Upregulation of Myocardial and Vascular Phosphodiesterase 9A in A Model of Atherosclerotic Cardiovascular Disease


 , , ,
, , ,
Abstract
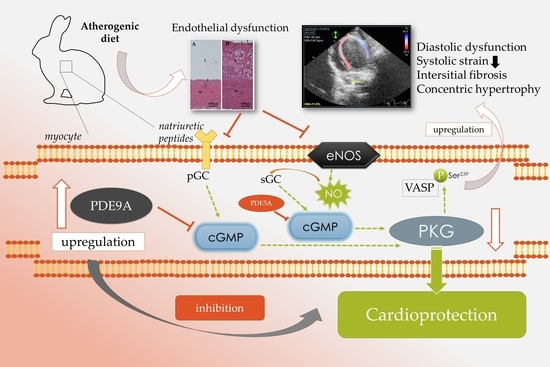
:
1. Introduction
2. Results
2.1. Atherogenic Diet Induces Cardiac Dysfunction
2.2. Increased Left Ventricle Mass and Bodyweight in HC Animals
2.3. Atherogenic Diet Elevates Serum Lipid Parameters
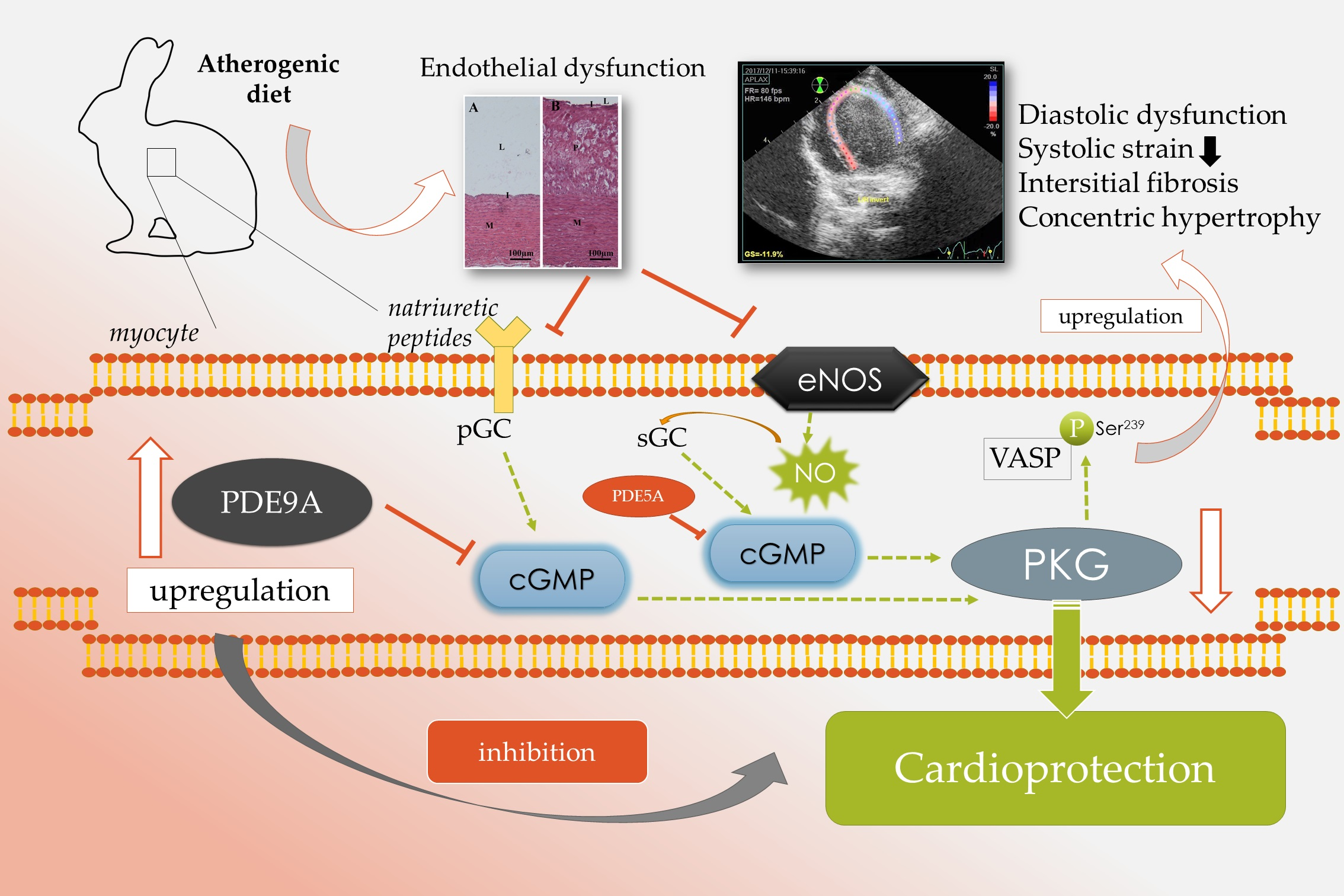
2.4. Atherogenic Diet and Endothelial Dysfunction with Increased Aortic PDE9A Expression
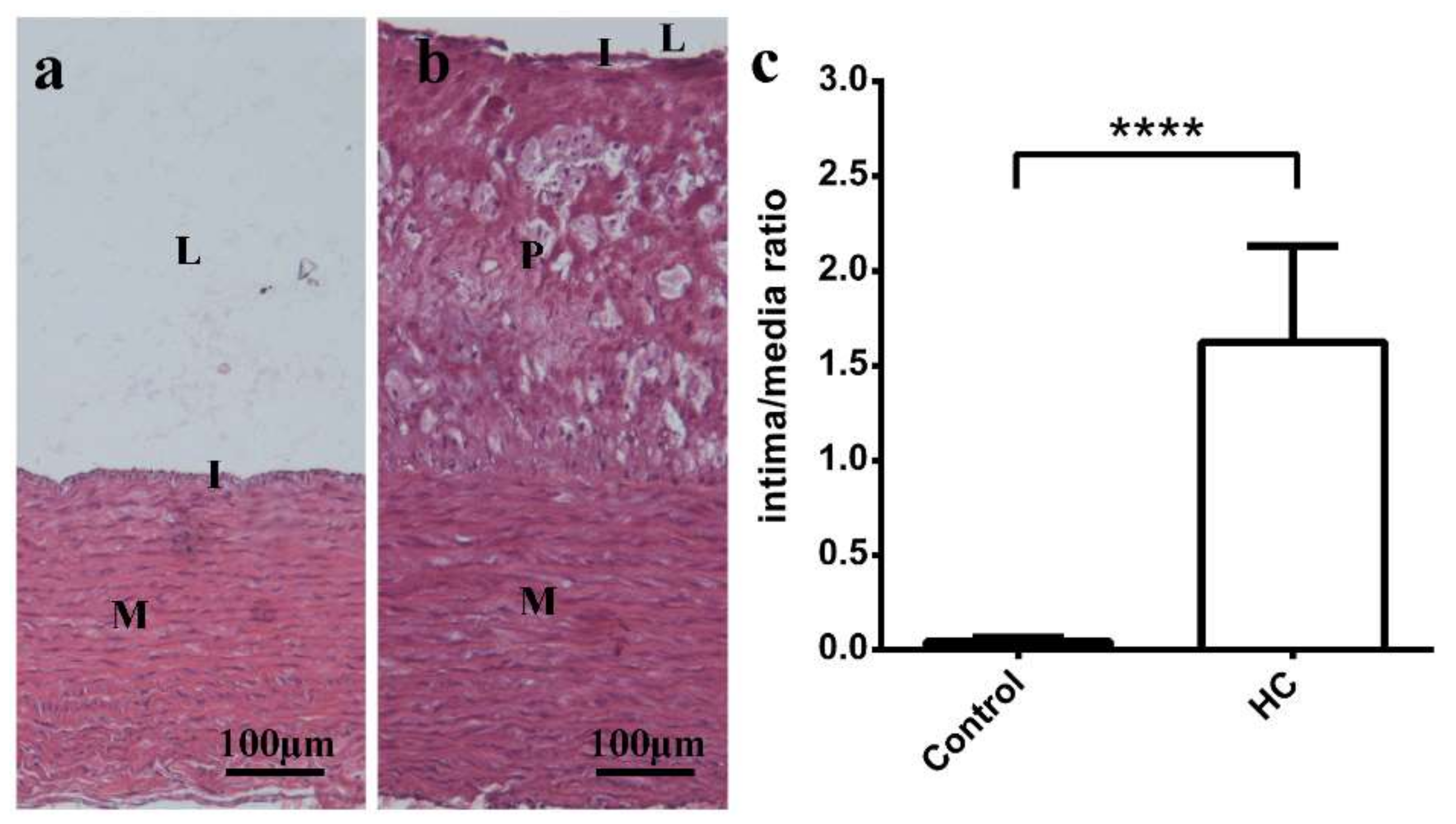
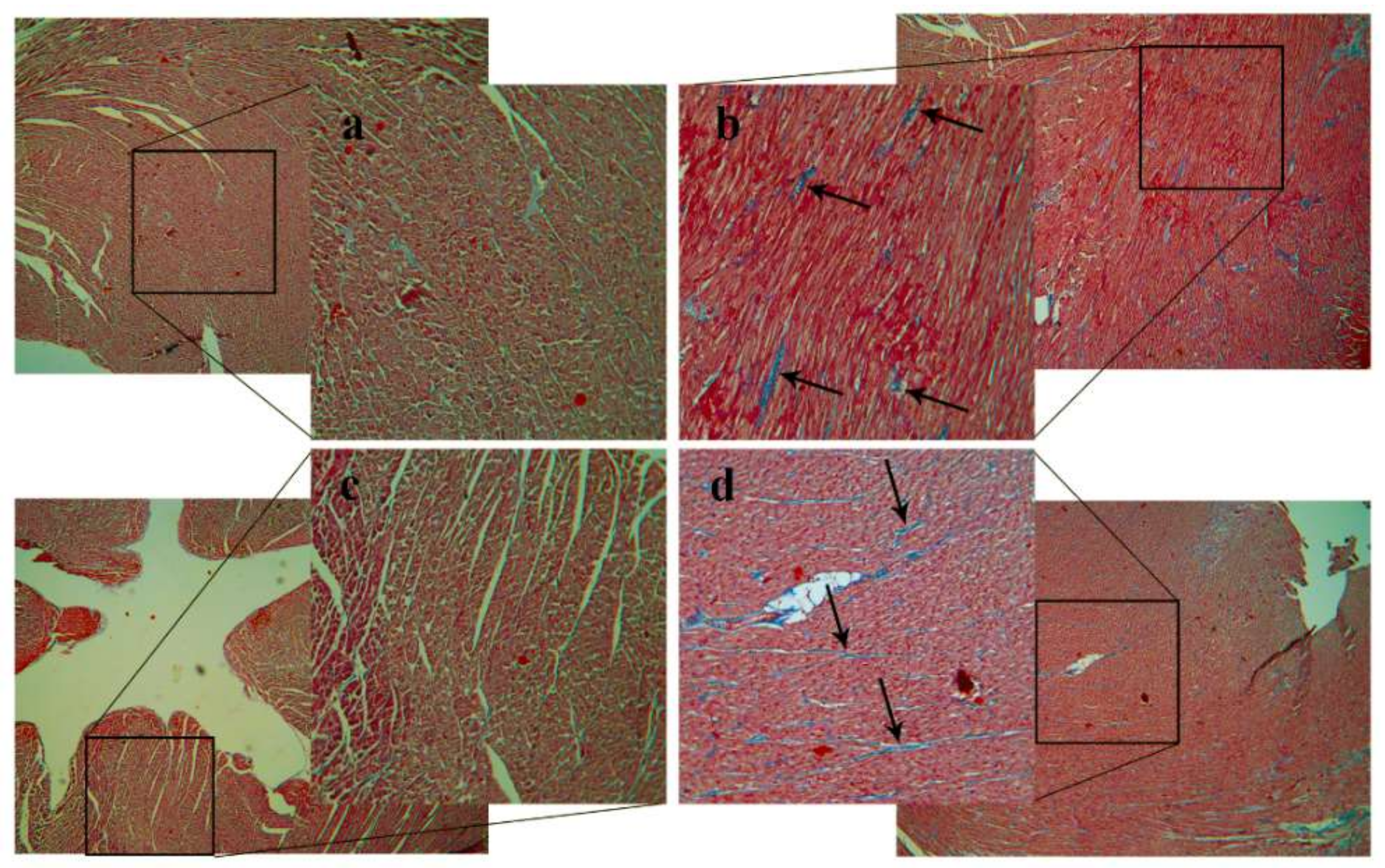
2.5. Plaque Coverage and Interstitial Fibrosis in HC Animals
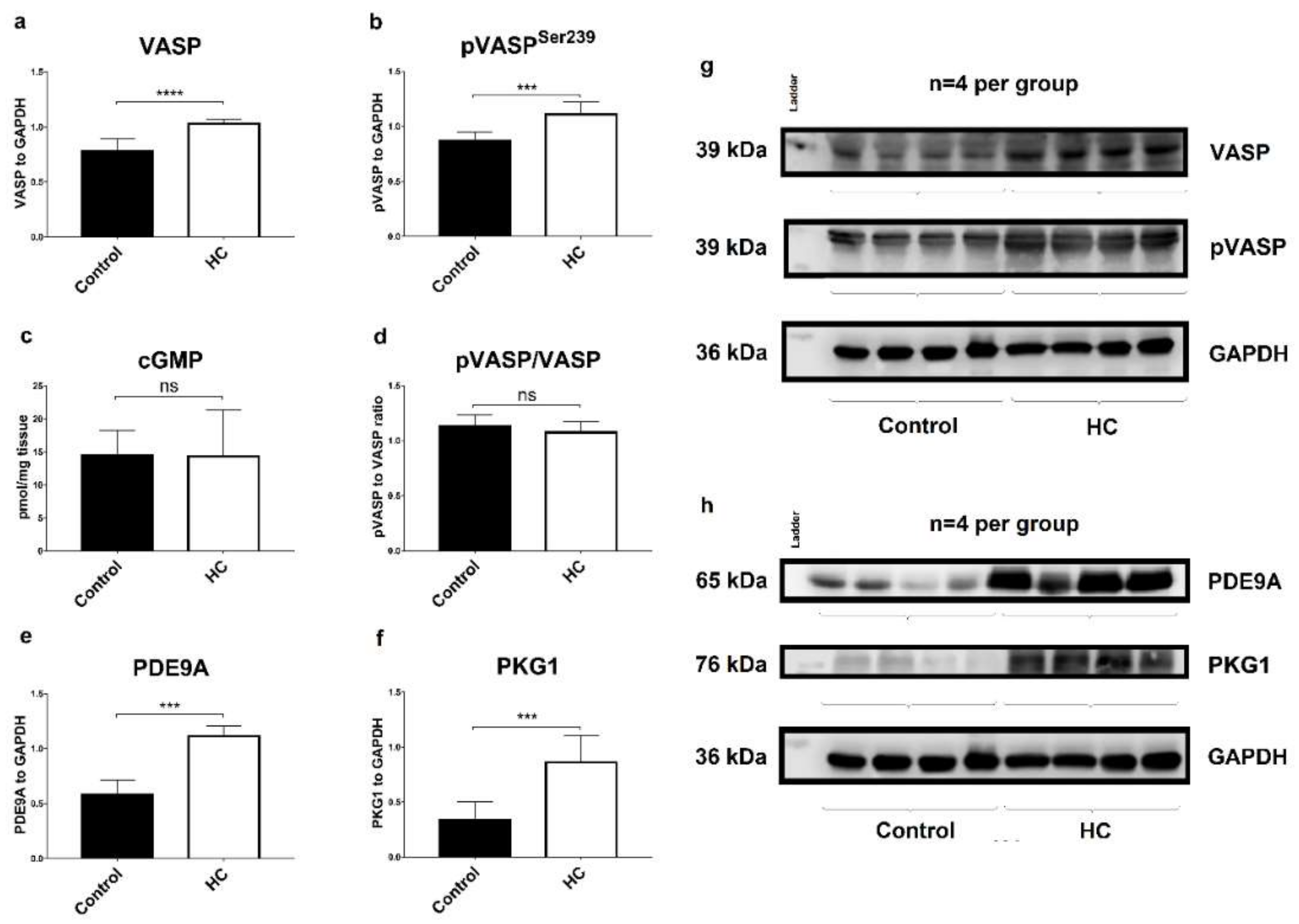
2.6. Atherogenic Diet Correlates with the Upregulation of Myocardial PDE9A and PKG1
3. Discussion
3.1. Cardiac Dysfunction
3.2. Dyslipidemia and Endothelial Dysfunction
3.3. Upregulation of Cardiac PDE9A in Correlation with Cardiac Dysfunction
3.4. Translational Aspects and Future Directions
4. Materials and Methods
4.1. Animal Model
4.2. Chemicals
4.3. Study Design
4.4. Echocardiographic Studies
4.5. Strain (Speckle Tracking) Analyses
4.6. Analysis of Serum Parameters
4.7. Morphometry
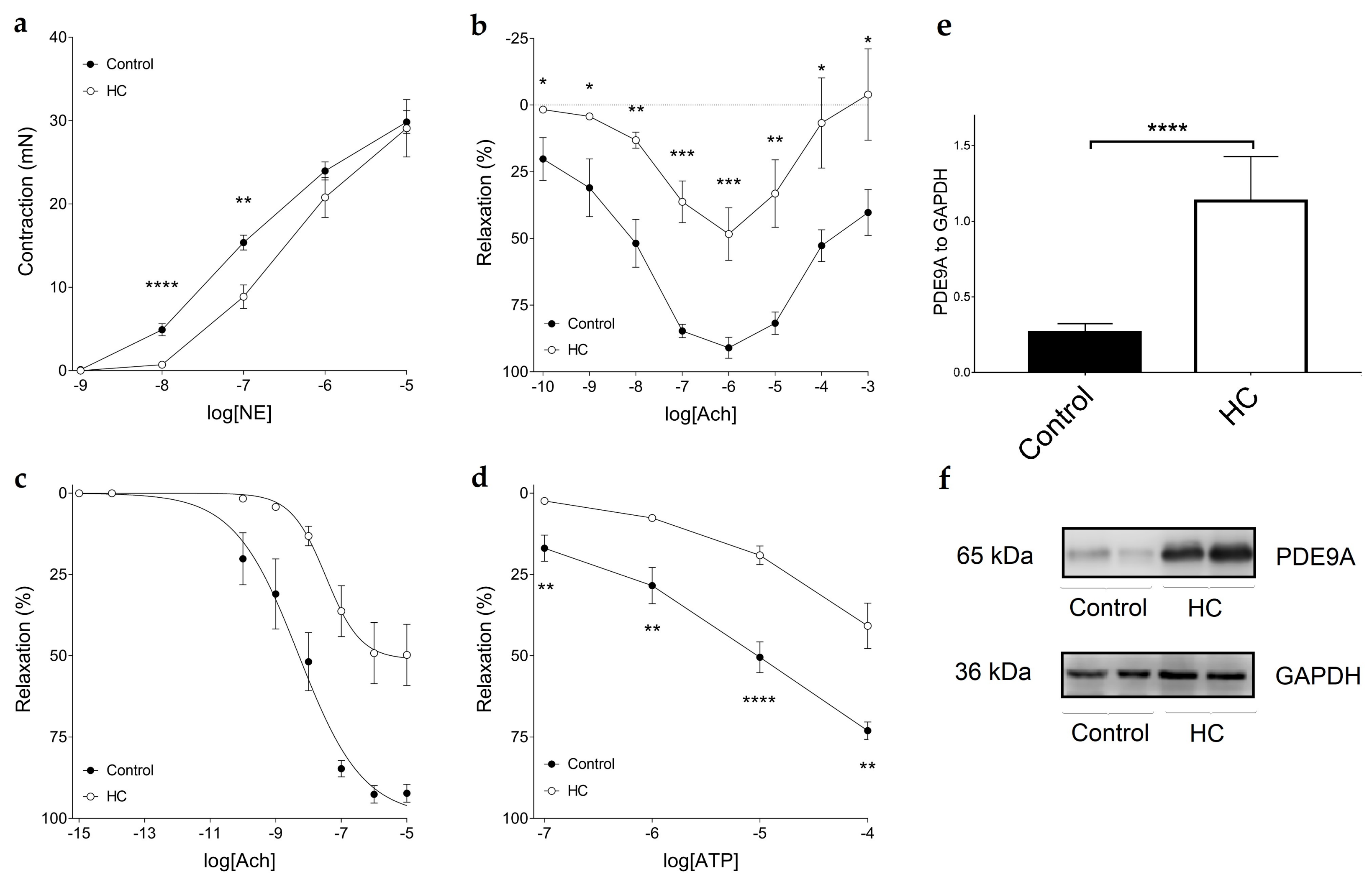
4.8. Functional Vascular Assays
4.9. Histology
4.10. Western Blot
4.11. cGMP Assay
4.12. Statistical Analyses
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ference, B.A.; Ginsberg, H.N.; Graham, I.; Ray, K.K.; Packard, C.J.; Bruckert, E.; Hegele, R.A.; Krauss, R.M.; Raal, F.J.; Schunkert, H.; et al. Low-density lipoproteins cause atherosclerotic cardiovascular disease. 1. Evidence from genetic, epidemiologic, and clinical studies. A consensus statement from the European atherosclerosis society consensus panel. Eur. Heart J. 2017, 38, 2459–2472. [Google Scholar] [CrossRef] [PubMed]
- Velagaleti, R.S.; Massaro, J.; Vasan, R.S.; Robins, S.J.; Kannel, W.B.; Levy, D. Relations of lipid concentrations to heart failure incidence: The framingham heart study. Circulation 2009, 120, 2345–2351. [Google Scholar] [CrossRef] [PubMed]
- Nelson, R.H. Hyperlipidemia as a risk factor for cardiovascular disease. Prim. Care 2013, 40, 195–211. [Google Scholar] [CrossRef] [PubMed]
- van Heerebeek, L.; Paulus, W.J. Understanding heart failure with preserved ejection fraction: Where are we today? Neth. Heart J. 2016, 24, 227–236. [Google Scholar] [CrossRef] [PubMed]
- Nishida, M.; Watanabe, K.; Sato, Y.; Nakaya, M.; Kitajima, N.; Ide, T.; Inoue, R.; Kurose, H. Phosphorylation of TRPC6 channels at Thr69 is required for anti-hypertrophic effects of phosphodiesterase 5 inhibition. J. Biol. Chem. 2010, 285, 13244–13253. [Google Scholar] [CrossRef] [PubMed]
- Fiedler, B.; Lohmann, S.M.; Smolenski, A.; Linnemuller, S.; Pieske, B.; Schroder, F.; Molkentin, J.D.; Drexler, H.; Wollert, K.C. Inhibition of calcineurin-NFAT hypertrophy signaling by cGMP-dependent protein kinase type I in cardiac myocytes. Proc. Natl. Acad. Sci. USA 2002, 99, 11363–11368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Heerebeek, L.; Hamdani, N.; Falcao-Pires, I.; Leite-Moreira, A.F.; Begieneman, M.P.; Bronzwaer, J.G.; van der Velden, J.; Stienen, G.J.; Laarman, G.J.; Somsen, A.; et al. Low myocardial protein kinase G activity in heart failure with preserved ejection fraction. Circulation 2012, 126, 830–839. [Google Scholar] [CrossRef] [PubMed]
- Valero-Munoz, M.; Backman, W.; Sam, F. Murine models of heart failure with preserved ejection fraction: A “fishing expedition”. JACC Basic Transl. Sci. 2017, 2, 770–789. [Google Scholar] [CrossRef] [PubMed]
- Pandey, K.N. Guanylyl cyclase/natriuretic peptide receptor-A signaling antagonizes phosphoinositide hydrolysis, Ca2+ release, and activation of protein kinase C. Front. Mol. Neurosci. 2014, 7, 75. [Google Scholar] [CrossRef] [PubMed]
- Madamanchi, C.; Alhosaini, H.; Sumida, A.; Runge, M.S. Obesity and natriuretic peptides, BNP and NT-proBNP: Mechanisms and diagnostic implications for heart failure. Int. J. Cardiol. 2014, 176, 611–617. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuhn, M. Cardiac actions of atrial natriuretic peptide: New visions of an old friend. Circ. Res. 2015, 116, 1278–1280. [Google Scholar] [CrossRef] [PubMed]
- Chirinos, J.A.; Segers, P.; Gupta, A.K.; Swillens, A.; Rietzschel, E.R.; De Buyzere, M.L.; Kirkpatrick, J.N.; Gillebert, T.C.; Wang, Y.; Keane, M.G.; et al. Time-varying myocardial stress and systolic pressure-stress relationship: Role in myocardial-arterial coupling in hypertension. Circulation 2009, 119, 2798–2807. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.I.; Zhu, G.; Sasaki, T.; Cho, G.S.; Hamdani, N.; Holewinski, R.; Jo, S.H.; Danner, T.; Zhang, M.; Rainer, P.P.; et al. Phosphodiesterase 9A controls nitric-oxide-independent cGMP and hypertrophic heart disease. Nature 2015, 519, 472–476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Solomon, S.D.; Rizkala, A.R.; Gong, J.; Wang, W.; Anand, I.S.; Ge, J.; Lam, C.S.P.; Maggioni, A.P.; Martinez, F.; Packer, M.; et al. Angiotensin receptor neprilysin inhibition in heart failure with preserved ejection fraction: Rationale and design of the PARAGON-HF trial. JACC Heart Fail. 2017, 5, 471–482. [Google Scholar] [CrossRef] [PubMed]
- Hasenfuss, G. Animal models of human cardiovascular disease, heart failure and hypertrophy. Cardiovasc. Res. 1998, 39, 60–76. [Google Scholar] [CrossRef] [Green Version]
- Conceicao, G.; Heinonen, I.; Lourenco, A.P.; Duncker, D.J.; Falcao-Pires, I. Animal models of heart failure with preserved ejection fraction. Neth. Heart J. 2016, 24, 275–286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Milani-Nejad, N.; Janssen, P.M. Small and large animal models in cardiac contraction research: Advantages and disadvantages. Pharmacol. Ther. 2014, 141, 235–249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bers, D.M. Cardiac Na/Ca exchange function in rabbit, mouse and man: What’s the difference? J. Mol. Cell. Cardiol. 2002, 34, 369–373. [Google Scholar] [CrossRef] [PubMed]
- Sanbe, A.; James, J.; Tuzcu, V.; Nas, S.; Martin, L.; Gulick, J.; Osinska, H.; Sakthivel, S.; Klevitsky, R.; Ginsburg, K.S.; et al. Transgenic rabbit model for human troponin I-based hypertrophic cardiomyopathy. Circulation 2005, 111, 2330–2338. [Google Scholar] [CrossRef] [PubMed]
- Fan, J.; Kitajima, S.; Watanabe, T.; Xu, J.; Zhang, J.; Liu, E.; Chen, Y.E. Rabbit models for the study of human atherosclerosis: From pathophysiological mechanisms to translational medicine. Pharmacol. Ther. 2015, 146, 104–119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagueh, S.F.; Smiseth, O.A.; Appleton, C.P.; Byrd, B.F.; Dokainish, H.; Edvardsen, T.; Flachskampf, F.A.; Gillebert, T.C.; Klein, A.L.; Lancellotti, P.; et al. Recommendations for the evaluation of left ventricular diastolic function by echocardiography: An update from the American society of echocardiography and the European association of cardiovascular imaging. J. Am. Soc. Echocardiogr. 2016, 29, 277–314. [Google Scholar] [CrossRef] [PubMed]
- Heinzel, F.R.; Hohendanner, F.; Jin, G.; Sedej, S.; Edelmann, F. Myocardial hypertrophy and its role in heart failure with preserved ejection fraction. J. Appl. Physiol. 2015, 119, 1233–1242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Westermann, D.; Lindner, D.; Kasner, M.; Zietsch, C.; Savvatis, K.; Escher, F.; von Schlippenbach, J.; Skurk, C.; Steendijk, P.; Riad, A.; et al. Cardiac inflammation contributes to changes in the extracellular matrix in patients with heart failure and normal ejection fraction. Circ. Heart Fail. 2011, 4, 44–52. [Google Scholar] [CrossRef] [PubMed]
- Pelosi, A.; St John, L.; Gaymer, J.; Ferguson, D.; Goyal, S.K.; Abela, G.S.; Rubinstein, J. Cardiac tissue Doppler and tissue velocity imaging in anesthetized New Zealand white rabbits. J. Am. Assoc. Lab. Anim. Sci. 2011, 50, 317–321. [Google Scholar] [PubMed]
- Liu, Y.W.; Tsai, W.C.; Su, C.T.; Lin, C.C.; Chen, J.H. Evidence of left ventricular systolic dysfunction detected by automated function imaging in patients with heart failure and preserved left ventricular ejection fraction. J. Card. Fail. 2009, 15, 782–789. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Luo, S.; Luo, J.; Liu, Y.; Huang, W.; Chen, J. Association between abdominal aortic plaque and coronary artery disease. Clin. Interv. Aging 2016, 11, 683–688. [Google Scholar] [CrossRef] [PubMed]
- Shah, R.V.; Chen-Tournoux, A.A.; Picard, M.H.; Januzzi, J.L. Association between troponin T and impaired left ventricular relaxation in patients with acute decompensated heart failure with preserved systolic function. Eur. J. Echocardiogr. 2009, 10, 765–768. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davignon, J.; Ganz, P. Role of endothelial dysfunction in atherosclerosis. Circulation 2004, 109, III27–III32. [Google Scholar] [CrossRef] [PubMed]
- Buchwalow, I.B.; Cacanyiova, S.; Neumann, J.; Samoilova, V.E.; Boecker, W.; Kristek, F. The role of arterial smooth muscle in vasorelaxation. Biochem. Biophys. Res. Commun. 2008, 377, 504–507. [Google Scholar] [CrossRef] [PubMed]
- Paulus, W.J.; Tschope, C. A novel paradigm for heart failure with preserved ejection fraction: Comorbidities drive myocardial dysfunction and remodeling through coronary microvascular endothelial inflammation. J. Am. Coll. Cardiol. 2013, 62, 263–271. [Google Scholar] [CrossRef] [PubMed]
- Altara, R.; Giordano, M.; Nordén, E.S.; Cataliotti, A.; Kurdi, M.; Bajestani, S.N.; Booz, G.W. Targeting obesity and diabetes to treat heart failure with preserved ejection fraction. Front. Endocrinol. 2017, 8, 160. [Google Scholar] [CrossRef] [PubMed]
- Eren, M.; Gorgulu, S.; Uslu, N.; Celik, S.; Dagdeviren, B.; Tezel, T. Relation between aortic stiffness and left ventricular diastolic function in patients with hypertension, diabetes, or both. Heart 2004, 90, 37–43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- deAlmeida, A.C.; van Oort, R.J.; Wehrens, X.H. Transverse aortic constriction in mice. J. Vis. Exp. 2010, 1729. [Google Scholar] [CrossRef] [PubMed]
- Hirt, M.N.; Sorensen, N.A.; Bartholdt, L.M.; Boeddinghaus, J.; Schaaf, S.; Eder, A.; Vollert, I.; Stohr, A.; Schulze, T.; Witten, A.; et al. Increased afterload induces pathological cardiac hypertrophy: A new in vitro model. Basic Res. Cardiol. 2012, 107, 307. [Google Scholar] [CrossRef] [PubMed]
- Rainer, P.P.; Kass, D.A. Old dog, new tricks: Novel cardiac targets and stress regulation by protein kinase G. Cardiovasc. Res. 2016, 111, 154–162. [Google Scholar] [CrossRef] [PubMed]
- Hoendermis, E.S.; Liu, L.C.; Hummel, Y.M.; van der Meer, P.; de Boer, R.A.; Berger, R.M.; van Veldhuisen, D.J.; Voors, A.A. Effects of sildenafil on invasive haemodynamics and exercise capacity in heart failure patients with preserved ejection fraction and pulmonary hypertension: A randomized controlled trial. Eur. Heart J. 2015, 36, 2565–2573. [Google Scholar] [CrossRef] [PubMed]
- Redfield, M.M.; Chen, H.H.; Borlaug, B.A.; Semigran, M.J.; Lee, K.L.; Lewis, G.; LeWinter, M.M.; Rouleau, J.L.; Bull, D.A.; Mann, D.L.; et al. Effect of phosphodiesterase-5 inhibition on exercise capacity and clinical status in heart failure with preserved ejection fraction: a randomized clinical trial. JAMA 2013, 309, 1268–1277. [Google Scholar] [CrossRef] [PubMed]
- Shah, S.J.; Kitzman, D.W.; Borlaug, B.A.; van Heerebeek, L.; Zile, M.R.; Kass, D.A.; Paulus, W.J. Phenotype-specific treatment of heart failure with preserved ejection fraction: A multiorgan roadmap. Circulation 2016, 134, 73–90. [Google Scholar] [CrossRef] [PubMed]
- Sartoretto, J.L.; Jin, B.Y.; Bauer, M.; Gertler, F.B.; Liao, R.; Michel, T. Regulation of VASP phosphorylation in cardiac myocytes: Differential regulation by cyclic nucleotides and modulation of protein expression in diabetic and hypertrophic heart. Am. J. Physiol. Heart Circ. Physiol. 2009, 297, H1697–H1710. [Google Scholar] [CrossRef] [PubMed]
- Benz, P.M.; Merkel, C.J.; Offner, K.; Abesser, M.; Ullrich, M.; Fischer, T.; Bayer, B.; Wagner, H.; Gambaryan, S.; Ursitti, J.A.; et al. Mena/VASP and αII-Spectrin complexes regulate cytoplasmic actin networks in cardiomyocytes and protect from conduction abnormalities and dilated cardiomyopathy. Cell Commun. Signal. 2013, 11, 56. [Google Scholar] [CrossRef] [PubMed]
- Smolenski, A.; Bachmann, C.; Reinhard, K.; Honig-Liedl, P.; Jarchau, T.; Hoschuetzky, H.; Walter, U. Analysis and regulation of vasodilator-stimulated phosphoprotein serine 239 phosphorylation in vitro and in intact cells using a phosphospecific monoclonal antibody. J. Biol. Chem. 1998, 273, 20029–20035. [Google Scholar] [CrossRef] [PubMed]
- Boerrigter, G.; Lapp, H.; Burnett, J.C. Modulation of cGMP in heart failure: A new therapeutic paradigm. Handb. Exp. Pharmacol. 2009, 485–506. [Google Scholar] [CrossRef]
- Inserte, J.; Garcia-Dorado, D. The cGMP/PKG pathway as a common mediator of cardioprotection: Translatability and mechanism. Br. J. Pharmacol. 2015, 172, 1996–2009. [Google Scholar] [CrossRef] [PubMed]
- Szilvassy, Z.; Ferdinandy, P.; Bor, P.; Jakab, I.; Szilvassy, J.; Nagy, I.; Lonovics, J.; Koltai, M. Loss of preconditioning in rabbits with vascular tolerance to nitroglycerin. Br. J. Pharmacol. 1994, 112, 999–1001. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Komajda, M.; Lam, C.S. Heart failure with preserved ejection fraction: A clinical dilemma. Eur. Heart J. 2014, 35, 1022–1032. [Google Scholar] [CrossRef] [PubMed]
- Kertesz, A.; Bombicz, M.; Priksz, D.; Balla, J.; Balla, G.; Gesztelyi, R.; Varga, B.; Haines, D.D.; Tosaki, A.; Juhasz, B. Adverse impact of diet-induced hypercholesterolemia on cardiovascular tissue homeostasis in a rabbit model: Time-dependent changes in cardiac parameters. Int. J. Mol. Sci. 2013, 14, 19086–19108. [Google Scholar] [CrossRef] [PubMed]
- Ponikowski, P.; Voors, A.A.; Anker, S.D.; Bueno, H.; Cleland, J.G.F.; Coats, A.J.S.; Falk, V.; Gonzalez-Juanatey, J.R.; Harjola, V.P.; Jankowska, E.A.; et al. 2016 ESC guidelines for the diagnosis and treatment of acute and chronic heart failure: The task force for the diagnosis and treatment of acute and chronic heart failure of the European Society of Cardiology (ESC) developed with the special contribution of the Heart Failure Association (HFA) of the ESC. Eur. Heart J. 2016, 37, 2129–2200. [Google Scholar] [PubMed]
- Maj, D.; Bieniek, J.; Łapa, P.; Sternstein, I. The effect of crossing New Zealand White with Californian rabbits on growth and slaughter traits. Arch. Anim. Breed. 2009, 52, 205–211. [Google Scholar] [CrossRef] [Green Version]
- Bombicz, M.; Priksz, D.; Varga, B.; Gesztelyi, R.; Kertesz, A.; Lengyel, P.; Balogh, P.; Csupor, D.; Hohmann, J.; Bhattoa, H.P.; et al. Anti-atherogenic properties of allium ursinum liophylisate: Impact on lipoprotein homeostasis and cardiac biomarkers in hypercholesterolemic rabbits. Int. J. Mol. Sci. 2016, 17, 1284. [Google Scholar] [CrossRef] [PubMed]
- Lang, R.M.; Badano, L.P.; Mor-Avi, V.; Afilalo, J.; Armstrong, A.; Ernande, L.; Flachskampf, F.A.; Foster, E.; Goldstein, S.A.; Kuznetsova, T.; et al. Recommendations for cardiac chamber quantification by echocardiography in adults: An update from the American society of echocardiography and the European association of cardiovascular imaging. Eur. Heart J. 2015, 16, 233–271. [Google Scholar]
- Smiseth, O.A.; Torp, H.; Opdahl, A.; Haugaa, K.H.; Urheim, S. Myocardial strain imaging: How useful is it in clinical decision making? Eur. Heart J. 2016, 37, 1196–1207. [Google Scholar] [CrossRef] [PubMed]
- Szabó, K.; Gesztelyi, R.; Lampé, N.; Kiss, R.; Remenyik, J.; Pesti-Asbóth, G.; Priksz, D.; Szilvássy, Z.; Juhász, B. Fenugreek (Trigonella foenum-graecum) seed flour and diosgenin preserve endothelium-dependent arterial relaxation in a rat model of early-stage metabolic syndrome. Int. J. Mol. Sci. 2018, 19, 798. [Google Scholar] [CrossRef] [PubMed]
- Gesztelyi, R.; Zsuga, J.; Kemeny-Beke, A.; Varga, B.; Juhasz, B.; Tosaki, A. The Hill equation and the origin of quantitative pharmacology. Arch. Hist. Exact Sci. 2012, 66, 427–438. [Google Scholar] [CrossRef]
- Bombicz, M.; Priksz, D.; Varga, B.; Kurucz, A.; Kertesz, A.; Takacs, A.; Posa, A.; Kiss, R.; Szilvassy, Z.; Juhasz, B. A novel therapeutic approach in the treatment of pulmonary arterial hypertension: Allium ursinum liophylisate alleviates symptoms comparably to sildenafil. Int. J. Mol. Sci. 2017, 18, 1436. [Google Scholar] [CrossRef] [PubMed]
- Juhasz, B.; Kertesz, A.; Balla, J.; Balla, G.; Szabo, Z.; Bombicz, M.; Priksz, D.; Gesztelyi, R.; Varga, B.; Haines, D.D.; et al. Cardioprotective effects of sour cherry seed extract (SCSE) on the hypercholesterolemic rabbit heart. Curr. Pharm. Des. 2013, 19, 6896–6905. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 2D and M-Mode | Baseline (n = 9) Start | Control (n = 9) Endpoint | HC (n = 9) Endpoint | p Value (HC vs. Control) |
| LA area (cm2) | 0.789 ± 0.054 | 0.833 ± 0.029 | 1.633 ± 0.065 **** | <0.0001 |
| IVSd (mm) | 2.153 ± 0.074 | 2.27 ± 0.139 | 2.882 ± 0.176 * | 0.0149 |
| LV mass (g) | 4.057 ± 0.292 | 4.651 ± 0.433 | 8.035 ± 0.655 *** | 0.0005 |
| RWT (%) | 0.260 ± 0.020 | 0.283 ± 0.017 | 0.362 ± 0.018 ** | 0.0050 |
| LV FS (%) | 32.1 ± 0.706 | 30.78 ± 0.434 | 28.44 ± 0.669 ** | 0.0099 |
| LV EF (%) | 63.9 ± 0.994 | 62.22 ± 0.619 | 58.33 ± 0.898 ** | 0.0026 |
| MAPSE (mm) | 5.263 ± 0.209 | 5.488 ± 0.093 | 5.661 ± 0.254 | 0.5147 |
| LVOT Parameters | Baseline | Control | HC | p Value |
| Vmax (m/s) | 0.884 ± 0.016 | 0.923 ± 0.018 | 1.204 ± 0.021 **** | <0.0001 |
| Vmean (m/s) | 0.542 ± 0.012 | 0.601 ± 0.018 | 0.701 ± 0.027 ** | 0.0076 |
| maxPG (mmHg) | 3.141 ± 0.117 | 3.453 ± 0.139 | 5.832 ± 0.199 **** | <0.0001 |
| meanPG (mmHg) | 1.478 ± 0.054 | 1.747 ± 0.104 | 2.637 ± 0.172 *** | 0.0004 |
| Doppler and TVI | Baseline (n = 9) Start | Control (n = 9) Endpoint | HC (n = 9) Endpoint | p Value (HC vs. Control) |
| MV E vel (m/s) | 0.707 ± 0.036 | 0.630 ± 0.022 | 0.698 ± 0.047 | 0.2049 |
| MV A vel (m/s) | 0.399 ± 0.023 | 0.391 ± 0.012 | 0.653 ± 0.042 **** | <0.0001 |
| E/A ratio | 1.836 ± 0.139 | 1.624 ± 0.068 | 1.069 ± 0.039 **** | <0.0001 |
| DecT (ms) | 58.30 ± 2.864 | 55.44 ± 2.001 | 71.22 ± 2.666 *** | 0.0002 |
| Sept e’/a’ ratio | 1.538 ± 0.078 | 1.233 ± 0.060 | 0.935 ± 0.054 ** | 0.0020 |
| E/e’ ratio | 8.311 ± 0.165 | 8.002 ± 0.164 | 13.20 ± 1.091 *** | 0.0002 |
| IVRT (ms) | 34.10 ± 1.690 | 30.78 ± 1.188 | 56.56 ± 2.588 **** | <0.0001 |
| Tei-index (MPI) | 0.560 ± 0.017 | 0.528 ± 0.013 | 0.8385 ± 0.049 **** | <0.0001 |
| Strain Data | Baseline | Control | HC | p Value |
| GLS | −22.50 ± 0.806 | −21.34 ± 0.664 | −16.07 ± 1.029 *** | 0.0007 |
| Parameter | Control (n = 9) | HC (n = 9) Endpoint |
|---|---|---|
| Bodyweight (g) | 3064 ± 87 | 4153 ± 86 *** |
| Heart weight | 7.795 ± 0.418 | 12.25 ± 0.535 *** |
| Heart weight to BW ratio | 0.00253 ± 0.0003 | 0.00294 ± 0.0002 |
| Left ventricle mass (g) | 6.22 ± 0.337 | 10.13 ± 0.534 *** |
| LV mass to tibial length | 0.605 ± 0.046 | 0.828 ± 0.038 ** |
| LV mass to BW ratio | 0.00087 ± 0.0003 | 0.0008 ± 0.0002 |
| Lung wet to dry ratio | 5.254 ± 0.147 | 5.118 ± 0.094 |
| Kidney wet to dry ratio | 3.421 ± 0.058 | 3.281 ± 0.191 |
| Serum Parameter | Baseline (n = 9) Start | Control (n = 9) Endpoint | HC (n = 9) Endpoint | p Value (HC vs. Control) |
|---|---|---|---|---|
| Total chol. (mmol/L) | 3.440 ± 0.199 | 1.753 ± 0.0187 | 32.360 ± 2.164 **** | <0.0001 |
| LDLc (mmol/L) | 2.759 ± 0.317 | 0.725 ± 0.312 | 32.030 ± 1.813 **** | <0.0001 |
| ApoB (mmol/L) | 0.057 ± 0.008 | 0.026 ± 0.005 | 0.154 ± 0.006 **** | <0.0001 |
| HDLc (mmol/L) | 1.188 ± 0.212 | 0.963 ± 0.092 | 6.428 ± 0.600 **** | <0.0001 |
| ApoA (mmol/L) | 0.112 ± 0.024 | 0.149 ± 0.011 | 0.034 ± 0.007 **** | <0.0001 |
| Atherogenic index | 2.457 ± 0.170 | 1.986 ± 0.391 | 5.499 ± 0.458 *** | 0.0001 |
| ApoB to ApoA ratio | 0.385 ± 0.046 | 0.188 ± 0.041 | 3.691 ± 0.492 **** | <0.0001 |
| Triglyceride (mmol/L) | 0.410 ± 0.066 | 1.139 ± 0.136 | 1.001 ± 0.225 | 0.6959 |
| AST (GOT) (U/L) | 25.000 ± 5.177 | 22.710 ± 6.781 | 33.47 ± 3.016 | 0.1056 |
| ALT (GPT) (U/L) | 50.830 ± 8.284 | 64.140 ± 19.660 | 57.730 ± 5.479 | 0.6813 |
| Creatinine (μmol/L) | 83.000 ± 6.033 | 71.430 ± 12.040 | 116.100 ± 5.984 ** | 0.0013 |
| CK-MB (U/L) | 1077.0 ± 384.7 | 1154.0 ± 276.9 | 785.6 ± 126.1 | 0.1754 |
| Insulin (µIU/mL) | 17.63 ± 0.41 | 23.70 ± 2.46 | 49.38 ± 6.70 ** | 0.0089 |
| Glucose (mmol/L) | 4.917 ± 0.202 | 7.100 ± 0.245 | 6.918 ± 0.278 | 0.7178 |
| CRP (mg/L) | 0.251 ± 0.075 | 0.079 ± 0.070 | 0.325 ± 0.073 * | 0.0359 |
| Troponin T (ng/L) | 5.528 ± 1.010 | 7.438 ± 1.199 | 20.21 ± 2.778 ** | 0.0092 |
| Parameter | Control | HC |
|---|---|---|
| Emax | 93.67 ± 2.66 | 50.9 ± 9.48 ** |
| logEC50 | −8.62 ± 0.36 | −7.59 ± 0.16 * |
| n | 0.91 ± 0.19 | 1 ± 0.11 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Priksz, D.; Bombicz, M.; Varga, B.; Kurucz, A.; Gesztelyi, R.; Balla, J.; Toth, A.; Papp, Z.; Szilvassy, Z.; Juhasz, B. Upregulation of Myocardial and Vascular Phosphodiesterase 9A in A Model of Atherosclerotic Cardiovascular Disease. Int. J. Mol. Sci. 2018, 19, 2882. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms19102882
Priksz D, Bombicz M, Varga B, Kurucz A, Gesztelyi R, Balla J, Toth A, Papp Z, Szilvassy Z, Juhasz B. Upregulation of Myocardial and Vascular Phosphodiesterase 9A in A Model of Atherosclerotic Cardiovascular Disease. International Journal of Molecular Sciences. 2018; 19(10):2882. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms19102882
Chicago/Turabian StylePriksz, Daniel, Mariann Bombicz, Balazs Varga, Andrea Kurucz, Rudolf Gesztelyi, Jozsef Balla, Attila Toth, Zoltan Papp, Zoltan Szilvassy, and Bela Juhasz. 2018. "Upregulation of Myocardial and Vascular Phosphodiesterase 9A in A Model of Atherosclerotic Cardiovascular Disease" International Journal of Molecular Sciences 19, no. 10: 2882. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms19102882