Nucleic Acid-Dependent Structural Transition of the Intrinsically Disordered N-Terminal Appended Domain of Human Lysyl-tRNA Synthetase

Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
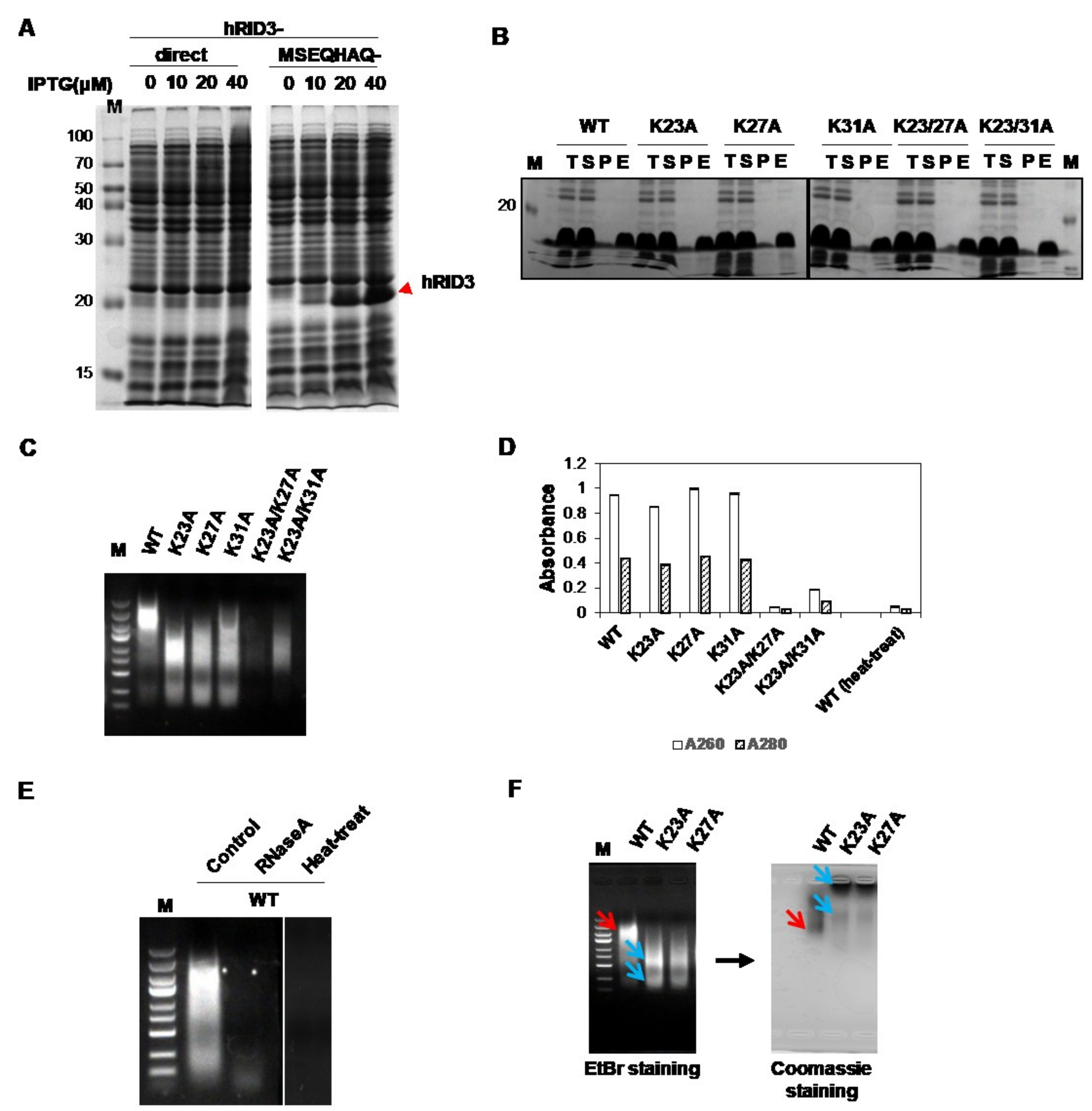
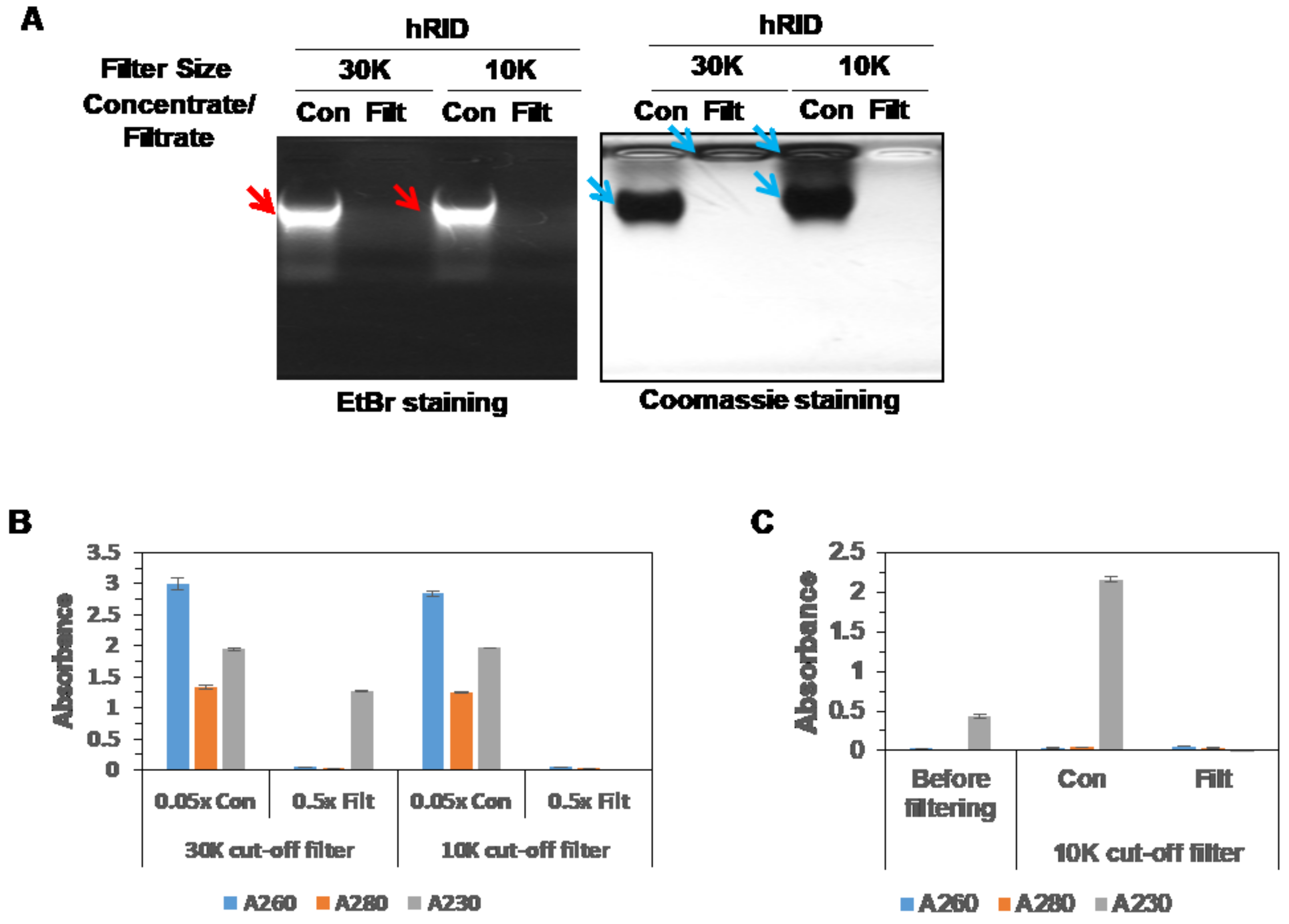
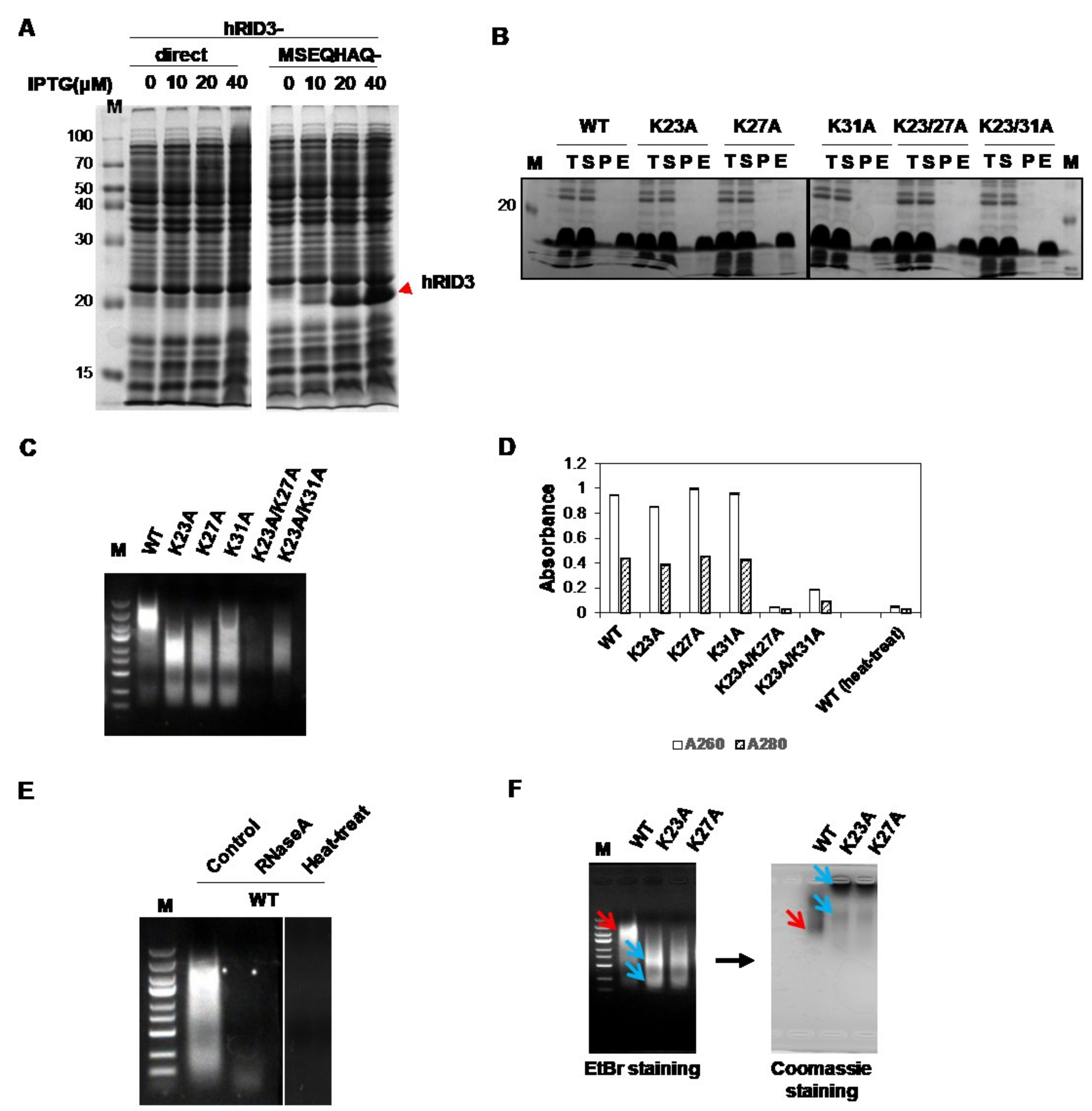
2.1. Expression and Purification of hRID and Its Mutants and Detection of Co-Purified NAs
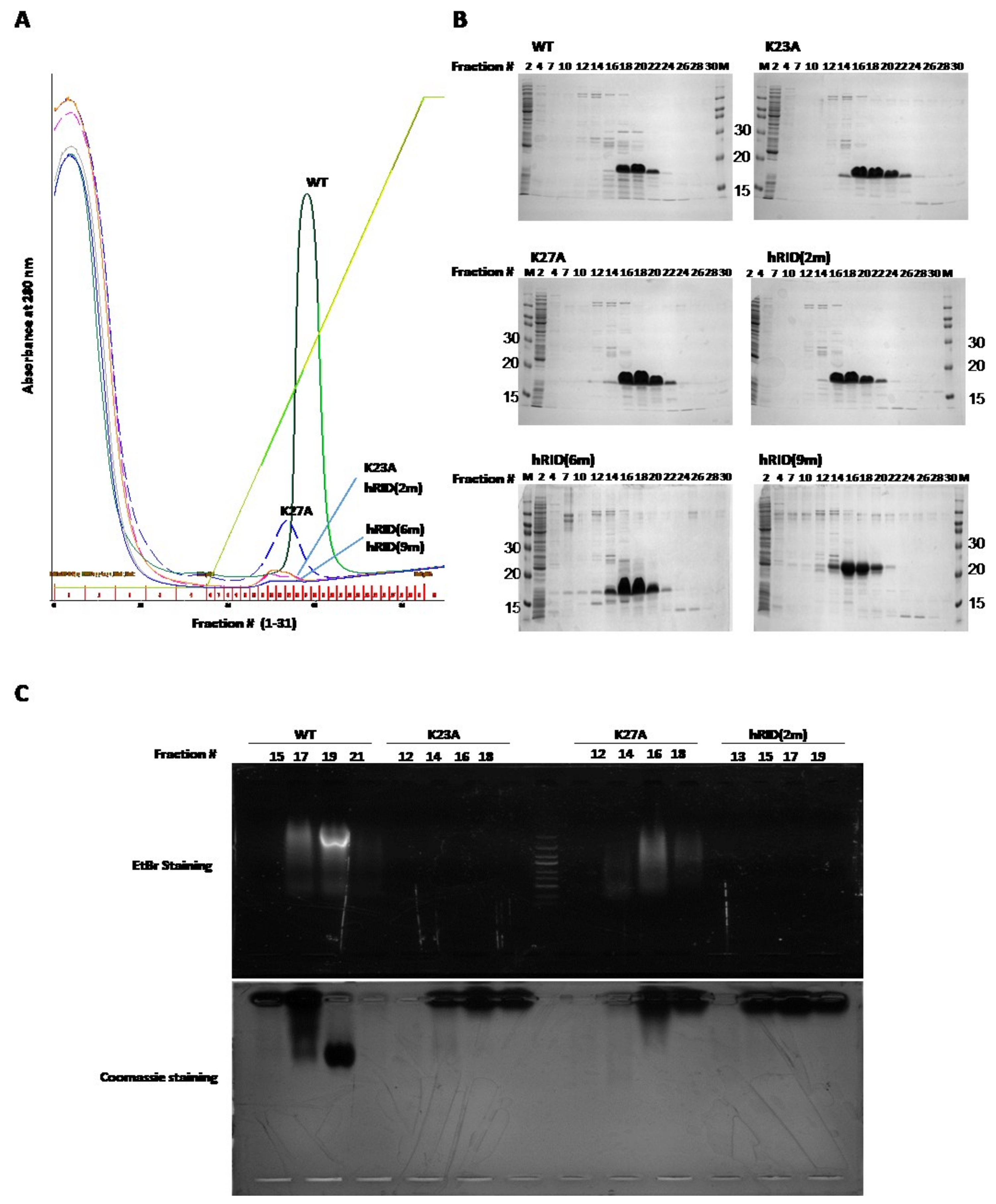
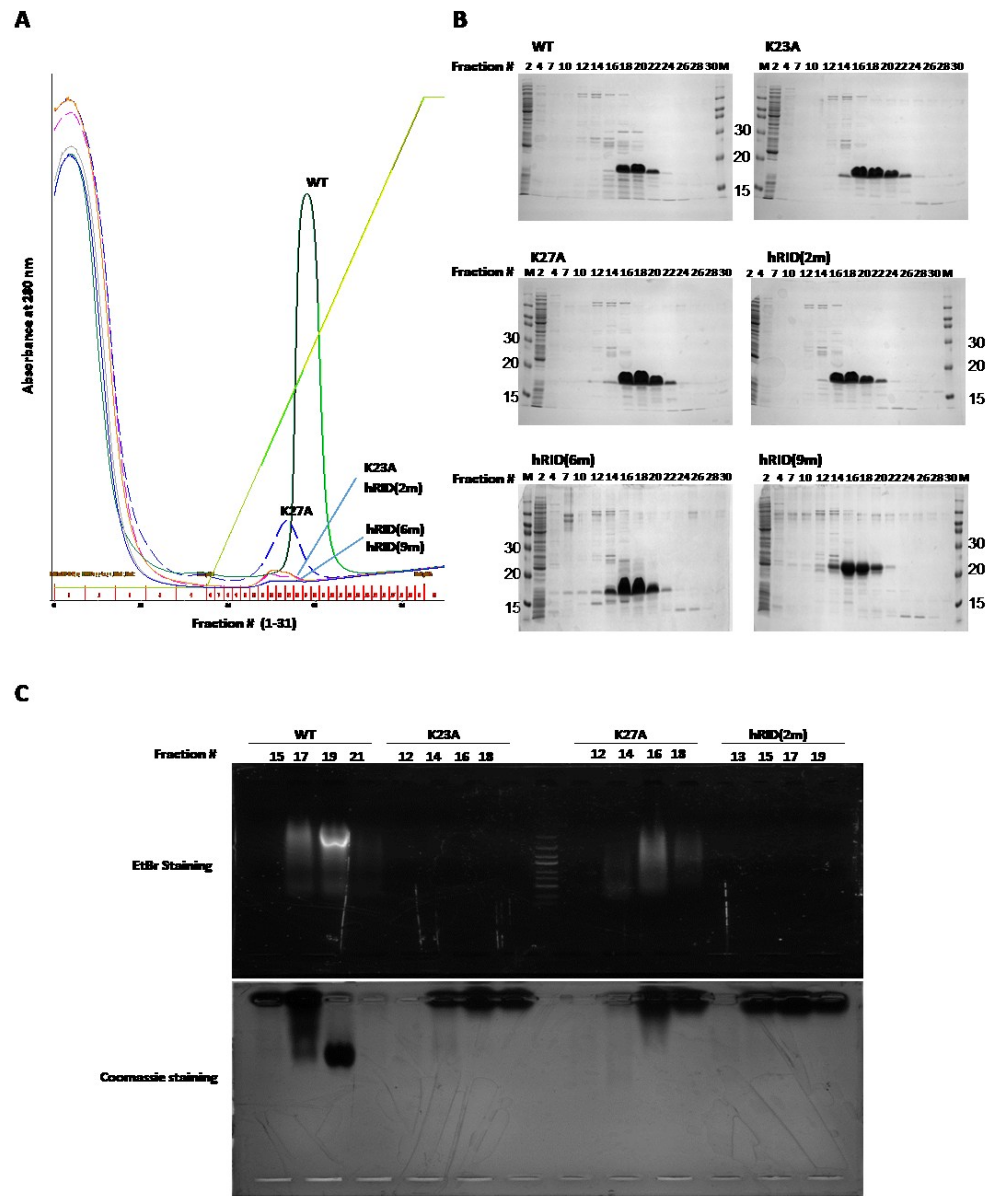
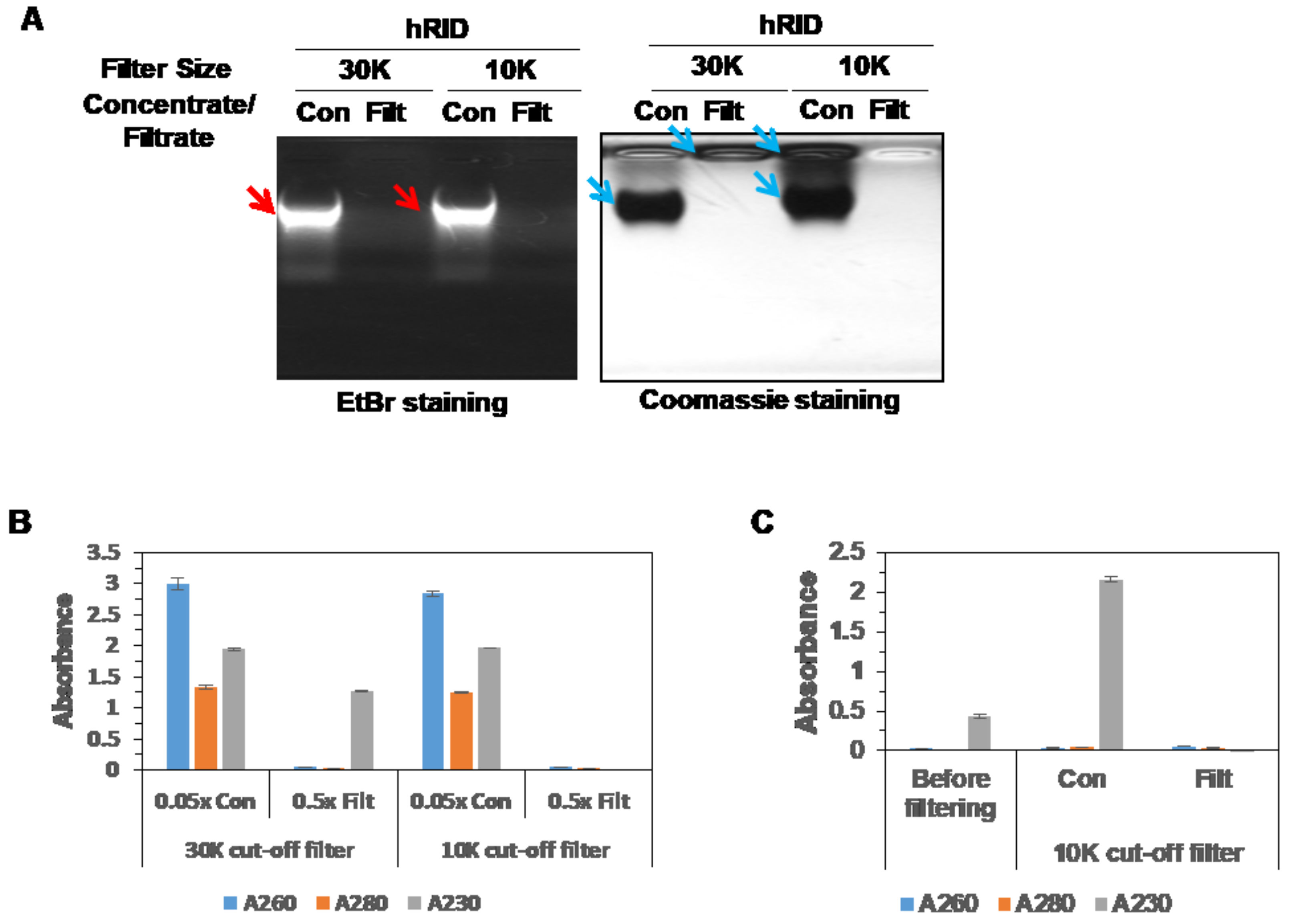
2.2. Separation of the Proteins with Co-Purified NAs
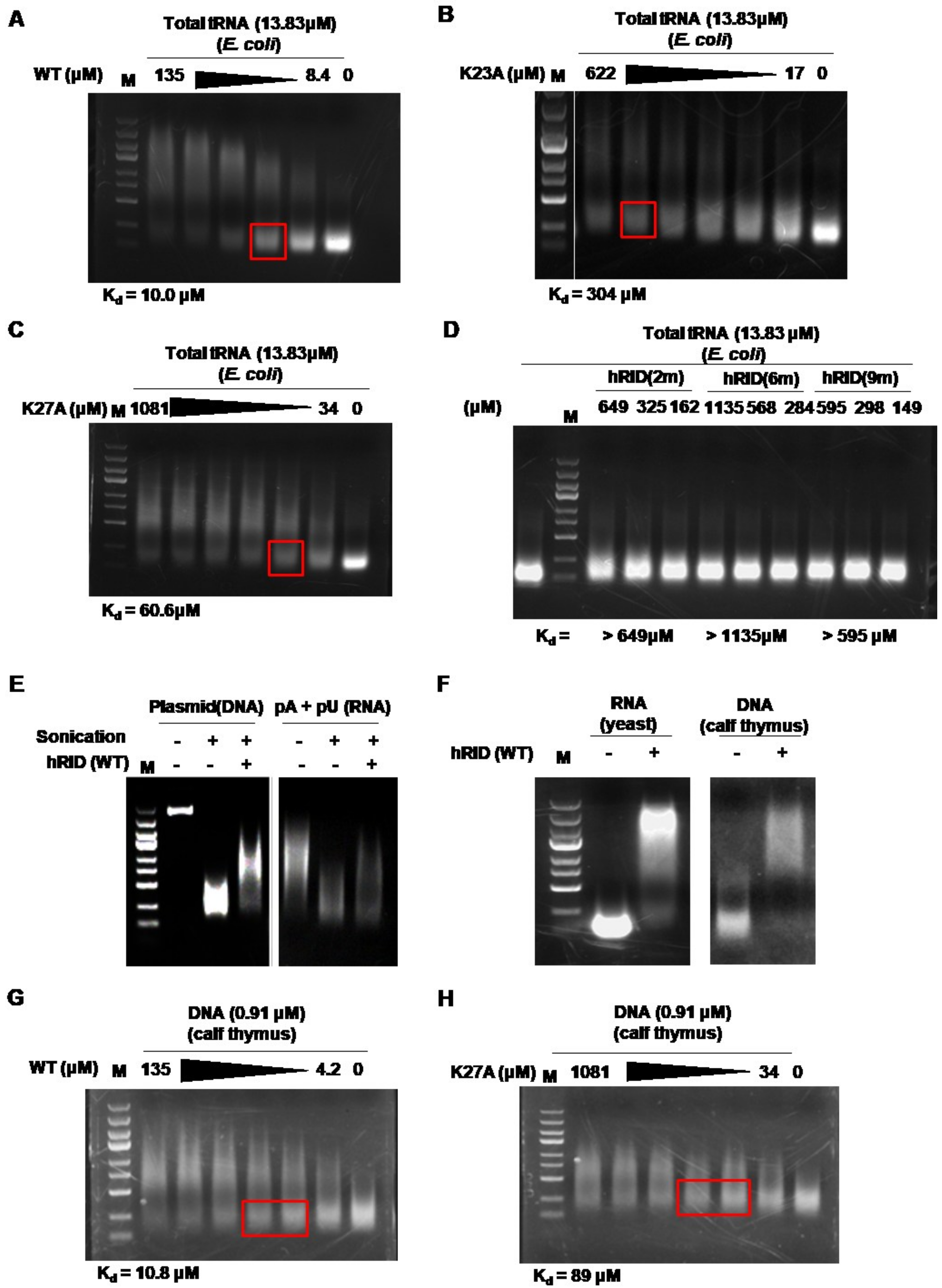
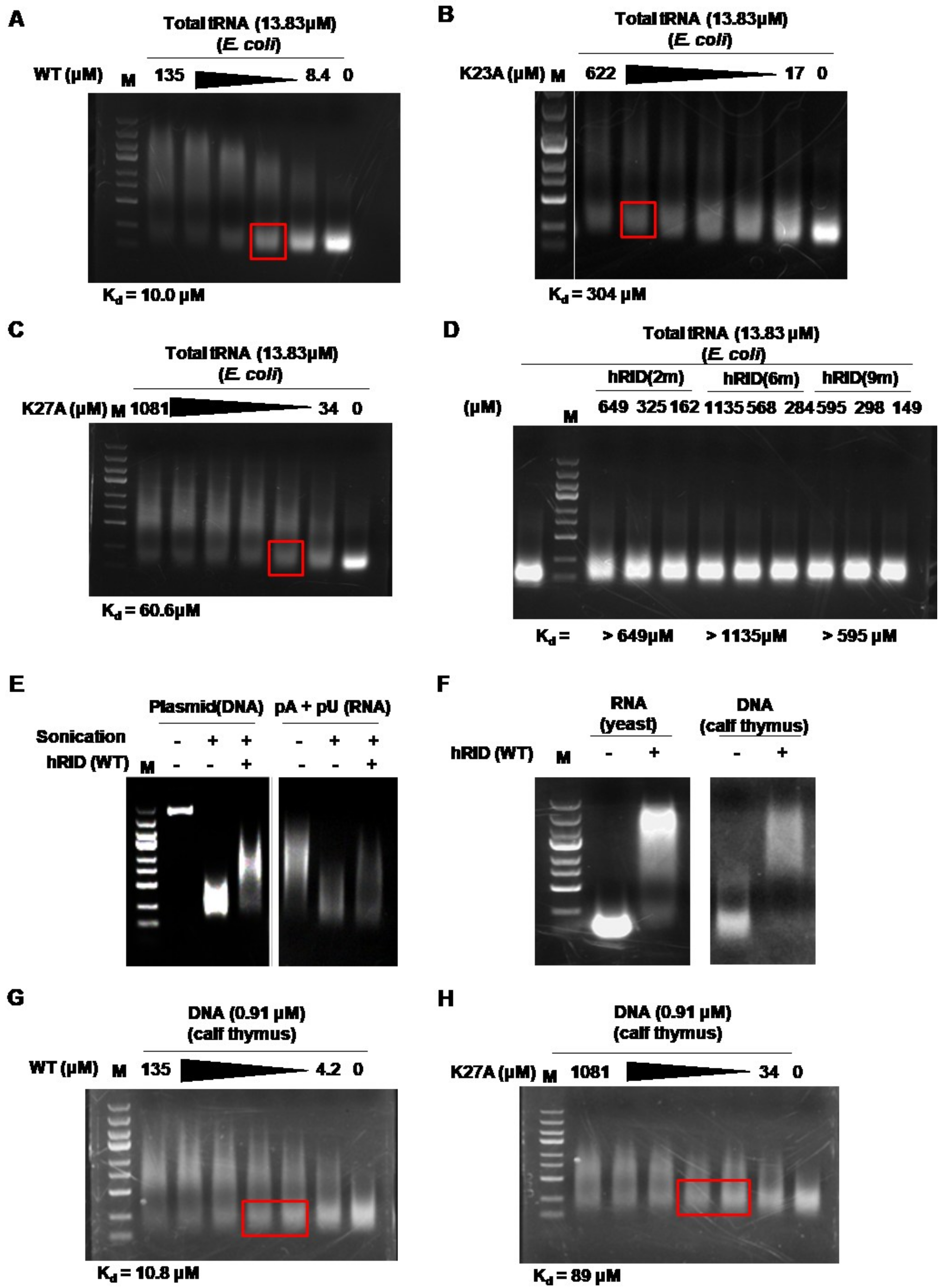
2.3. Binding Affinity of hRID and Its Mutants
2.4. hRID Can Bind to NAs Non-Specifically
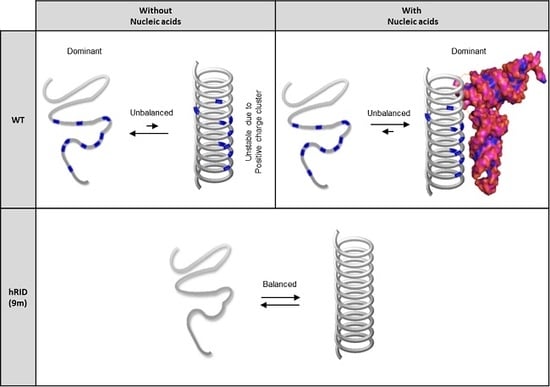
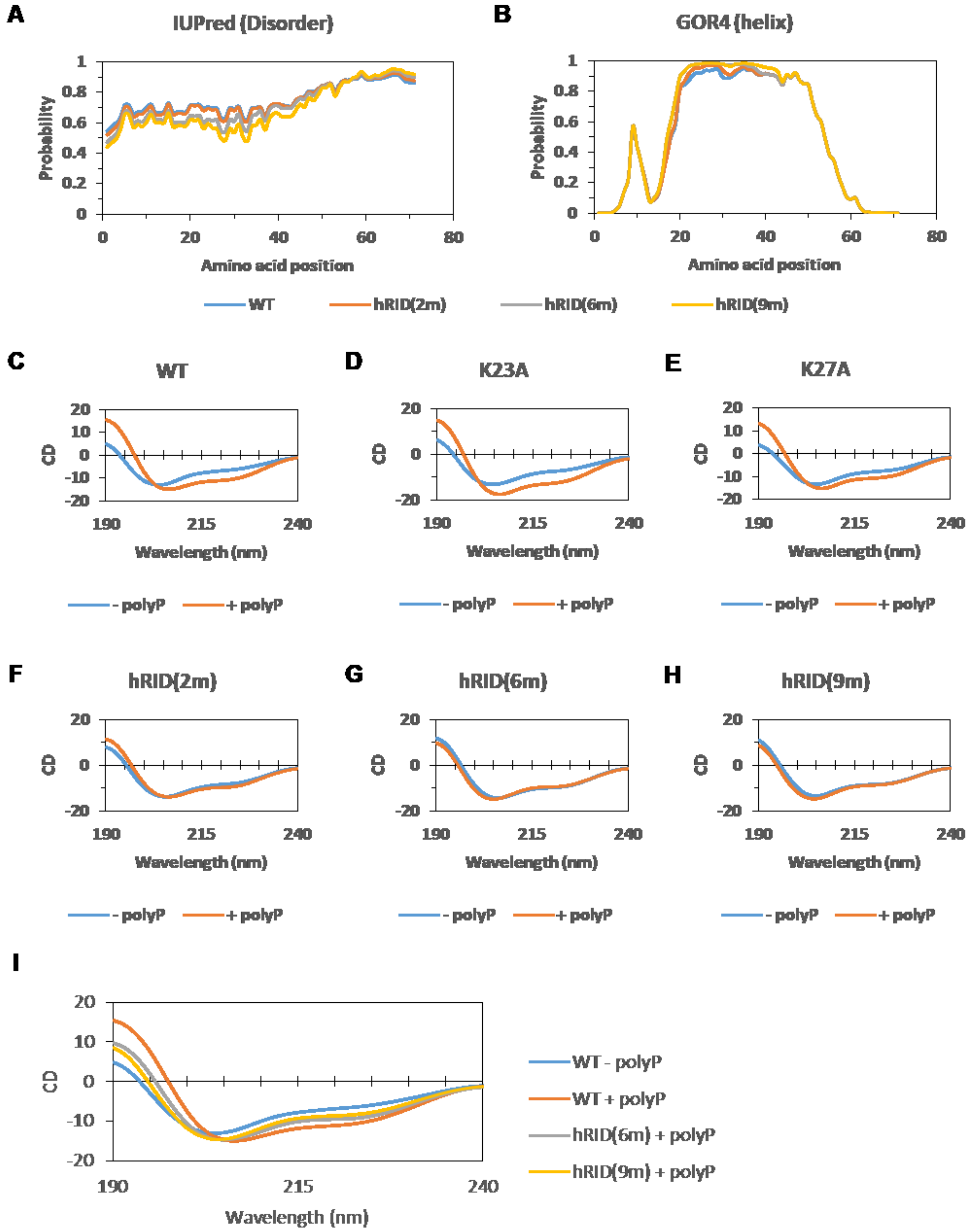
2.5. Secondary Structure of WT and Mutant hRID Constructs
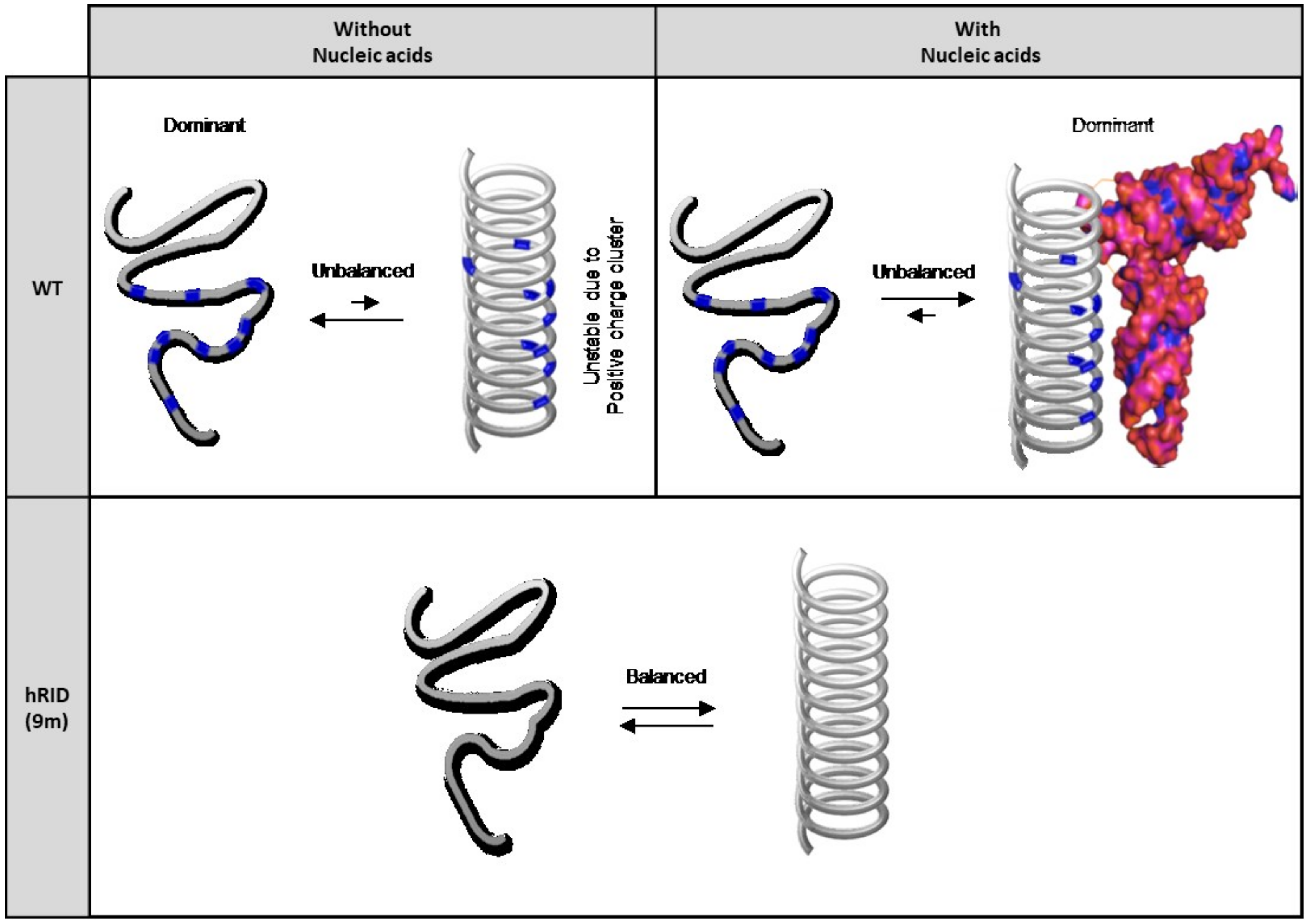
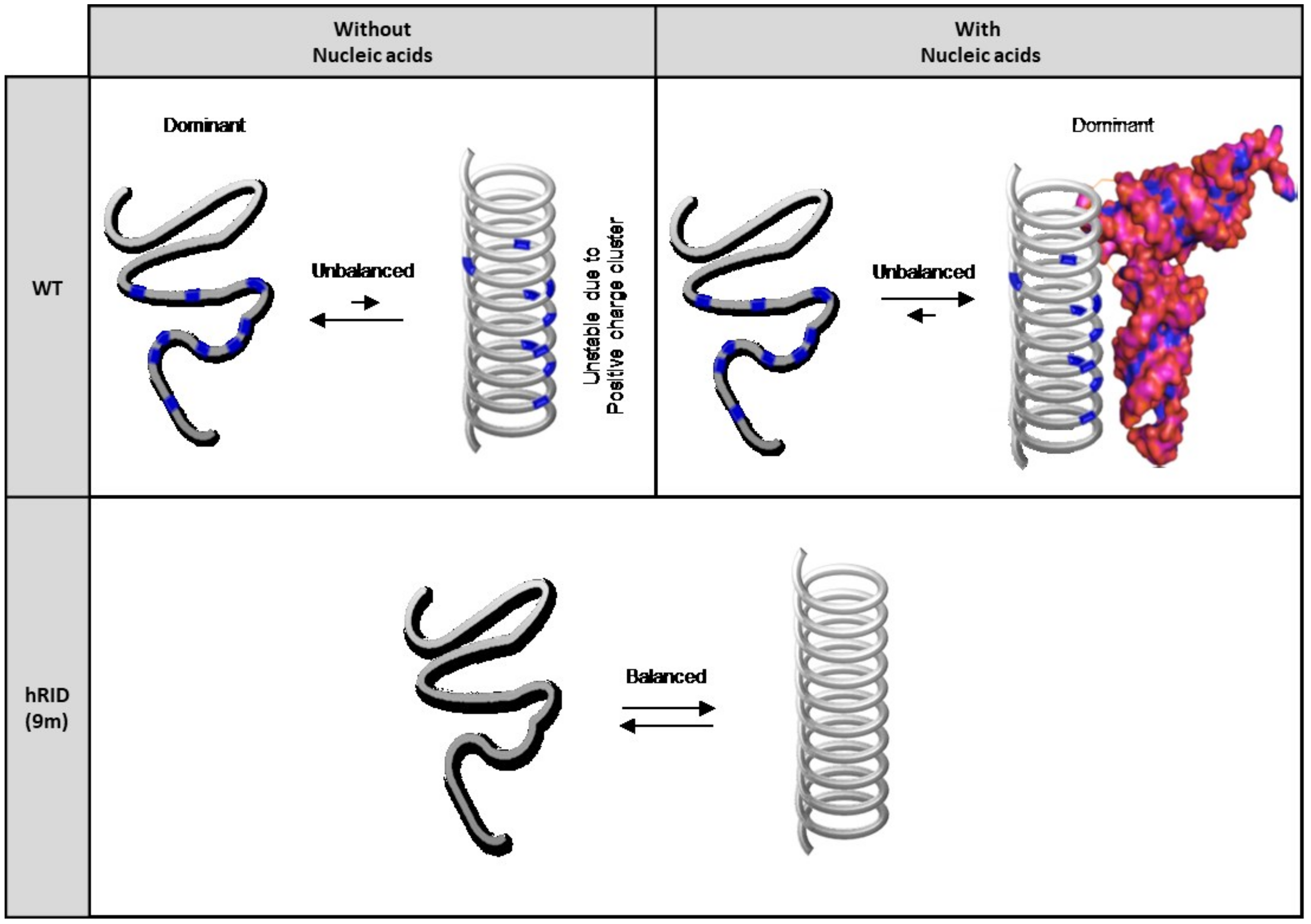
3. Discussion
4. Materials and Methods
4.1. Expression Vector Construction
4.2. Protein Expression and Purification
4.3. Large Culture and Purification
4.4. Electrophoresis Mobility Shift Assay
4.5. Prediction of the Intrinsically Disordered Regions and the Protein Secondary Structure
4.6. Circular Dichroism
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Mirande, M. Aminoacyl-tRNA synthetase family from prokaryotes and eukaryotes: Structural domains and their implications. Prog. Nucleic Acid Res. Mol. Biol. 1991, 40, 95–142. [Google Scholar] [PubMed]
- Brevet, A.; Chen, J.; Leveque, F.; Blanquet, S.; Plateau, P. Comparison of the enzymatic properties of the two Escherichia coli lysyl-tRNA synthetase species. J. Biol. Chem. 1995, 270, 14439–14444. [Google Scholar] [CrossRef] [PubMed]
- Commans, S.; Plateau, P.; Blanquet, S.; Dardel, F. Solution structure of the anticodon-binding domain of Escherichia coli lysyl-tRNA synthetase and studies of its interaction with tRNA(Lys). J. Mol. Biol. 1995, 253, 100–113. [Google Scholar] [CrossRef] [PubMed]
- Guo, M.; Ignatov, M.; Musier-Forsyth, K.; Schimmel, P.; Yang, X.L. Crystal structure of tetrameric form of human lysyl-tRNA synthetase: Implications for multisynthetase complex formation. Proc. Natl. Acad. Sci. USA 2008, 105, 2331–2336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Motzik, A.; Nechushtan, H.; Foo, S.Y.; Razin, E. Non-canonical roles of lysyl-tRNA synthetase in health and disease. Trend. Mol. Med. 2013, 19, 726–731. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.S.; Kwon, H.; Nguyen, L.T.; Lee, E.Y.; Lee, C.Y.; Choi, S.H.; Kim, M.H. Shiga toxins trigger the secretion of Lysyl-tRNA synthetase to enhance proinflammatory responses. J. Microbiol. Biotechnol. 2016, 26, 432–439. [Google Scholar] [CrossRef] [PubMed]
- Francin, M.; Kaminska, M.; Kerjan, P.; Mirande, M. The N-terminal domain of mammalian Lysyl-tRNA synthetase is a functional tRNA-binding domain. J. Biol. Chem. 2002, 277, 1762–1769. [Google Scholar] [CrossRef] [PubMed]
- Francin, M.; Mirande, M. Functional dissection of the eukaryotic-specific tRNA-interacting factor of lysyl-tRNA synthetase. J. Biol. Chem. 2003, 278, 1472–1479. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.P.; Lin, G.; Chen, S.J.; Chiu, W.C.; Chen, W.H.; Wang, C.C. Promoting the formation of an active synthetase/tRNA complex by a nonspecific tRNA-binding domain. J. Biol. Chem. 2008, 283, 30699–30706. [Google Scholar] [CrossRef] [PubMed]
- Agou, F.; Yang, Y.; Gesquiere, J.C.; Waller, J.P.; Guittet, E. Polyanion-induced alpha-helical structure of a synthetic 23-residue peptide representing the lysine-rich segment of the N-terminal extension of yeast cytoplasmic aspartyl-tRNA synthetase. Biochemistry 1995, 34, 569–576. [Google Scholar] [CrossRef] [PubMed]
- Kwon, S.B.; Kim, P.; Woo, H.S.; Kim, T.Y.; Kim, J.Y.; Lee, H.M.; Jang, Y.S.; Kim, E.M.; Yong, T.S.; Seong, B.L. Recombinant adenylate kinase 3 from liver fluke Clonorchis sinensis for histochemical analysis and serodiagnosis of clonorchiasis. Parasitology 2018, 145, 1531–1539. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.J.; Yu, J.E.; Kim, P.; Lee, J.Y.; Cheong, Y.C.; Lee, Y.J.; Chang, J.; Seong, B.L. Eliciting unnatural immune responses by activating cryptic epitopes in viral antigens. FASEB J. 2018. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.W.; Jang, Y.H.; Kwon, S.B.; Lee, Y.J.; Chae, W.; Byun, Y.H.; Kim, P.; Park, C.; Lee, Y.J.; Kim, C.K.; et al. Harnessing an RNA-mediated chaperone for the assembly of influenza hemagglutinin in an immunologically relevant conformation. FASEB J. 2018, 32, 2658–2675. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.S.; Son, A.; Kim, J.; Kwon, S.B.; Kim, M.H.; Kim, P.; Kim, J.; Byun, Y.H.; Sung, J.; Lee, J.; et al. Chaperna-mediated assembly of ferritin-based middle east respiratory syndrome-Coronavirus nanoparticles. Front. Immunol. 2018, 9, 1093. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.I.; Han, K.S.; Kim, C.W.; Ryu, K.S.; Kim, B.H.; Kim, K.H.; Kim, S.I.; Kang, T.H.; Shin, H.C.; Lim, K.H.; et al. Protein solubility and folding enhancement by interaction with RNA. PLoS ONE 2008, 3, e2677. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.I.; Ryu, K.; Seong, B.L. RNA-mediated chaperone type for de novo protein folding. RNA Biol. 2009, 6, 21–24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, H.K.; Choi, S.I.; Seong, B.L. 5S rRNA-assisted DnaK refolding. Biochem. Biophys. Res. Commun. 2010, 391, 1177–1181. [Google Scholar] [CrossRef] [PubMed]
- Horowitz, S.; Bardwell, J.C. RNAs as chaperones. RNA Biol. 2016, 13, 1228–1231. [Google Scholar] [CrossRef] [PubMed]
- Das, D.; Das, A.; Samanta, D.; Ghosh, J.; Dasgupta, S.; Bhattacharya, A.; Basu, A.; Sanyal, S.; Das Gupta, C. Role of the ribosome in protein folding. Biotechnol. J. 2008, 3, 999–1009. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.W.; Cho, B.H.; Park, S.G.; Kim, S. Aminoacyl-tRNA synthetase complexes: Beyond translation. J. Cell Sci. 2004, 117, 3725–3734. [Google Scholar] [CrossRef] [PubMed]
- Glasel, J.A. Validity of nucleic acid purities monitored by 260nm/280nm absorbance ratios. Biotechniques 1995, 18, 62–63. [Google Scholar] [PubMed]
- Tompa, P. Intrinsically unstructured proteins. Trends Biochem. Sci. 2002, 27, 527–533. [Google Scholar] [CrossRef]
- Wang, C.C.; Schimmel, P. Species barrier to RNA recognition overcome with nonspecific RNA binding domains. J. Biol. Chem. 1999, 274, 16508–16512. [Google Scholar] [CrossRef] [PubMed]
- Docter, B.E.; Horowitz, S.; Gray, M.J.; Jakob, U.; Bardwell, J.C. Do nucleic acids moonlight as molecular chaperones? Nucleic Acids Res. 2016, 44, 4835–4845. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bremer, H.; Dennis, P.P. Modulation of chemical composition and other parameters of the cell at different exponential growth rates. EcoSal Plus 2008, 3. [Google Scholar] [CrossRef] [PubMed]
- Guo, M.; Yang, X.L.; Schimmel, P. New functions of aminoacyl-tRNA synthetases beyond translation. Nat. Rev. Mol. Cell Biol. 2010, 11, 668–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yao, P.; Fox, P.L. Aminoacyl-tRNA synthetases in medicine and disease. EMBO Mol. Med. 2013, 5, 332–343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, C.W.; Han, K.S.; Ryu, K.S.; Kim, B.H.; Kim, K.H.; Choi, S.I.; Seong, B.L. N-terminal domains of native multidomain proteins have the potential to assist de novo folding of their downstream domains in vivo by acting as solubility enhancers. Protein Sci. 2007, 16, 635–643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grana-Montes, R.; Marinelli, P.; Reverter, D.; Ventura, S. N-terminal protein tails act as aggregation protective entropic bristles: The SUMO case. Biomacromolecules 2014, 15, 1194–1203. [Google Scholar] [CrossRef] [PubMed]
- Santner, A.A.; Croy, C.H.; Vasanwala, F.H.; Uversky, V.N.; Van, Y.Y.; Dunker, A.K. Sweeping away protein aggregation with entropic bristles: Intrinsically disordered protein fusions enhance soluble expression. Biochemistry 2012, 51, 7250–7262. [Google Scholar] [CrossRef] [PubMed]
- Yannay-Cohen, N.; Carmi-Levy, I.; Kay, G.; Yang, C.M.; Han, J.M.; Kemeny, D.M.; Kim, S.; Nechushtan, H.; Razin, E. LysRS serves as a key signaling molecule in the immune response by regulating gene expression. Mol. Cell 2009, 34, 603–611. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.N.; Razin, E. Nonconventional involvement of LysRS in the molecular mechanism of USF2 transcriptional activity in FcepsilonRI-activated mast cells. Mol. Cell. Biol. 2005, 25, 8904–8912. [Google Scholar] [CrossRef] [PubMed]
- Nam, S.H.; Kim, D.; Lee, M.S.; Lee, D.; Kwak, T.K.; Kang, M.; Ryu, J.; Kim, H.J.; Song, H.E.; Choi, J.; et al. Noncanonical roles of membranous lysyl-tRNA synthetase in transducing cell-substrate signaling for invasive dissemination of colon cancer spheroids in 3D collagen I gels. Oncotarget 2015, 6, 21655–21674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, S.; Zhang, D.; Wang, M.; Cui, W.; Chen, K.; Du, G.; Chen, J.; Zhou, Z. The order of expression is a key factor in the production of active transglutaminase in Escherichia coli by co-expression with its pro-peptide. Microb. Cell Fact. 2011, 10, 112. [Google Scholar] [CrossRef] [PubMed]
- Son, A.; Choi, S.I.; Han, G.; Seong, B.L. M1 RNA is important for the in-cell solubility of its cognate C5 protein: Implications for RNA-mediated protein folding. RNA Biol. 2015, 12, 1198–1208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.M.; Choi, H.S.; Seong, B.L. The folding competence of HIV-1 Tat mediated by interaction with TAR RNA. RNA Biol. 2017, 14, 926–937. [Google Scholar] [CrossRef] [PubMed]
- Shiber, A.; Doring, K.; Friedrich, U.; Klann, K.; Merker, D.; Zedan, M.; Tippmann, F.; Kramer, G.; Bukau, B. Cotranslational assembly of protein complexes in eukaryotes revealed by ribosome profiling. Nature 2018, 561, 268–272. [Google Scholar] [CrossRef] [PubMed]
- Park, S.G.; Kim, H.J.; Min, Y.H.; Choi, E.C.; Shin, Y.K.; Park, B.J.; Lee, S.W.; Kim, S. Human lysyl-tRNA synthetase is secreted to trigger proinflammatory response. Proc. Natl. Acad. Sci. USA. 2005, 102, 6356–6361. [Google Scholar] [CrossRef] [PubMed]
- Halwani, R.; Cen, S.; Javanbakht, H.; Saadatmand, J.; Kim, S.; Shiba, K.; Kleiman, L. Cellular distribution of Lysyl-tRNA synthetase and its interaction with Gag during human immunodeficiency virus type 1 assembly. J. Virol. 2004, 78, 7553–7564. [Google Scholar] [CrossRef] [PubMed]
- Ofir-Birin, Y.; Fang, P.; Bennett, S.P.; Zhang, H.M.; Wang, J.; Rachmin, I.; Shapiro, R.; Song, J.; Dagan, A.; Pozo, J.; et al. Structural switch of lysyl-tRNA synthetase between translation and transcription. Mol. Cell 2013, 49, 30–42. [Google Scholar] [CrossRef] [PubMed]
- Nechushtan, H.; Kim, S.; Kay, G.; Razin, E. Chapter 1: The physiological role of lysyl tRNA synthetase in the immune system. Adv. Immunol. 2009, 103, 1–27. [Google Scholar] [PubMed]
- Theillet, F.X.; Binolfi, A.; Frembgen-Kesner, T.; Hingorani, K.; Sarkar, M.; Kyne, C.; Li, C.; Crowley, P.B.; Gierasch, L.; Pielak, G.J.; et al. Physicochemical properties of cells and their effects on intrinsically disordered proteins (IDPs). Chem. Rev. 2014, 114, 6661–6714. [Google Scholar] [CrossRef] [PubMed]
- Qing, G.; Xia, B.; Inouye, M. Enhancement of translation initiation by A/T-rich sequences downstream of the initiation codon in Escherichia coli. J. Mol. Microbiol. Biotechnol. 2003, 6, 133–144. [Google Scholar] [CrossRef] [PubMed]
- Dosztanyi, Z.; Csizmok, V.; Tompa, P.; Simon, I. The pairwise energy content estimated from amino acid composition discriminates between folded and intrinsically unstructured proteins. J. Mol. Biol. 2005, 347, 827–839. [Google Scholar] [CrossRef] [PubMed]
- Dosztanyi, Z.; Csizmok, V.; Tompa, P.; Simon, I. IUPred: Web server for the prediction of intrinsically unstructured regions of proteins based on estimated energy content. Bioinformatics 2005, 21, 3433–3434. [Google Scholar] [CrossRef] [PubMed]
- Combet, C.; Blanchet, C.; Geourjon, C.; Deleage, G. NPS@: Network protein sequence analysis. Trends Biochem. Sci. 2000, 25, 147–150. [Google Scholar] [CrossRef]

© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kwon, S.B.; Yu, J.E.; Park, C.; Lee, J.; Seong, B.L. Nucleic Acid-Dependent Structural Transition of the Intrinsically Disordered N-Terminal Appended Domain of Human Lysyl-tRNA Synthetase. Int. J. Mol. Sci. 2018, 19, 3016. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms19103016
Kwon SB, Yu JE, Park C, Lee J, Seong BL. Nucleic Acid-Dependent Structural Transition of the Intrinsically Disordered N-Terminal Appended Domain of Human Lysyl-tRNA Synthetase. International Journal of Molecular Sciences. 2018; 19(10):3016. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms19103016
Chicago/Turabian StyleKwon, Soon Bin, Ji Eun Yu, Chan Park, Jiseop Lee, and Baik L. Seong. 2018. "Nucleic Acid-Dependent Structural Transition of the Intrinsically Disordered N-Terminal Appended Domain of Human Lysyl-tRNA Synthetase" International Journal of Molecular Sciences 19, no. 10: 3016. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms19103016