Transcription of the Human 5-Hydroxytryptamine Receptor 2B (HTR2B) Gene Is under the Regulatory Influence of the Transcription Factors NFI and RUNX1 in Human Uveal Melanoma


Abstract
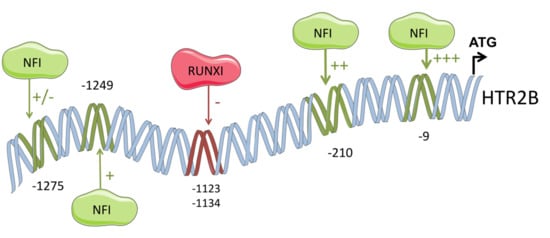
:
1. Introduction
2. Results
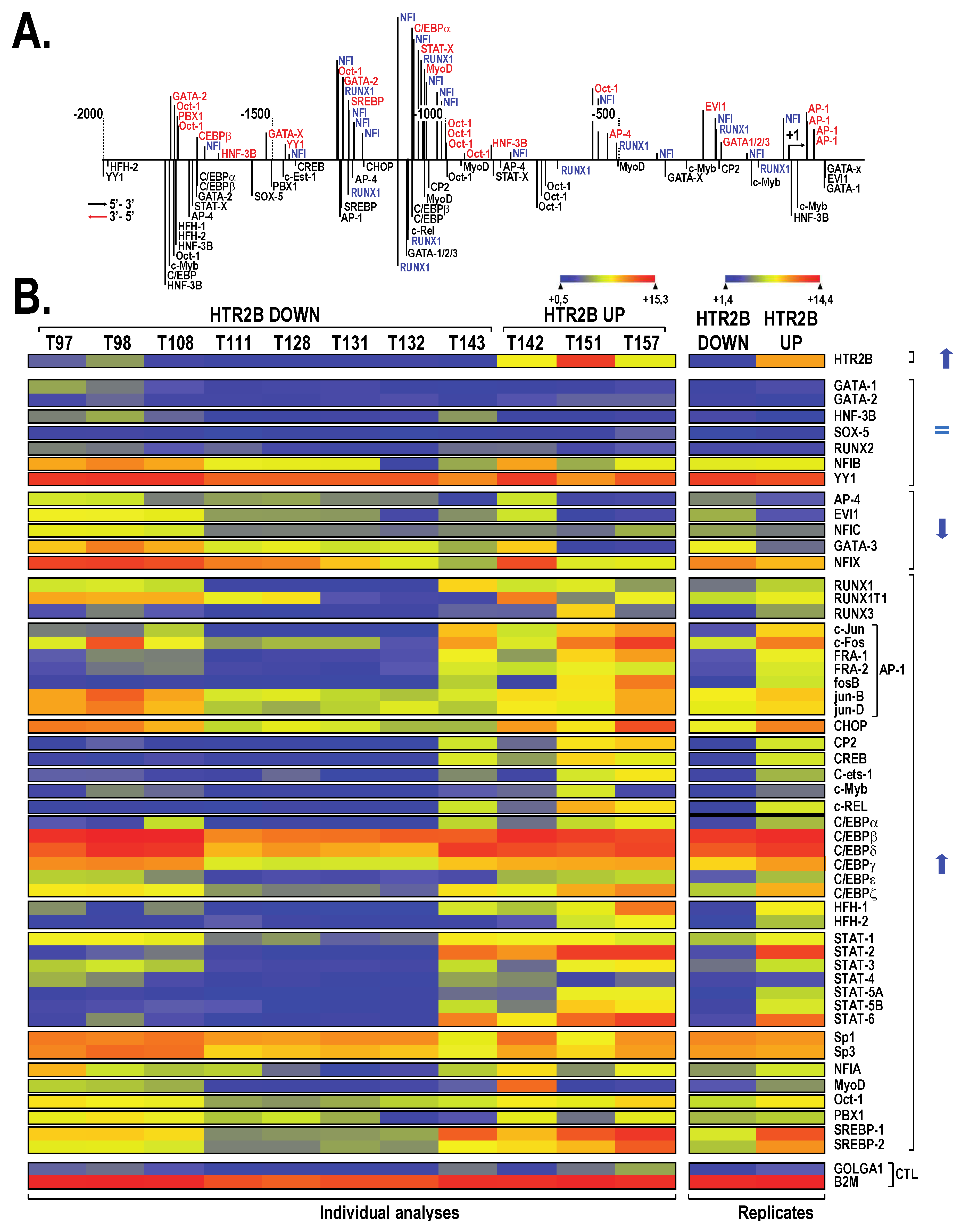
2.1. HTR2B Expression in Human Uveal Melanoma Cell Lines
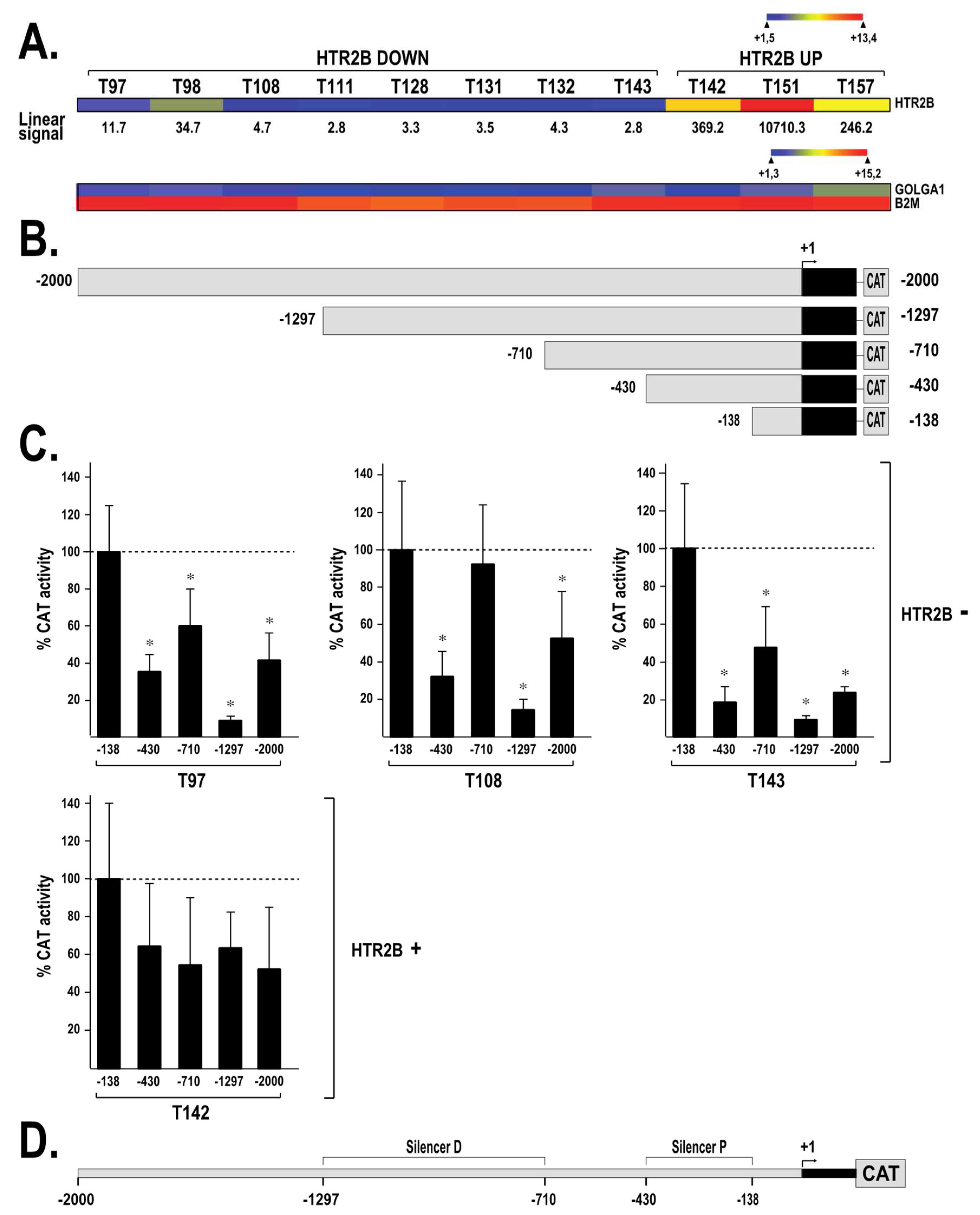
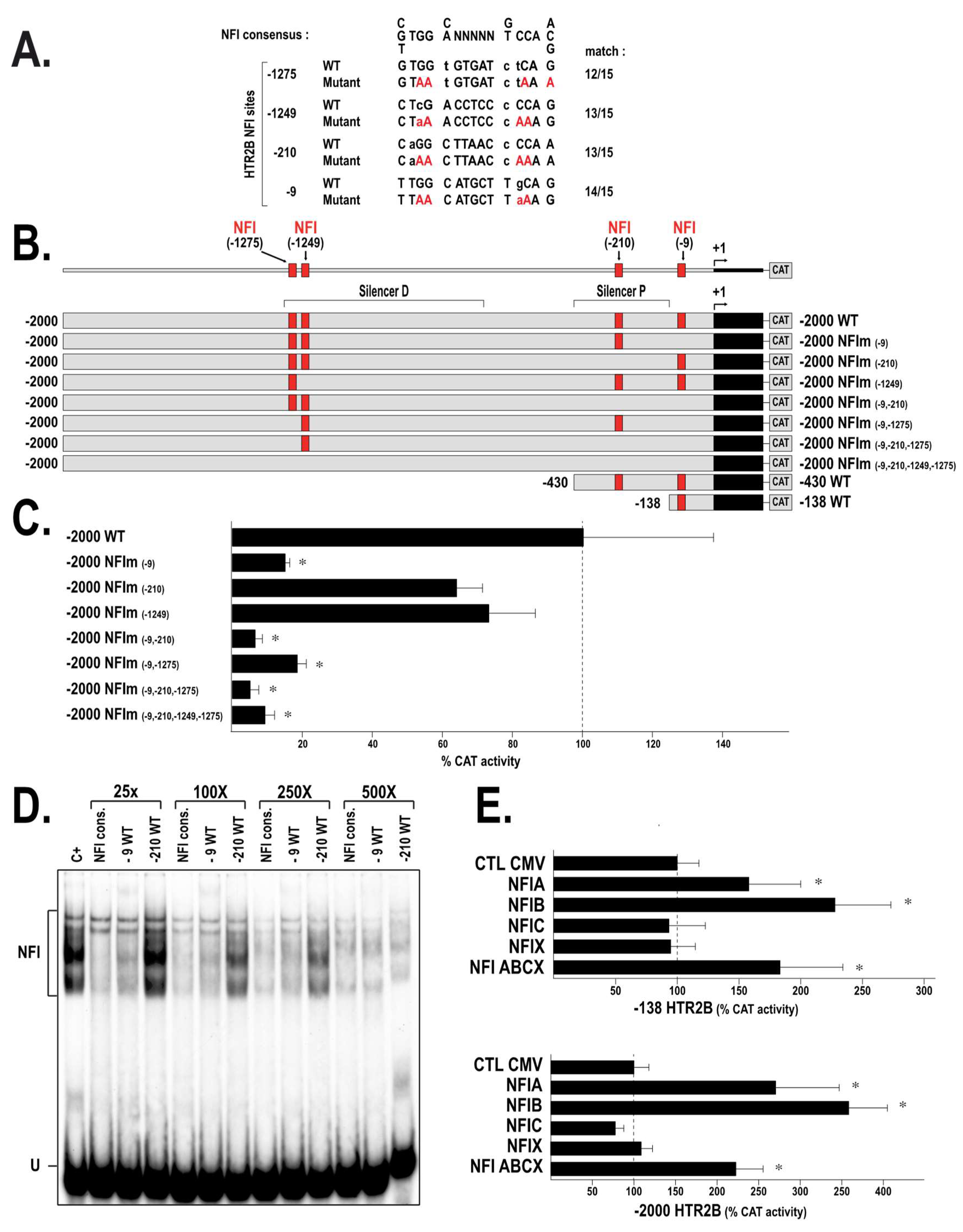
2.2. HTR2B Gene Transcription Is Modulated by Both Positive and Negative Regulatory Elements
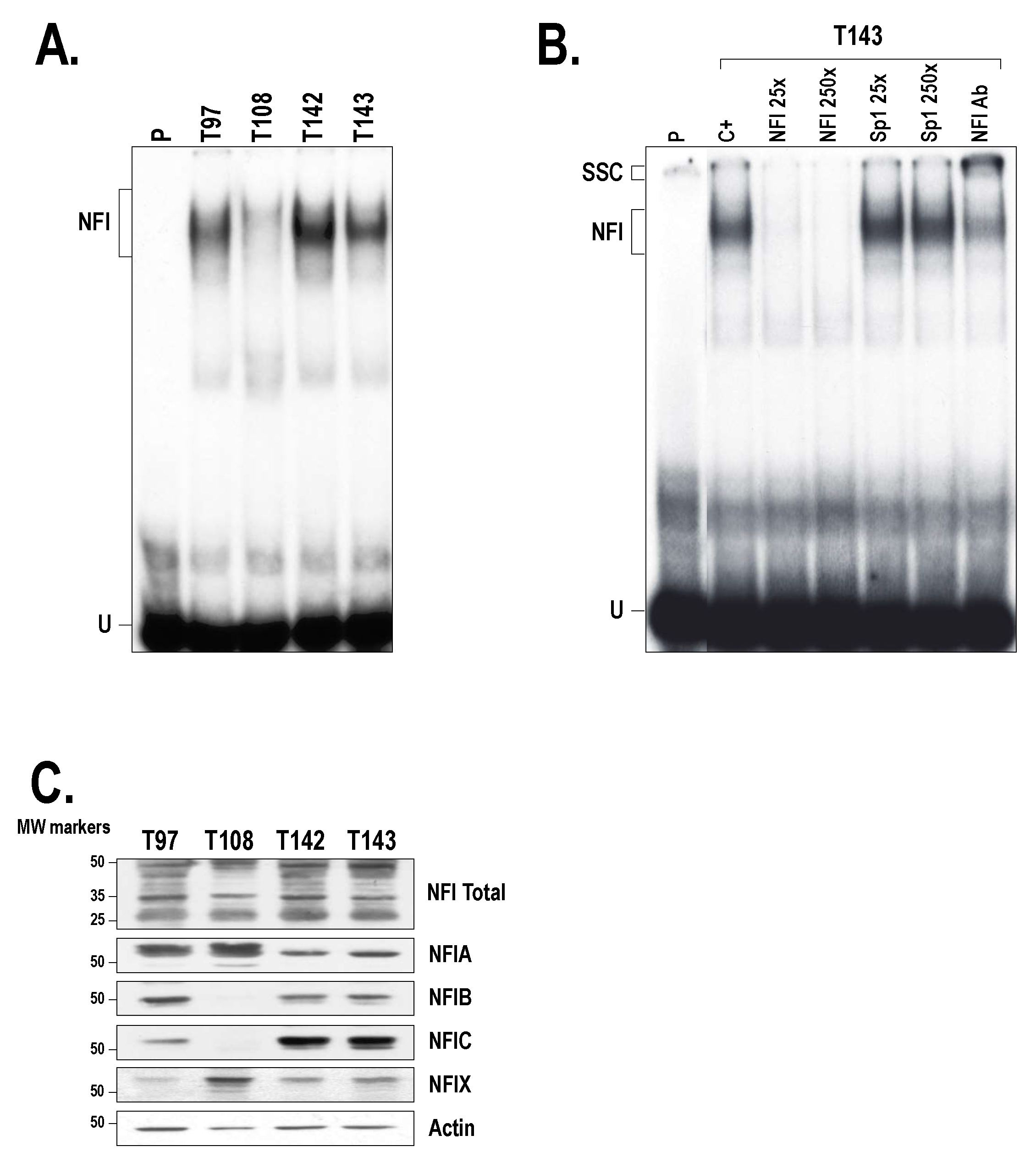
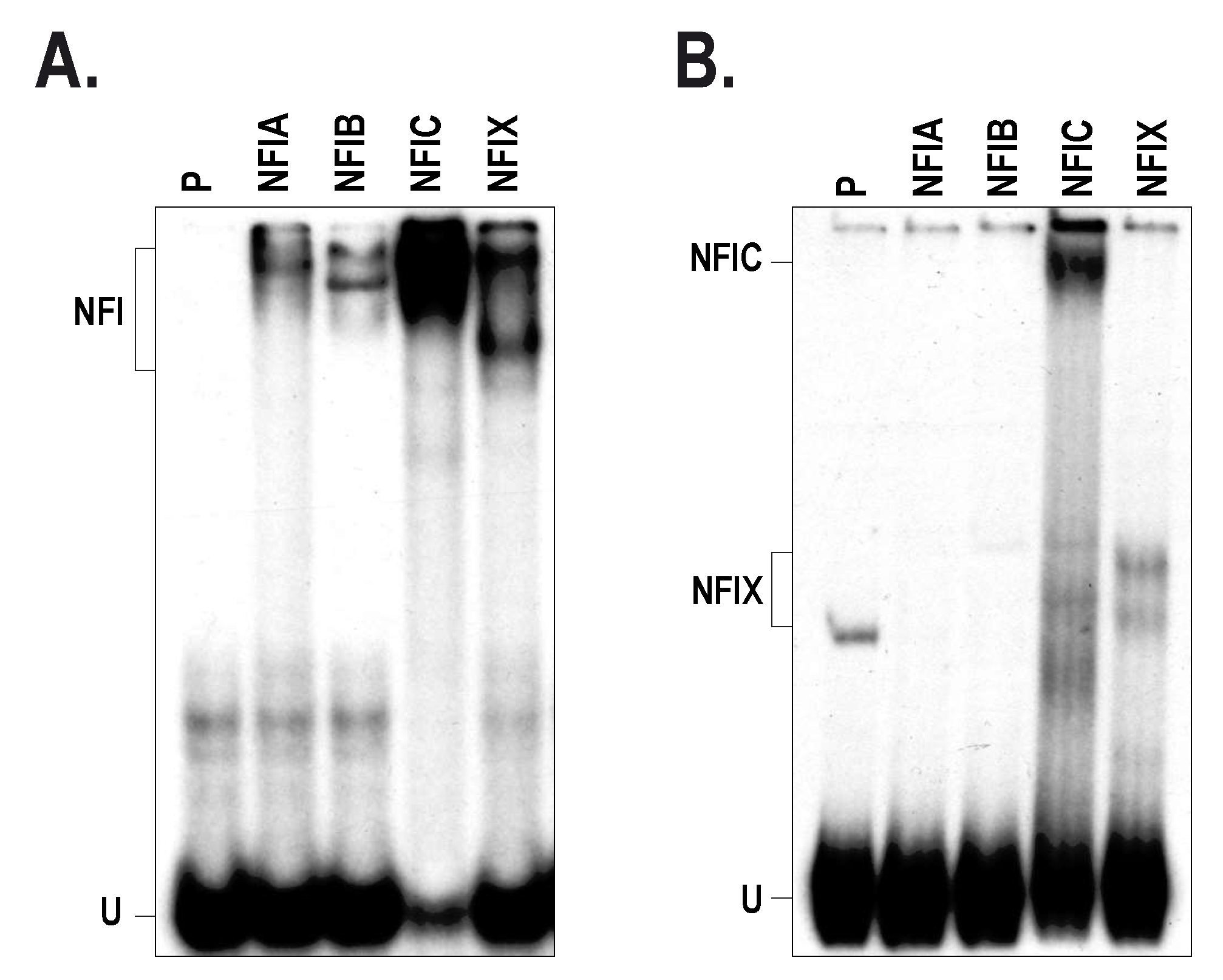
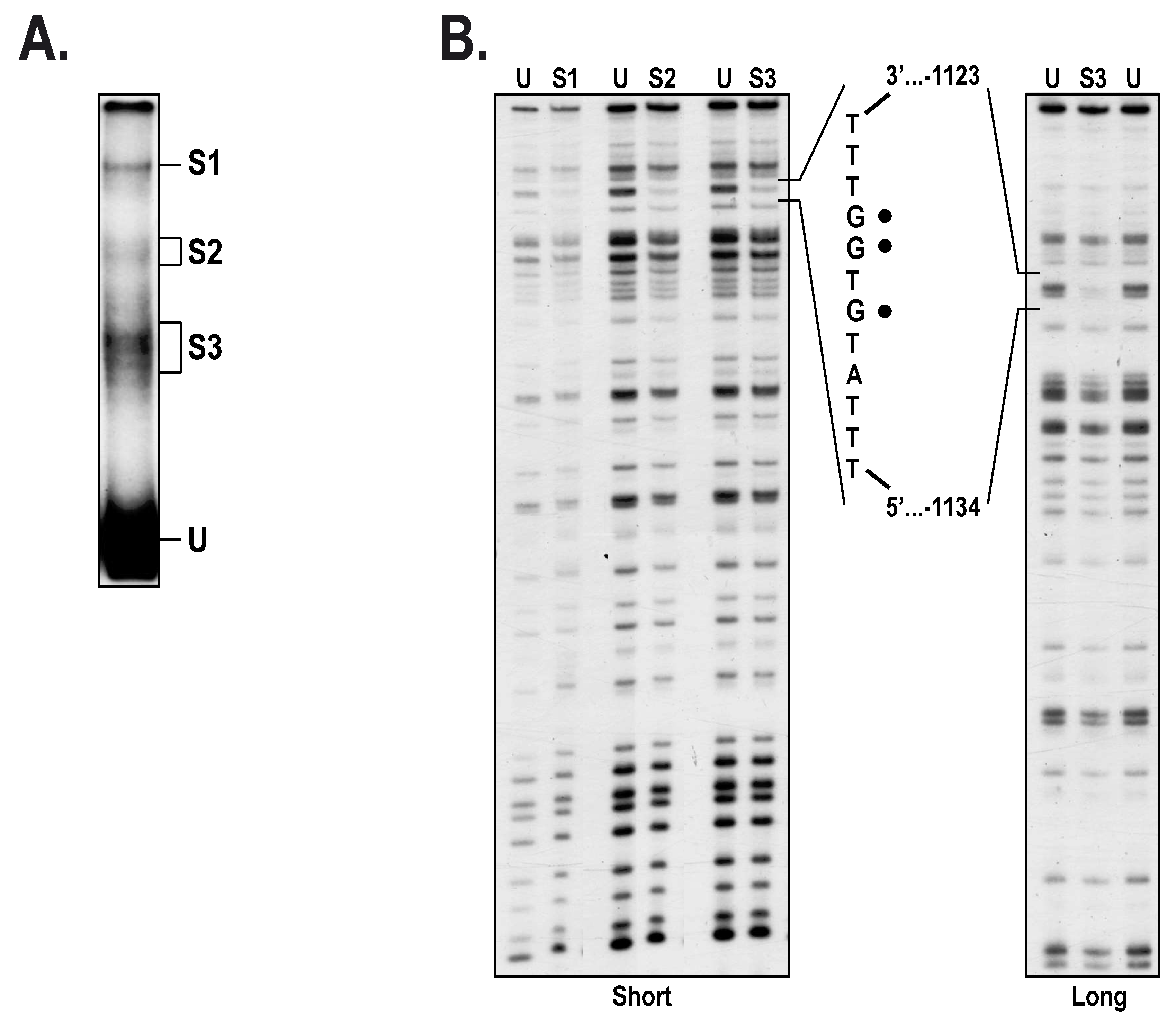
2.3. Members from the NFI Family Bind to the HTR2B Promoter and Positively Regulate Its Transcriptional Activity in Uveal Melanoma
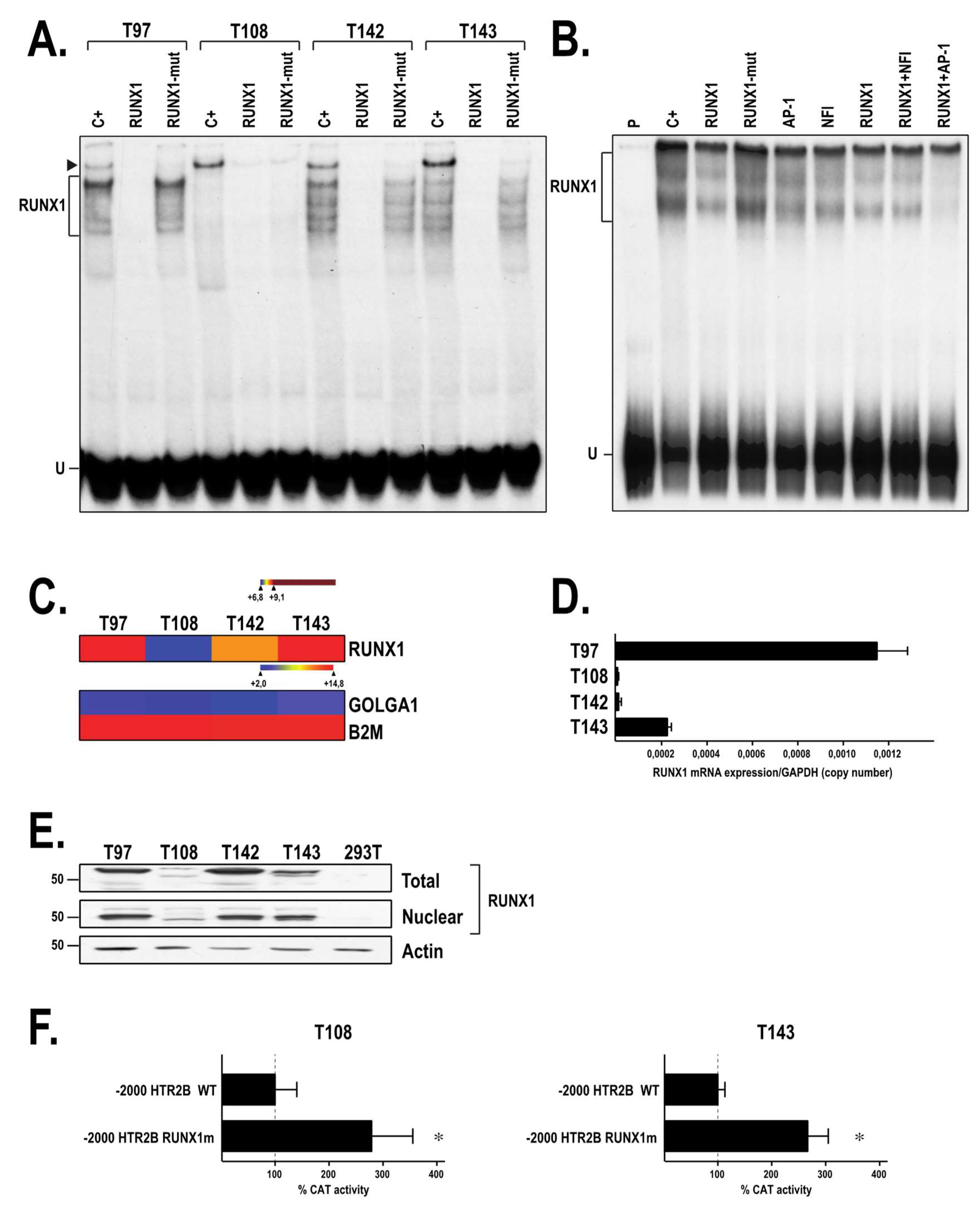
2.4. The Transcription Factor RUNX1 Binds to the HTR2B Promoter In Vitro
2.5. Both RUNX1 and NFI Binds In Vivo to the HTR2B Promoter in Uveal Melanoma
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Indirect Immunofluorescence
4.3. Plasmid Constructs and Oligonucleotides
4.4. Expression of the Human Recombinant NFI Isoforms
4.5. Transient Transfections and CAT Assays
4.6. Preparation of Nuclear Extracts and EMSA
4.7. Methylation Interference Footprinting
4.8. Western Blots
4.9. Gene Expression Profiling
4.10. Chromatin Immunoprecipitation Assays (ChIP)-qPCR
4.11. Quantitative PCR (qPCR)
4.12. Statistical Analyses
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Singh, A.D.; Topham, A. Incidence of uveal melanoma in the United States: 1973–1997. Ophthalmology 2003, 110, 956–961. [Google Scholar] [CrossRef]
- Kujala, E.; Makitie, T.; Kivela, T. Very long-term prognosis of patients with malignant uveal melanoma. Investig. Ophthalmol. Vis. Sci. 2003, 44, 4651–4659. [Google Scholar] [CrossRef]
- Onken, M.D.; Worley, L.A.; Ehlers, J.P.; Harbour, J.W. Gene expression profiling in uveal melanoma reveals two molecular classes and predicts metastatic death. Cancer Res. 2004, 64, 7205–7209. [Google Scholar] [CrossRef] [PubMed]
- Onken, M.D.; Worley, L.A.; Tuscan, M.D.; Harbour, J.W. An accurate, clinically feasible multi-gene expression assay for predicting metastasis in uveal melanoma. J. Mol. Diagn. 2010, 12, 461–468. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Yang, Y.; Chen, L.; Zhang, J. Expression analysis of genes and pathways associated with liver metastases of the uveal melanoma. BMC Med. Genet. 2014, 15, 29. [Google Scholar] [CrossRef] [PubMed]
- Raymond, J.R.; Mukhin, Y.V.; Gelasco, A.; Turner, J.; Collinsworth, G.; Gettys, T.W.; Grewal, J.S.; Garnovskaya, M.N. Multiplicity of mechanisms of serotonin receptor signal transduction. Pharmacol. Ther. 2001, 92, 179–212. [Google Scholar] [CrossRef]
- Harbour, J.W.; Onken, M.D.; Roberson, E.D.; Duan, S.; Cao, L.; Worley, L.A.; Council, M.L.; Matatall, K.A.; Helms, C.; Bowcock, A.M. Frequent mutation of BAP1 in metastasizing uveal melanomas. Science 2010, 330, 1410–1413. [Google Scholar] [CrossRef] [PubMed]
- Van Raamsdonk, C.D.; Bezrookove, V.; Green, G.; Bauer, J.; Gaugler, L.; O’Brien, J.M.; Simpson, E.M.; Barsh, G.S.; Bastian, B.C. Frequent somatic mutations of GNAQ in uveal melanoma and blue naevi. Nature 2009, 457, 599–602. [Google Scholar] [CrossRef] [PubMed]
- Banes, A.K.; Shaw, S.M.; Tawfik, A.; Patel, B.P.; Ogbi, S.; Fulton, D.; Marrero, M.B. Activation of the JAK/STAT pathway in vascular smooth muscle by serotonin. Am. J. Physiol. Cell Physiol. 2005, 288, C805–C812. [Google Scholar] [CrossRef] [PubMed]
- Naito, K.; Tanaka, C.; Mitsuhashi, M.; Moteki, H.; Kimura, M.; Natsume, H.; Ogihara, M. Signal Transduction Mechanism for Serotonin 5-HT2B Receptor-Mediated DNA Synthesis and Proliferation in Primary Cultures of Adult Rat Hepatocytes. Biol. Pharmaceut. Bull. 2016, 39, 121–129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, D.S.; Ward, S.J.; Messaddeq, N.; Launay, J.M.; Maroteaux, L. 5-HT2B receptor-mediated serotonin morphogenetic functions in mouse cranial neural crest and myocardiac cells. Development 1997, 124, 1745–1755. [Google Scholar] [PubMed]
- Vicaut, E.; Laemmel, E.; Stucker, O. Impact of serotonin on tumour growth. Ann. Med. 2000, 32, 187–194. [Google Scholar] [CrossRef] [PubMed]
- Siddiqui, E.J.; Thompson, C.S.; Mikhailidis, D.P.; Mumtaz, F.H. The role of serotonin in tumour growth (review). Oncol. Rep. 2005, 14, 1593–1597. [Google Scholar] [CrossRef] [PubMed]
- Svejda, B.; Kidd, M.; Giovinazzo, F.; Eltawil, K.; Gustafsson, B.I.; Pfragner, R.; Modlin, I.M. The 5-HT(2B) receptor plays a key regulatory role in both neuroendocrine tumor cell proliferation and the modulation of the fibroblast component of the neoplastic microenvironment. Cancer 2010, 116, 2902–2912. [Google Scholar] [CrossRef] [PubMed]
- Soll, C.; Jang, J.H.; Riener, M.O.; Moritz, W.; Wild, P.J.; Graf, R.; Clavien, P.A. Serotonin promotes tumor growth in human hepatocellular cancer. Hepatology 2010, 51, 1244–1254. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Ranvier, G.G.; Weng, J.; Yeh, R.F.; Khanafshar, E.; Suh, I.; Barker, C.; Duh, Q.Y.; Clark, O.H.; Kebebew, E. Identification of biomarkers of adrenocortical carcinoma using genomewide gene expression profiling. Arch. Surg. 2008, 143, 841–846. [Google Scholar] [CrossRef] [PubMed]
- Dizeyi, N.; Bjartell, A.; Hedlund, P.; Tasken, K.A.; Gadaleanu, V.; Abrahamsson, P.A. Expression of serotonin receptors 2B and 4 in human prostate cancer tissue and effects of their antagonists on prostate cancer cell lines. Eur. Urol. 2005, 47, 895–900. [Google Scholar] [CrossRef] [PubMed]
- Henriksen, R.; Dizeyi, N.; Abrahamsson, P.A. Expression of serotonin receptors 5-HT1A, 5-HT1B, 5-HT2B and 5-HT4 in ovary and in ovarian tumours. AntiCancer Res. 2012, 32, 1361–1366. [Google Scholar] [PubMed]
- Reisoli, E.; De Lucchini, S.; Anelli, T.; Biagioni, S.; Nardi, I.; Ori, M. Overexpression of 5-HT2B receptor results in retinal dysplasia and defective ocular morphogenesis in Xenopus embryos. Brain Res. 2008, 1244, 32–39. [Google Scholar] [CrossRef] [PubMed]
- Gaudreault, M.; Vigneault, F.; Gingras, M.E.; Leclerc, S.; Carrier, P.; Germain, L.; Guerin, S.L. Transcriptional regulation of the human alpha6 integrin gene by the transcription factor NFI during corneal wound healing. Investig. Ophthalmol. Vis. Sci. 2008, 49, 3758–3767. [Google Scholar] [CrossRef] [PubMed]
- Gingras, M.E.; Masson-Gadais, B.; Zaniolo, K.; Leclerc, S.; Drouin, R.; Germain, L.; Guerin, S.L. Differential binding of the transcription factors Sp1, AP-1, and NFI to the promoter of the human alpha5 integrin gene dictates its transcriptional activity. Investig. Ophthalmol. Vis. Sci. 2009, 50, 57–67. [Google Scholar] [CrossRef] [PubMed]
- Laniel, M.A.; Poirier, G.G.; Guerin, S.L. Nuclear factor 1 interferes with Sp1 binding through a composite element on the rat poly(ADP-ribose) polymerase promoter to modulate its activity in vitro. J. Biol. Chem. 2001, 276, 20766–20773. [Google Scholar] [CrossRef] [PubMed]
- Brettingham-Moore, K.H.; Taberlay, P.C.; Holloway, A.F. Interplay between Transcription Factors and the Epigenome: Insight from the Role of RUNX1 in Leukemia. Front. Immunol. 2015, 6, 499. [Google Scholar] [CrossRef] [PubMed]
- Kuvardina, O.N.; Herglotz, J.; Kolodziej, S.; Kohrs, N.; Herkt, S.; Wojcik, B.; Oellerich, T.; Corso, J.; Behrens, K.; Kumar, A.; et al. RUNX1 represses the erythroid gene expression program during megakaryocytic differentiation. Blood 2015, 125, 3570–3579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roulet, E.; Bucher, P.; Schneider, R.; Wingender, E.; Dusserre, Y.; Werner, T.; Mermod, N. Experimental analysis and computer prediction of CTF/NFI transcription factor DNA binding sites. J. Mol. Biol. 2000, 297, 833–848. [Google Scholar] [CrossRef] [PubMed]
- Roulet, E.; Busso, S.; Camargo, A.A.; Simpson, A.J.; Mermod, N.; Bucher, P. High-throughput SELEX SAGE method for quantitative modeling of transcription-factor binding sites. Nat. Biotechnol. 2002, 20, 831–835. [Google Scholar] [CrossRef] [PubMed]
- Malho, P.; Dunn, K.; Donaldson, D.; Dubielzig, R.R.; Birand, Z.; Starkey, M. Investigation of prognostic indicators for human uveal melanoma as biomarkers of canine uveal melanoma metastasis. J. Small Anim. Pract. 2013, 54, 584–593. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gronostajski, R.M. Roles of the NFI/CTF gene family in transcription and development. Gene 2000, 249, 31–45. [Google Scholar] [CrossRef]
- Steffensen, K.R.; Holter, E.; Tobin, K.A.; Leclerc, S.; Gustafsson, J.A.; Guerin, S.L.; Eskild, W. Members of the nuclear factor 1 family reduce the transcriptional potential of the nuclear receptor LXRα promoter. Biochem. Biophys. Res. Commun. 2001, 289, 1262–1267. [Google Scholar] [CrossRef] [PubMed]
- Nilsson, J.; Bjursell, G.; Kannius-Janson, M. Nuclear Jak2 and transcription factor NF1-C2: A novel mechanism of prolactin signaling in mammary epithelial cells. Mol. Cell. Biol. 2006, 26, 5663–5674. [Google Scholar] [CrossRef] [PubMed]
- Duval, C.; Gaudreault, M.; Vigneault, F.; Touzel-Deschenes, L.; Rochette, P.J.; Masson-Gadais, B.; Germain, L.; Guerin, S.L. Rescue of the transcription factors Sp1 and NFI in human skin keratinocytes through a feeder-layer-dependent suppression of the proteasome activity. J. Mol. Biol. 2012, 418, 281–299. [Google Scholar] [CrossRef] [PubMed]
- Xia, S.S.; Zhang, G.J.; Liu, Z.L.; Tian, H.P.; He, Y.; Meng, C.Y.; Li, L.F.; Wang, Z.W.; Zhou, T. MicroRNA-22 suppresses the growth, migration and invasion of colorectal cancer cells through a Sp1 negative feedback loop. Oncotarget 2017, 8, 36266–36278. [Google Scholar] [CrossRef] [PubMed]
- Ren, D.; Wang, M.; Guo, W.; Huang, S.; Wang, Z.; Zhao, X.; Du, H.; Song, L.; Peng, X. Double-negative feedback loop between ZEB2 and miR-145 regulates epithelial-mesenchymal transition and stem cell properties in prostate cancer cells. Cell Tissue Res. 2014, 358, 763–778. [Google Scholar] [CrossRef] [PubMed]
- Ohno, Y.; Saeki, K.; Yasunaga, S.; Kurogi, T.; Suzuki-Takedachi, K.; Shirai, M.; Mihara, K.; Yoshida, K.; Voncken, J.W.; Ohtsubo, M.; et al. Transcription of the Geminin gene is regulated by a negative-feedback loop. Mol. Biol. Cell 2014, 25, 1374–1383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murtagh, J.; Martin, F.; Gronostajski, R.M. The Nuclear Factor I (NFI) gene family in mammary gland development and function. J. Mammary Gland Biol. Neoplasia 2003, 8, 241–254. [Google Scholar] [CrossRef] [PubMed]
- Kim, W.Y.; Sieweke, M.; Ogawa, E.; Wee, H.J.; Englmeier, U.; Graf, T.; Ito, Y. Mutual activation of Ets-1 and AML1 DNA binding by direct interaction of their autoinhibitory domains. EMBO J. 1999, 18, 1609–1620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, D.E.; Hetherington, C.J.; Meyers, S.; Rhoades, K.L.; Larson, C.J.; Chen, H.M.; Hiebert, S.W.; Tenen, D.G. CCAAT enhancer-binding protein (C/EBP) and AML1 (CBF alpha2) synergistically activate the macrophage colony-stimulating factor receptor promoter. Mol. Cell. Biol. 1996, 16, 1231–1240. [Google Scholar] [CrossRef] [PubMed]
- Hernandez-Munain, C.; Krangel, M.S. c-Myb and core-binding factor/PEBP2 display functional synergy but bind independently to adjacent sites in the T-cell receptor delta enhancer. Mol. Cell. Biol. 1995, 15, 3090–3099. [Google Scholar] [CrossRef] [PubMed]
- Pencovich, N.; Jaschek, R.; Tanay, A.; Groner, Y. Dynamic combinatorial interactions of RUNX1 and cooperating partners regulates megakaryocytic differentiation in cell line models. Blood 2011, 117, e1–e4. [Google Scholar] [CrossRef] [PubMed]
- Bertrand-Philippe, M.; Ruddell, R.G.; Arthur, M.J.; Thomas, J.; Mungalsingh, N.; Mann, D.A. Regulation of tissue inhibitor of metalloproteinase 1 gene transcription by RUNX1 and RUNX2. J. Biol. Chem. 2004, 279, 24530–24539. [Google Scholar] [CrossRef] [PubMed]
- Bowers, S.R.; Calero-Nieto, F.J.; Valeaux, S.; Fernandez-Fuentes, N.; Cockerill, P.N. Runx1 binds as a dimeric complex to overlapping Runx1 sites within a palindromic element in the human GM-CSF enhancer. Nucleic Acids Res. 2010, 38, 6124–6134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Umansky, K.B.; Gruenbaum-Cohen, Y.; Tsoory, M.; Feldmesser, E.; Goldenberg, D.; Brenner, O.; Groner, Y. Runx1 Transcription Factor Is Required for Myoblasts Proliferation during Muscle Regeneration. PLoS Genet. 2015, 11, e1005457. [Google Scholar] [CrossRef] [PubMed]
- Ichikawa, M.; Yoshimi, A.; Nakagawa, M.; Nishimoto, N.; Watanabe-Okochi, N.; Kurokawa, M. A role for RUNX1 in hematopoiesis and myeloid leukemia. Int. J. Hematol. 2013, 97, 726–734. [Google Scholar] [CrossRef] [PubMed]
- Slattery, M.L.; Lundgreen, A.; Herrick, J.S.; Caan, B.J.; Potter, J.D.; Wolff, R.K. Associations between genetic variation in RUNX1, RUNX2, RUNX3, MAPK1 and eIF4E and riskof colon and rectal cancer: Additional support for a TGF-beta-signaling pathway. Carcinogenesis 2011, 32, 318–326. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.P.; Lan, Y.H.; Lu, T.L.; Pao, J.B.; Chang, T.Y.; Lee, H.Z.; Yang, W.H.; Hsieh, C.J.; Chen, L.M.; Huang, L.C.; et al. Clinical significance of runt-related transcription factor 1 polymorphism in prostate cancer. BJU Int. 2011, 107, 486–492. [Google Scholar] [CrossRef] [PubMed]
- Planaguma, J.; Gonzalez, M.; Doll, A.; Monge, M.; Gil-Moreno, A.; Baro, T.; Garcia, A.; Xercavins, J.; Alameda, F.; Abal, M.; et al. The up-regulation profiles of p21WAF1/CIP1 and RUNX1/AML1 correlate with myometrial infiltration in endometrioid endometrial carcinoma. Hum. Pathol. 2006, 37, 1050–1057. [Google Scholar] [CrossRef] [PubMed]
- Scheitz, C.J.; Lee, T.S.; McDermitt, D.J.; Tumbar, T. Defining a tissue stem cell-driven Runx1/Stat3 signalling axis in epithelial cancer. EMBO J. 2012, 31, 4124–4139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Recouvreux, M.S.; Grasso, E.N.; Echeverria, P.C.; Rocha-Viegas, L.; Castilla, L.H.; Schere-Levy, C.; Tocci, J.M.; Kordon, E.C.; Rubinstein, N. RUNX1 and FOXP3 interplay regulates expression of breast cancer related genes. Oncotarget 2016, 7, 6552–6565. [Google Scholar] [CrossRef] [PubMed]
- Duval, C.; Zaniolo, K.; Leclerc, S.; Salesse, C.; Guerin, S.L. Characterization of the human α9 integrin subunit gene: Promoter analysis and transcriptional regulation in ocular cells. Exp. Eye Res. 2015, 135, 146–163. [Google Scholar] [CrossRef] [PubMed]
- Landreville, S.; Vigneault, F.; Bergeron, M.A.; Leclerc, S.; Gaudreault, M.; Morcos, M.; Mouriaux, F.; Salesse, C.; Guerin, S.L. Suppression of α5 gene expression is closely related to the tumorigenic properties of uveal melanoma cell lines. Pigment Cell Melanoma Res. 2011, 24, 643–655. [Google Scholar] [CrossRef] [PubMed]
- Molloy-Simard, V.; St-Laurent, J.F.; Vigneault, F.; Gaudreault, M.; Dargis, N.; Guerin, M.C.; Leclerc, S.; Morcos, M.; Black, D.; Molgat, Y.; et al. Altered expression of the poly(ADP-ribosyl)ation enzymes in uveal melanoma and regulation of PARG gene expression by the transcription factor ERM. Investig. Ophthalmol. Vis. Sci. 2012, 53, 6219–6231. [Google Scholar] [CrossRef] [PubMed]
- Mouriaux, F.; Zaniolo, K.; Bergeron, M.A.; Weidmann, C.; De La Fouchardiere, A.; Fournier, F.; Droit, A.; Morcos, M.W.; Landreville, S.; Guerin, S.L. Effects of Long-term Serial Passaging on the Characteristics and Properties of Cell Lines Derived From Uveal Melanoma Primary Tumors. Investig. Ophthalmol. Vis. Sci. 2016, 57, 5288–5301. [Google Scholar] [CrossRef] [PubMed]
- Larouche, K.; Leclerc, S.; Salesse, C.; Guerin, S.L. Expression of the alpha 5 integrin subunit gene promoter is positively regulated by the extracellular matrix component fibronectin through the transcription factor Sp1 in corneal epithelial cells in vitro. J. Biol. Chem. 2000, 275, 39182–39192. [Google Scholar] [CrossRef] [PubMed]
- Pothier, F.; Ouellet, M.; Julien, J.P.; Guerin, S.L. An improved CAT assay for promoter analysis in either transgenic mice or tissue culture cells. DNA Cell Biol. 1992, 11, 83–90. [Google Scholar] [CrossRef] [PubMed]
- Roy, R.J.; Gosselin, P.; Guerin, S.L. A short protocol for micro-purification of nuclear proteins from whole animal tissue. Biotechniques 1991, 11, 770–777. [Google Scholar] [PubMed]
- Gaudreault, M.; Gingras, M.E.; Lessard, M.; Leclerc, S.; Guerin, S.L. Electrophoretic mobility shift assays for the analysis of DNA-protein interactions. Methods Mol. Biol. 2009, 543, 15–35. [Google Scholar] [PubMed]
- De Vries, E.; van Driel, W.; van den Heuvel, S.J.; van der Vliet, P.C. Contactpoint analysis of the HeLa nuclear factor I recognition site reveals symmetrical binding at one side of the DNA helix. EMBO J. 1987, 6, 161–168. [Google Scholar] [CrossRef] [PubMed]
- Baldwin, A.S.J. Current Protocols in Molecular Biology. In Current Protocols in Molecular Biology; Struhl, K., Ed.; Wiley: New York, NY, USA, 1989. [Google Scholar]
- Harvey, M.; Brisson, I.; Guerin, S.L. A simple apparatus for fast and inexpensive recovery of DNA from polyacrylamide gels. Biotechniques 1993, 14, 942–948. [Google Scholar] [PubMed]
- Gaudreault, M.; Vigneault, F.; Leclerc, S.; Guerin, S.L. Laminin reduces expression of the human alpha6 integrin subunit gene by altering the level of the transcription factors Sp1 and Sp3. Investig. Ophthalmol. Vis. Sci. 2007, 48, 3490–3505. [Google Scholar] [CrossRef] [PubMed]
- Gingras, M.E.; Larouche, K.; Larouche, N.; Leclerc, S.; Salesse, C.; Guerin, S.L. Regulation of the integrin subunit alpha5 gene promoter by the transcription factors Sp1/Sp3 is influenced by the cell density in rabbit corneal epithelial cells. Investig. Ophthalmol. Vis. Sci. 2003, 44, 3742–3755. [Google Scholar] [CrossRef]
- Couture, C.; Zaniolo, K.; Carrier, P.; Lake, J.; Patenaude, J.; Germain, L.; Guerin, S.L. The tissue-engineered human cornea as a model to study expression of matrix metalloproteinases during corneal wound healing. Biomaterials 2016, 78, 86–101. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| UM Cell Line | T97 | T108 | T142 | T143 | ||||
|---|---|---|---|---|---|---|---|---|
| Immunoprecipitated TF * | NFI | RUNX1 | NFI | RUNX1 | NFI | RUNX1 | NFI | RUNX1 |
| HTR2B region | ||||||||
| −123/+83 (−9 NFI) | 2,766,666 | 158 | 1082 | 8 | 5260 | 6 | 554,945,054 | 51 |
| 387/−133 (−210 NFI) | 19,814 | 43 | 14,938,271 | 93 | 1682 | 515 | 5,586,956 | 63 |
| −1420/−1229 (−1249/−1275 NFI) | 3533 | 1,596,666 | 132,564 | 4 | 28,220 | 8458 | 12 | 1713 |
| −1234/−1022 (−1127 RUNX1) | 1,882,926 | 760,975,609 | 1067 | 1 | 66 | 2,368,750 | 34,982 | 1,269,624 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Benhassine, M.; Guérin, S.L. Transcription of the Human 5-Hydroxytryptamine Receptor 2B (HTR2B) Gene Is under the Regulatory Influence of the Transcription Factors NFI and RUNX1 in Human Uveal Melanoma. Int. J. Mol. Sci. 2018, 19, 3272. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms19103272
Benhassine M, Guérin SL. Transcription of the Human 5-Hydroxytryptamine Receptor 2B (HTR2B) Gene Is under the Regulatory Influence of the Transcription Factors NFI and RUNX1 in Human Uveal Melanoma. International Journal of Molecular Sciences. 2018; 19(10):3272. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms19103272
Chicago/Turabian StyleBenhassine, Manel, and Sylvain L. Guérin. 2018. "Transcription of the Human 5-Hydroxytryptamine Receptor 2B (HTR2B) Gene Is under the Regulatory Influence of the Transcription Factors NFI and RUNX1 in Human Uveal Melanoma" International Journal of Molecular Sciences 19, no. 10: 3272. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms19103272