Construction of A High-Density Genetic Map and Mapping of Fruit Traits in Watermelon (Citrullus Lanatus L.) Based on Whole-Genome Resequencing

 ,
,
Abstract
:1. Introduction
2. Results
2.1. Phenotypic Analysis of the Recombinant Inbred Line (RIL) Population
2.2. Analysis of Whole-Genome Resequencing (WGR) Data and Single-Nucleotide Polymorphism (SNP) Markers
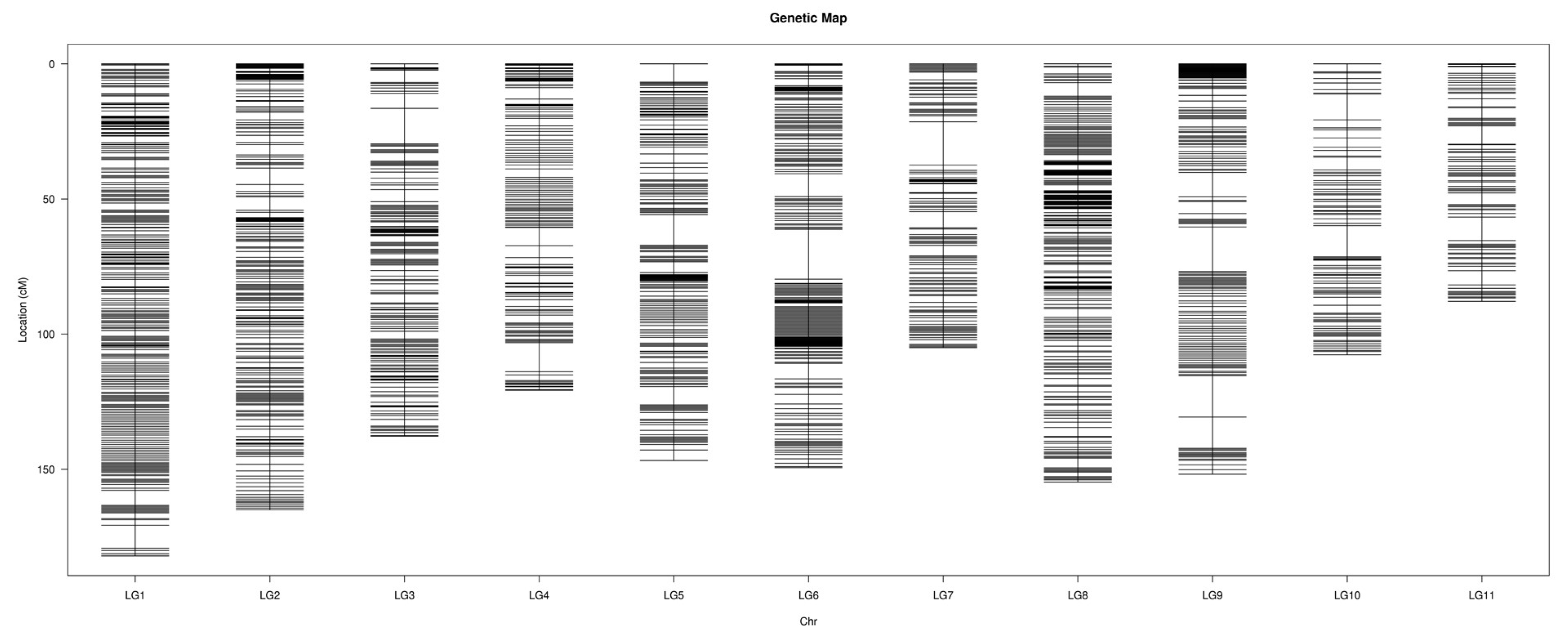
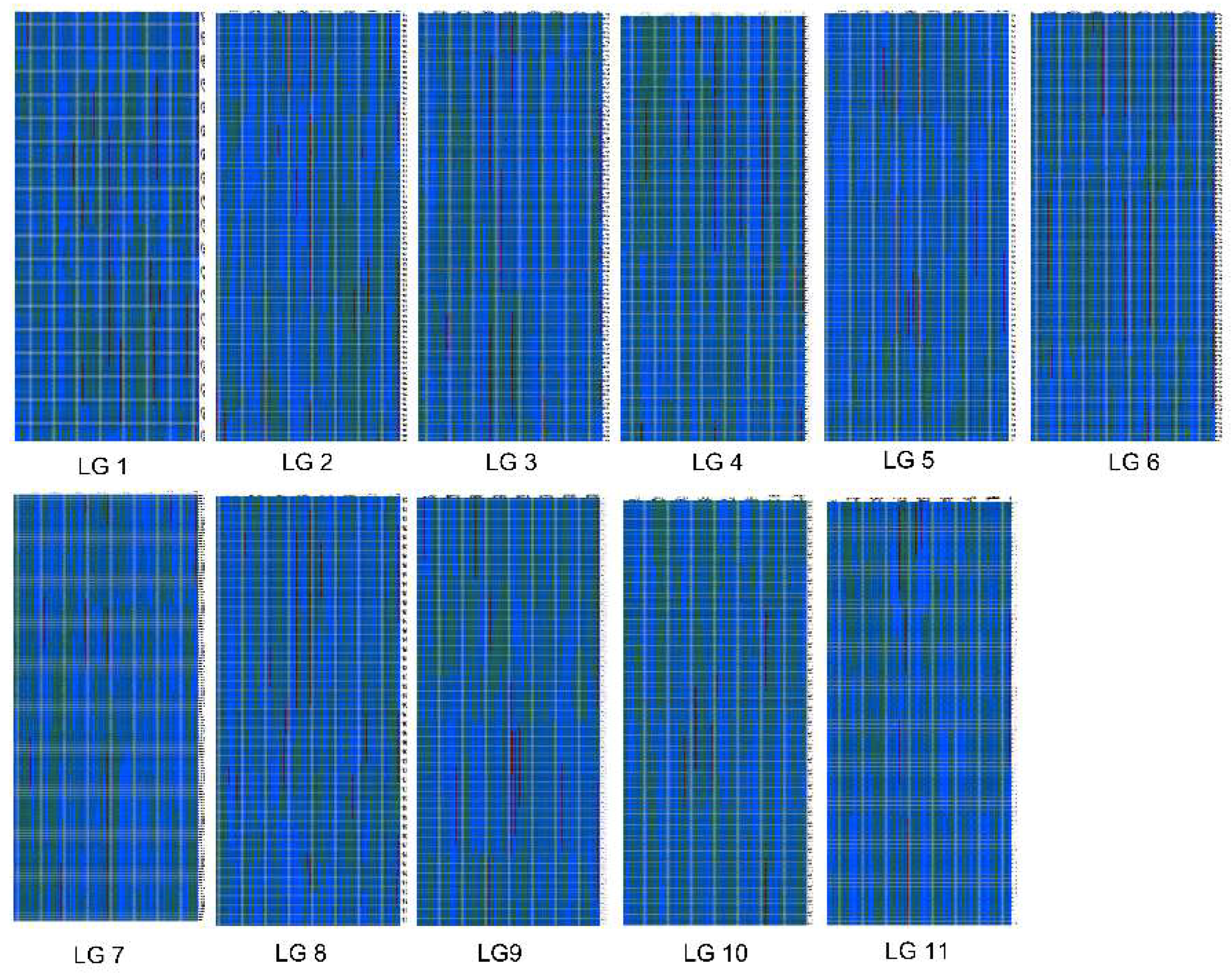
2.3. Construction of the Genetic Map
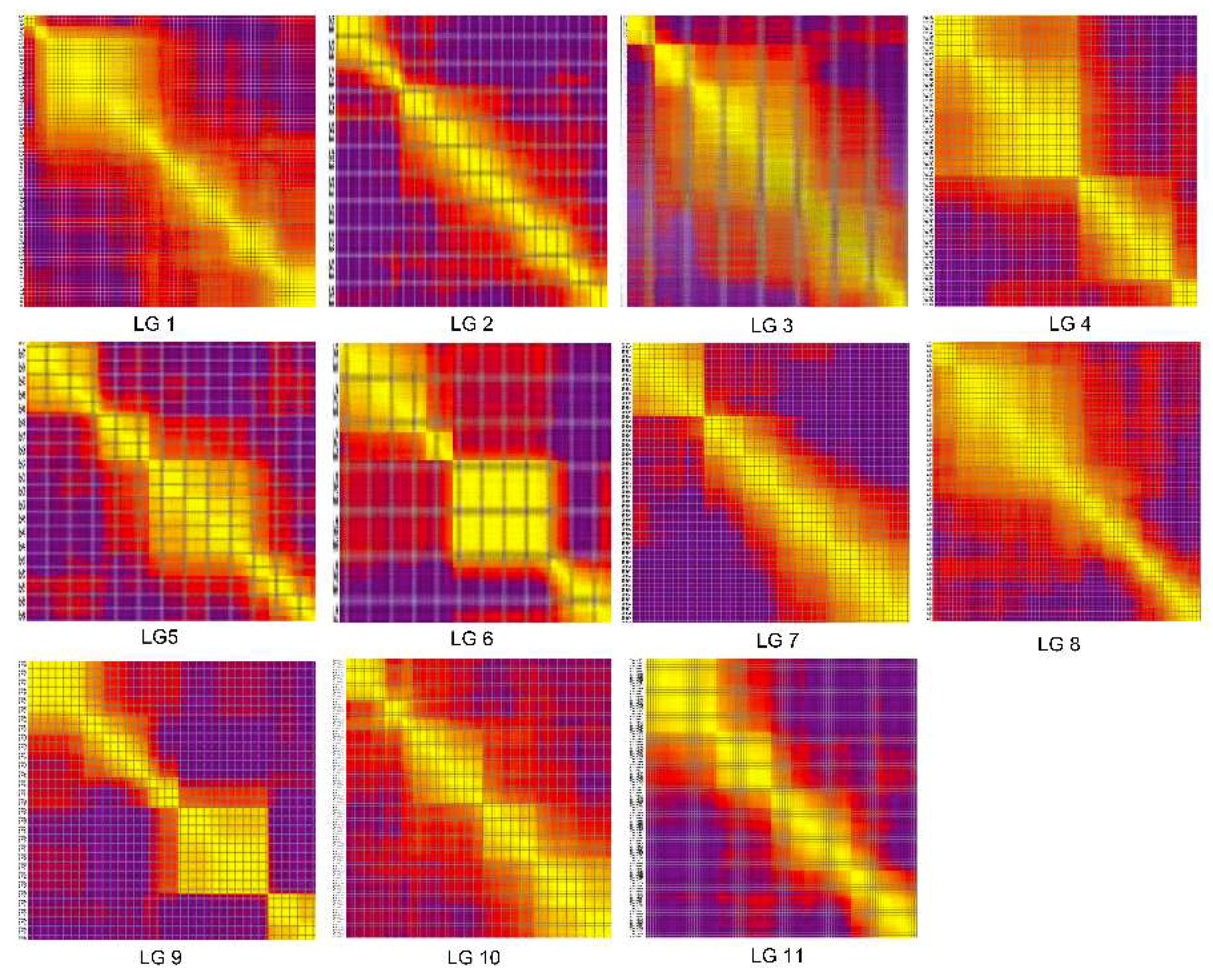
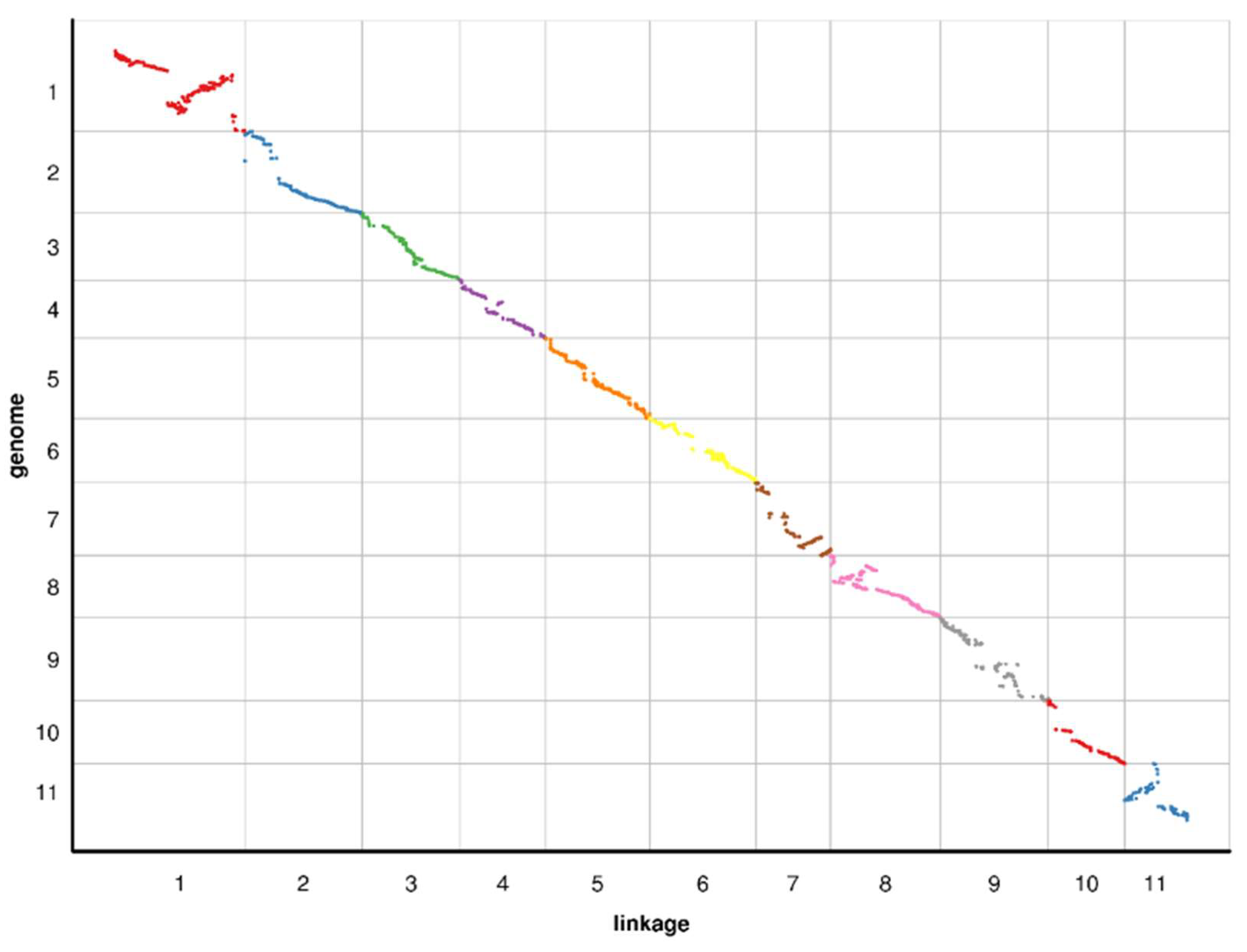
2.4. Collinearity of the Genetic and Physical Maps
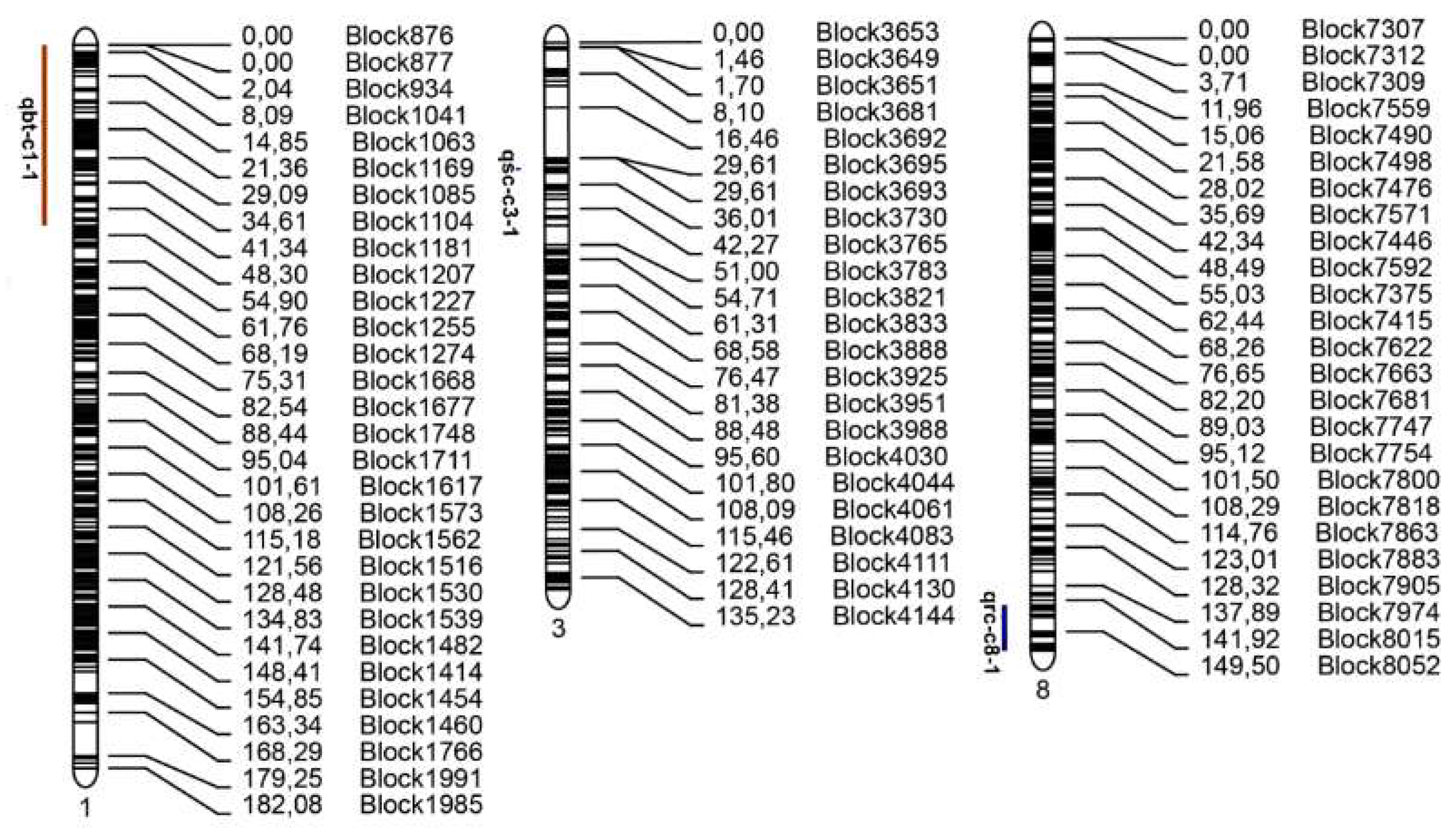
2.5. Detection of Candidate Regions Associated with Fruit Traits
2.5.1. Identification of Candidate Region Associated with Fruit Bitterness
2.5.2. Identification of Candidate Region Associated with Rind Color
2.5.3. Identification of Candidate Region Associated with Seed Coat Color
2.6. Annotation of Candidate Genes
3. Discussion
3.1. Characteristics of the WGR Approach for Developing Markers and Candidate Region Analysis
3.2. Features of the High-Density Genetic Map of Watermelon
3.3. The Identification of Loci of Fruit Bitterness, Rind and Seed Coat Color
3.4. Preliminary Analysis of the Potential Functions of Candidate Genes
4. Materials and Methods
4.1. Plant Material and DNA Extraction
4.2. Evaluation of Phenotypes
4.3. Sequencing Library Construction and High-Throughput Sequencing
4.4. SNP Identification and Genotyping
4.5. Genetic Map Construction and Evaluation
4.6. Relationship between the Genetic and Physical Maps
4.7. Gene Mapping and Candidate Genes Prediction
4.8. Candidate Regions Naming
5. Conclusion
Supplementary Materials
Author Contributions
Funding
Acknowledgements
Conflicts of Interest
Abbreviations
| WGR | Whole-genome resequencing |
| SNP | Single-nucleotide polymorphism |
| RIL | Recombinant inbred lines |
| LGs | Linkage groups |
| MAS | Marker-assisted selection |
| QTL | Quantitative trait loci |
| NGS | Next-generation sequencing |
References
- Paris, H.S.; Daunay, M.C.; Janick, J. Medieval iconography of watermelons in Mediterranean Europe. Ann. Bot. 2013, 112, 867–879. [Google Scholar] [CrossRef] [PubMed]
- Arumuganathan, K.; Earle, E. Nuclear DNA content of some important plant species. Plant Mol. Biol. Rep. 1991, 9, 208–218. [Google Scholar] [CrossRef]
- Navot, N.; Zamir, D. Isozyme and seed protein phylogeny of the genus Citrullus (Cucurbitaceae). Plant Syst. Evol. 1987, 156, 61–67. [Google Scholar] [CrossRef]
- Zamir, D.; Navot, N.; Rudich, J. Enzyme polymorphism in Citrullus lanatus and C. colocynthis in Israel and Sinai. Plant Syst. Evol. 1984, 146, 163–170. [Google Scholar] [CrossRef]
- Hashizume, T.; Shimamoto, I.; Harushima, Y.; Yui, M.; Sato, T.; Imai, T.; Hirai, M. Construction of a linkage map for watermelon (Citrullus lanatus (Thunb.) Matsum & Nakai) using random amplified polymorphic DNA (RAPD). Euphytica 1996, 90, 265–273. [Google Scholar]
- Hawkins, L.K.; Dane, F.; Kubisiak, T.L. Molecular markers associated with morphological traits in watermelon. HortScience 2001, 36, 1318–1322. [Google Scholar]
- Levi, A.; Thomas, C.; Trebitsh, T.; Salman, A.; King, J.; Karalius, J.; Newman, M.; Reddy, O.; Xu, Y.; Zhang, X. An extended linkage map for watermelon based on SRAP, AFLP, SSR, ISSR, and RAPD markers. J. Am. Soc. Hortic. Sci. 2006, 131, 393–402. [Google Scholar]
- Ren, Y.; Zhao, H.; Kou, Q.; Jiang, J.; Guo, S.; Zhang, H.; Hou, W.; Zou, X.; Sun, H.; Gong, G. A high resolution genetic map anchoring scaffolds of the sequenced watermelon genome. PLoS ONE 2012, 7, e29453. [Google Scholar] [CrossRef] [PubMed]
- Prothro, J.; Sandlin, K.; Abdel-Haleem, H.; Bachlava, E.; White, V.; Knapp, S.; McGregor, C. Main and epistatic quantitative trait loci associated with seed size in watermelon. J. Am. Soc. Hortic. Sci. 2012, 137, 452–457. [Google Scholar]
- Prothro, J.; Sandlin, K.; Gill, R.; Bachlava, E.; White, V.; Knapp, S.J.; McGregor, C. Mapping of the Egusi seed trait locus (eg) and quantitative trait Loci associated with seed oil percentage in watermelon. J. Am. Soc. Hortic. Sci. 2012, 137, 311–315. [Google Scholar]
- Ren, Y.; McGregor, C.; Zhang, Y.; Gong, G.; Zhang, H.; Guo, S.; Sun, H.; Cai, W.; Zhang, J.; Xu, Y. An integrated genetic map based on four mapping populations and quantitative trait loci associated with economically important traits in watermelon (Citrullus lanatus). BMC Plant Biol. 2014, 14, 33. [Google Scholar] [CrossRef] [PubMed]
- Ren, R.; Ray, R.; Li, P.; Xu, J.; Zhang, M.; Liu, G.; Yao, X.; Kilian, A.; Yang, X. Construction of a high-density DArTseq SNP-based genetic map and identification of genomic regions with segregation distortion in a genetic population derived from a cross between feral and cultivated-type watermelon. Mol. Genet. Genom. 2015, 290, 1457–1470. [Google Scholar] [CrossRef] [PubMed]
- Davey, J.W.; Hohenlohe, P.A.; Etter, P.D.; Boone, J.Q.; Catchen, J.M.; Blaxter, M.L. Genome-wide genetic marker discovery and genotyping using next-generation sequencing. Nat. Rev. Genet. 2011, 12, 499. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Wang, A.; Huang, X.; Zhao, Q.; Dong, G.; Qian, Q.; Sang, T.; Han, B. Mapping 49 quantitative trait loci at high resolution through sequencing-based genotyping of rice recombinant inbred lines. Theor. Appl. Genet. 2011, 122, 327–340. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Li, H.; Wang, J.; Zhang, H.; Hu, Z.; Chu, S.; Lv, H.; Yu, D. High-density genetic mapping identifies new major loci for tolerance to low-phosphorus stress in soybean. Front. Plant Sci. 2016, 7. [Google Scholar] [CrossRef] [PubMed]
- Mei, J.; Li, F.; Liu, X.; Hu, G.; Fu, Y.; Liu, W. Newly identified CSP41b gene localized in chloroplasts affects leaf color in rice. Plant Sci. 2017, 256, 39–45. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.; Su, T.; Zhi, S.; Zhang, F.; Wang, W.; Zhang, D.; Zhao, X.; Yu, Y. Construction of a sequence-based bin map and mapping of QTLs for downy mildew resistance at four developmental stages in Chinese cabbage (Brassica rapa L. ssp. pekinensis). Mol. Breed. 2016, 36, 44. [Google Scholar] [CrossRef]
- Zhang, Z.; Shang, H.; Shi, Y.; Huang, L.; Li, J.; Ge, Q.; Gong, J.; Liu, A.; Chen, T.; Wang, D. Construction of a high-density genetic map by specific locus amplified fragment sequencing (SLAF-seq) and its application to Quantitative Trait Loci (QTL) analysis for boll weight in upland cotton (Gossypium hirsutum.). BMC Plant Biol. 2016, 16, 79. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.; Luan, F.; Wang, X.; Gao, P.; Zhu, Z.; Liu, S.; Baloch, A.M.; Zhang, Y. Construction of a genetic linkage map of watermelon (Citrullus lanatus) using CAPS and SSR markers and QTL analysis for fruit quality traits. Sci. Hort. 2016, 202, 25–31. [Google Scholar] [CrossRef]
- Shang, J.; Li, N.; Xu, Y.; Ma, S.; Wang, J. Construction of a high-density genetic map for watermelon (Citrullus lanatus L.) based on large-scale SNP discovery by specific length amplified fragment sequencing (SLAF-seq). Sci. Hort. 2016, 203, 38–46. [Google Scholar] [CrossRef]
- Liu, S.; Gao, P.; Zhu, Q.; Luan, F.; Davis, A.R.; Wang, X. Development of cleaved amplified polymorphic sequence markers and a CAPS-based genetic linkage map in watermelon (Citrullus lanatus [Thunb.] Matsum. and Nakai) constructed using whole-genome re-sequencing data. Breed. Sci. 2016, 66, 244–259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, P.; Zhou, G.; Yu, H.; Yu, S. Fine mapping a major QTL for flag leaf size and yield-related traits in rice. Theor. Appl. Genet. 2011, 123, 1319–1330. [Google Scholar] [CrossRef] [PubMed]
- Lim, J.H.; Yang, H.J.; Jung, K.H.; Yoo, S.C.; Paek, N.C. Quantitative trait locus mapping and candidate gene analysis for plant architecture traits using whole genome re-sequencing in rice. Mol. Cells 2014, 37, 149. [Google Scholar] [CrossRef] [PubMed]
- Balkema-Boomstra, A.; Zijlstra, S.; Verstappen, F.; Inggamer, H.; Mercke, P.; Jongsma, M.; Bouwmeester, H. Role of cucurbitacin C in resistance to spider mite (Tetranychus urticae) in cucumber (Cucumis sativus L.). J. Chem. Ecol. 2003, 29, 225–235. [Google Scholar] [CrossRef] [PubMed]
- Rice, C.; Rymal, K.; Chambliss, O.; Johnson, F. Chromatographic and mass spectral analysis of cucurbitacins of three Cucumis sativus cultivars. J. Agric. Food Chem. 1981, 29, 194–196. [Google Scholar] [CrossRef]
- Blaskovich, M.A.; Sun, J.; Cantor, A.; Turkson, J.; Jove, R.; Sebti, S.M. Discovery of JSI-124 (cucurbitacin I), a selective Janus kinase/signal transducer and activator of transcription 3 signaling pathway inhibitor with potent antitumor activity against human and murine cancer cells in mice. Cancer Res. 2003, 63, 1270–1279. [Google Scholar] [PubMed]
- Thoennissen, N.H.; Iwanski, G.B.; Doan, N.B.; Okamoto, R.; Lin, P.; Abbassi, S.; Song, J.H.; Yin, D.; Toh, M.; Xie, W.D. Cucurbitacin B induces apoptosis by inhibition of the JAK/STAT pathway and potentiates antiproliferative effects of gemcitabine on pancreatic cancer cells. Cancer Res. 2009, 69, 5876–5884. [Google Scholar] [CrossRef] [PubMed]
- Qi, J.; Liu, X.; Shen, D.; Miao, H.; Xie, B.; Li, X.; Zeng, P.; Wang, S.; Shang, Y.; Gu, X. A genomic variation map provides insights into the genetic basis of cucumber domestication and diversity. Nat. Genet. 2013, 45, 1510–1515. [Google Scholar] [CrossRef] [PubMed]
- Shang, Y.; Ma, Y.; Zhou, Y.; Zhang, H.; Duan, L.; Chen, H.; Zeng, J.; Zhou, Q.; Wang, S.; Gu, W. Biosynthesis, regulation, and domestication of bitterness in cucumber. Science 2014, 346, 1084–1088. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Ma, Y.; Zeng, J.; Duan, L.; Xue, X.; Wang, H.; Lin, T.; Liu, Z.; Zeng, K.; Zhong, Y. Convergence and divergence of bitterness biosynthesis and regulation in Cucurbitaceae. Nat. Plants 2016, 2, 16183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huh, J.; Kang, B.; Nahm, S.; Kim, S.; Ha, K.; Lee, M.; Kim, B. A candidate gene approach identified phytoene synthase as the locus for mature fruit color in red pepper (Capsicum spp.). Theor. Appl. Genet. 2001, 102, 524–530. [Google Scholar] [CrossRef]
- Liu, H.; Meng, H.; Pan, Y.; Liang, X.; Jiao, J.; Li, Y.; Chen, S.; Cheng, Z. Fine genetic mapping of the white immature fruit color gene w to a 33.0-kb region in cucumber (Cucumis sativus L.). Theor. Appl. Genet. 2015, 128, 2375–2385. [Google Scholar] [CrossRef] [PubMed]
- Jiang, B.; Liu, W.; Xie, D.; Peng, Q.; He, X.; Lin, Y.e.; Liang, Z. High-density genetic map construction and gene mapping of pericarp color in wax gourd using specific-locus amplified fragment (SLAF) sequencing. BMC Genom. 2015, 16, 1035. [Google Scholar] [CrossRef] [PubMed]
- Weetman, L.M. Inheritance and correlation of shape, size and color in the watermelon, Citrullus Vulgaris Schrad. Res. Bull. IAHEES 1937, 20, 1. [Google Scholar]
- Kumar, R.; Wehner, T.C. Discovery of second gene for solid dark green versus light green rind pattern in watermelon. J. Hered. 2011, 102, 489–493. [Google Scholar] [CrossRef] [PubMed]
- Park, S.W.; Kim, K.T.; Kang, S.C.; Yang, H.B. Rapid and practical molecular marker development for rind traits in watermelon. Hortic. Environ. Biotechnol. 2016, 57, 385–391. [Google Scholar] [CrossRef]
- Yang, K.K.; Liang, Z.H.; Wu, C.J. Analysis on differentially expressed genes in watermelon rind color based on RNA–Seq. J. Cent. South Univ. 2016, 23, 2818–2826. [Google Scholar] [CrossRef]
- El-Hamid Sayid El-Bramawy, M.; El-Hendawy, S.; Amin Shaban, W. Assessing the suitability of morphological and phenological traits to screen Sesame genotypes for Fusarium wilt and charcoal rot disease resistance. J. Plant Prot. Res. 2008, 48, 397–410. [Google Scholar] [CrossRef]
- McKay, J. Factor interaction in citrullus: Seed-coat color, fruit shape and markings show evidence of mendelian inheritance in watermelon crosses. J. Hered. 1936, 27, 110–112. [Google Scholar] [CrossRef]
- Chi, Y.Y.; Peng, G.; Zhu, Z.C.; Luan, F.S.; Guiying, L.I.; Peng, Y.U. The QTL analysis of fruit and seed associated traits in watermelon based on CAPS markers. Sci. Agric. Sin. 2017. [Google Scholar]
- Nimmakayala, P.; Abburi, V.L.; Bhandary, A.; Abburi, L.; Vajja, V.G.; Reddy, R.; Malkaram, S.; Venkatramana, P.; Wijeratne, A.; Tomason, Y.R. Use of VeraCode 384-plex assays for watermelon diversity analysis and integrated genetic map of watermelon with single nucleotide polymorphisms and simple sequence repeats. Mol. Breed. 2014, 34, 537–548. [Google Scholar] [CrossRef]
- Reddy, U.K.; Nimmakayala, P.; Levi, A.; Abburi, V.L.; Saminathan, T.; Tomason, Y.R.; Vajja, G.; Reddy, R.; Abburi, L.; Wehner, T.C. High-resolution genetic map for understanding the effect of genome-wide recombination rate on nucleotide diversity in watermelon. G3 (Bethesda) 2014, 4, 2219–2230. [Google Scholar] [CrossRef] [PubMed]
- Jansen, R.C. Interval mapping of multiple quantitative trait loci. Genetics 1993, 135, 205–211. [Google Scholar] [PubMed]
- Lander, E.S.; Botstein, D. Mapping mendelian factors underlying quantitative traits using RFLP linkage maps. Genetics 1989, 121, 185–199. [Google Scholar] [PubMed]
- Qi, X.; Stam, P.; Lindhout, P. Use of locus-specific AFLP markers to construct a high-density molecular map in barley. Theor. Appl. Genet. 1998, 96, 376–384. [Google Scholar] [CrossRef] [PubMed]
- Levi, A.; Thomas, C.E.; Keinath, A.P.; Wehner, T.C. Genetic diversity among watermelon (Citrullus lanatus and Citrullus colocynthis) accessions. Genet. Resour. Crop. Evol. 2001, 48, 559–566. [Google Scholar] [CrossRef]
- Guo, S.; Zhang, J.; Sun, H.; Salse, J.; Lucas, W.J.; Zhang, H.; Zheng, Y.; Mao, L.; Ren, Y.; Wang, Z. The draft genome of watermelon (Citrullus lanatus) and resequencing of 20 diverse accessions. Nat. Genet. 2013, 45, 51. [Google Scholar] [CrossRef] [PubMed]
- Ariyadasa, R.; Mascher, M.; Nussbaumer, T.; Schulte, D.; Frenkel, Z.; Poursarebani, N.; Zhou, R.; Steuernagel, B.; Gundlach, H.; Taudien, S. A sequence-ready physical map of barley anchored genetically by two million single-nucleotide polymorphisms. Plant Physiol. 2014, 164, 412–423. [Google Scholar] [CrossRef] [PubMed]
- Trick, M.; Long, Y.; Meng, J.; Bancroft, I. Single nucleotide polymorphism (SNP) discovery in the polyploid Brassica napus using Solexa transcriptome sequencing. Plant. Biotechnol. J. 2009, 7, 334–346. [Google Scholar] [CrossRef] [PubMed]
- Sandlin, K.; Prothro, J.; Heesacker, A.; Khalilian, N.; Okashah, R.; Xiang, W.; Bachlava, E.; Caldwell, D.G.; Taylor, C.A.; Seymour, D.K. Comparative mapping in watermelon [Citrullus lanatus (Thunb.) Matsum. et Nakai]. Theor. Appl. Genet. 2012, 125, 1603–1618. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Zeng, L.; Tao, Y.; Vuong, T.; Wan, J.; Boerma, R.; Noe, J.; Li, Z.; Finnerty, S.; Pathan, S.M. Pinpointing genes underlying the quantitative trait loci for root-knot nematode resistance in palaeopolyploid soybean by whole genome resequencing. Proc. Natl. Acad. Sci. USA 2013, 110, 13469–13474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poland, J.A.; Rife, T.W. Genotyping-by-sequencing for plant breeding and genetics. Plant Genome 2012, 5, 92–102. [Google Scholar] [CrossRef]
- Zhang, Q.; Liu, C.; Liu, Y.; VanBuren, R.; Yao, X.; Zhong, C.; Huang, H. High-density interspecific genetic maps of kiwifruit and the identification of sex-specific markers. DNA Res. 2015, 22, 367–375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamamoto, T.; Terakami, S.; Takada, N.; Nishio, S.; Onoue, N.; Nishitani, C.; Kunihisa, M.; Inoue, E.; Iwata, H.; Hayashi, T. Identification of QTLs controlling harvest time and fruit skin color in Japanese pear (Pyrus pyrifolia Nakai). Breed. Sci. 2014, 64, 351–361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Badani, A.G.; Snowdon, R.; Baetzel, R.; Horn, R.; Friedt, W. QTL Mapping for Yellow Seed Colour in Oilseed Rape (Brassica Napus). In Proceeding of the 11th International Rapeseed Congress, Copenhagen, Denmark, 2003; Royal Veterinary and Agricultural University: Copenhagen, Denmark, 2003; pp. 85–87. [Google Scholar]
- Wang, L.; Xia, Q.; Zhang, Y.; Zhu, X.; Zhu, X.; Li, D.; Ni, X.; Gao, Y.; Xiang, H.; Wei, X. Updated sesame genome assembly and fine mapping of plant height and seed coat color QTLs using a new high-density genetic map. BMC Genom. 2016, 17, 31. [Google Scholar] [CrossRef] [PubMed]
- Itle, R.A.; Kabelka, E.A. Correlation between L* a* b* color space values and carotenoid content in pumpkins and squash (Cucurbita spp.). HortScience 2009, 44, 633–637. [Google Scholar]
- Dixon, R.A.; Sumner, L.W. Legume natural products: Understanding and manipulating complex pathways for human and animal health. Plant Physiol. 2003, 131, 878–885. [Google Scholar] [CrossRef] [PubMed]
- Murray, M.G.; Thompson, W.F. Rapid isolation of high molecular weight plant DNA. Nucleic Acids Res. 1980, 8, 4321–4326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, S.P.; Miao, H.; Cheng, Z.C.; Liu, M.M.; Zhang, Z.H.; Wang, X.W.; Sun, R.F.; Gu, X.F. Genetic Mapping of the Fruit Bitterness Gene(Bt) in Cucumber(Cucumis sativus L.). Acta Hortic. Sin. 2011, 38, 709–716. [Google Scholar]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed]
- DePristo, M.A.; Banks, E.; Poplin, R.; Garimella, K.V.; Maguire, J.R.; Hartl, C.; Philippakis, A.A.; Del Angel, G.; Rivas, M.A.; Hanna, M. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet. 2011, 43, 491–498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, X.; Feng, Q.; Qian, Q.; Zhao, Q.; Wang, L.; Wang, A.; Guan, J.; Fan, D.; Weng, Q.; Huang, T. High-throughput genotyping by whole-genome resequencing. Genome Res. 2009, 19, 1068–1076. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, D.; Ma, C.; Hong, W.; Huang, L.; Liu, M.; Liu, H.; Zeng, H.; Deng, D.; Xin, H.; Song, J. Construction and analysis of high-density linkage map using high-throughput sequencing data. PLoS ONE 2014, 9, e98855. [Google Scholar] [CrossRef] [PubMed]
- Van Os, H.; Stam, P.; Visser, R.G.; Van Eck, H.J. SMOOTH: A statistical method for successful removal of genotyping errors from high-density genetic linkage data. Theor. Appl. Genet. 2005, 112, 187–194. [Google Scholar] [CrossRef] [PubMed]
- Tang, H.; Zhang, X.; Miao, C.; Zhang, J.; Ming, R.; Schnable, J.C.; Schnable, P.S.; Lyons, E.; Lu, J. Allmaps: Robust scaffold ordering based on multiple maps. Genome Biol. 2015, 16, 3. [Google Scholar] [CrossRef] [PubMed]
- Kao, C.H.; Zeng, Z.B.; Teasdale, R.D. Multiple interval mapping for quantitative trait loci. Genetics 1999, 152, 1203–1216. [Google Scholar] [PubMed]
- Van Ooijen, J.W.; Boer, M.; Jansen, R.; Maliepaard, C. MapQTL 4.0: Software for the Calculation of QTL Positions on Genetic Maps (User Manual); Research Report, Plant Research International: Wageningen, Dutch, 2000. [Google Scholar]
- Churchill, G.A.; Doerge, R.W. Empirical threshold values for quantitative trait mapping. Genetics 1994, 138, 963–971. [Google Scholar] [PubMed]
- Doerge, R.W.; Churchill, G.A. Permutation tests for multiple loci affecting a quantitative character. Genetics 1996, 142, 285–294. [Google Scholar] [PubMed]
- Sun, F.D.; Zhang, J.H.; Wang, S.F.; Gong, W.K.; Shi, Y.Z.; Liu, A.Y.; Li, J.W.; Gong, J.W.; Shang, H.H.; Yuan, Y.L. QTL mapping for fiber quality traits across multiple generations and environments in upland cotton. Mol. Breed. 2012, 30, 569–582. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| LGs | Total Bin Markers | Total Distance (cM) | Average Distance (cM) | Max Gap (cM) | Gap < 5 cM (%) |
|---|---|---|---|---|---|
| LG01 | 294 | 182.08 | 0.62 | 8.49 | 99.32% |
| LG02 | 229 | 165.00 | 0.72 | 6.10 | 99.56% |
| LG03 | 218 | 137.08 | 0.63 | 13.15 | 99.08% |
| LG04 | 137 | 120.79 | 0.88 | 10.74 | 98.53% |
| LG05 | 230 | 146.77 | 0.64 | 11.24 | 98.69% |
| LG06 | 216 | 149.42 | 0.69 | 18.45 | 98.60% |
| LG07 | 150 | 105.01 | 0.70 | 16.03 | 98.66% |
| LG08 | 266 | 154.74 | 0.58 | 5.04 | 99.62% |
| LG09 | 179 | 151.82 | 0.85 | 16.43 | 97.75% |
| LG10 | 114 | 107.63 | 0.94 | 11.52 | 98.23% |
| LG11 | 99 | 87.88 | 0.89 | 8.74 | 96.94% |
| Total | 2132 | 1508.94 | 0.74 | 98.63% |
| LG ID | Spearman |
|---|---|
| LG01 | 0.8017 |
| LG02 | 0.9950 |
| LG03 | 0.9948 |
| LG04 | 0.9834 |
| LG05 | 0.9976 |
| LG06 | 0.9867 |
| LG07 | 0.8905 |
| LG08 | 0.8722 |
| LG09 | 0.9822 |
| LG10 | 0.9990 |
| LG11 | 0.8002 |
| Name | Chromosome | Marker Interval | Position (cM) | LOD | PVE | ADD |
|---|---|---|---|---|---|---|
| qbt-c1-1 | 1 | Block876–Block1188 | 0–45.91 | 58.361 | 82.927 | −0.465 |
| qrc-c8-1 | 8 | Block8000–Block8110 | 142.728–154.742 | 18.353 | 49.942 | −0.358 |
| qsc-c3-1 | 3 | Block3708–Block3722 | 31.705–32.505 | 26.852 | 58.685 | −0.625 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, B.; Lu, X.; Dou, J.; Aslam, A.; Gao, L.; Zhao, S.; He, N.; Liu, W. Construction of A High-Density Genetic Map and Mapping of Fruit Traits in Watermelon (Citrullus Lanatus L.) Based on Whole-Genome Resequencing. Int. J. Mol. Sci. 2018, 19, 3268. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms19103268
Li B, Lu X, Dou J, Aslam A, Gao L, Zhao S, He N, Liu W. Construction of A High-Density Genetic Map and Mapping of Fruit Traits in Watermelon (Citrullus Lanatus L.) Based on Whole-Genome Resequencing. International Journal of Molecular Sciences. 2018; 19(10):3268. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms19103268
Chicago/Turabian StyleLi, Bingbing, Xuqiang Lu, Junling Dou, Ali Aslam, Lei Gao, Shengjie Zhao, Nan He, and Wenge Liu. 2018. "Construction of A High-Density Genetic Map and Mapping of Fruit Traits in Watermelon (Citrullus Lanatus L.) Based on Whole-Genome Resequencing" International Journal of Molecular Sciences 19, no. 10: 3268. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms19103268