Identification of a Novel PPAR-γ Agonist through a Scaffold Tuning Approach

Abstract
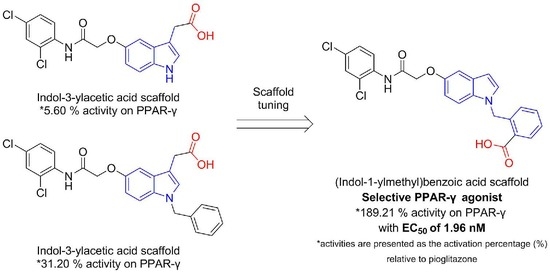
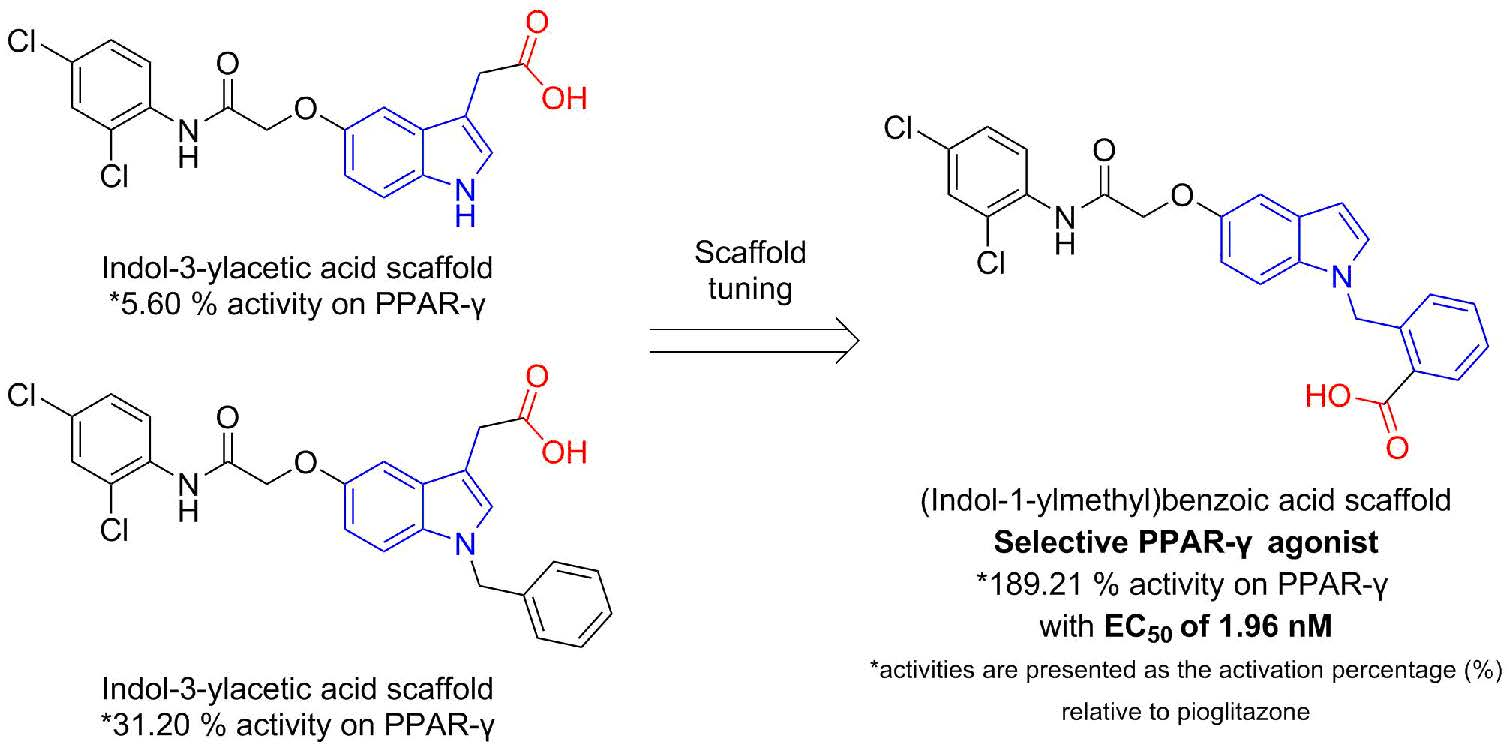
:
1. Introduction
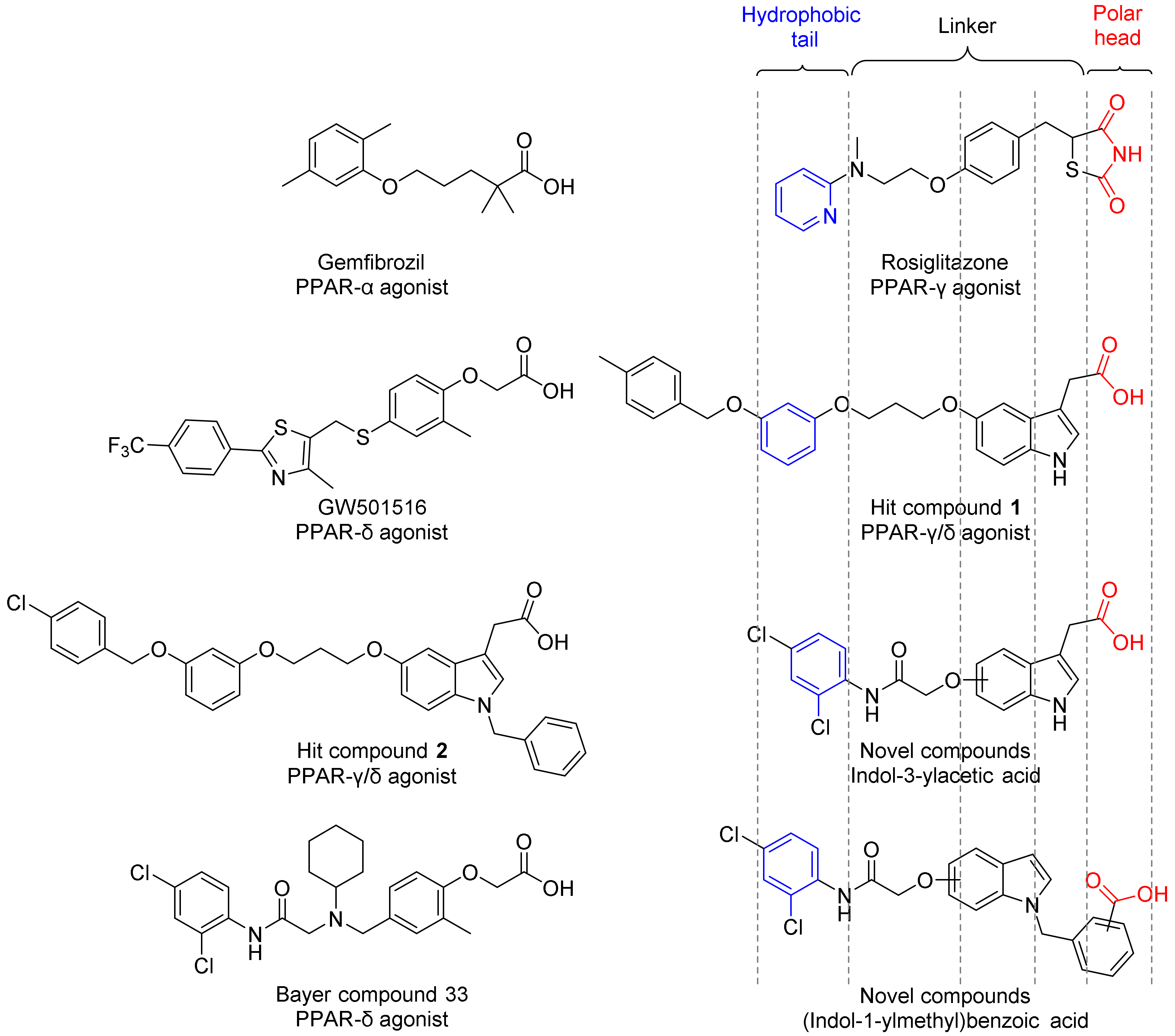
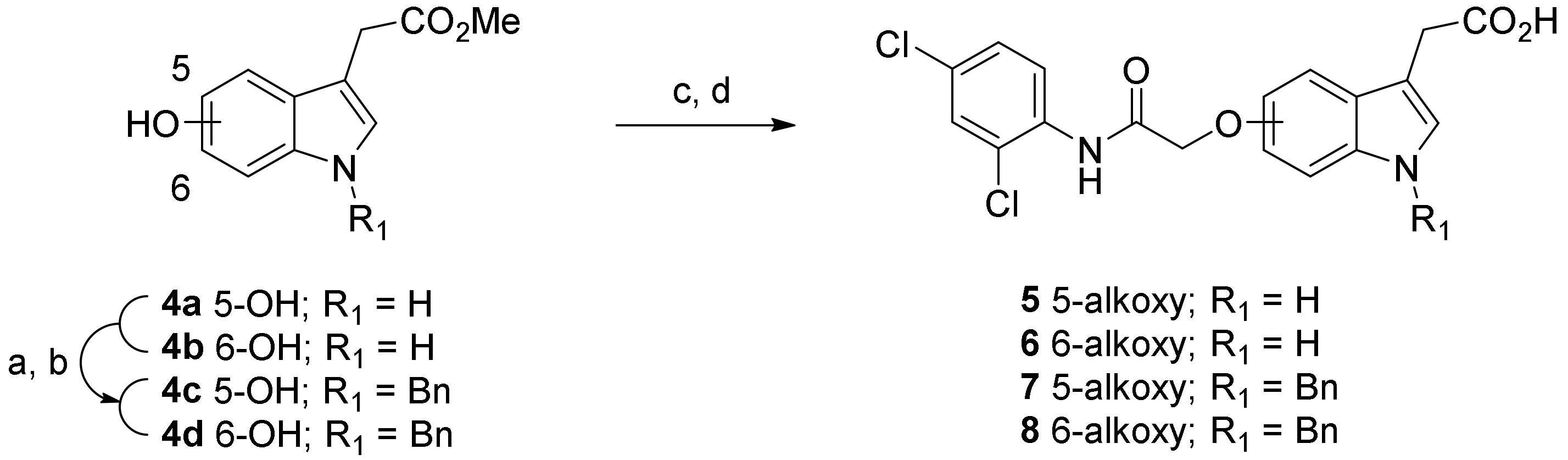
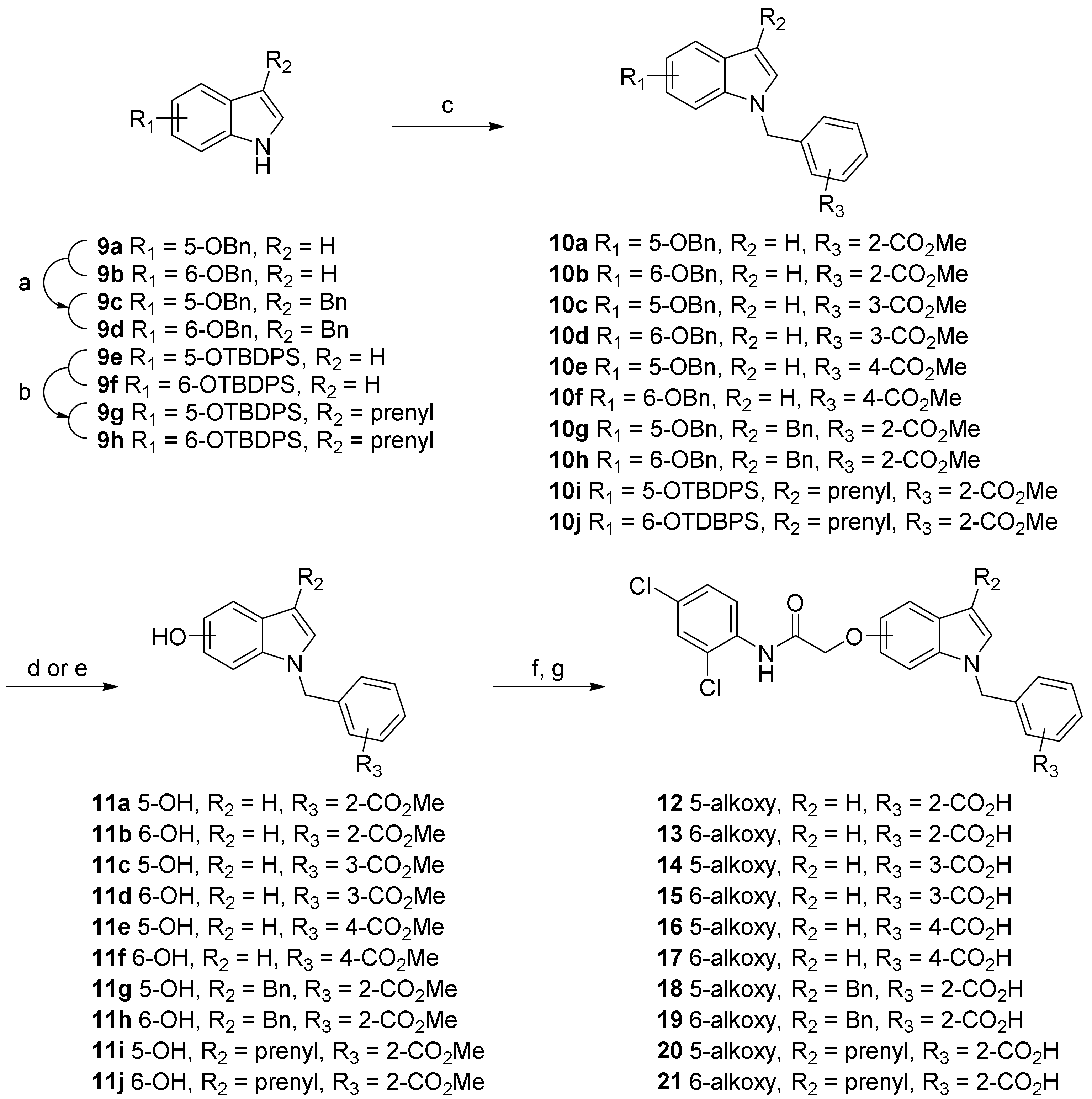
2. Result and Discussion
3. Materials and Methods
3.1. General Information
3.2. Experimental Procedures and Analytical Data of Compounds
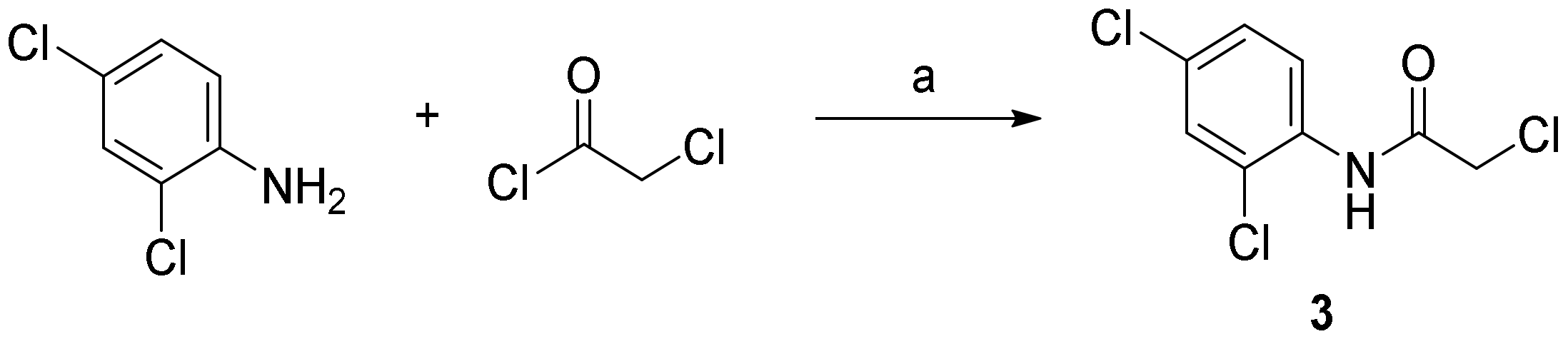
3.2.1. 2-Chloro-N-(2,4-Dichlorophenyl)Acetamide (3)
3.2.2. Synthetic Procedure for O-Alkylation of 5- or 6-Hydroxyindole Compounds
3.2.3. Synthetic Procedure for Hydrolysis of the Methyl Esters to Yield the Target Acid Compounds
3.3. In Vitro PPAR Transactivation Assay
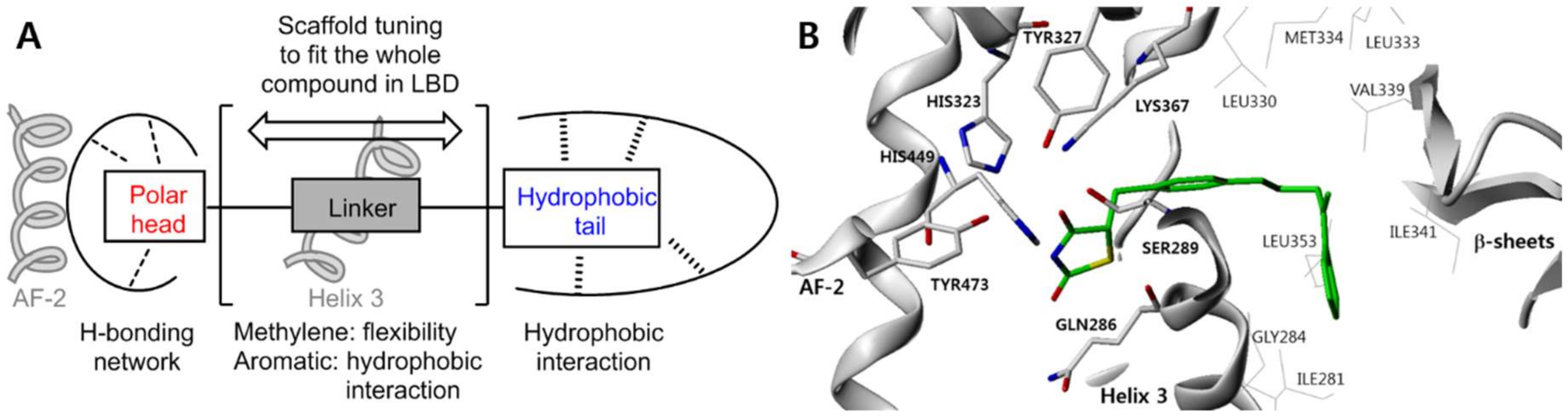
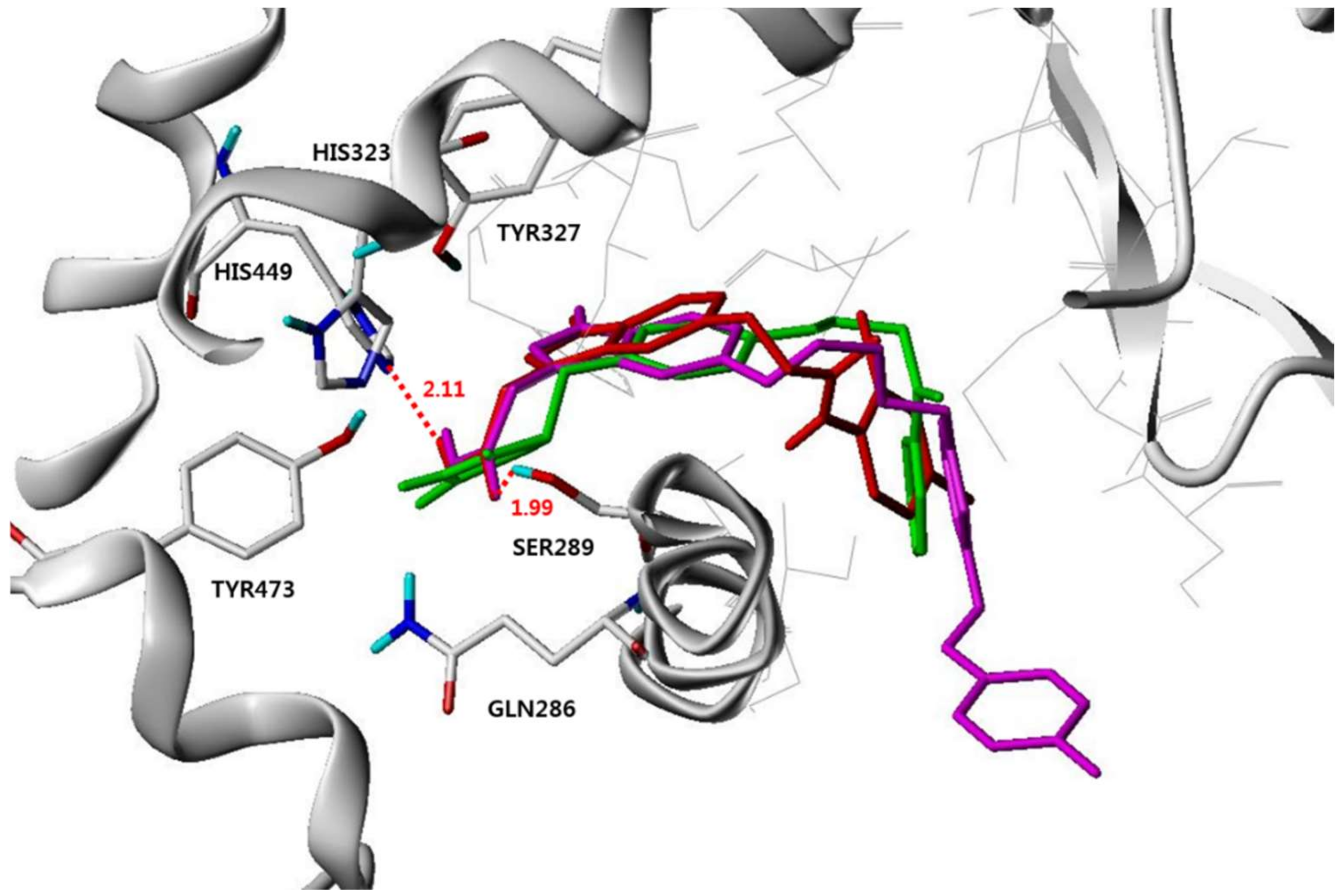
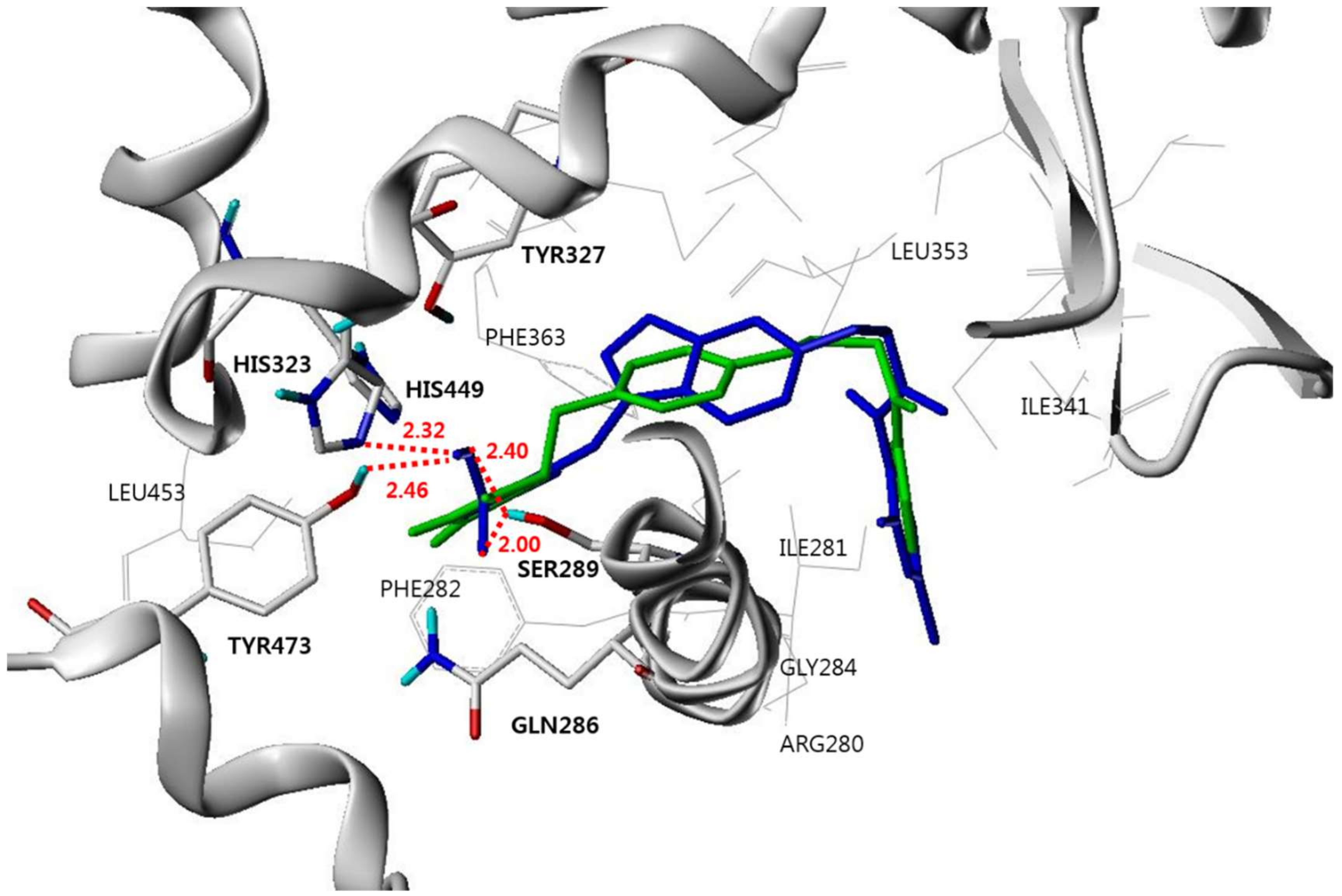
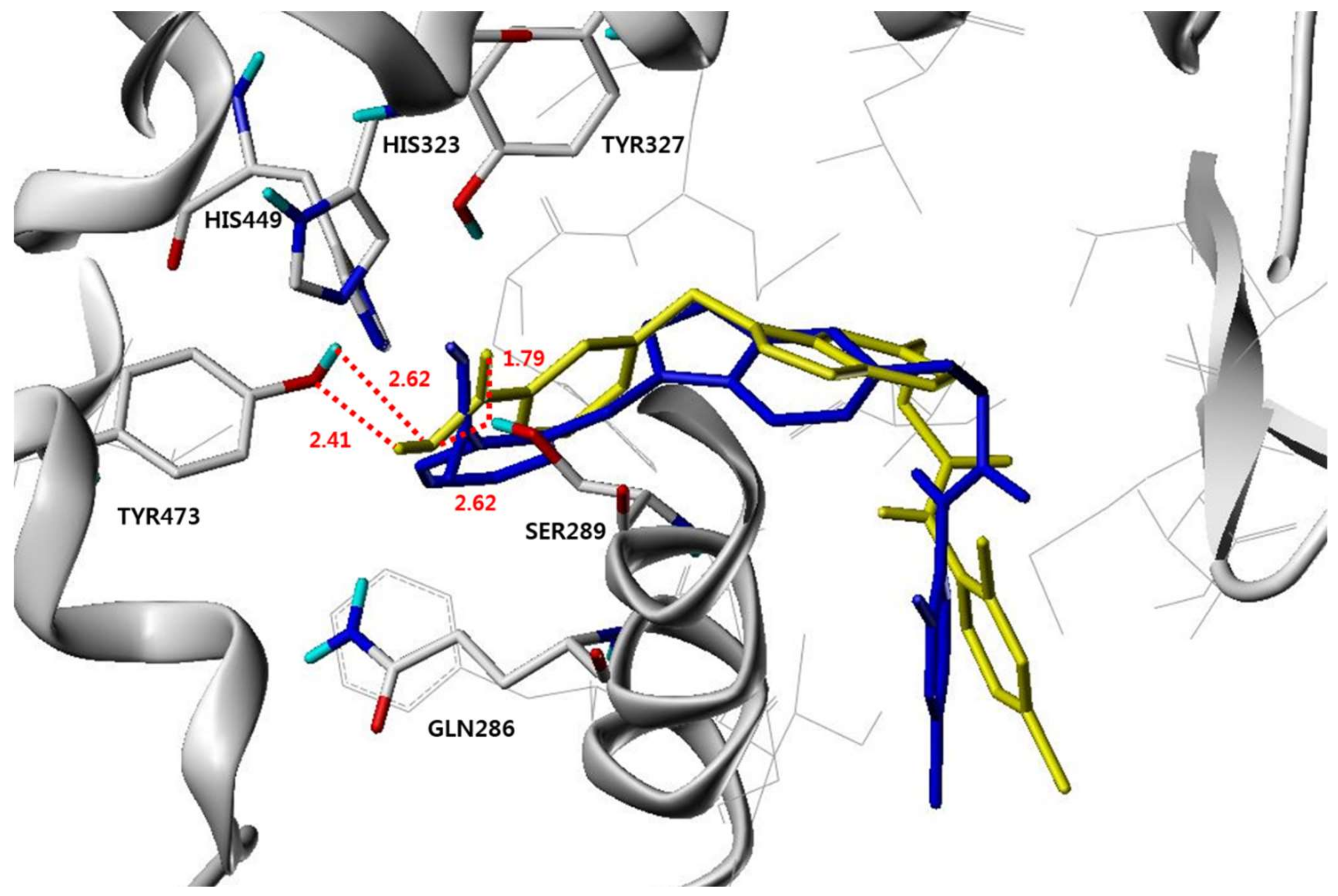
3.4. Molecular Modeling
3.5. In vitro PPAR Coactivator Recruit Assay
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Stales, B.; Dallongeville, J.; Auwerx, J.; Schoonjans, K.; Leitersdorf, E.; Fruchart, J.-C. Mechanism of action of fibrates on lipid and lipoprotein metabolism. Circulation 1998, 98, 2088–2093. [Google Scholar] [CrossRef]
- Berger, J.; Moller, D.E. The mechanisms of action of PPARs. Annu. Rev. Med. 2002, 53, 409–435. [Google Scholar] [CrossRef] [PubMed]
- Semple, R.K.; Chatterjee, V.K.K.; O’Rahilly, S. PPAR-γ and human metabolic disease. J. Clin. Investig. 2006, 116, 581–589. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.-X. PPARs: Diverse regulators in energy metabolism and metabolic diseases. Cell Res. 2010, 20, 124–137. [Google Scholar] [CrossRef] [PubMed]
- Ahmadian, M.; Suh, J.M.; Hah, N.; Liddle, C.; Atkins, A.R.; Downes, M.; Evans, R.M. PPAR-γ signaling and metabolism: The good, the bad and the future. Nat. Med. 2013, 19, 557–566. [Google Scholar] [CrossRef] [PubMed]
- Vamecq, J.; Latruffe, N. Medical significance of peroxisome proliferator-activated receptors. Lancet 1999, 354, 141–148. [Google Scholar] [CrossRef]
- Kersten, S.; Desvergne, B.; Wahli, W. Roles of PPARs in health and disease. Nature 2000, 405, 421–424. [Google Scholar] [CrossRef] [PubMed]
- Berger, J.P.; Akiyama, T.E.; Meinke, P.T. PPARs: Therapeutic targets for the metabolic disease. Trends Pharmacol. Sci. 2005, 26, 244–251. [Google Scholar] [CrossRef] [PubMed]
- Jay, M.A.; Ren, J. Peroxisome proliferator-activated receptor (PPAR) in metabolic syndrome and Type 2 diabetes. Curr. Diabetes Rev. 2007, 3, 33–39. [Google Scholar] [CrossRef] [PubMed]
- Tain, Y.-L.; Hsu, C.-N.; Chan, J.Y.H. PPARs link early life nutritional insults to later programmed hypertension and metabolic syndrome. Int. J. Mol. Sci. 2016, 17, 20–30. [Google Scholar] [CrossRef] [PubMed]
- Gross, B.; Pawlak, M.; Lefebvre, P.; Staels, B. PPARs in obesity-induced T2DM, dyslipidemia and NAFLD. Nat. Rev. Endocrinol. 2017, 13, 36–49. [Google Scholar] [CrossRef] [PubMed]
- Delerive, P.; Fruchart, J.-C.; Staels, B. Peroxisome proliferator-activated receptors in inflammation control. J. Endocrinol. 2001, 169, 453–459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lefebvre, P.; Chinetti, G.; Fruchart, J.-C.; Staels, B. Sorting out the roles of PPAR-α in energy metabolism and vascular homeostasis. J. Clin. Investig. 2006, 116, 571–580. [Google Scholar] [CrossRef] [PubMed]
- Moraes, L.A.; Piqueras, L.; Bishop-Bailey, D. Peroxisome proliferator-activated receptors and inflammation. Pharmacol. Ther. 2006, 110, 371–385. [Google Scholar] [CrossRef] [PubMed]
- Biscetti, F.; Straface, G.; Pitocco, D.; Zaccardi, F.; Ghirlanda, G.; Flex, A. Peroxisome proliferator-activated receptors and angiogenesis. Nutr. Metab. Cardiovasc. Dis. 2009, 19, 751–759. [Google Scholar] [CrossRef] [PubMed]
- Lagana, A.S.; Vitale, S.G.; Nigro, A.; Sofo, V.; Salmeri, F.M.; Rossetti, P.; Rapisarda, A.M.C.; La Vignera, S.; Condorelli, R.A.; Rizzo, G.; et al. Pleiotropic actions of peroxisome proliferator-activated receptors (PPARs) in dysregulated metabolic homeostasis, inflammation and cancer: Current evidence and future perspectives. Int. J. Mol. Sci. 2016, 17, 999–1008. [Google Scholar] [CrossRef] [PubMed]
- Ivanova, E.A.; Parolari, A.; Myasoedova, V.; Melnichenko, A.A.; Bobryshev, Y.V.; Orekhov, A.N. Peroxisome proliferator-activated receptor (PPAR) gamma in cardiovascular disorders and cardiovascular surgery. J. Cardiol. 2015, 66, 271–278. [Google Scholar] [CrossRef] [PubMed]
- Han, L.; Shen, W.-J.; Bittner, S.; Kraemer, F.B.; Azhar, S. PPARs: Regulators of metabolism and as therapeutic targets in cardiovascular disease. Part I: PPAR-α. Future Cardiol. 2017, 13, 259–278. [Google Scholar] [CrossRef] [PubMed]
- Han, L.; Shen, W.-J.; Bittner, S.; Kraemer, F.B.; Azhar, S. PPARs: Regulators of metabolism and as therapeutic targets in cardiovascular disease. Part II: PPAR-β/δ and PPAR-γ. Future Cardiol. 2017, 13, 279–296. [Google Scholar] [CrossRef] [PubMed]
- Copland, J.A.; Marlow, L.A.; Kurakata, S.; Fujiwara, K.; Wong, A.K.C.; Kreinest, P.A.; Williams, S.F.; Haugen, B.R.; Klopper, J.P.; Smallridge, R.C. Novel high-affinity PPARγ agonist alone and in combination with paclitaxel inhibits human anaplastic thyroid carcinoma tumor growth via p21WAF1/CIP1. Oncogene 2006, 25, 2304–2317. [Google Scholar] [CrossRef] [PubMed]
- Shimazaki, N.; Togashi, N.; Hanai, M.; Isoyama, T.; Wada, K.; Fujita, T.; Fujiwara, K.; Kurakata, S. Anti-tumour activity of CS-7017, a selective peroxisome proliferator-activated receptor gamma agonist of thiazolidinedione class, in human tumour xenografts and a syngeneic tumour implant model. Eur. J. Cancer 2008, 44, 1734–1743. [Google Scholar] [CrossRef] [PubMed]
- Serizawa, M.; Murakami, H.; Watanabe, M.; Takahashi, T.; Yamamoto, N.; Koh, Y. Peroxisome proliferator-activated receptor γ agonist efatutazone impairs transforming growth factor β2-induced motility of epidermal growth factor receptor tyrosine kinase inhibitor-resistant lung cancer cells. Cancer Sci. 2014, 105, 683–689. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prost, S.; Relouzat, F.; Spentchian, M.; Ouzegdouh, Y.; Saliba, J.; Massonnet, G.; Beressi, J.-P.; Verhoeyen, E.; Raggueneau, V.; Maneglier, B.; et al. Erosion of the chronic myeloid leukaemia stem cell pool by PPARγ agonists. Nature 2015, 525, 380–383. [Google Scholar] [CrossRef] [PubMed]
- Winger, B.A.; Neil, P.S. PPARγ: Welcoming the new kid on the CML stem cell block. Cancer Cell 2015, 28, 409–411. [Google Scholar]
- Yousefi, B.; Samadi, N.; Baradaran, B.; Shafiei-Irannejad, V.; Zarghami, N. Peroxisome proliferator-activated receptor ligands and their role in chronic myeloid leukemia: Therapeutic strategies. Chem. Biol. Drug Des. 2016, 88, 17–25. [Google Scholar] [CrossRef] [PubMed]
- Werner, A.L.; Travaglini, M.T. A review of rosiglitazone in type 2 diabetes mellitus. Pharmacotherapy 2001, 21, 1082–1099. [Google Scholar] [CrossRef] [PubMed]
- Nesto, R.W.; Bell, D.; Bonow, R.O.; Fonseca, V.; Grundy, S.M.; Horton, E.S.; Le Winter, M.; Porte, D.; Semenkovich, C.F.; Smith, S.; et al. Thiazolidinedione use, fluid retention, and congestive heart failure. Circulation 2003, 108, 2941–2949. [Google Scholar] [CrossRef] [PubMed]
- Marceille, J.R.; Goins, J.A.; Soni, R.; Biery, J.C.; Lee, T.A. Chronic heart failure-related interventions after starting rosiglitazone in patients receiving insulin. Pharmacotherapy 2004, 24, 1317–1322. [Google Scholar] [CrossRef] [PubMed]
- Henke, B.R.; Adkison, K.K.; Blanchard, S.G.; Leesnitzer, L.M.; Mook, R.A. Jr.; Plunket, K.D.; Ray, J.A.; Roberson, C.; Unwalla, R.; Willson, T.M. Synthesis and biological activity of a novel series of indole-derived PPARγ agonists. Bioorg. Med. Chem. Lett. 1999, 9, 3329–3334. [Google Scholar] [CrossRef]
- Hopkins, C.R.; O’Neil, S.V.; Laufersweiler, M.C.; Wang, Y.; Pokross, M.; Mekel, M.; Evdokimov, A.; Walter, R.; Kontoyianni, M.; Petrey, M.E.; et al. Design and synthesis of novel N-sulfonyl-2-indole carboxamides as potent PPAR-γ binding agents with potential application to the treatment of osteoporosis. Bioorg. Med. Chem. Lett. 2006, 16, 5659–5663. [Google Scholar] [CrossRef] [PubMed]
- Le Naour, M.; Leclerc, V.; Farce, A.; Caignard, D.H.; Hennuyer, N.; Staels, B.; Audinot-Bouchez, V.; Boutin, J.A.; Lonchampt, M.; Dacquet, C.; et al. Effect of oxime ether incorporation in acyl indole derivatives on PPAR subtype selectivity. ChemMedChem 2012, 7, 2179–2193. [Google Scholar] [CrossRef] [PubMed]
- Tsakovska, I.; Al Sharif, M.; Alov, P.; Diukendjieva, A.; Fioravanzo, E.; Cronin, M.T.D.; Pajeva, I. Molecular modelling study of the PPARγ receptor in relation to the mode of action/adverse outcome pathway framework for liver steatosis. Int. J. Mol. Sci. 2014, 15, 7651–7666. [Google Scholar] [CrossRef] [PubMed]
- Jian, Y.; He, Y.; Yang, J.; Han, W.; Zhai, X.; Zhao, Y.; Li, Y. Molecular modeling study for the design of novel peroxisome proliferator-activated receptor gamma agonists using 3D-QSAR and molecular docking. Int. J. Mol. Sci. 2018, 19, 630–644. [Google Scholar] [CrossRef] [PubMed]
- Nolte, R.T.; Wisely, G.B.; Westin, S.; Cobb, J.E.; Lambert, M.H.; Kurokawa, R.; Rosenfeld, M.G.; Willson, T.M.; Glass, C.K.; Milburn, M.V. Ligand binding and co-activator assembly of the peroxisome proliferator-activated receptor-γ. Nature 1998, 395, 137–143. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.E.; Lambert, M.H.; Montana, V.G.; Plunket, K.D.; Moore, L.B.; Collins, J.L.; Oplinger, J.A.; Kliewer, S.A.; Gampe, R.T., Jr.; McKee, D.D.; et al. Structural determinants of ligand binding selectivity between the peroxisome proliferator-activated receptors. Proc. Natl. Acad. Sci. USA 2001, 98, 13919–13924. [Google Scholar] [CrossRef] [PubMed]
- Gim, H.J.; Cheon, Y.-J.; Ryu, J.-H.; Jeon, R. Design and synthesis of benzoxazole containing indole analogs as peroxisome proliferator-activated receptor-γ/δ dual agonists. Bioorg. Med. Chem. Lett. 2011, 21, 3057–3061. [Google Scholar] [CrossRef] [PubMed]
- Gim, H.J.; Li, H.; Lee, E.; Ryu, J.-H.; Jeon, R. Design and synthesis of alkoxyindolyl-3-acetic acid analogs as peroxisome proliferator-activated receptor-γ/δ agonists. Bioorg. Med. Chem. Lett. 2013, 23, 513–517. [Google Scholar] [CrossRef] [PubMed]
- Gim, H.J.; Li, H.; Jeong, J.H.; Lee, S.J.; Sung, M.-K.; Song, M.-Y.; Park, B.-H.; Oh, S.J.; Ryu, J.-H.; Jeon, R. Design, synthesis, and biological evaluation of a series of alkoxy-3-indolylacetic acids as peroxisome proliferator-activated receptor γ/δ agonists. Bioorg. Med. Chem. 2015, 23, 3322–3336. [Google Scholar] [CrossRef] [PubMed]
- Weigand, S.; Bischoff, H.; Dittrich-Wengenroth, E.; Heckroth, H.; Lang, D.; Vaupel, A.; Woltering, M. Minor structural modifications convert a selective PPARα agonist into a potent, highly selective PPARα agonist. Bioorg. Med. Chem. Lett. 2005, 15, 4619–4623. [Google Scholar] [CrossRef] [PubMed]
- Luckhurst, C.A.; Stein, L.A.; Furber, M.; Webb, N.; Ratcliffe, M.J.; Allenby, G.; Botterell, S.; Tomlinson, W.; Martin, B.; Walding, A. Discovery of isoindoline and tetrahydroisoquinoline derivatives as potent, selective PPARδ agonists. Bioorg. Med. Chem. Lett. 2011, 21, 492–496. [Google Scholar] [CrossRef] [PubMed]
- Luckhurst, C.A.; Ratcliffe, M.; Stein, L.; Furber, M.; Botterell, S.; Laughton, D.; Tomlinson, W.; Weaver, R.; Chohan, K.; Walding, A. Synthesis and biological evaluation of N-alkylated 8-oxybenz[c]azepine derivatives as selective PPARδ agonists. Bioorg. Med. Chem. Lett. 2011, 21, 531–536. [Google Scholar] [CrossRef] [PubMed]
- Brown, P.J.; Stuart, L.W.; Hurley, K.P.; Lewis, M.C.; Winegar, D.A.; Wilson, J.G.; Wilkison, W.O.; Ittoop, O.R.; Willson, T.M. Identification of a subtype selective human PPARα agonist through parallel-array synthesis. Bioorg. Med. Chem. Lett. 2001, 11, 1225–1227. [Google Scholar] [CrossRef]
- Sznaidman, M.L.; Haffner, C.D.; Maloney, P.R.; Fivush, A.; Chao, E.; Goreham, D.; Sierra, M.L.; LeGrumelec, C.; Xu, H.E.; Montana, V.G.; et al. Novel selective small molecule agonists for peroxisome proliferator-activated receptor δ (PPARδ)-synthesis and biological activity. Bioorg. Med. Chem. Lett. 2003, 13, 1517–1521. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | |||||
|---|---|---|---|---|---|
| Cpd. No. | Alkoxy position | R1 | % max b | ||
| PPAR-α | PPAR-γ | PPAR-δ | |||
| 5 | 5 | H | −38.13 ± 0.06 | 5.60 ± 0.14 | 11.84 ± 0.35 |
| 6 | 6 | H | 1.12 ± 0.24 | 8.66 ± 0.23 | 8.27 ± 0.33 |
| 7 | 5 | benzyl | 21.41 ± 0.21 | 31.20 ± 0.38 | 44.35 ± 0.77 |
| 8 | 6 | benzyl | 31.68 ± 0.22 | 71.87 ± 0.91 | 65.41 ± 0.70 |
 | ||||||
|---|---|---|---|---|---|---|
| Cpd. No. | Alkoxy position | R2 | R3 | % max b | ||
| PPAR-α | PPAR-γ | PPAR-δ | ||||
| 12 | 5 | H | 2-CO2H | 105.08 ± 0.41 | 189.21 ± 1.67 | 94.86 ± 0.20 |
| 13 | 6 | H | 2-CO2H | 51.84 ± 0.25 | 74.54 ± 0.94 | 86.70 ± 0.53 |
| 14 | 5 | H | 3-CO2H | 10.33 ± 0.07 | 42.01 ± 0.52 | 23.85 ± 0.51 |
| 15 | 6 | H | 3-CO2H | 78.15 ± 0.04 | 126.55 ± 0.92 | 86.09 ± 0.31 |
| 16 | 5 | H | 4-CO2H | -19.98 ± 0.08 | 0.29 ± 0.06 | -1.07 ± 0.14 |
| 17 | 6 | H | 4-CO2H | -14.44 ± 0.16 | -0.84 ± 0.16 | 4.87 ± 0.24 |
| 18 | 5 | benzyl | 2-CO2H | 23.79 ± 0.11 | 93.20 ± 0.59 | 75.88 ± 0.35 |
| 19 | 6 | benzyl | 2-CO2H | 7.25 ± 0.12 | 19.03 ± 0.13 | 19.40 ± 0.54 |
| 20 | 5 | prenyl | 2-CO2H | -5.93 ± 0.14 | 5.10 ± 0.20 | 8.92 ± 0.48 |
| 21 | 6 | prenyl | 2-CO2H | -2.30 ± 0.19 | 9.55 ± 0.32 | 22.16 ± 0.59 |
 | ||||||
|---|---|---|---|---|---|---|
| Cpd. No. | Alkoxy position | R2 | R3 | EC50 (μM) | ||
| PPAR-α | PPAR-γ | PPAR-δ | ||||
| 12 | 5 | H | 2-CO2H | 3.87 | 0.00196 | 32.6 |
| 13 | 6 | H | 2-CO2H | 16.9 | 0.571 | >100 |
| 15 | 6 | H | 3-CO2H | >100 | 0.0326 | >100 |
| 18 | 5 | benzyl | 2-CO2H | >100 | >11.1 | 28.1 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gim, H.J.; Choi, Y.-S.; Li, H.; Kim, Y.-J.; Ryu, J.-H.; Jeon, R. Identification of a Novel PPAR-γ Agonist through a Scaffold Tuning Approach. Int. J. Mol. Sci. 2018, 19, 3032. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms19103032
Gim HJ, Choi Y-S, Li H, Kim Y-J, Ryu J-H, Jeon R. Identification of a Novel PPAR-γ Agonist through a Scaffold Tuning Approach. International Journal of Molecular Sciences. 2018; 19(10):3032. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms19103032
Chicago/Turabian StyleGim, Hyo Jin, Yong-Sung Choi, Hua Li, Yoon-Jung Kim, Jae-Ha Ryu, and Raok Jeon. 2018. "Identification of a Novel PPAR-γ Agonist through a Scaffold Tuning Approach" International Journal of Molecular Sciences 19, no. 10: 3032. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms19103032