Metabolomic Profiling of Portal Blood and Bile Reveals Metabolic Signatures of Primary Sclerosing Cholangitis

 , ,
, ,
Abstract
:1. Introduction
2. Results
2.1. Patient Characteristics
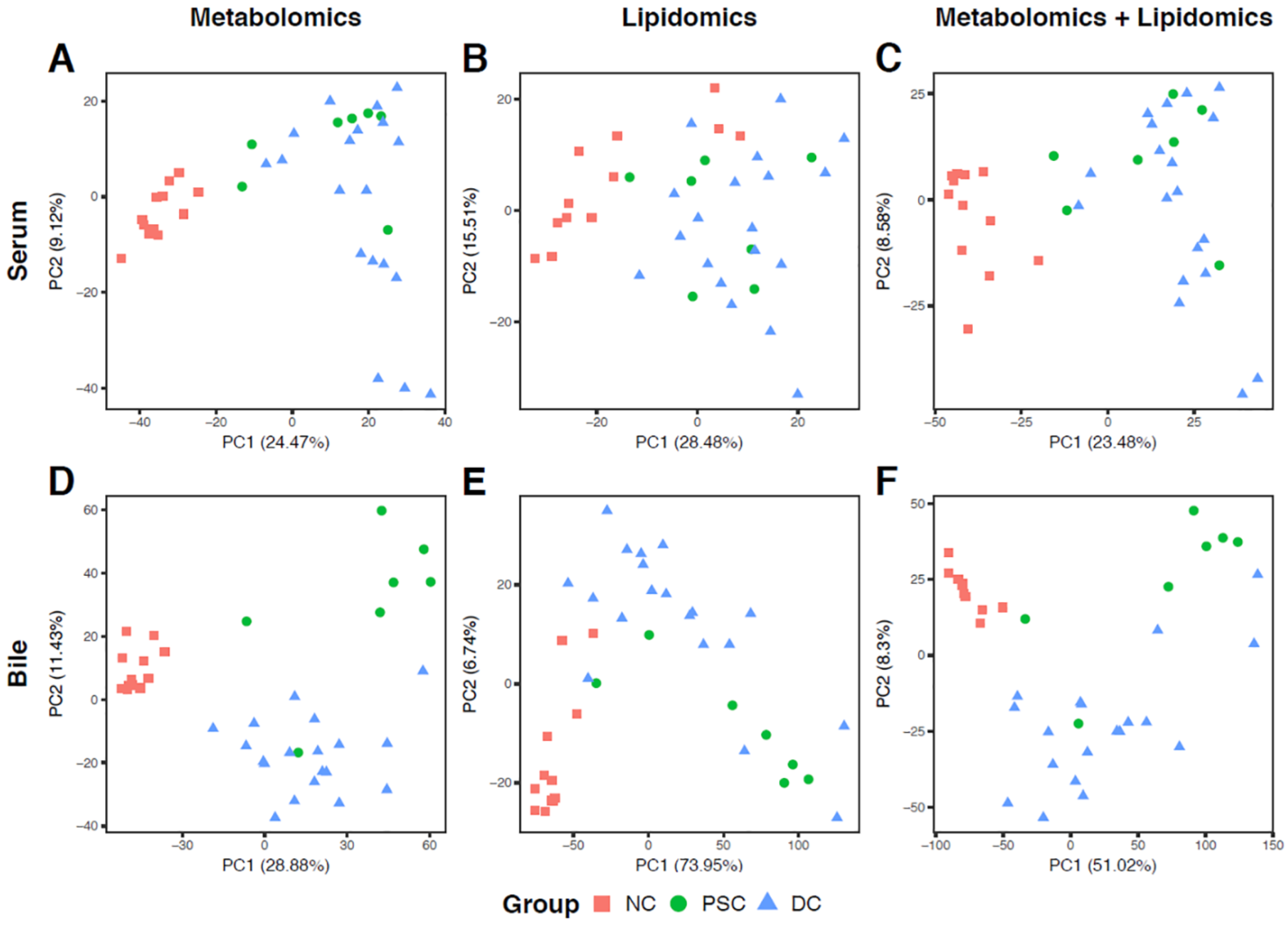
2.2. Global Metabolomic Profiling to Identify Phenotype-Specific Chemical Species and Enriched Categories
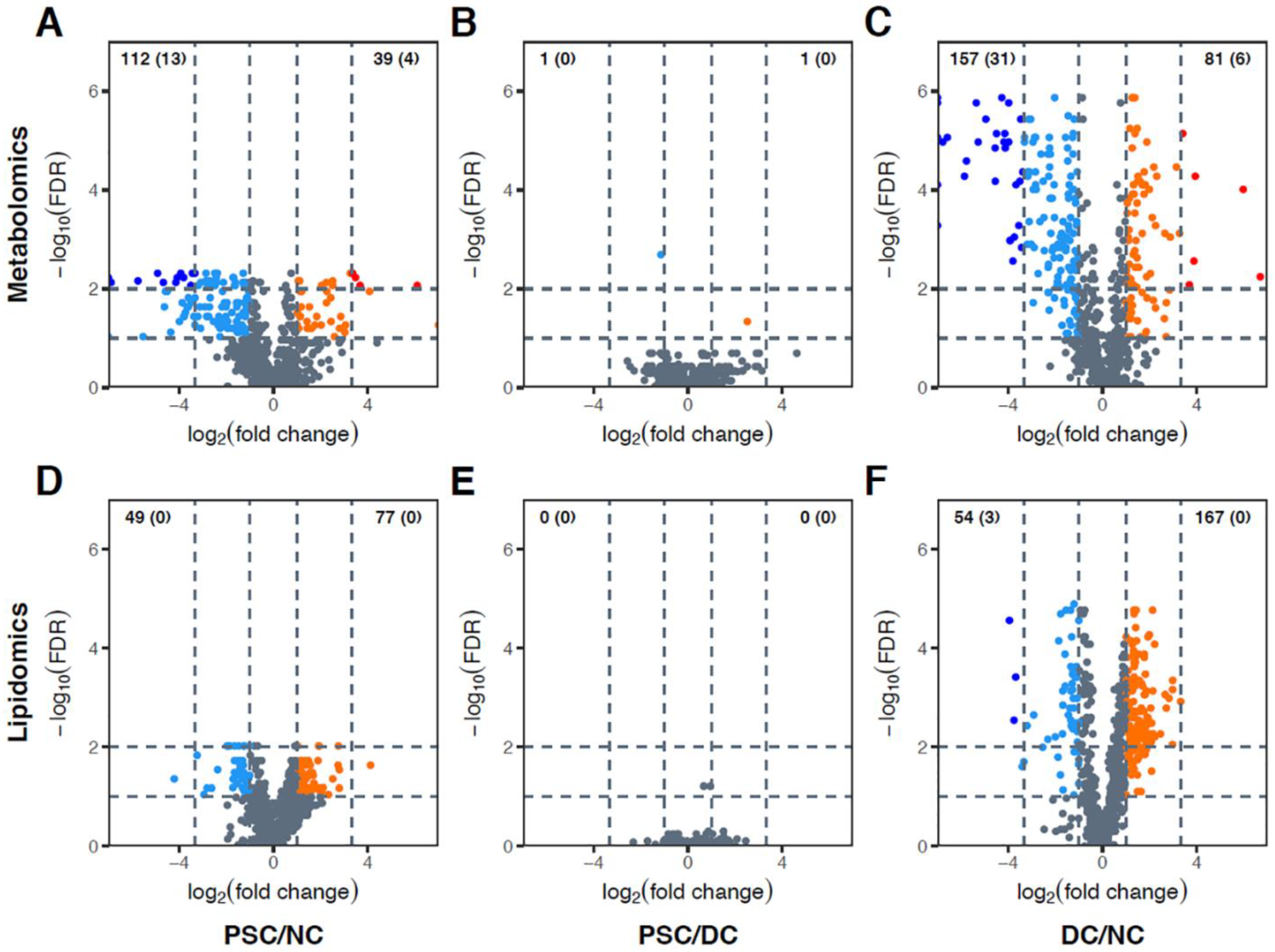
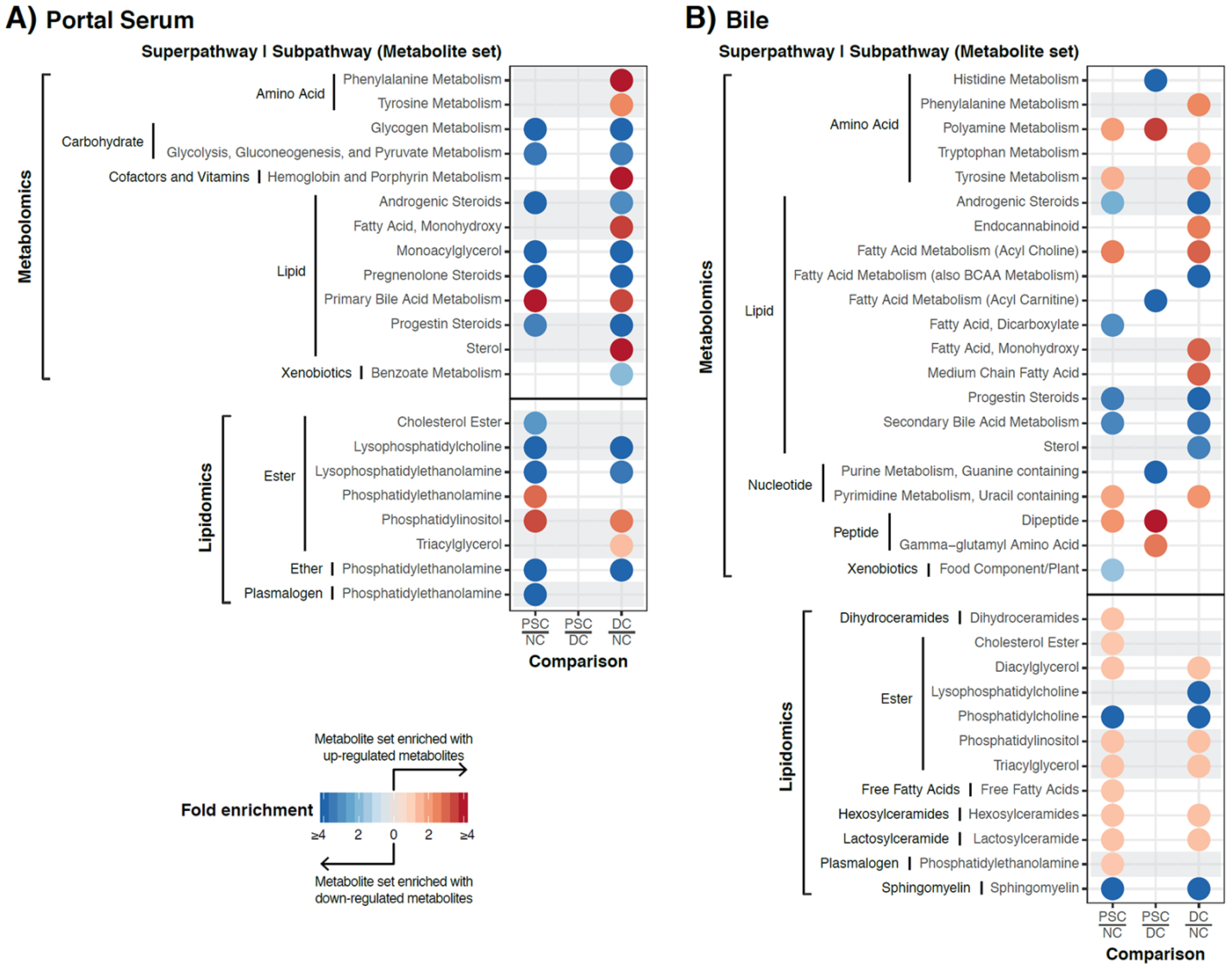
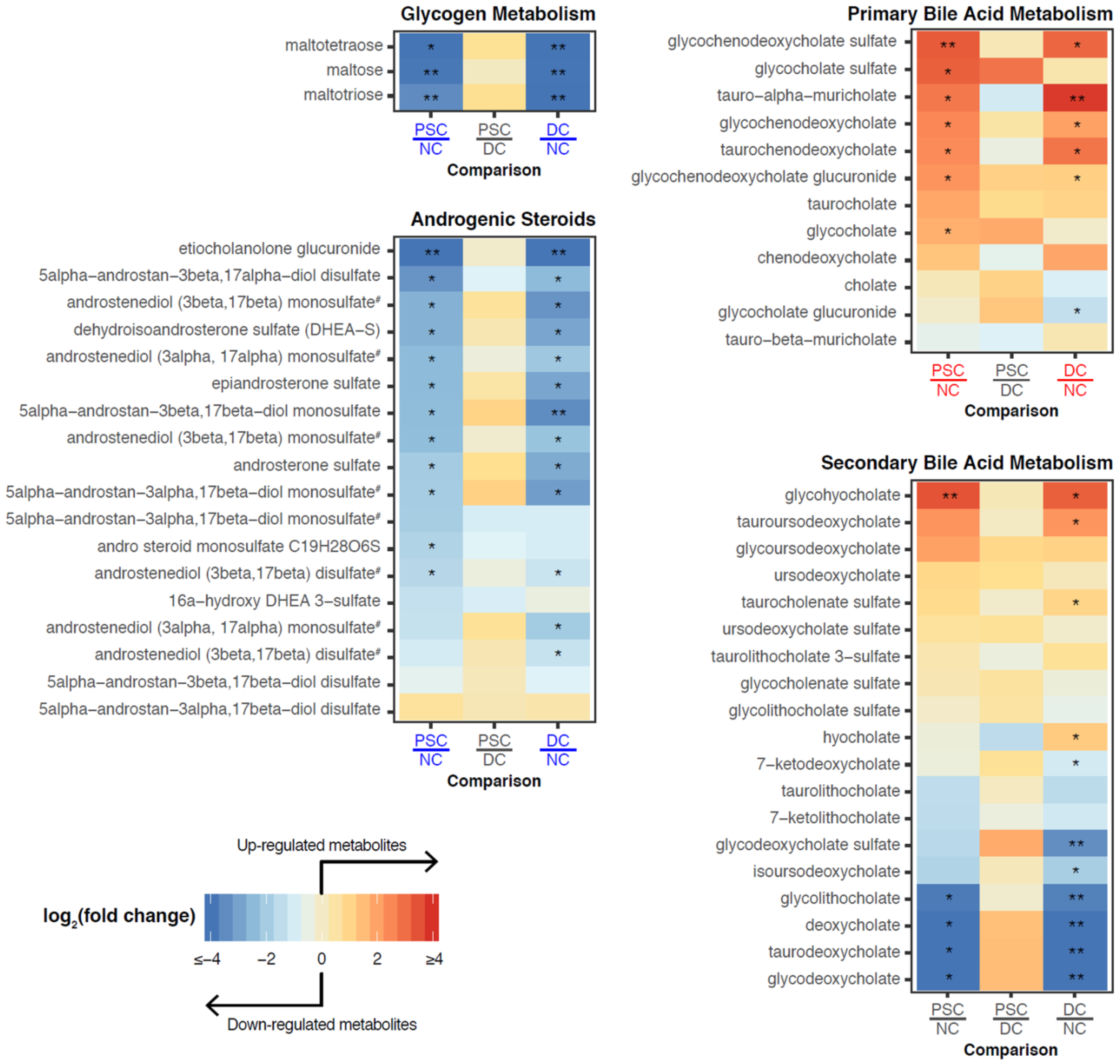
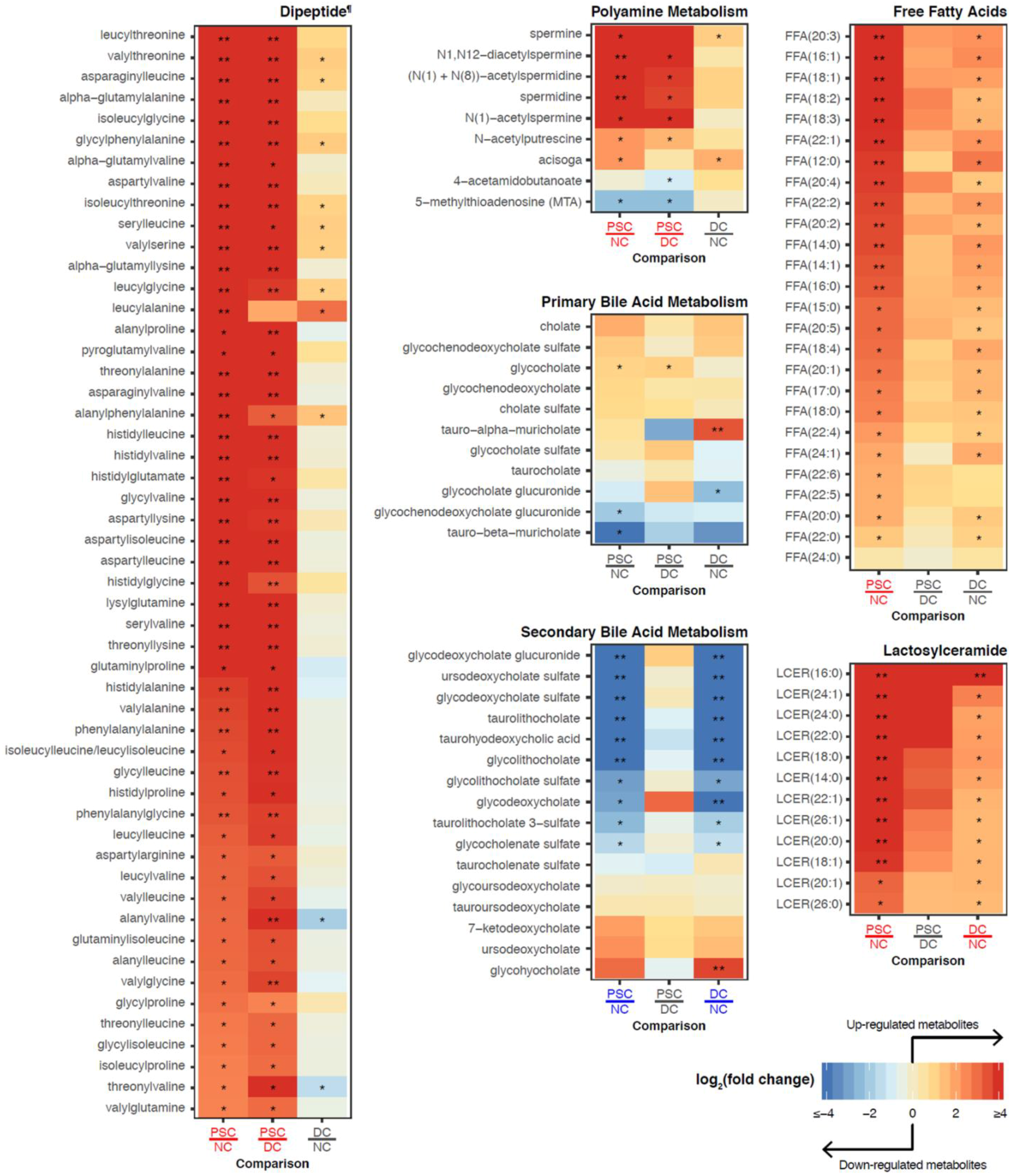
2.3. Identification of Differentially Abundant Metabolites or Lipids within Enriched Metabolite Sets
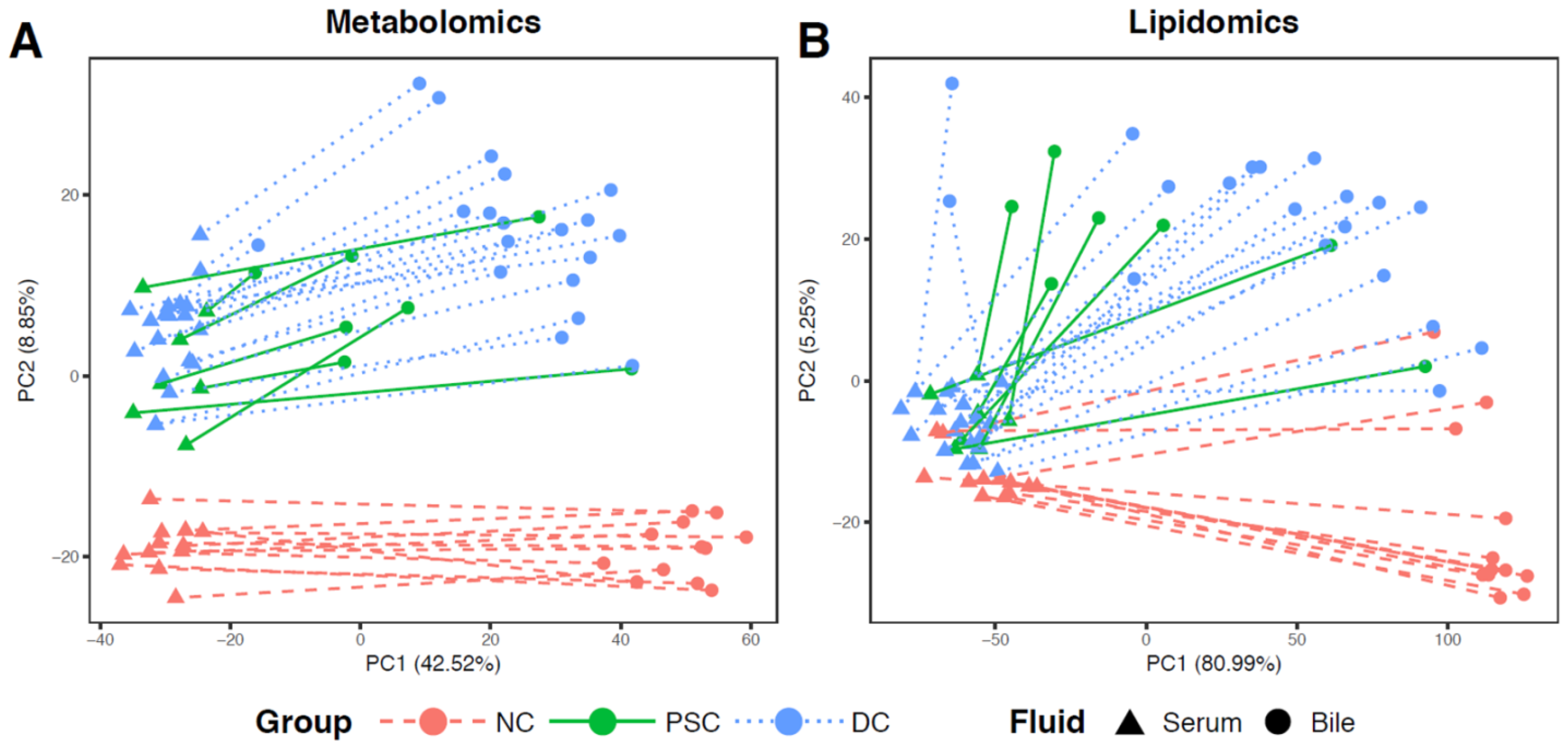
2.4. Comparisons of Patient-Matched Serum and Bile Samples
3. Discussion
4. Materials and Methods
4.1. Study Design and Population
4.2. Sample Accessioning and Preparation
4.3. Sample Analysis
4.3.1. UPLC-MS/MS
4.3.2. Complex Lipids Platform
4.4. Quality Control
4.5. Data Extraction and Compound Identification
4.6. Statistical Calculations
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Karlsen, T.H.; Folseraas, T.; Thorburn, D.; Vesterhus, M. Primary sclerosing cholangitis—A comprehensive review. J. Hepatol. 2017, 67, 1298–1323. [Google Scholar] [CrossRef] [PubMed]
- Lazaridis, K.N.; LaRusso, N.F. Primary Sclerosing Cholangitis. N. Engl. J. Med. 2016, 375, 1161–1170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bjoro, K.; Brandsaeter, B.; Foss, A.; Schrumpf, E. Liver transplantation in primary sclerosing cholangitis. Semin. Liver Dis. 2006, 26, 69–79. [Google Scholar] [CrossRef] [PubMed]
- Karlsen, T.H. Primary sclerosing cholangitis: 50 years of a gut-liver relationship and still no love? Gut 2016, 65, 1579–1581. [Google Scholar] [CrossRef] [PubMed]
- Rappaport, A.M. Hepatic blood flow: Morphologic aspects and physiologic regulation. Int. Rev. Physiol. 1980, 21, 1–63. [Google Scholar] [PubMed]
- Vollmar, B.; Menger, M.D. The hepatic microcirculation: mechanistic contributions and therapeutic targets in liver injury and repair. Physiol. Rev. 2009, 89, 1269–1339. [Google Scholar] [CrossRef] [PubMed]
- Boonstra, K.; van Erpecum, K.J.; van Nieuwkerk, K.M.; Drenth, J.P.; Poen, A.C.; Witteman, B.J.; Tuynman, H.A.; Beuers, U.; Ponsioen, C.Y. Primary sclerosing cholangitis is associated with a distinct phenotype of inflammatory bowel disease. Inflamm. Bowel Dis. 2012, 18, 2270–2276. [Google Scholar] [CrossRef] [PubMed]
- Chapman, R.; Fevery, J.; Kalloo, A.; Nagorney, D.M.; Boberg, K.M.; Shneider, B.; Gores, G.J. Diagnosis and management of primary sclerosing cholangitis. Hepatology 2010, 51, 660–678. [Google Scholar] [CrossRef] [PubMed]
- Bajer, L.; Kverka, M.; Kostovcik, M.; Macinga, P.; Dvorak, J.; Stehlikova, Z.; Brezina, J.; Wohl, P.; Spicak, J.; Drastich, P. Distinct gut microbiota profiles in patients with primary sclerosing cholangitis and ulcerative colitis. World J. Gastroenterol. 2017, 23, 4548–4558. [Google Scholar] [CrossRef] [PubMed]
- Kummen, M.; Holm, K.; Anmarkrud, J.A.; Nygard, S.; Vesterhus, M.; Hoivik, M.L.; Troseid, M.; Marschall, H.U.; Schrumpf, E.; Moum, B.; et al. The gut microbial profile in patients with primary sclerosing cholangitis is distinct from patients with ulcerative colitis without biliary disease and healthy controls. Gut 2017, 66, 611–619. [Google Scholar] [CrossRef] [PubMed]
- Quraishi, M.N.; Sergeant, M.; Kay, G.; Iqbal, T.; Chan, J.; Constantinidou, C.; Trivedi, P.; Ferguson, J.; Adams, D.H.; Pallen, M.; et al. The gut-adherent microbiota of PSC-IBD is distinct to that of IBD. Gut 2017, 66, 386–388. [Google Scholar] [CrossRef] [PubMed]
- Ruhlemann, M.C.; Heinsen, F.A.; Zenouzi, R.; Lieb, W.; Franke, A.; Schramm, C. Faecal microbiota profiles as diagnostic biomarkers in primary sclerosing cholangitis. Gut 2017, 66, 753–754. [Google Scholar] [CrossRef] [PubMed]
- Sabino, J.; Vieira-Silva, S.; Machiels, K.; Joossens, M.; Falony, G.; Ballet, V.; Ferrante, M.; van Assche, G.; van der Merwe, S.; Vermeire, S.; et al. Primary sclerosing cholangitis is characterised by intestinal dysbiosis independent from IBD. Gut 2016, 65, 1681–1689. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Damman, J.L.; Rodriguez, E.A.; Ali, A.H.; Buness, C.W.; Cox, K.L.; Carey, E.J.; Lindor, K.D. Review article: The evidence that vancomycin is a therapeutic option for primary sclerosing cholangitis. Aliment. Pharmacol. Ther. 2018, 47, 886–895. [Google Scholar] [CrossRef] [PubMed]
- Tabibian, J.H.; Weeding, E.; Jorgensen, R.A.; Petz, J.L.; Keach, J.C.; Talwalkar, J.A.; Lindor, K.D. Randomised clinical trial: Vancomycin or metronidazole in patients with primary sclerosing cholangitis—A pilot study. Aliment. Pharmacol. Ther. 2013, 37, 604–612. [Google Scholar] [CrossRef] [PubMed]
- O’Hara, S.P.; Splinter, P.L.; Gajdos, G.B.; Trussoni, C.E.; Fernandez-Zapico, M.E.; Chen, X.M.; LaRusso, N.F. NFkappaB p50-CCAAT/enhancer-binding protein beta (C/EBPbeta)-mediated transcriptional repression of microRNA let-7i following microbial infection. J. Biol. Chem. 2010, 285, 216–225. [Google Scholar] [CrossRef] [PubMed]
- O’Hara, S.P.; Splinter, P.L.; Trussoni, C.E.; Gajdos, G.B.; Lineswala, P.N.; LaRusso, N.F. Cholangiocyte N-Ras protein mediates lipopolysaccharide-induced interleukin 6 secretion and proliferation. J. Biol. Chem. 2011, 286, 30352–30360. [Google Scholar] [CrossRef] [PubMed]
- Trussoni, C.E.; Tabibian, J.H.; Splinter, P.L.; O’Hara, S.P. Lipopolysaccharide (LPS)-Induced Biliary Epithelial Cell NRas Activation Requires Epidermal Growth Factor Receptor (EGFR). PLoS ONE 2015, 10, e0125793. [Google Scholar] [CrossRef] [PubMed]
- Tabibian, J.H.; O’Hara, S.P.; Trussoni, C.E.; Tietz, P.S.; Splinter, P.L.; Mounajjed, T.; Hagey, L.R.; LaRusso, N.F. Absence of the intestinal microbiota exacerbates hepatobiliary disease in a murine model of primary sclerosing cholangitis. Hepatology 2016, 63, 185–196. [Google Scholar] [CrossRef] [PubMed]
- Bell, L.N.; Wulff, J.; Comerford, M.; Vuppalanchi, R.; Chalasani, N. Serum metabolic signatures of primary biliary cirrhosis and primary sclerosing cholangitis. Liver Int. 2015, 35, 263–274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trottier, J.; Bialek, A.; Caron, P.; Straka, R.J.; Heathcote, J.; Milkiewicz, P.; Barbier, O. Metabolomic profiling of 17 bile acids in serum from patients with primary biliary cirrhosis and primary sclerosing cholangitis: A pilot study. Dig. Liver Dis. 2012, 44, 303–310. [Google Scholar] [CrossRef] [PubMed]
- Bugianesi, E.; Kalhan, S.; Burkett, E.; Marchesini, G.; McCullough, A. Quantification of gluconeogenesis in cirrhosis: Response to glucagon. Gastroenterology 1998, 115, 1530–1540. [Google Scholar] [CrossRef]
- Petersen, K.F.; Krssak, M.; Navarro, V.; Chandramouli, V.; Hundal, R.; Schumann, W.C.; Landau, B.R.; Shulman, G.I. Contributions of net hepatic glycogenolysis and gluconeogenesis to glucose production in cirrhosis. Am. J. Physiol. 1999, 276, E529–E535. [Google Scholar] [CrossRef] [PubMed]
- Charlton, M.; Angulo, P.; Chalasani, N.; Merriman, R.; Viker, K.; Charatcharoenwitthaya, P.; Sanderson, S.; Gawrieh, S.; Krishnan, A.; Lindor, K. Low circulating levels of dehydroepiandrosterone in histologically advanced nonalcoholic fatty liver disease. Hepatology 2008, 47, 484–492. [Google Scholar] [CrossRef] [PubMed]
- Mendoza-Milla, C.; Valero Jimenez, A.; Rangel, C.; Lozano, A.; Morales, V.; Becerril, C.; Chavira, R.; Ruiz, V.; Barrera, L.; Montano, M.; et al. Dehydroepiandrosterone has strong antifibrotic effects and is decreased in idiopathic pulmonary fibrosis. Eur. Respir. J. 2013, 42, 1309–1321. [Google Scholar] [CrossRef] [PubMed]
- Janicko, M.; Drazilova, S.; Pella, D.; Fedacko, J.; Jarcuska, P. Pleiotropic effects of statins in the diseases of the liver. World J. Gastroenterol. 2016, 22, 6201–6213. [Google Scholar] [CrossRef] [PubMed]
- Kamal, S.; Khan, M.A.; Seth, A.; Cholankeril, G.; Gupta, D.; Singh, U.; Kamal, F.; Howden, C.W.; Stave, C.; Nair, S.; et al. Beneficial Effects of Statins on the Rates of Hepatic Fibrosis, Hepatic Decompensation, and Mortality in Chronic Liver Disease: A Systematic Review and Meta-Analysis. Am. J. Gastroenterol. 2017, 112, 1495–1505. [Google Scholar] [CrossRef] [PubMed]
- Argo, C.K.; Loria, P.; Caldwell, S.H.; Lonardo, A. Statins in liver disease: A molehill, an iceberg, or neither? Hepatology 2008, 48, 662–669. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Milkiewicz, M.; Klak, M.; Kempinska-Podhorodecka, A.; Wiechowska-Kozlowska, A.; Urasinska, E.; Blatkiewicz, M.; Wunsch, E.; Elias, E.; Milkiewicz, P. Impaired Hepatic Adaptation to Chronic Cholestasis induced by Primary Sclerosing Cholangitis. Sci. Rep. 2016, 6, 39573. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Urdaneta, V.; Casadesus, J. Interactions between Bacteria and Bile Salts in the Gastrointestinal and Hepatobiliary Tracts. Front. Med. (Lausanne) 2017, 4, 163. [Google Scholar] [CrossRef] [PubMed]
- Bechmann, L.P.; Kocabayoglu, P.; Sowa, J.P.; Sydor, S.; Best, J.; Schlattjan, M.; Beilfuss, A.; Schmitt, J.; Hannivoort, R.A.; Kilicarslan, A.; et al. Free fatty acids repress small heterodimer partner (SHP) activation and adiponectin counteracts bile acid-induced liver injury in superobese patients with nonalcoholic steatohepatitis. Hepatology 2013, 57, 1394–1406. [Google Scholar] [CrossRef] [PubMed]
- Zweers, S.J.; de Vries, E.M.; Lenicek, M.; Tolenaars, D.; de Waart, D.R.; Koelfat, K.V.; Groen, A.K.; Olde Damink, S.W.; Beuers, U.; Ponsioen, C.; et al. Prolonged fibroblast growth factor 19 response in patients with primary sclerosing cholangitis after an oral chenodeoxycholic acid challenge. Hepatol. Int. 2017, 11, 132–140. [Google Scholar] [CrossRef] [PubMed]
- Cariou, B.; Staels, B. FXR: A promising target for the metabolic syndrome? Trends Pharmacol. Sci. 2007, 28, 236–243. [Google Scholar] [CrossRef] [PubMed]
- Chiang, J.Y. Bile acid metabolism and signaling. Compr. Physiol. 2013, 3, 1191–1212. [Google Scholar] [CrossRef] [PubMed]
- Vitek, L.; Zelenka, J.; Zadinova, M.; Malina, J. The impact of intestinal microflora on serum bilirubin levels. J. Hepatol. 2005, 42, 238–243. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, S.; Pandey, A. The Yin and Yang of lactosylceramide metabolism: implications in cell function. Biochim. Biophys. Acta 2008, 1780, 370–382. [Google Scholar] [CrossRef] [PubMed]
- Spirli, C.; Fabris, L.; Duner, E.; Fiorotto, R.; Ballardini, G.; Roskams, T.; Larusso, N.F.; Sonzogni, A.; Okolicsanyi, L.; Strazzabosco, M. Cytokine-stimulated nitric oxide production inhibits adenylyl cyclase and cAMP-dependent secretion in cholangiocytes. Gastroenterology 2003, 124, 737–753. [Google Scholar] [CrossRef] [PubMed]
- Trauner, M. When bile ducts say NO: The good, the bad, and the ugly. Gastroenterology 2003, 124, 847–851. [Google Scholar] [CrossRef] [PubMed]
- Won, J.S.; Singh, A.K.; Singh, I. Lactosylceramide: A lipid second messenger in neuroinflammatory disease. J. Neurochem. 2007, 103 (Suppl. 1), 180–191. [Google Scholar] [CrossRef]
- Hall, C.; Sato, K.; Wu, N.; Zhou, T.; Kyritsi, K.; Meng, F.; Glaser, S.; Alpini, G. Regulators of Cholangiocyte Proliferation. Gene Expr. 2017, 17, 155–171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiao, Y.; Wang, J.; Chen, Y.; Zhou, K.; Wen, J.; Wang, Y.; Zhou, Y.; Pan, W.; Cai, W. Up-regulation of miR-200b in biliary atresia patients accelerates proliferation and migration of hepatic stallate cells by activating PI3K/Akt signaling. Cell Signal. 2014, 26, 925–932. [Google Scholar] [CrossRef] [PubMed]
- Iwabuchi, K.; Nagaoka, I. Lactosylceramide-enriched glycosphingolipid signaling domain mediates superoxide generation from human neutrophils. Blood 2002, 100, 1454–1464. [Google Scholar] [PubMed]
- Walkley, S.U.; Vanier, M.T. Secondary lipid accumulation in lysosomal disease. Biochim. Biophys. Acta 2009, 1793, 726–736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boyer, J.L. Bile formation and secretion. Compr. Physiol. 2013, 3, 1035–1078. [Google Scholar] [CrossRef] [PubMed]
- Esteller, A. Physiology of bile secretion. World J. Gastroenterol. 2008, 14, 5641–5649. [Google Scholar] [CrossRef] [PubMed]
- Pang, K.S.; Wang, P.J.; Chung, A.Y.; Wolkoff, A.W. The modified dipeptide, enalapril, an angiotensin-converting enzyme inhibitor, is transported by the rat liver organic anion transport protein. Hepatology 1998, 28, 1341–1346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petzinger, E.; Wickboldt, A.; Pagels, P.; Starke, D.; Kramer, W. Hepatobiliary transport of bile acid amino acid, bile acid peptide, and bile acid oligonucleotide conjugates in rats. Hepatology 1999, 30, 1257–1268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stieger, B.; Hagenbuch, B. Organic anion-transporting polypeptides. Curr. Top. Membr. 2014, 73, 205–232. [Google Scholar] [CrossRef] [PubMed]
- Takeshita, T.; Okochi, M.; Kato, R.; Kaga, C.; Tomita, Y.; Nagaoka, S.; Honda, H. Screening of peptides with a high affinity to bile acids using peptide arrays and a computational analysis. J. Biosci. Bioeng. 2011, 112, 92–97. [Google Scholar] [CrossRef] [PubMed]
- Casero, R.A.; Pegg, A.E. Polyamine catabolism and disease. Biochem. J. 2009, 421, 323–338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pegg, A.E. Functions of Polyamines in Mammals. J. Biol. Chem. 2016, 291, 14904–14912. [Google Scholar] [CrossRef] [PubMed]
- Uemura, T.; Yerushalmi, H.F.; Tsaprailis, G.; Stringer, D.E.; Pastorian, K.E.; Hawel, L., 3rd; Byus, C.V.; Gerner, E.W. Identification and characterization of a diamine exporter in colon epithelial cells. J. Biol. Chem. 2008, 283, 26428–26435. [Google Scholar] [CrossRef] [PubMed]
- Zahedi, K.; Lentsch, A.B.; Okaya, T.; Barone, S.; Sakai, N.; Witte, D.P.; Arend, L.J.; Alhonen, L.; Jell, J.; Janne, J.; et al. Spermidine/spermine-N1-acetyltransferase ablation protects against liver and kidney ischemia-reperfusion injury in mice. Am. J. Physiol. Gastrointest. Liver Physiol. 2009, 296, G899–G909. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anstee, Q.M.; Day, C.P. S-Adenosylmethionine (SAMe) therapy in liver disease: A review of current evidence and clinical utility. J. Hepatol. 2012, 57, 1097–1109. [Google Scholar] [CrossRef] [PubMed]
- Bigaud, E.; Corrales, F.J. Methylthioadenosine (MTA) Regulates Liver Cells Proteome and Methylproteome: Implications in Liver Biology and Disease. Mol. Cell. Proteom. 2016, 15, 1498–1510. [Google Scholar] [CrossRef] [PubMed]
- Shah, P.; Swiatlo, E. A multifaceted role for polyamines in bacterial pathogens. Mol. Microbiol. 2008, 68, 4–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- North, J.A.; Miller, A.R.; Wildenthal, J.A.; Young, S.J.; Tabita, F.R. Microbial pathway for anaerobic 5′-methylthioadenosine metabolism coupled to ethylene formation. Proc. Natl. Acad. Sci. USA 2017, 114, E10455–E10464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Payne, J.W. The characterization of dipeptidases from Escherichia coli. J. Gen. Microbiol. 1972, 71, 267–279. [Google Scholar] [CrossRef] [PubMed]
- Saito, H.E.; Harp, J.R.; Fozo, E.M. Incorporation of exogenous fatty acids protects Enterococcus faecalis from membrane-damaging agents. Appl. Environ. Microbiol. 2014, 80, 6527–6538. [Google Scholar] [CrossRef] [PubMed]
- Yao, J.; Rock, C.O. Exogenous fatty acid metabolism in bacteria. Biochimie 2017, 141, 30–39. [Google Scholar] [CrossRef] [PubMed]
- Xia, X.; Francis, H.; Glaser, S.; Alpini, G.; LeSage, G. Bile acid interactions with cholangiocytes. World J. Gastroenterol. 2006, 12, 3553–3563. [Google Scholar] [CrossRef] [PubMed]
- Manautou, J.E.; Campion, S.N.; Aleksunes, L.M. Regulation of hepatobiliary transporters during liver injury. In Comprehensive Toxicology; McQueen, C.A., Ed.; Elsevier: New York, NY, USA, 2010; Volume 9, pp. 175–220. [Google Scholar]
- Zweers, S.J.; Shiryaev, A.; Komuta, M.; Vesterhus, M.; Hov, J.R.; Perugorria, M.J.; de Waart, D.R.; Chang, J.C.; Tol, S.; Te Velde, A.A.; et al. Elevated interleukin-8 in bile of patients with primary sclerosing cholangitis. Liver Int. 2016, 36, 1370–1377. [Google Scholar] [CrossRef] [PubMed]
- Said, K.; Glaumann, H.; Bergquist, A. Gallbladder disease in patients with primary sclerosing cholangitis. J. Hepatol. 2008, 48, 598–605. [Google Scholar] [CrossRef] [PubMed]
- Xia, J.; Wishart, D.S. MSEA: A web-based tool to identify biologically meaningful patterns in quantitative metabolomic data. Nucleic Acids Res. 2010, 38, W71–W77. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primary Sclerosing Cholangitis | Non-Diseased Controls | Disease Controls | |
|---|---|---|---|
| (PSC, n = 7) | NC, n = 12) | (DC, n = 19) | |
| Demographics | |||
| Age (years), median (range) | 37 (20–61) | 34 (22–54) | 58 (22–71) |
| Female (%) | 43 | 25 | 16 |
| BMI, median (range) | 24 (20–26) | 26 (22–30) | 28 (20–43) |
| Diabetes (%) | 0 | 0 | 21 |
| IBD (%) | 57 | 0 | 0 |
| Liver Biochemistries | |||
| ALT (U/L), median (range) | 60 (20–742) | 21 (13–68) | 42 (22–745) |
| AST (U/L), median (range) | 70 (15–884) | 21 (15–157) | 62 (34–1730) |
| Alk Phos (U/L), median (range) | 329 (186–1626) *,# | 65 (45–95) | 118 (34–255) * |
| Bilirubin total (mmol/L), median (range) | 1.40 (0.4–12.2) * | 0.7 (0.2–1.8) | 3.5 (0.4–38.7) * |
| Other Parameters | |||
| Leukocyte count (×103/mL), median (range) | 6.7 (1.9–9.0) | 5.7 (3.5–9.0) | 3.6 (1.7–7.9) |
| Colectomy (%) | 0 | 0 | 0 |
| ABO blood type | 71% (O), 29% (AB) | 91% (O), 9% (A) | 32% (O), 53% (A), 10% (B) |
| MELD Score, median (range) | 25 (13–31) # | N/A | 31 (19–40) |
| Medications at TX | |||
| UDCA (%) | 29 | 0 | 10 |
| Antibiotics (%) | 14 | 0 | 21 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tietz-Bogert, P.S.; Kim, M.; Cheung, A.; Tabibian, J.H.; Heimbach, J.K.; Rosen, C.B.; Nandakumar, M.; Lazaridis, K.N.; LaRusso, N.F.; Sung, J.; et al. Metabolomic Profiling of Portal Blood and Bile Reveals Metabolic Signatures of Primary Sclerosing Cholangitis. Int. J. Mol. Sci. 2018, 19, 3188. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms19103188
Tietz-Bogert PS, Kim M, Cheung A, Tabibian JH, Heimbach JK, Rosen CB, Nandakumar M, Lazaridis KN, LaRusso NF, Sung J, et al. Metabolomic Profiling of Portal Blood and Bile Reveals Metabolic Signatures of Primary Sclerosing Cholangitis. International Journal of Molecular Sciences. 2018; 19(10):3188. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms19103188
Chicago/Turabian StyleTietz-Bogert, Pamela S., Minsuk Kim, Angela Cheung, James H. Tabibian, Julie K. Heimbach, Charles B. Rosen, Madhumitha Nandakumar, Konstantinos N. Lazaridis, Nicholas F. LaRusso, Jaeyun Sung, and et al. 2018. "Metabolomic Profiling of Portal Blood and Bile Reveals Metabolic Signatures of Primary Sclerosing Cholangitis" International Journal of Molecular Sciences 19, no. 10: 3188. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms19103188