholar] [CrossRef] [PubMed]
- Simonet, W.S.; Hughes, T.M.; Nguyen, H.Q.; Trebasky, L.D.; Danilenko, D.M.; Medlock, E.S. Long-term impaired neutrophil migration in mice overexpressing human interleukin-8. J. Clin. Investig. 1994, 94, 1310–1319. [Google Scholar] [CrossRef] [PubMed]
- Hechtman, D.H.; Cybulsky, M.I.; Fuchs, H.J.; Baker, J.B.; Gimbrone, M.A., Jr. Intravascular IL-8. Inhibitor of polymorphonuclear leukocyte accumulation at sites of acute inflammation. J. Immunol. 1991, 147, 883–892. [Google Scholar] [PubMed]
- Boles, U.; Zhao, Y.; David, S.; Eriksson, P.; Henein, M.Y. Pure coronary ectasia differs from atherosclerosis: Morphological and risk factors analysis. Int. J. Cardiol. 2012, 155, 321–323. [Google Scholar] [CrossRef] [PubMed]
- Nicoll, R.; Wiklund, U.; Zhao, Y.; Diederichsen, A.; Mickley, H.; Ovrehus, K.; Zamorano, P.; Gueret, P.; Schmermund, A.; Maffei, E.; et al. The coronary calcium score is a more accurate predictor of significant coronary stenosis than conventional risk factors in symptomatic patients: Euro-CCAD study. Int. J. Cardiol. 2016, 207, 13–19. [Google Scholar] [CrossRef] [PubMed]
- Kocaman, S.A.; Tacoy, G.; Sahinarslan, A.; Cengel, A. Relationship between total and differential leukocyte counts and isolated coronary artery ectasia. Coron. Artery Dis. 2008, 19, 307–310. [Google Scholar] [CrossRef] [PubMed]
- Tedgui, A.; Mallat, Z. Cytokines in atherosclerosis: Pathogenic and regulatory pathways. Physiol. Rev. 2006, 86, 515–581. [Google Scholar] [CrossRef] [PubMed]
- Triantafyllis, A.S.; Kalogeropoulos, A.S.; Rigopoulos, A.G.; Sakadakis, E.A.; Toumpoulis, I.K.; Tsikrikas, S.; Kremastinos, D.T.; Rizos, I. Coronary artery ectasia and inflammatory cytokines: Link with a predominant Th-2 immune response? Cytokine 2013, 64, 427–432. [Google Scholar] [CrossRef] [PubMed]
- Adiloglu, A.K.; Ocal, A.; Tas, T.; Onal, S.; Kapan, S.; Aridogan, B. Increased expression of CD11a and CD45 on leukocytes and decreased serum TNF-alpha levels in patients with isolated coronary artery ectasia. Clin. Lab. 2011, 57, 703–709. [Google Scholar] [PubMed]
- Ait-Oufella, H.; Taleb, S.; Mallat, Z.; Tedgui, A. Recent advances on the role of cytokines in atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 969–979. [Google Scholar] [CrossRef] [PubMed]
- Elhage, R.; Clamens, S.; Besnard, S.; Mallat, Z.; Tedgui, A.; Arnal, J.; Maret, A.; Bayard, F. Involvement of interleukin-6 in atherosclerosis but not in the prevention of fatty streak formation by 17beta-estradiol in apolipoprotein E-deficient mice. Atherosclerosis 2001, 156, 315–320. [Google Scholar] [CrossRef]
- Xing, Z.; Gauldie, J.; Cox, G.; Baumann, H.; Jordana, M.; Lei, X.F.; Achong, M.K. IL-6 is an antiinflammatory cytokine required for controlling local or systemic acute inflammatory responses. J. Clin. Investig. 1998, 101, 311–320. [Google Scholar] [CrossRef] [PubMed]
- Liao, H.S.; Matsumoto, A.; Itakura, H.; Doi, T.; Honda, M.; Kodama, T.; Geng, Y.J. Transcriptional inhibition by interleukin-6 of the class A macrophage scavenger receptor in macrophages derived from human peripheral monocytes and the THP-1 monocytic cell line. Arterioscler. Thromb. Vasc. Biol. 1999, 19, 1872–1880. [Google Scholar] [CrossRef] [PubMed]
- El Bakry, S.A.; Fayez, D.; Morad, C.S.; Abdel-Salam, A.M.; Abdel-Salam, Z.; ElKabarity, R.H.; Al Hussein, M. Ischemic heart disease and rheumatoid arthritis: Do inflammatory cytokines have a role? Cytokine 2017, 96, 228–233. [Google Scholar] [CrossRef] [PubMed]
- Wilson, H.M. Macrophages heterogeneity in atherosclerosis—Implications for therapy. J. Cell. Mol. Med. 2010, 14, 2055–2065. [Google Scholar] [CrossRef] [PubMed]
- Mosser, D.M.; Edwards, J.P. Exploring the full spectrum of macrophage activation. Nat. Rev. Immunol. 2008, 8, 958–969. [Google Scholar] [CrossRef] [PubMed]
- Sherry, B.; Horii, Y.; Manogue, K.R.; Widmer, U.; Cerami, A. Macrophage inflammatory proteins 1 and 2: An overview. Cytokines 1992, 4, 117–130. [Google Scholar] [PubMed]
- Caligiuri, G.; Liuzzo, G.; Biasucci, L.M.; Maseri, A. Immune system activation follows inflammation in unstable angina: Pathogenetic implications. J. Am. Coll. Cardiol. 1998, 32, 1295–1304. [Google Scholar] [CrossRef]
- Caligiuri, G.; Paulsson, G.; Nicoletti, A.; Maseri, A.; Hansson, G.K. Evidence for antigen-driven T-cell response in unstable angina. Circulation 2000, 102, 1114–1119. [Google Scholar] [CrossRef] [PubMed]
- Simon, A.D.; Yazdani, S.; Wang, W.; Schwartz, A.; Rabbani, L.E. Elevated plasma levels of interleukin-2 and soluble IL-2 receptor in ischemic heart disease. Clin. Cardiol. 2001, 24, 253–256. [Google Scholar] [CrossRef] [PubMed]
- Liuzzo, G.; Kopecky, S.L.; Frye, R.L.; O’Fallon, W.M.; Maseri, A.; Goronzy, J.J.; Weyand, C.M. Perturbation of the T-cell repertoire in patients with unstable angina. Circulation 1999, 100, 2135–2139. [Google Scholar] [CrossRef] [PubMed]
- Antoniadis, A.P.; Chatzizisis, Y.S.; Giannoglou, G.D. Pathogenetic mechanisms of coronary ectasia. Int. J. Cardiol. 2008, 130, 335–343. [Google Scholar] [CrossRef] [PubMed]
- Li, J.J.; Li, Z.; Li, J. Is any link between inflammation and coronary artery ectasia? Med. Hypotheses 2007, 69, 678–683. [Google Scholar] [CrossRef] [PubMed]
- Finkelstein, A.; Michowitz, Y.; Abashidze, A.; Miller, H.; Keren, G.; George, J. Temporal association between circulating proteolytic, inflammatory and neurohormonal markers in patients with coronary ectasia. Atherosclerosis 2005, 179, 353–359. [Google Scholar] [CrossRef] [PubMed]
- Savino, M.; Parisi, Q.; Biondi-Zoccai, G.G.; Pristipino, C.; Cianflone, D.; Crea, F. New insights into molecular mechanisms of diffuse coronary ectasiae: A possible role for VEGF. Int. J. Cardiol. 2006, 106, 307–312. [Google Scholar] [CrossRef] [PubMed]
- Weber, C.; Zernecke, A.; Libby, P. The multifaceted contributions of leukocyte subsets to atherosclerosis: Lessons from mouse models. Nat. Rev. Immunol. 2008, 8, 802–815. [Google Scholar] [CrossRef] [PubMed]
- AC, N. The Th2-type cytokine IL-4 inhibits the production of most MMPs from macrophages. Arterioscler. Thromb. Vasc. Biol. 2008, 28, 2108–2114. [Google Scholar]


{kind=link}
{kind=link}
| CAE Patients (n = 16) | Controls (n = 140) | p-Value CAE vs. Control | CAD Patients (n = 69) | p-Value CAE vs. CAD | |
|---|---|---|---|---|---|
| Gender (Female, %) | 6 (38%) | 81 (58%) | NS | 41 (59%) | NS |
| Age (mean ± SD) years | 64.9 ± 7.3 | 58.6 ± 4.1 | <0.011 | 64.5 ± 8.7 | NS |
| Hypertension (N, %) | 9 (56.2%) | 34 (25%) | <0.01 | 49 (71%) | NS |
| Diabetes Mellitus (N, %) | 4 (25%) | 22 (15.7%) | NS | 11 (15.9%) | NS |
| Hyperlipidaemia (N, %) | 9 (56%) | 18 (25%) | <0.001 | 51 (74%) | NS |
| BMI (mean ± SD) | 25.8 ± 4.5 | 27 ± 4.4 | NS | 27.2 ± 5.6 | NS |
| Family history of IHD (N, %) | 7 (43%) | 57 (40.7%) | NS | 48 (69.6%) | NS |
| Smoking (N, %) | 7 (43%) | 47 (33.8%) | NS | 40 (58%) | NS |
| Cytokine | Controls (n = 140) | CAE Patients (n = 16) | p-Value CAE vs. Control | CAD (n = 69) | p-Value CAD vs. CAE | p-Value Kruskal-Wallis |
|---|---|---|---|---|---|---|
| I-Equal Levels (median and interquartile) | ||||||
| IL-10 (pg./mL) | 0.25 (0.15) | 0.26 (0.11) | - | 0.29 (0.21) | - | 0.07 |
| IL-12p (sub-unit) IL-12 and IL-23 (pg./mL) | 0.09 (0.11) | 0.08 (0.08) | - | 0.09 (0.09) | - | 0.41 |
| IL-13 (pg./mL) | 0.26 (0.60) | 0.14 (0.44) | - | 0.28 (0.66) | - | 0.46 |
| II-Reduced levels (median and interquartile) | ||||||
| IL-2 (pg./mL) | 0.25 (0.08) | 0.12 (0.05) | <0.001 | 0.26 (0.11) | <0.001 | <0.001 |
| IL-4 (pg./mL) | 0.04 (0.03) | 0.004 (0.006) | <0.001 | 0.04 (0.03) | <0.001 | <0.001 |
| III-Increased levels (median and interquartile) | ||||||
| IL-6 (pg./mL) | 0.64 (0.44) | 0.98 (0.60) | 0.049 | 0.73 (0.48) | 0.25 | 0.046 |
| IL-8 (pg./mL) | 3.62 (2.18) | 5.59 (3.65) | 0.007 | 3.82 (2.30) | 0.023 | 0.009 |
| IFN-γ (pg./mL) | 3.88 (2.55) | 5.45 (3.33) | 0.032 | 4.72 (4.58) | 0.74 | 0.006 |
| IL-1β (pg./mL) | 0.001 (0.01) | 0.17 (0.24) | <0.001 | 0.00 (0.05) | <0.001 | <0.001 |
| TNF-α (pg./mL) | 1.82 (0.71) | 2.37 (0.74) | 0.002 | 1.96 (0.66) | <0.12 | <0.001 |
| Cytokines | Mixed Ectasia (n = 10) | Pure Ectasia (n = 6) | p-Value |
|---|---|---|---|
| IL-10 (pg./mL) | 0.27 (0.12) | 0.25 (0.19) | 0.79 |
| IL-12p (sub-unit IL-12) and IL-23 (pg./mL) | 0.07 (0.07) | 0.11 (0.12) | 0.64 |
| IL-13 (pg./mL) | 0.18 (0.57) | 0.14 (0.37) | 0.71 |
| IL-2 (pg./mL) | 0.15 (0.07) | 0.12 (0.03) | 0.15 |
| IL-4 (pg./mL) | 0.004 (0.004) | 0.007 (0.012) | 0.06 |
| IL-6 (pg./mL) | 0.99 (1.06) | 0.94 (0.51) | 0.49 |
| IL-8 (pg./mL) | 5.67 (3.49) | 4.79 (3.67) | 0.37 |
| IFN-γ (pg./mL) | 5.39 (3.10) | 5.96 (6.25) | 0.26 |
| IL-1β (pg./mL) | 0.22 (0.37) | 0.13 (0.17) | 0.49 |
| TNF-α (pg./mL) | 2.37 (0.73) | 2.22 (0.97) | 0.49 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Boles, U.; Johansson, A.; Wiklund, U.; Sharif, Z.; David, S.; McGrory, S.; Henein, M.Y. Cytokine Disturbances in Coronary Artery Ectasia Do Not Support Atherosclerosis Pathogenesis. Int. J. Mol. Sci. 2018, 19, 260. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms19010260
Boles U, Johansson A, Wiklund U, Sharif Z, David S, McGrory S, Henein MY. Cytokine Disturbances in Coronary Artery Ectasia Do Not Support Atherosclerosis Pathogenesis. International Journal of Molecular Sciences. 2018; 19(1):260. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms19010260
Chicago/Turabian StyleBoles, Usama, Anders Johansson, Urban Wiklund, Zain Sharif, Santhosh David, Siobhan McGrory, and Michael Y. Henein. 2018. "Cytokine Disturbances in Coronary Artery Ectasia Do Not Support Atherosclerosis Pathogenesis" International Journal of Molecular Sciences 19, no. 1: 260. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms19010260