Human Mesenchymal Stem Cell Secretome from Bone Marrow or Adipose-Derived Tissue Sources for Treatment of Hypoxia-Induced Pulmonary Epithelial Injury


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
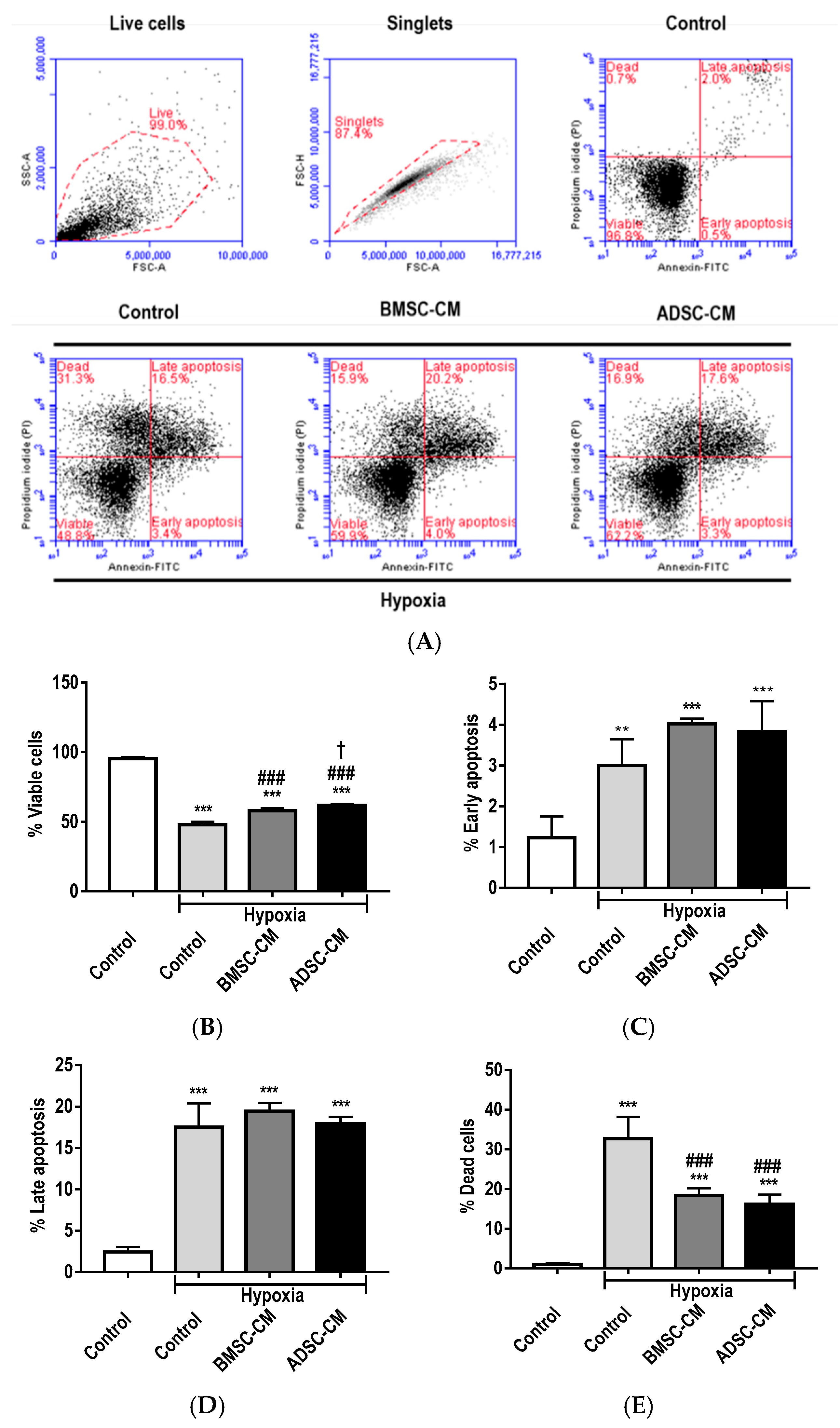
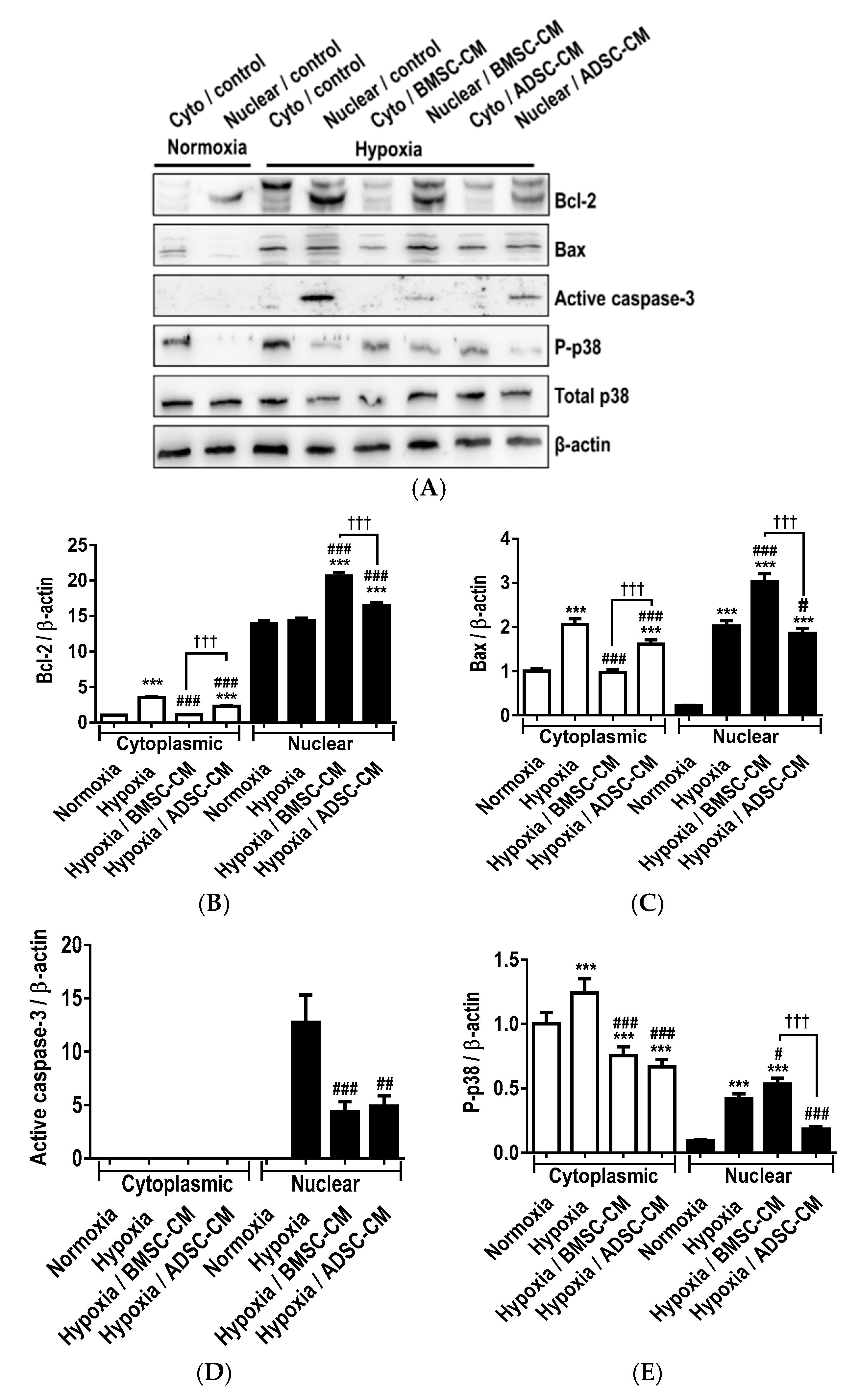
2.1. Anti-Apoptotic Effects of MSC Secretome
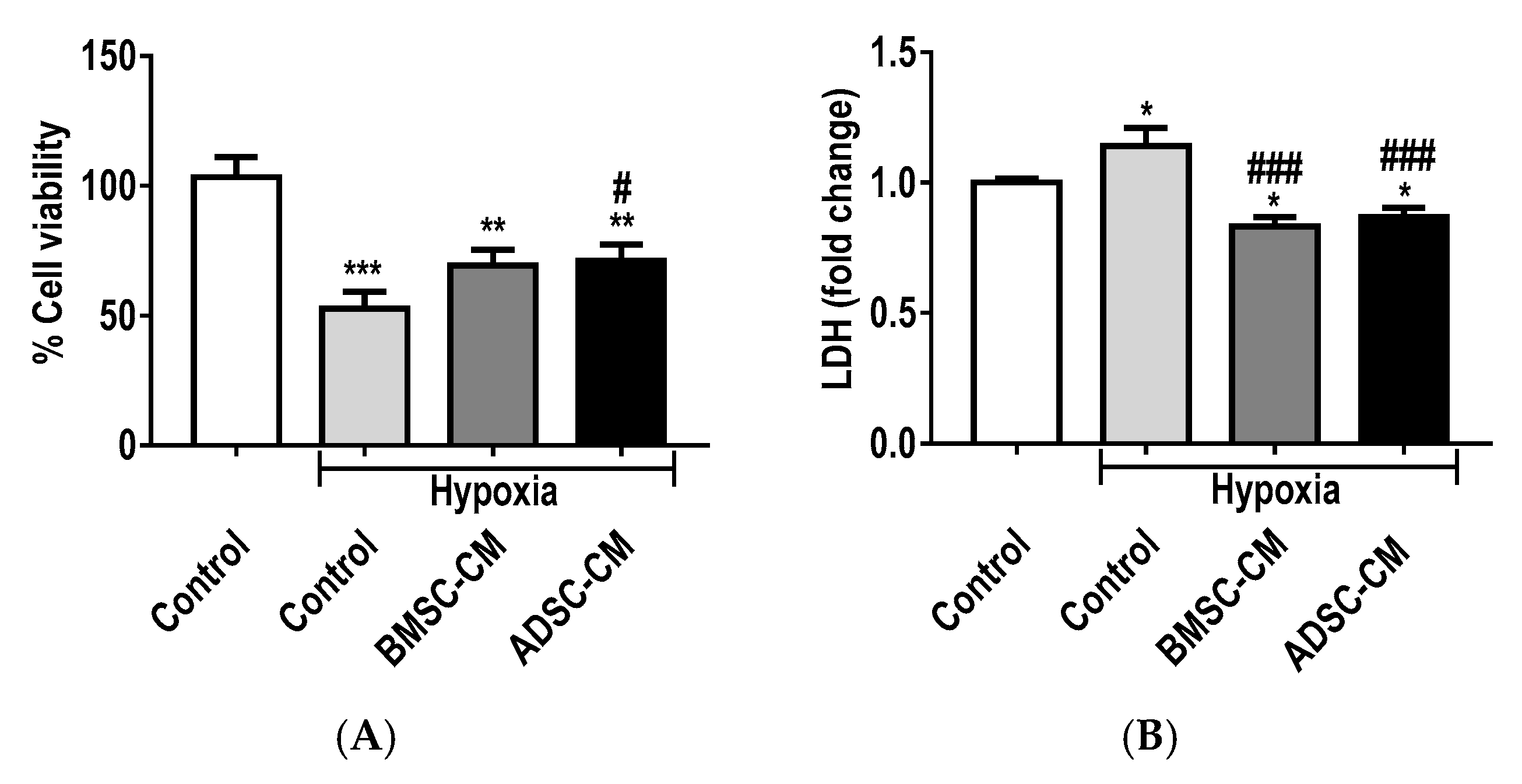
2.2. Protective Effects of MSC Secretome
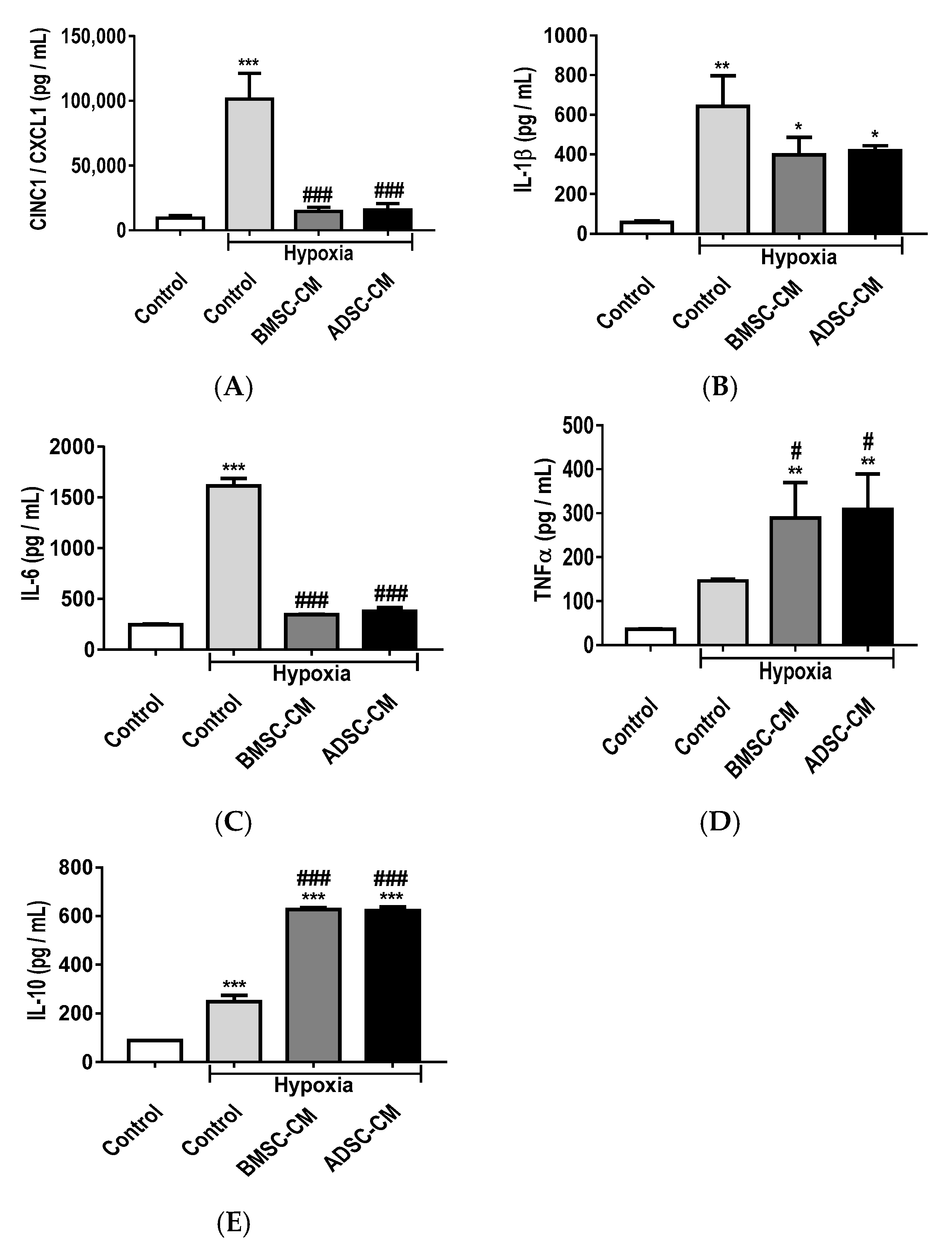
2.3. Anti-Inflammatory Effects of MSC Secretome
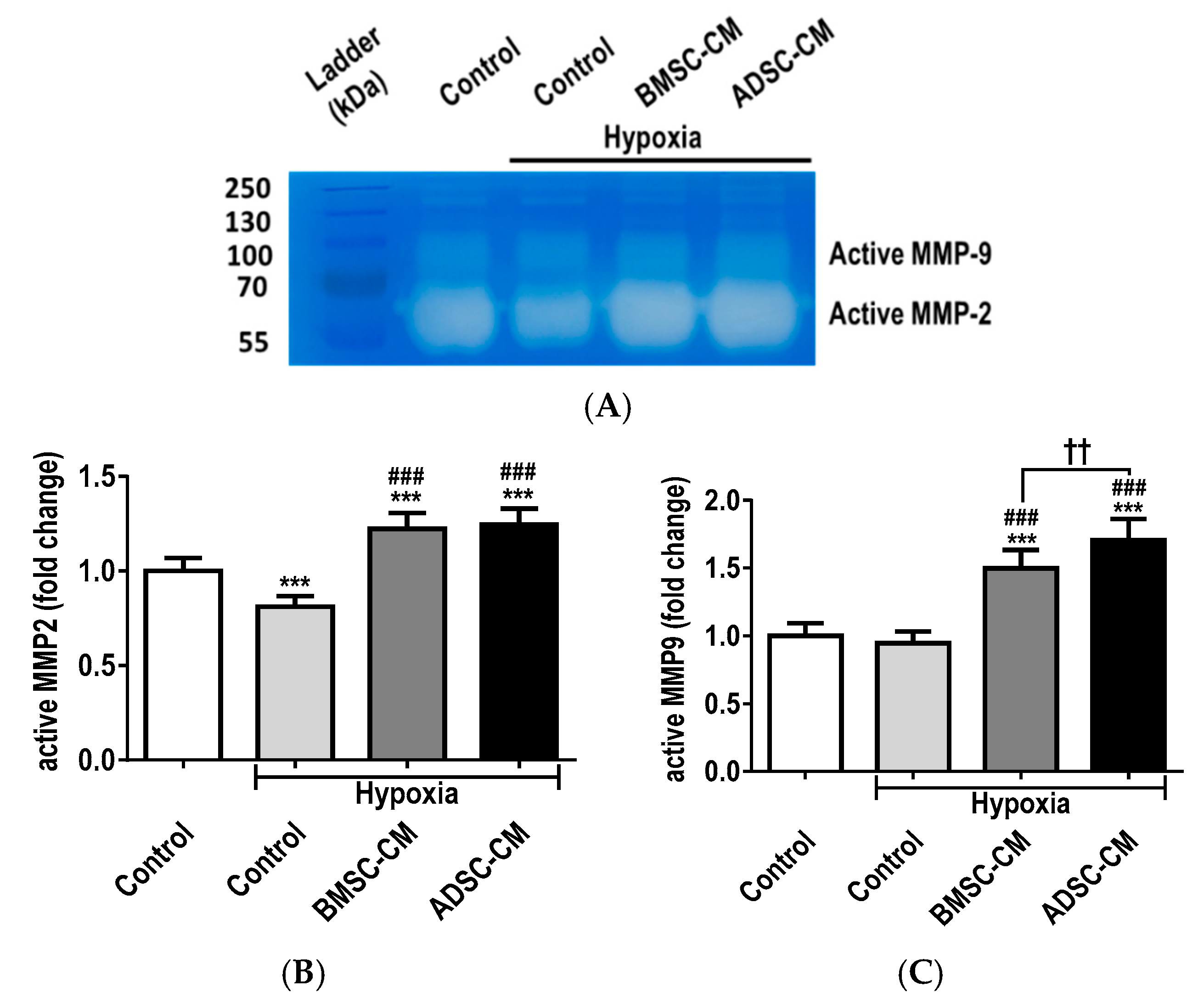
2.4. MSC Secretome Restores MMP Function during Cellular Injury
2.5. Cytoprotective Effects of MSC Secretome
2.6. Enhancement of Protective GRPs by MSC Secretome
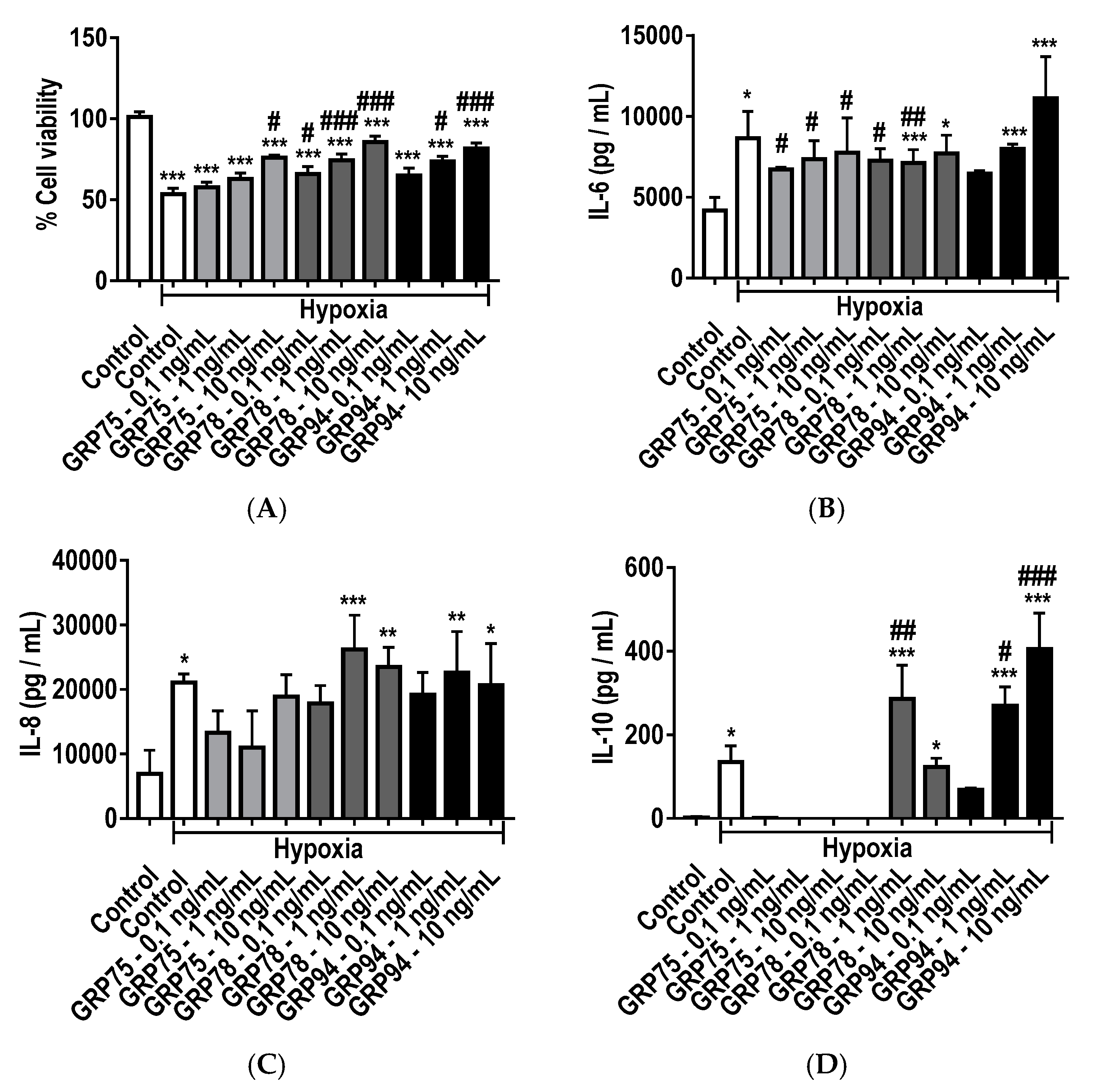
2.7. Protective and Immunomodulatory Effects of Recombinant GRPs
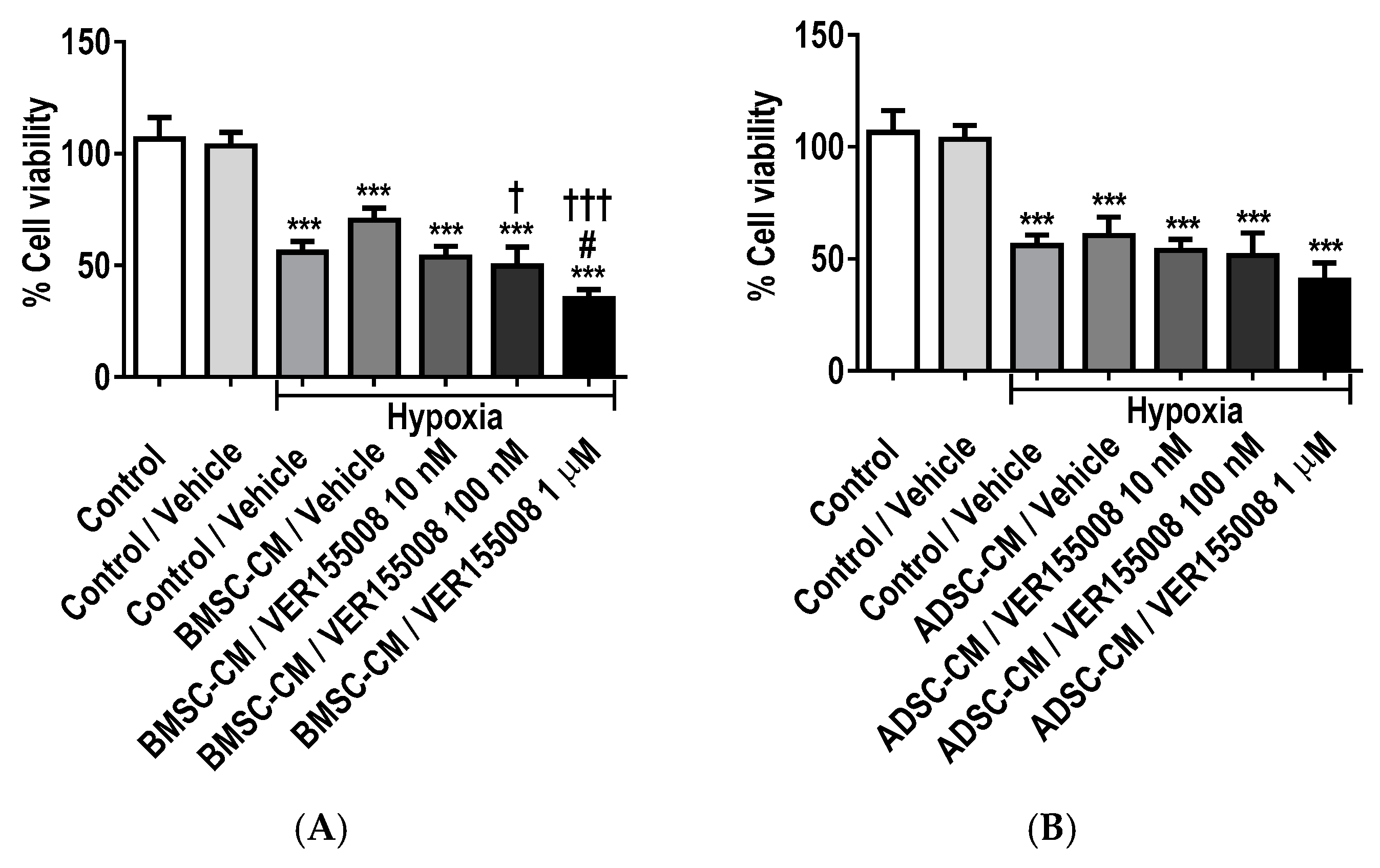
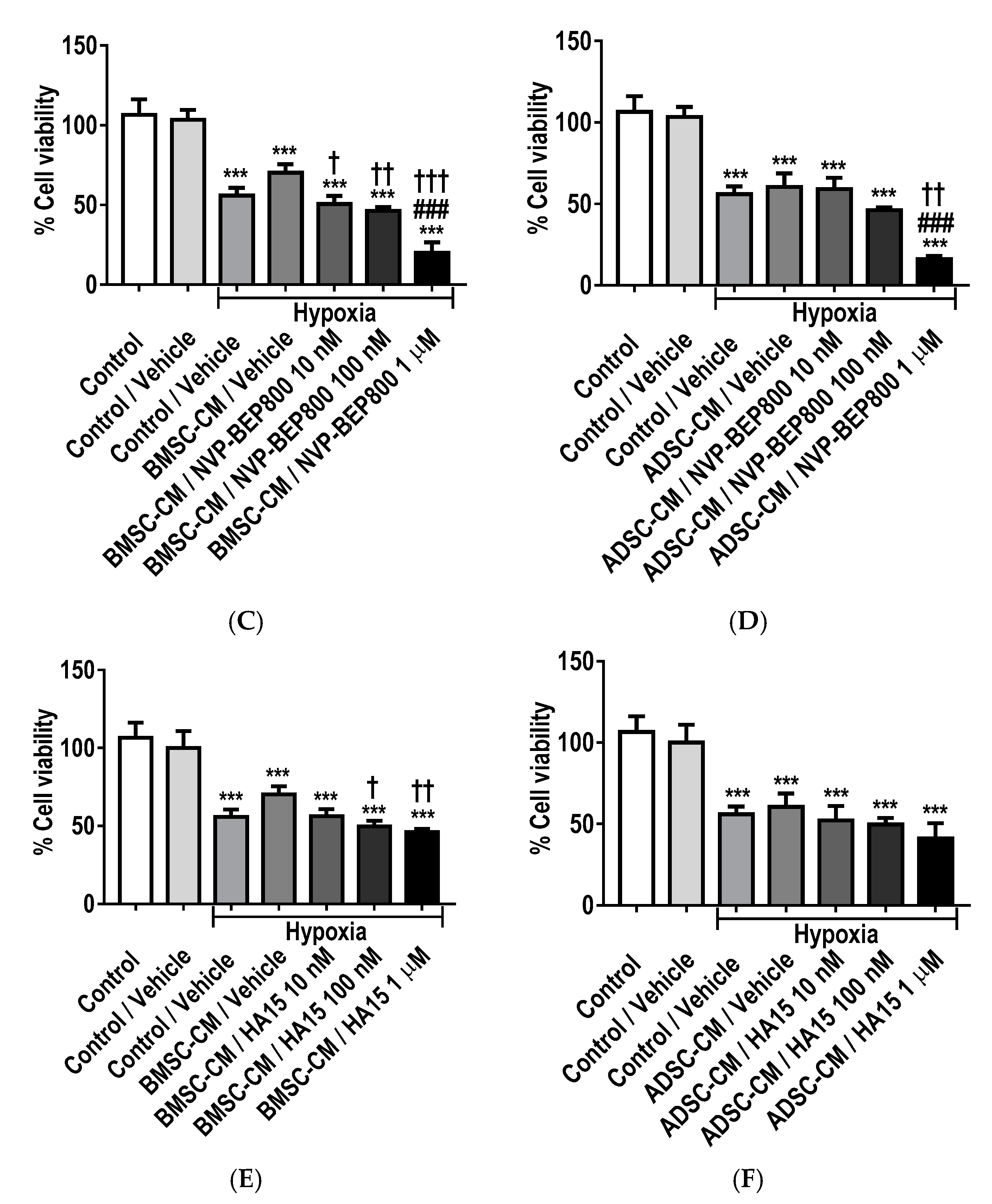
2.8. Inhibition of GRPs Reduces the Protective Effects of MSC Secretome
3. Discussion
4. Materials and Methods
4.1. Material
4.2. Cell Culture
4.3. Conditioned Medium
4.4. Primary Cell Isolation and Culture
4.5. Exposure to Hypoxia
4.6. Flow Cytometric Analysis of Apoptosis
4.7. Cell Viability and Proliferation Assay
4.8. Lactate Dehydrogenase (LDH) Assay
4.9. Gelatin Zymography
4.10. Western Blot Analysis
4.11. Enzyme-Linked Immunosorbent Assay (ELISA)
4.12. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| A549 | Transformed type II alveolar epithelial cell |
| AEC | Alveolar epithelial cell |
| ADSC | Adipose-derived stem cell |
| Bax | Bcl-2-associated X protein |
| Bcl-2 | B-cell lymphoma 2 |
| BMSC | Bone marrow-derived mesenchymal stem cell |
| CM | Conditioned medium |
| CINC-1 | Cytokine-induced neutrophil chemoattractant 1 |
| CXCL-1 | Chemokine (C-X-C motif) ligand 1 |
| GRP | Glucose-regulated protein |
| IL | Interleukin |
| IRI | Ischemia-reperfusion injury |
| MAPK p38 | Mitogen-activated protein kinase |
| MMP | Matrix metalloproteinases |
| MSC | Mesenchymal stem cell |
| TNF-α | Tumour necrosis factor alpha |
References
- Antunes, M.A.; Laffey, J.G.; Pelosi, P.; Rocco, P.R. Mesenchymal stem cell trials for pulmonary diseases. J. Cell. Biochem. 2014, 115, 1023–1032. [Google Scholar] [CrossRef] [PubMed]
- Chamberlain, G.; Fox, J.; Ashton, B.; Middleton, J. Concise review: Mesenchymal stem cells: Their phenotype, differentiation capacity, immunological features, and potential for homing. Stem Cells 2007, 25, 2739–2749. [Google Scholar] [CrossRef] [PubMed]
- Garcia, J.; Wright, K.; Roberts, S.; Kuiper, J.H.; Mangham, C.; Richardson, J.; Mennan, C. Characterisation of synovial fluid and infrapatellar fat pad derived mesenchymal stromal cells: The influence of tissue source and inflammatory stimulus. Sci. Rep. 2016, 6, 24295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singer, N.G.; Caplan, A.I. Mesenchymal stem cells: Mechanisms of inflammation. Annu. Rev. Pathol. 2011, 6, 457–478. [Google Scholar] [CrossRef] [PubMed]
- Chase, M.A.; Wheeler, D.S.; Lierl, K.M.; Hughes, V.S.; Wong, H.R.; Page, K. Hsp72 Induces Inflammation and Regulates Cytokine Production in Airway Epithelium through a TLR4- and NF-κB-Dependent Mechanism. J. Immunol. 2007, 179, 6318–6324. [Google Scholar] [CrossRef] [PubMed]
- Gao, F.; Hu, X.Y.; Xie, X.J.; Xu, Q.Y.; Wang, Y.P.; Liu, X.B.; Xiang, M.X.; Sun, Y.; Wang, J.A. Heat shock protein 90 protects rat mesenchymal stem cells against hypoxia and serum deprivation-induced apoptosis via the PI3K/Akt and ERK1/2 pathways. J. Zhejiang Univ. Sci. B 2010, 11, 608–617. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, A.S. Glucose-regulated proteins in cancer: Molecular mechanisms and therapeutic potential. Nat. Rev. Cancer 2014, 14, 263–276. [Google Scholar] [CrossRef] [PubMed]
- Trulock, E.P.; Christie, J.D.; Edwards, L.B.; Boucek, M.M.; Aurora, P.; Taylor, D.O.; Dobbels, F.; Rahmel, A.O.; Keck, B.M.; Hertz, M.I. Registry of the international society for heart and lung transplantation: Twenty-fourth official adult lung and heart-lung transplantation report-2007. J. Heart Lung Transplant. 2007, 26, 782–795. [Google Scholar] [CrossRef] [PubMed]
- Lama, V.N.; Belperio, J.A.; Christie, J.D.; El-Chemaly, S.; Fishbein, M.C.; Gelman, A.E.; Hancock, W.W.; Keshavjee, S.; Kreisel, D.; Laubach, V.E.; et al. Models of lung transplant research: A consensus statement from the National Heart, Lung, and Blood Institute workshop. JCI Insight 2017, 2, e93121. [Google Scholar] [CrossRef] [PubMed]
- Vohwinkel, C.U.; Hoegl, S.; Eltzschig, H.K. Hypoxia signaling during acute lung injury. J. Appl. Physiol. 2015, 119, 1157–1163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kosieradzki, M.; Rowinski, W. Ischemia/reperfusion injury in kidney transplantation: Mechanisms and prevention. Transpl. Proc. 2008, 40, 3279–3288. [Google Scholar] [CrossRef] [PubMed]
- Irwin, D.C.; Baek, J.H.; Hassell, K.; Nuss, R.; Eigenberger, P.; Lisk, C.; Loomis, Z.; Maltzahn, J.; Stenmark, K.R.; Nozik-Grayck, E.; et al. Hemoglobin induced lung vascular oxidation, inflammation, and remodeling contributes to the progression of hypoxic pulmonary hypertension and is attenuated in rats with repeat dose haptoglobin administration. Free Radic. Biol. Med. 2015, 82, 50–62. [Google Scholar] [CrossRef] [PubMed]
- Ueda, M.; Fuchs, S.; Nakamura, T.; Schafer, U.F.; Lehr, C.M.; Menger, M.D.; Schafers, H.J. Reoxygenation results in cell death of human alveolar epithelial cells. J. Heart Lung Transplant. 2004, 23, 1198–1204. [Google Scholar] [CrossRef] [PubMed]
- Casiraghi, M.; Tatreau, J.R.; Abano, J.B.; Blackwell, J.W.; Watson, L.; Burridge, K.; Randell, S.H.; Egan, T.M. In vitro modeling of nonhypoxic cold ischemia-reperfusion simulating lung transplantation. J. Thorac. Cardiovasc. Surg. 2009, 138, 760–767. [Google Scholar] [CrossRef] [PubMed]
- Ferng, A.S.; Schipper, D.; Connell, A.M.; Marsh, K.M.; Knapp, S.; Khalpey, Z. Novel vs. clinical organ preservation solutions: Improved cardiac mitochondrial protection. J. Cardiothorac. Surg. 2017, 12, 7. [Google Scholar] [CrossRef] [PubMed]
- Tan, J.; Liu, D.; Lv, X.; Wang, L.; Zhao, C.; Che, Y.; Xie, Q.; Cui, X. MAPK mediates inflammatory response and cell death in rat pulmonary microvascular endothelial cells in an ischemia–reperfusion model of lung transplantation. J. Heart Lung Transplant. 2013, 32, 823–831. [Google Scholar] [CrossRef] [PubMed]
- Fröhlich, S.; Boylan, J.; McLoughlin, P. Hypoxia-induced inflammation in the lung: A potential therapeutic target in acute lung injury? Am. J. Respir. Cell Mol. Biol. 2013, 48, 271–279. [Google Scholar] [CrossRef] [PubMed]
- Eltzschig, H.K.; Carmeliet, P. Hypoxia and inflammation. N. Engl. J. Med. 2011, 364, 656–665. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.Y.; Nunez, G. Sterile inflammation: Sensing and reacting to damage. Nat. Rev. Immunol. 2010, 10, 826–837. [Google Scholar] [CrossRef] [PubMed]
- Hung, Y.C.; Parolini, O.; Deng, L. Should hypoxia preconditioning become the standardized procedure for bone marrow MSCs preparation for clinical use? Stem Cells 2016, 34, 1992–1993. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.H.; Yoon, Y.M.; Lee, S.H. Hypoxic preconditioning promotes the bioactivities of mesenchymal stem cells via the HIF-1alpha-GRP78-Akt axis. Int. J. Mol. Sci. 2017, 18, 1320. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.S.; Lee, J.H.; Yoon, Y.M.; Yun, C.W.; Noh, H.; Lee, S.H. Hypoxia-induced expression of cellular prion protein improves the therapeutic potential of mesenchymal stem cells. Cell Death Dis. 2016, 7, e2395. [Google Scholar] [CrossRef] [PubMed]
- Hwang, B.; Liles, W.C.; Waworuntu, R.; Mulligan, M.S. Pretreatment with Bone Marrow Derived Mesenchymal Stromal Cell Conditioned Media Confers Pulmonary Ischemic Tolerance. J. Thorac. Cardiovasc. Surg. 2016, 151, 841–849. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Chen, X.; Wu, X.; Wei, S.; Han, W.; Lin, J.; Kang, M.; Chen, L. Hepatocyte growth factor-modified mesenchymal stem cells improve ischemia/reperfusion-induced acute lung injury in rats. Gene Ther. 2017, 24, 3–11. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.Y.; Chiang, C.H.; Hung, S.C.; Chian, C.F.; Tsai, C.L.; Chen, W.C.; Zhang, H. Hypoxia-preconditioned mesenchymal stem cells ameliorate ischemia/reperfusion-induced lung injury. PLoS ONE 2017, 12, e0187637. [Google Scholar] [CrossRef] [PubMed]
- Ito, A.; Mukaiyama, A.; Itoh, Y.; Nagase, H.; Thøgersen, I.B.; Enghild, J.J.; Sasaguri, Y.; Mori, Y. Degradation of interleukin 1β by matrix metalloproteinases. J. Biol. Chem. 1996, 271, 14657–14660. [Google Scholar] [CrossRef] [PubMed]
- Schönbeck, U.; Mach, F.; Libby, P. Generation of biologically active IL-1β by matrix metalloproteinases: A novel Caspase-1-independent pathway of IL-1β processing. J. Immunol. 1998, 161, 3340–3346. [Google Scholar] [PubMed]
- English, W.R.; Puente, X.S.; Freije, J.M.; Knauper, V.; Amour, A.; Merryweather, A.; Lopez-Otin, C.; Murphy, G. Membrane type 4 matrix metalloproteinase (MMP17) has tumor necrosis factor-alpha convertase activity but does not activate pro-MMP2. J. Biol. Chem. 2000, 275, 14046–14055. [Google Scholar] [CrossRef] [PubMed]
- Mohan, M.J.; Seaton, T.; Mitchell, J.; Howe, A.; Blackburn, K.; Burkhart, W.; Moyer, M.; Patel, I.; Waitt, G.M.; Becherer, J.D.; et al. The tumor necrosis factor-alpha converting enzyme (TACE): A unique metalloproteinase with highly defined substrate selectivity. Biochemistry 2002, 41, 9462–9469. [Google Scholar] [CrossRef] [PubMed]
- Chipuk, J.E.; Green, D.R. How do BCL-2 proteins induce mitochondrial outer membrane permeabilization? Trends Cell Biol. 2008, 18, 157–164. [Google Scholar] [CrossRef] [PubMed]
- Salah-eldin, A.; Inoue, S.; Tsuda, M.; Matsuura, A. Abnormal intracellular localization of Bax with a normal membrane anchor domain in human lung cancer cell lines. Jpn. J. Cancer Res. 2000, 91, 1269–1277. [Google Scholar] [CrossRef] [PubMed]
- Brayer, S.; Joannes, A.; Jaillet, M.; Gregianin, E.; Mahmoudi, S.; Marchal Somme, J.; Fabre, A.; Mordant, P.; Cazes, A.; Crestani, B.; et al. The pro-apoptotic BAX protein influences cell growth and differentiation from the nucleus in healthy interphasic cells. Cell Cycle 2017, 16, 2108–2118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamada, S.; Kikkawa, U.; Tsujimoto, Y.; Hunter, T. Nuclear translocation of caspase-3 Is dependent on its proteolytic activation and recognition of a substrate-like protein(s). J. Biol. Chem. 2005, 280, 857–860. [Google Scholar] [CrossRef] [PubMed]
- Okada, M.; Yamane, M.; Iga, N.; Nishikawa, H.; Yamamoto, S.; Otani, S.; Waki, N.; Hirayama, S.; Miyoshi, K.; Sugimoto, S.; et al. MAPK/ERK pathway activation leads to severe ischemia reperfusion induced lung injury. J. Heart Lung Transplant. 2013, 32, S138. [Google Scholar] [CrossRef]
- Hashimoto, N.; Takeyoshi, I.; Yoshinari, D.; Tsutsumi, H.; Tokumine, M.; Totsuka, O.; Sunose, Y.; Ohwada, S.; Matsumoto, K.; Morishita, Y. Effects of a p38 mitogen-activated protein kinase inhibitor as an additive to Euro-Collins solution on reperfusion injury in canine lung transplantation. Transplantation 2002, 74, 320–326. [Google Scholar] [CrossRef] [PubMed]
- Sakiyama, S.; dePerrot, M.; Han, B.; Waddell, T.K.; Keshavjee, S.; Liu, M. Ischemia–reperfusion decreases protein tyrosine phosphorylation and p38 mitogen-activated protein kinase phosphorylation in rat lung transplants. J. Heart Lung Transplant. 2003, 22, 338–346. [Google Scholar] [CrossRef]
- Zhang, X.; Shan, P.; Alam, J.; Davis, R.J.; Flavell, R.A.; Lee, P.J. Carbon monoxide modulates Fas/Fas ligand, caspases, and Bcl-2 family proteins via the p38alpha mitogen-activated protein kinase pathway during ischemia-reperfusion lung injury. J. Biol. Chem. 2003, 278, 22061–22070. [Google Scholar] [CrossRef] [PubMed]
- Lu, Q.; Jankowich, M.; Newton, J.; Harrington, E.O.; Rounds, S. Alterations in molecular chaperones and eIF2α during lung endothelial cell apoptosis. Am. J. Physiol. Lung Cell. Mol. Physiol. 2010, 298, L501–L508. [Google Scholar] [CrossRef] [PubMed]
- Luo, S.; Mao, C.; Lee, B.; Lee, A.S. GRP78/BiP is required for cell proliferation and protecting the inner cell mass from apoptosis during early mouse embryonic development. Mol. Cell. Biol. 2006, 26, 5688–5697. [Google Scholar] [CrossRef] [PubMed]
- Rozhkova, E.; Yurinskaya, M.; Zatsepina, O.; Garbuz, D.; Karpov, V.; Surkov, S.; Murashev, A.; Ostrov, V.; Margulis, B.; Evgen’ev, M.; et al. Exogenous mammalian extracellular HSP70 reduces endotoxin manifestations at the cellular and organism levels. Ann. N. Y. Acad. Sci. 2010, 1197, 94–107. [Google Scholar] [CrossRef] [PubMed]
- Ni, M.; Zhou, H.; Wey, S.; Baumeister, P.; Lee, A.S. Regulation of PERK signaling and leukemic cell survival by a novel cytosolic isoform of the UPR regulator GRP78/BiP. PLoS ONE 2009, 4, e6868. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.-T.; Lee, A.S. Chapter Thirteen—Measurement and Modification of the Expression Level of the Chaperone Protein and Signaling Regulator GRP78/BiP in Mammalian Cells. In Methods in Enzymology; Conn, P.M., Ed.; Academic Press: Cambridge, MA, USA, 2011; Volume 490, pp. 217–233. [Google Scholar]
- Rondas, D.; Crevecoeur, I.; D’Hertog, W.; Ferreira, G.B.; Staes, A.; Garg, A.D.; Eizirik, D.L.; Agostinis, P.; Gevaert, K.; Overbergh, L.; et al. Citrullinated glucose-regulated protein 78 is an autoantigen in type 1 diabetes. Diabetes 2015, 64, 573–586. [Google Scholar] [CrossRef] [PubMed]
- Ni, M.; Zhang, Y.; Lee, A.S. Beyond the endoplasmic reticulum: Atypical GRP78 in cell viability, signaling and therapeutic targeting. Biochem. J. 2011, 434, 181–188. [Google Scholar] [CrossRef] [PubMed]
- Qian, Y.; Zheng, Y.; Ramos, K.S.; Tiffany-Castiglioni, E. The involvement of copper transporter in lead-induced oxidative stress in astroglia. Neurochem. Res. 2005, 30, 429–438. [Google Scholar] [CrossRef] [PubMed]
- Little, E.; Lee, A.S. Generation of a mammalian cell line deficient in glucose-regulated protein stress induction through targeted ribozyme driven by a stress-inducible promoter. J. Biol. Chem. 1995, 270, 9526–9534. [Google Scholar] [CrossRef] [PubMed]
- McCormick, T.S.; McColl, K.S.; Distelhorst, C.W. Mouse lymphoma cells destined to undergo apoptosis in response to thapsigargin treatment fail to generate a calcium-mediated grp78/grp94 stress response. J. Biol. Chem. 1997, 272, 6087–6092. [Google Scholar] [CrossRef] [PubMed]
- Reddy, R.K.; Lu, J.; Lee, A.S. The endoplasmic reticulum chaperone glycoprotein GRP94 with Ca2+-binding and antiapoptotic properties is a novel proteolytic target of calpain during etoposide-induced apoptosis. J. Biol. Chem. 1999, 274, 28476–28483. [Google Scholar] [CrossRef] [PubMed]
- Barker, S.; Weinfeld, M.; Zheng, J.; Li, L.; Murray, D. Identification of mammalian proteins cross-linked to DNA by ionizing radiation. J. Biol. Chem. 2005, 280, 33826–33838. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Liu, R.; Ni, M.; Gill, P.; Lee, A.S. Cell surface relocalization of the endoplasmic reticulum chaperone and unfolded protein response regulator GRP78/BiP. J. Biol. Chem. 2010, 285, 15065–15075. [Google Scholar] [CrossRef] [PubMed]
- Asea, A.; Kraeft, S.K.; Kurt-Jones, E.A.; Stevenson, M.A.; Chen, L.B.; Finberg, R.W.; Koo, G.C.; Calderwood, S.K. HSP70 stimulates cytokine production through a CD14-dependant pathway, demonstrating its dual role as a chaperone and cytokine. Nat. Med. 2000, 6, 435–442. [Google Scholar] [CrossRef] [PubMed]
- Moroi, Y.; Mayhew, M.; Trcka, J.; Hoe, M.H.; Takechi, Y.; Hartl, F.U.; Rothman, J.E.; Houghton, A.N. Induction of cellular immunity by immunization with novel hybrid peptides complexed to heat shock protein 70. Proc. Natl. Acad. Sci. USA 2000, 97, 3485. [Google Scholar] [CrossRef] [PubMed]
- Asea, A.; Rehli, M.; Kabingu, E.; Boch, J.A.; Bare, O.; Auron, P.E.; Stevenson, M.A.; Calderwood, S.K. Novel signal transduction pathway utilized by extracellular HSP70: Role of toll-like receptor (TLR) 2 and TLR4. J. Biol. Chem. 2002, 277, 15028–15034. [Google Scholar] [CrossRef] [PubMed]
- Becker, T.; Hartl, F.U.; Wieland, F. CD40, an extracellular receptor for binding and uptake of Hsp70-peptide complexes. J. Cell Biol. 2002, 158, 1277–1285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Kelly, C.G.; Karttunen, J.T.; Whittall, T.; Lehner, P.J.; Duncan, L.; MacAry, P.; Younson, J.S.; Singh, M.; Oehlmann, W.; et al. CD40 is a cellular receptor mediating mycobacterial heat shock protein 70 stimulation of CC-chemokines. Immunity 2001, 15, 971–983. [Google Scholar] [CrossRef]
- Basu, S.; Binder, R.J.; Ramalingam, T.; Srivastava, P.K. CD91 is a common receptor for heat shock proteins gp96, hsp90, hsp70, and calreticulin. Immunity 2001, 14, 303–313. [Google Scholar] [CrossRef]
- Floto, R.A.; MacAry, P.A.; Boname, J.M.; Mien, T.S.; Kampmann, B.; Hair, J.R.; Huey, O.S.; Houben, E.N.; Pieters, J.; Day, C.; et al. Dendritic cell stimulation by mycobacterial Hsp70 is mediated through CCR5. Science 2006, 314, 454–458. [Google Scholar] [CrossRef] [PubMed]
- Rao, R.V.; Peel, A.; Logvinova, A.; del Rio, G.; Hermel, E.; Yokota, T.; Goldsmith, P.C.; Ellerby, L.M.; Ellerby, H.M.; Bredesen, D.E. Coupling endoplasmic reticulum stress to the cell death program: Role of the ER chaperone GRP78. FEBS Lett. 2002, 514, 122–128. [Google Scholar] [CrossRef]
- Reddy, R.K.; Mao, C.; Baumeister, P.; Austin, R.C.; Kaufman, R.J.; Lee, A.S. Endoplasmic reticulum chaperone protein GRP78 protects cells from apoptosis induced by topoisomerase inhibitors: Role of ATP binding site in suppression of caspase-7 activation. J. Biol. Chem. 2003, 278, 20915–20924. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Zhang, Y.; Fu, Y.; Chan, L.; Lee, A.S. Novel mechanism of anti-apoptotic function of 78-kDa glucose-regulated protein (GRP78): Endocrine resistance factor in breast cancer, through release of B-cell lymphoma 2 (BCL-2) from BCL-2-interacting killer (BIK). J. Biol. Chem. 2011, 286, 25687–25696. [Google Scholar] [CrossRef] [PubMed]
- Somensi, N.; Brum, P.O.; de Miranda Ramos, V.; Gasparotto, J.; Zanotto-Filho, A.; Rostirolla, D.C.; da Silva Morrone, M.; Moreira, J.C.F.; Pens Gelain, D. Extracellular HSP70 activates ERK1/2, NF-κB and pro-inflammatory gene transcription through binding with RAGE in A549 human lung cancer cells. Cell Physiol. Biochem. 2017, 42, 2507–2522. [Google Scholar] [CrossRef] [PubMed]
- Aksoy, M.O.; Kim, V.; Cornwell, W.D.; Rogers, T.J.; Kosmider, B.; Bahmed, K.; Barrero, C.; Merali, S.; Shetty, N.; Kelsen, S.G. Secretion of the endoplasmic reticulum stress protein, GRP78, into the BALF is increased in cigarette smokers. Respir. Res. 2017, 18, 78. [Google Scholar] [CrossRef] [PubMed]
- Vega, V.L.; Rodriguez-Silva, M.; Frey, T.; Gehrmann, M.; Diaz, J.C.; Steinem, C.; Multhoff, G.; Arispe, N.; De Maio, A. Hsp70 translocates into the plasma membrane after stress and is released into the extracellular environment in a membrane-associated form that activates macrophages. J. Immunol. 2008, 180, 4299–4307. [Google Scholar] [CrossRef] [PubMed]
- Qin, K.; Ma, S.; Li, H.; Wu, M.; Sun, Y.; Fu, M.; Guo, Z.; Zhu, H.; Gong, F.; Lei, P.; et al. GRP78 impairs production of lipopolysaccharide-induced cytokines by interaction with CD14. Front. Immunol. 2017, 8, 579. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.M.; Li, S.J.; Wu, C.H.; Hu, C.M.; Cheng, H.W.; Chang, J.S. Transient knock down of Grp78 reveals roles in serum ferritin mediated pro-inflammatory cytokine secretion in rat primary activated hepatic stellate cells. Asian Pac. J. Cancer Prev. 2014, 15, 605–610. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Chen, Z.; Narasaraju, T.; Jin, N.; Liu, L. Isolation of highly pure alveolar epithelial type I and type II cells from rat lungs. Lab. Investig. 2004, 84, 727–735. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bernard, O.; Jeny, F.; Uzunhan, Y.; Dondi, E.; Terfous, R.; Label, R.; Sutton, A.; Larghero, J.; Vanneaux, V.; Nunes, H.; et al. Mesenchymal stem cells reduce hypoxia-induced apoptosis in alveolar epithelial cells by modulating HIF and ROS hypoxic signaling. Am. J. Physiol. Lung Cell Mol. Physiol. 2018, 314, L360–L371. [Google Scholar] [CrossRef] [PubMed]
- Krick, S.; Eul, B.G.; Hanze, J.; Savai, R.; Grimminger, F.; Seeger, W.; Rose, F. Role of hypoxia-inducible factor-1alpha in hypoxia-induced apoptosis of primary alveolar epithelial type II cells. Am. J. Respir. Cell Mol. Biol. 2005, 32, 395–403. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Lee, S.W.; Baek, K.M.; Park, J.S.; Min, J.H. Continuous hypoxia attenuates paraquat-induced cytotoxicity in the human A549 lung carcinoma cell line. Exp. Mol. Med. 2011, 43, 494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, Y.; Lv, J.; Feng, D.; Jiang, F.; Fan, X.; Zhang, Z.; Yin, R.; Xu, L. Restoration of alveolar type II cell function contributes to simvastatin-induced attenuation of lung ischemia-reperfusion injury. Int. J. Mol. Med. 2012, 30, 1294–1306. [Google Scholar] [CrossRef] [PubMed]









© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shologu, N.; Scully, M.; Laffey, J.G.; O’Toole, D. Human Mesenchymal Stem Cell Secretome from Bone Marrow or Adipose-Derived Tissue Sources for Treatment of Hypoxia-Induced Pulmonary Epithelial Injury. Int. J. Mol. Sci. 2018, 19, 2996. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms19102996
Shologu N, Scully M, Laffey JG, O’Toole D. Human Mesenchymal Stem Cell Secretome from Bone Marrow or Adipose-Derived Tissue Sources for Treatment of Hypoxia-Induced Pulmonary Epithelial Injury. International Journal of Molecular Sciences. 2018; 19(10):2996. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms19102996
Chicago/Turabian StyleShologu, Nala, Michael Scully, John G. Laffey, and Daniel O’Toole. 2018. "Human Mesenchymal Stem Cell Secretome from Bone Marrow or Adipose-Derived Tissue Sources for Treatment of Hypoxia-Induced Pulmonary Epithelial Injury" International Journal of Molecular Sciences 19, no. 10: 2996. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms19102996