Type I Interferon Signaling Is Required for Dacryoadenitis in the Nonobese Diabetic Mouse Model of Sjögren Syndrome

 , , and
, , and
Abstract
:1. Introduction
2. Results
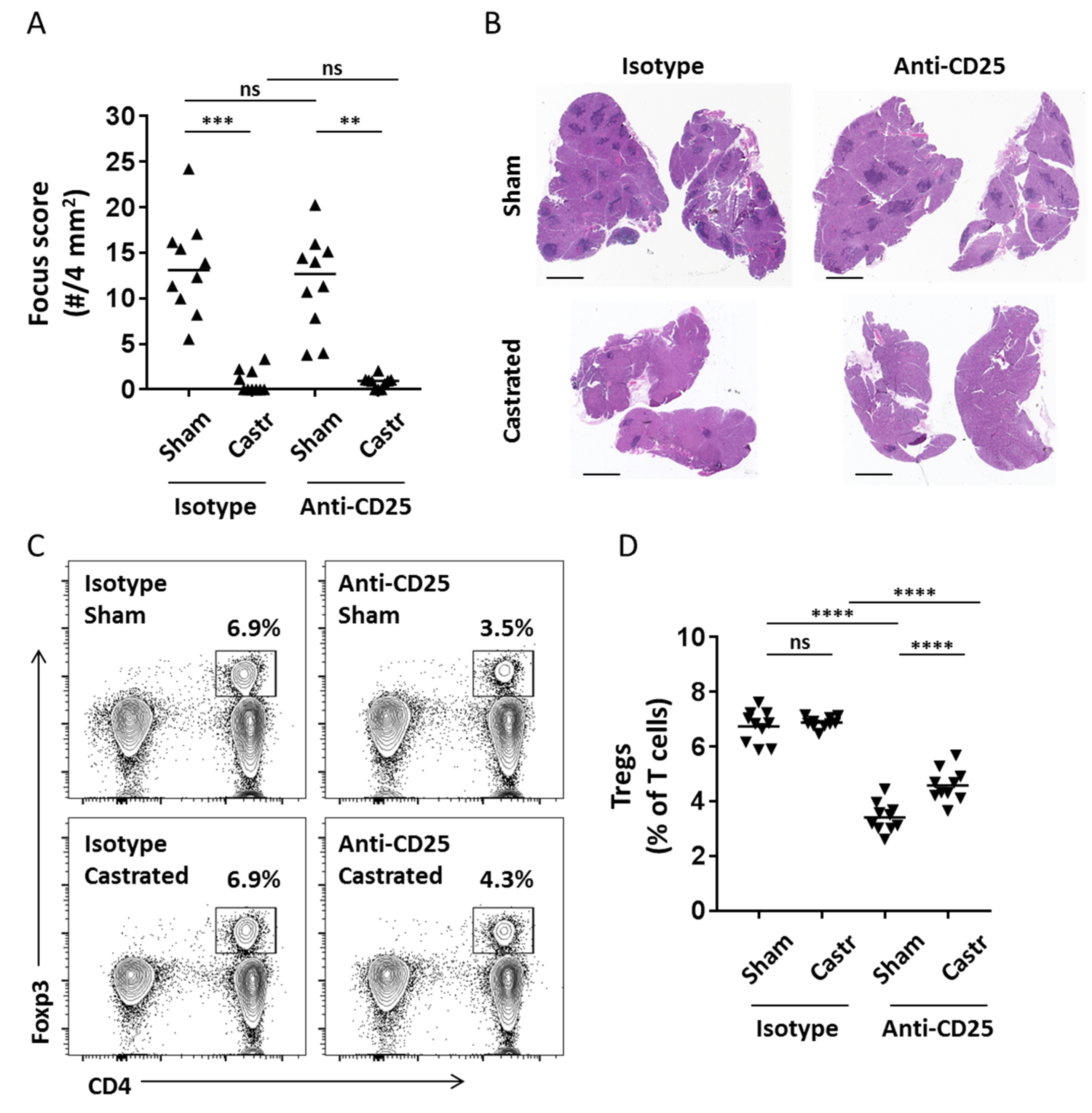
2.1. Transient Treg Depletion Is Not Sufficient to Drive Dacryoadenitis in Castrated Male NOD Mice
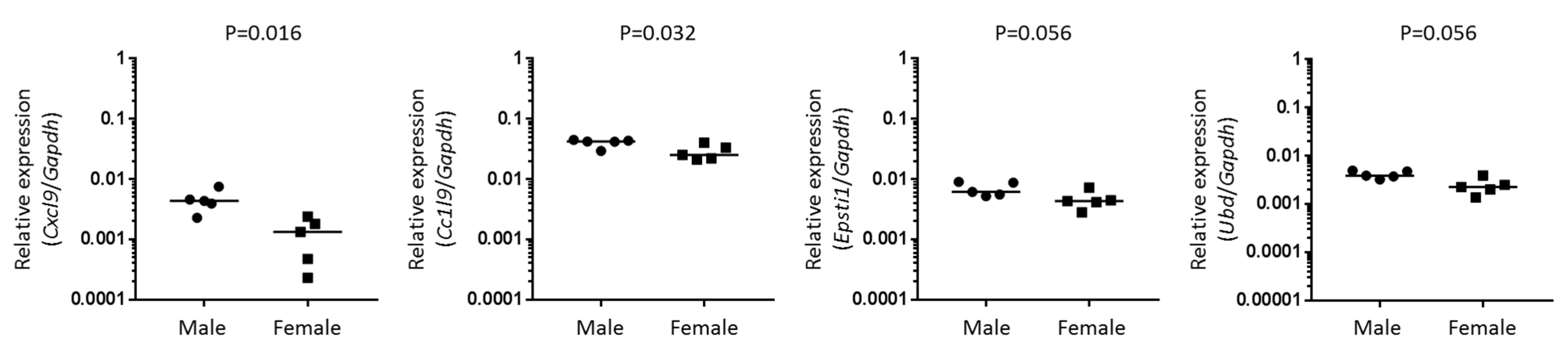
2.2. Identification and Validation of Disease-Relevant Genes Upregulated in Lacrimal Glands of Male NOD Mice
2.3. Disease-Relevant Chemokine Genes Are Significantly Upregulated in Lymphocyte-Deficient NOD-SCID Mice
2.4. Type I Interferon Signaling Affects Disease-Relevant Gene Expression and Is Required for Dacryoadenitis
3. Discussion
4. Materials and Methods
4.1. Mice
4.2. In Vivo Treg Depletion
4.3. Histological Characterization of Lacrimal Gland Inflammation
4.4. Lymphocyte Isolation and Flow Cytometry
4.5. Analysis of Publicly Available Microarray Data
4.6. Quantitative RT-PCR
4.7. Statistical Analysis
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| H&E | Hematoxylin and eosin |
| IFN | Interferon |
| MRL/lpr | Murphy Roths Large/lymphoproliferation |
| NOD | Nonobese diabetic |
| SCID | Severe combined immunodeficiency |
| Tregs | Regulatory T cells |
Appendix

References
- Jonsson, R.; Theander, E.; Sjostrom, B.; Brokstad, K.; Henriksson, G. Autoantibodies present before symptom onset in primary Sjogren syndrome. JAMA 2013, 310, 1854–1855. [Google Scholar] [CrossRef] [PubMed]
- Theander, E.; Jonsson, R.; Sjostrom, B.; Brokstad, K.; Olsson, P.; Henriksson, G. Prediction of Sjogren’s Syndrome Years Before Diagnosis and Identification of Patients with Early Onset and Severe Disease Course by Autoantibody Profiling. Arthritis Rheumatol. 2015, 67, 2427–2436. [Google Scholar] [CrossRef] [PubMed]
- Maria, N.I.; Vogelsang, P.; Versnel, M.A. The clinical relevance of animal models in Sjogren’s syndrome: The interferon signature from mouse to man. Arthritis Res. Ther. 2015, 17, 172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Blokland, S.C.; Versnel, M.A. Pathogenesis of Sjogren’s syndrome: Characteristics of different mouse models for autoimmune exocrinopathy. Clin. Immunol. 2002, 103, 111–124. [Google Scholar] [CrossRef] [PubMed]
- Toda, I.; Sullivan, B.D.; Rocha, E.M.; da Silveira, L.A.; Wickham, L.A.; Sullivan, D.A. Impact of gender on exocrine gland inflammation in mouse models of Sjogren’s syndrome. Exp. Eye Res. 1999, 69, 355–366. [Google Scholar] [CrossRef] [PubMed]
- Barr, J.Y.; Wang, X.; Kreiger, P.A.; Lieberman, S.M. Salivary-gland-protective regulatory T-cell dysfunction underlies female-specific sialadenitis in the non-obese diabetic mouse model of Sjogren syndrome. Immunology 2018, 155, 225–237. [Google Scholar] [CrossRef] [PubMed]
- Hunger, R.E.; Carnaud, C.; Vogt, I.; Mueller, C. Male gonadal environment paradoxically promotes dacryoadenitis in nonobese diabetic mice. J. Clin. Investig. 1998, 101, 1300–1309. [Google Scholar] [CrossRef] [PubMed]
- Lieberman, S.M.; Kreiger, P.A.; Koretzky, G.A. Reversible lacrimal gland-protective regulatory T-cell dysfunction underlies male-specific autoimmune dacryoadenitis in the non-obese diabetic mouse model of Sjogren syndrome. Immunology 2015, 145, 232–241. [Google Scholar] [CrossRef] [PubMed]
- Mikulowska-Mennis, A.; Xu, B.; Berberian, J.M.; Michie, S.A. Lymphocyte migration to inflamed lacrimal glands is mediated by vascular cell adhesion molecule-1/α4β1 integrin, peripheral node addressin/l-selectin, and lymphocyte function-associated antigen-1 adhesion pathways. Am. J. Pathol. 2001, 159, 671–681. [Google Scholar] [CrossRef]
- Takahashi, M.; Ishimaru, N.; Yanagi, K.; Haneji, N.; Saito, I.; Hayashi, Y. High incidence of autoimmune dacryoadenitis in male non-obese diabetic (NOD) mice depending on sex steroid. Clin. Exp. Immunol. 1997, 109, 555–561. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szczerba, B.M.; Rybakowska, P.D.; Dey, P.; Payerhin, K.M.; Peck, A.B.; Bagavant, H.; Deshmukh, U.S. Type I interferon receptor deficiency prevents murine Sjogren’s syndrome. J. Dent. Res. 2013, 92, 444–449. [Google Scholar] [CrossRef] [PubMed]
- Thorlacius, G.E.; Wahren-Herlenius, M.; Ronnblom, L. An update on the role of type I interferons in systemic lupus erythematosus and Sjogren’s syndrome. Curr. Opin. Rheumatol. 2018, 30, 471–481. [Google Scholar] [PubMed]
- Rocha, F.J.; Wickham, L.A.; Pena, J.D.; Gao, J.; Ono, M.; Lambert, R.W.; Kelleher, R.S.; Sullivan, D.A. Influence of gender and the endocrine environment on the distribution of androgen receptors in the lacrimal gland. J. Steroid Biochem. Mol. Biol. 1993, 46, 737–749. [Google Scholar] [CrossRef]
- Richards, S.M.; Jensen, R.V.; Liu, M.; Sullivan, B.D.; Lombardi, M.J.; Rowley, P.; Schirra, F.; Treister, N.S.; Suzuki, T.; Steagall, R.J.; et al. Influence of sex on gene expression in the mouse lacrimal gland. Exp. Eye Res. 2006, 82, 13–23. [Google Scholar] [CrossRef] [PubMed]
- Richards, S.M.; Liu, M.; Jensen, R.V.; Schirra, F.; Yamagami, H.; Lombardi, M.J.; Rowley, P.; Treister, N.S.; Suzuki, T.; Sullivan, B.D.; et al. Androgen regulation of gene expression in the mouse lacrimal gland. J. Steroid Biochem. Mol. Biol. 2005, 96, 401–413. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, D.A.; Block, L.; Pena, J.D. Influence of androgens and pituitary hormones on the structural profile and secretory activity of the lacrimal gland. Acta Ophthalmol. Scand. 1996, 74, 421–435. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, D.A.; Jensen, R.V.; Suzuki, T.; Richards, S.M. Do sex steroids exert sex-specific and/or opposite effects on gene expression in lacrimal and meibomian glands? Mol. Vis. 2009, 15, 1553–1572. [Google Scholar] [PubMed]
- Tellefsen, S.; Kaurstad, M.; Richards, S.M.; Lieberman, S.M.; Darabad, R.R.; Kam, W.R.; Sullivan, D.A. Sex effects on gene expression in autoimmune lacrimal glands. Invest. Ophthalmol. Vis. Sci. 2018, submitted. [Google Scholar]
- Kaurstad, M.; Tellefsen, S.; Richards, S.M.; Lieberman, S.M.; Darabad, R.R.; Kam, W.R.; Sullivan, D.A. Androgen influence on gene expression in autoimmune lacrimal glands. Unpublished material.
- Hjelmervik, T.O.; Lindqvist, A.K.; Petersen, K.; Johannesson, M.; Stavrum, A.K.; Johansson, A.; Jonsson, R.; Holmdahl, R.; Bolstad, A.I. The influence of the NOD Nss1/Idd5 loci on sialadenitis and gene expression in salivary glands of congenic mice. Arthritis Res. Ther. 2007, 9, R99. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, J.; Kawai, T.; Yu, Q. Pathogenic role of endogenous TNF-α in the development of Sjogren’s-like sialadenitis and secretory dysfunction in non-obese diabetic mice. Lab. Investig. 2017, 97, 458–467. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Yu, Q. Disruption of CXCR3 function impedes the development of Sjogren’s syndrome-like xerostomia in non-obese diabetic mice. Lab. Investig. 2018, 98, 620–628. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Yu, Q. Anti-IL-7 receptor-α treatment ameliorates newly established Sjogren’s-like exocrinopathy in non-obese diabetic mice. Biochim. Biophys. Acta 2018, 1864, 2438–2447. [Google Scholar] [CrossRef] [PubMed]
- Ushio, A.; Arakaki, R.; Eguchi, H.; Hotta, F.; Yamada, A.; Kudo, Y.; Ishimaru, N. Pathological Analysis of Ocular Lesions in a Murine Model of Sjogren’s Syndrome. Int. J. Mol. Sci. 2017, 18, 1209. [Google Scholar] [CrossRef] [PubMed]
- Lisi, S.; Sisto, M.; Lofrumento, D.D.; D’Amore, M. Sjogren’s syndrome autoantibodies provoke changes in gene expression profiles of inflammatory cytokines triggering a pathway involving TACE/NF-κB. Lab. Investig. 2012, 92, 615–624. [Google Scholar] [CrossRef] [PubMed]
- Kawasaki, S.; Kawamoto, S.; Yokoi, N.; Connon, C.; Minesaki, Y.; Kinoshita, S.; Okubo, K. Up-regulated gene expression in the conjunctival epithelium of patients with Sjogren’s syndrome. Exp. Eye Res. 2003, 77, 17–26. [Google Scholar] [CrossRef]
- Yoon, K.C.; Park, C.S.; You, I.C.; Choi, H.J.; Lee, K.H.; Im, S.K.; Park, H.Y.; Pflugfelder, S.C. Expression of CXCL9, -10, -11, and CXCR3 in the tear film and ocular surface of patients with dry eye syndrome. Investig. Ophthalmol. Vis. Sci. 2010, 51, 643–650. [Google Scholar] [CrossRef] [PubMed]
- Khuder, S.A.; Al-Hashimi, I.; Mutgi, A.B.; Altorok, N. Identification of potential genomic biomarkers for Sjogren’s syndrome using data pooling of gene expression microarrays. Rheumatol. Int. 2015, 35, 829–836. [Google Scholar] [CrossRef] [PubMed]
- Song, G.G.; Kim, J.H.; Seo, Y.H.; Choi, S.J.; Ji, J.D.; Lee, Y.H. Meta-analysis of differentially expressed genes in primary Sjogren’s syndrome by using microarray. Hum. Immunol. 2014, 75, 98–104. [Google Scholar] [CrossRef] [PubMed]
- Fava, R.A.; Kennedy, S.M.; Wood, S.G.; Bolstad, A.I.; Bienkowska, J.; Papandile, A.; Kelly, J.A.; Mavragani, C.P.; Gatumu, M.; Skarstein, K.; et al. Lymphotoxin-β receptor blockade reduces CXCL13 in lacrimal glands and improves corneal integrity in the NOD model of Sjogren’s syndrome. Arthritis Res. Ther. 2011, 13, R182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Samarajiwa, S.A.; Forster, S.; Auchettl, K.; Hertzog, P.J. INTERFEROME: The database of interferon regulated genes. Nucleic Acids Res. 2009, 37, D852–D857. [Google Scholar] [CrossRef] [PubMed]
- Cha, S.; Brayer, J.; Gao, J.; Brown, V.; Killedar, S.; Yasunari, U.; Peck, A.B. A dual role for interferon-gamma in the pathogenesis of Sjogren’s syndrome-like autoimmune exocrinopathy in the nonobese diabetic mouse. Scand. J. Immunol. 2004, 60, 552–565. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, C.Q.; Sharma, A.; She, J.X.; McIndoe, R.A.; Peck, A.B. Differential gene expressions in the lacrimal gland during development and onset of keratoconjunctivitis sicca in Sjogren’s syndrome (SJS)-like disease of the C57BL/6.NOD-Aec1Aec2 mouse. Exp. Eye Res. 2009, 88, 398–409. [Google Scholar] [CrossRef] [PubMed]
- Peck, A.B.; Saylor, B.T.; Nguyen, L.; Sharma, A.; She, J.X.; Nguyen, C.Q.; McIndoe, R.A. Gene expression profiling of early-phase Sjogren’s syndrome in C57BL/6.NOD-Aec1Aec2 mice identifies focal adhesion maturation associated with infiltrating leukocytes. Investig. Ophthalmol. Vis. Sci. 2011, 52, 5647–5655. [Google Scholar] [CrossRef] [PubMed]
- Jin, J.O.; Shinohara, Y.; Yu, Q. Innate immune signaling induces interleukin-7 production from salivary gland cells and accelerates the development of primary Sjogren’s syndrome in a mouse model. PLoS ONE 2013, 8, e77605. [Google Scholar] [CrossRef] [PubMed]
- Ishii, T.; Onda, H.; Tanigawa, A.; Ohshima, S.; Fujiwara, H.; Mima, T.; Katada, Y.; Deguchi, H.; Suemura, M.; Miyake, T.; et al. Isolation and expression profiling of genes upregulated in the peripheral blood cells of systemic lupus erythematosus patients. DNA Res. 2005, 12, 429–439. [Google Scholar] [CrossRef] [PubMed]
- Raterman, H.G.; Vosslamber, S.; de Ridder, S.; Nurmohamed, M.T.; Lems, W.F.; Boers, M.; van de Wiel, M.; Dijkmans, B.A.; Verweij, C.L.; Voskuyl, A.E. The interferon type I signature towards prediction of non-response to rituximab in rheumatoid arthritis patients. Arthritis Res. Ther. 2012, 14, R95. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.H.; Lee, J.R.; Hahn, M.J. Regulation of inflammatory gene expression in macrophages by epithelial-stromal interaction 1 (Epsti1). Biochem. Biophys. Res. Commun. 2018, 496, 778–783. [Google Scholar] [CrossRef] [PubMed]
- Raasi, S.; Schmidtke, G.; de Giuli, R.; Groettrup, M. A ubiquitin-like protein which is synergistically inducible by interferon-γ and tumor necrosis factor-α. Eur. J. Immunol. 1999, 29, 4030–4036. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.G.; Ren, J.; Cheong, I.S.; Ban, K.H.; Ooi, L.L.; Yong Tan, S.; Kan, A.; Nuchprayoon, I.; Jin, R.; Lee, K.H.; et al. Expression of the FAT10 gene is highly upregulated in hepatocellular carcinoma and other gastrointestinal and gynecological cancers. Oncogene 2003, 22, 2592–2603. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.C.; Pan, J.; Zhang, C.; Fan, W.; Collinge, M.; Bender, J.R.; Weissman, S.M. A MHC-encoded ubiquitin-like protein (FAT10) binds noncovalently to the spindle assembly checkpoint protein MAD2. Proc. Natl. Acad. Sci. USA 1999, 96, 4313–4318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Canaan, A.; Yu, X.; Booth, C.J.; Lian, J.; Lazar, I.; Gamfi, S.L.; Castille, K.; Kohya, N.; Nakayama, Y.; Liu, Y.C.; et al. FAT10/diubiquitin-like protein-deficient mice exhibit minimal phenotypic differences. Mol. Cell. Biol 2006, 26, 5180–5189. [Google Scholar] [CrossRef] [PubMed]
- Lukasiak, S.; Schiller, C.; Oehlschlaeger, P.; Schmidtke, G.; Krause, P.; Legler, D.F.; Autschbach, F.; Schirmacher, P.; Breuhahn, K.; Groettrup, M. Proinflammatory cytokines cause FAT10 upregulation in cancers of liver and colon. Oncogene 2008, 27, 6068–6074. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, N.T.; Now, H.; Kim, W.J.; Kim, N.; Yoo, J.Y. Ubiquitin-like modifier FAT10 attenuates RIG-I mediated antiviral signaling by segregating activated RIG-I from its signaling platform. Sci. Rep. 2016, 6, 23377. [Google Scholar] [CrossRef] [PubMed]
- Ding, C.; MacVeigh, M.; Pidgeon, M.; da Costa, S.R.; Wu, K.; Hamm-Alvarez, S.F.; Schechter, J.E. Unique ultrastructure of exorbital lacrimal glands in male NOD and BALB/c mice. Curr. Eye Res. 2006, 31, 13–22. [Google Scholar] [CrossRef] [PubMed]
- Da Costa, S.R.; Wu, K.; Veigh, M.M.; Pidgeon, M.; Ding, C.; Schechter, J.E.; Hamm-Alvarez, S.F. Male NOD mouse external lacrimal glands exhibit profound changes in the exocytotic pathway early in postnatal development. Exp. Eye Res. 2006, 82, 33–45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schenke-Layland, K.; Xie, J.; Angelis, E.; Starcher, B.; Wu, K.; Riemann, I.; MacLellan, W.R.; Hamm-Alvarez, S.F. Increased degradation of extracellular matrix structures of lacrimal glands implicated in the pathogenesis of Sjogren’s syndrome. Matrix Biol. 2008, 27, 53–66. [Google Scholar] [CrossRef] [PubMed]
- Wu, K.; Joffre, C.; Li, X.; MacVeigh-Aloni, M.; Hom, M.; Hwang, J.; Ding, C.; Gregoire, S.; Bretillon, L.; Zhong, J.F.; et al. Altered expression of genes functioning in lipid homeostasis is associated with lipid deposition in NOD mouse lacrimal gland. Exp. Eye Res. 2009, 89, 319–332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barr, J.Y.; Wang, X.; Meyerholz, D.K.; Lieberman, S.M. CD8 T cells contribute to lacrimal gland pathology in the nonobese diabetic mouse model of Sjogren syndrome. Immunol. Cell Biol. 2017, 95, 684–694. [Google Scholar] [CrossRef] [PubMed]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oliveros, J.C.; Venny. An Interactive Tool for Coparing Lists with Venn’s Diagrams. In 2007–2015. Available online: http://bioinfogp.cnb.csic.es/tools/venny/index.html (accessed on 3 January 2018).
- Clutter, S.D.; Wilson, D.C.; Marinov, A.D.; Hirsch, R. Follistatin-like protein 1 promotes arthritis by up-regulating IFN-γ. J. Immunol. 2009, 182, 234–239. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene ID | Gene Identifier | Gene Name | Ratios 1 |
|---|---|---|---|
| Cxcl9 | NM_008599 | Chemokine (C-X-C motif) ligand 9 | 7.65 (2.53–28.95) |
| Ubd | NM_023137 | Ubiquitin D | 7.75 (3.01–20.37) |
| Ifi47 | NM_008330 | Interferon γ inducible protein 47 | 4.40 (2.75–8.2) |
| Tgtp | NM_011579 | T-cell specific GTPase | 5 (3.01–7.56) |
| Epsti1 | AK017174 | Epithelial stromal interaction 1 | 4.1 (2.6–6.79) |
| Ccl19 | NM_011888 | Chemokine (C-C motif) ligand 19 | 3.37 (2.05–5.51) |
| Clec7a | NM_020008 | C-type lectin domain family 7, member a | 3.13 (2.9–5) |
| Gene ID | Relative Expression 1 | p Value 2 | Ratio 3 | |
|---|---|---|---|---|
| Male NOD | Female NOD | |||
| Cxcl9 | 0.419 (0.352–0.895) | 0.013 (0.003–0.016) | 0.006 | 32.2 |
| Ubd | 0.024 (0.023–0.027) | 0.0004 (0.0001–0.002) | 0.001 | 60 |
| Epsti1 | 0.134 (0.075–0.172) | 0.029 (0.007–0.038) | 0.0003 | 4.6 |
| Ccl19 | 0.099 (0.078–0.193) | 0.0063 (0.0057–0.023) | 0.001 | 15.7 |
| Clec7a | 0.059 (0.029–0.156) | 0.016 (0.01–0.208) | 0.463 | 3.7 |
| Gene ID | Relative Expression 1 | p Value 2 | Ratio 3 | |
|---|---|---|---|---|
| Sham-Castrated | Castrated | |||
| Cxcl9 | 0.520 (0.407–0.589) | 0.051 (0.035–0.071) | <0.01 | 10.2 |
| Ubd | 0.021 (0.019–0.030) | 0.0012 (0.001–0.0017) | <0.01 | 17.5 |
| Epsti1 | 0.079 (0.061–0.108) | 0.028 (0.024–0.031) | <0.01 | 2.8 |
| Ccl19 | 0.107 (0.088–0.114) | 0.017 (0.015–0.024) | <0.01 | 6.3 |
| Clec7a | 0.082 (0.081–0.1) | 0.038 (0.031–0.044) | <0.01 | 2.2 |
| Gene ID | Forward (5′–3′) | Reverse (5′–3′) |
|---|---|---|
| Cxcl9 | CCGAGGCACGATCCACTACA | CGAGTCCGGATCTAGGCAGGT |
| Ubd | ACCAGATCCTTCTGCTAGACT | GGGTAAGGTGGATAGTGGTTTC |
| Epsti1 | GAGAGCAGAAGGCAGAAGATAC | CGTTGTGGCACCAAGTAGA |
| Ccl19 | ATGTGAATCACTCTGGCCCAGGAA | AAGCGGCTTTATTGGAAGCTCTGC |
| Clec7a | ATCAGCATTCTTCCCCAACTCG | CAGTTCCTTCTCACAGATACTGTATGA |
| Gapdh | TTCACCACCATGGAGAAGGC | GGCATGGACTGTGGTCATGA |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chaly, Y.; Barr, J.Y.; Sullivan, D.A.; Thomas, H.E.; Brodnicki, T.C.; Lieberman, S.M. Type I Interferon Signaling Is Required for Dacryoadenitis in the Nonobese Diabetic Mouse Model of Sjögren Syndrome. Int. J. Mol. Sci. 2018, 19, 3259. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms19103259
Chaly Y, Barr JY, Sullivan DA, Thomas HE, Brodnicki TC, Lieberman SM. Type I Interferon Signaling Is Required for Dacryoadenitis in the Nonobese Diabetic Mouse Model of Sjögren Syndrome. International Journal of Molecular Sciences. 2018; 19(10):3259. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms19103259
Chicago/Turabian StyleChaly, Yury, Jennifer Y. Barr, David A. Sullivan, Helen E. Thomas, Thomas C. Brodnicki, and Scott M. Lieberman. 2018. "Type I Interferon Signaling Is Required for Dacryoadenitis in the Nonobese Diabetic Mouse Model of Sjögren Syndrome" International Journal of Molecular Sciences 19, no. 10: 3259. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms19103259