Co-Evolution of Intrinsically Disordered Proteins with Folded Partners Witnessed by Evolutionary Couplings


Abstract
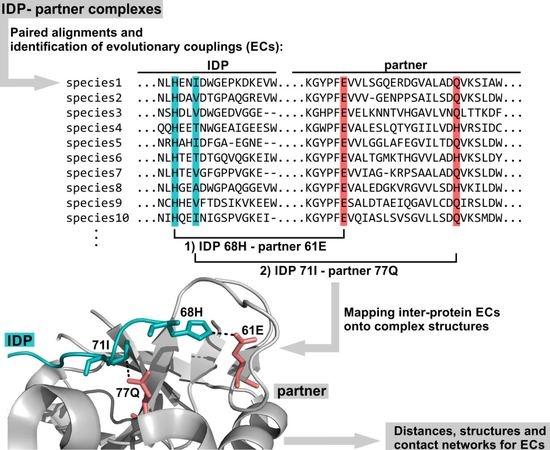
:
1. Introduction
2. Results
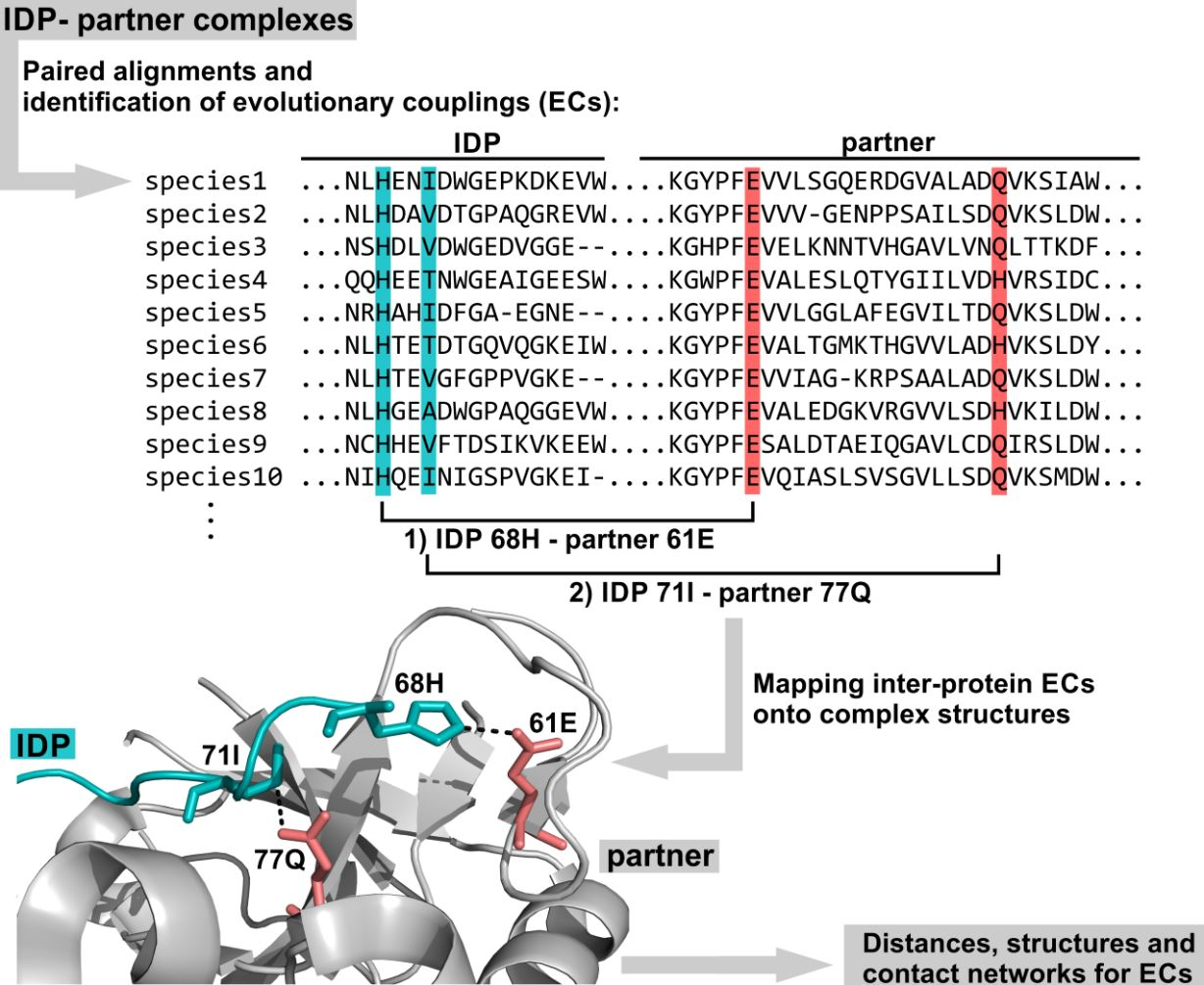
2.1. ECs Were Detected Outside the Sequence Ranges with PDB Coordinates, but Visible ECs Reside on the Interfaces
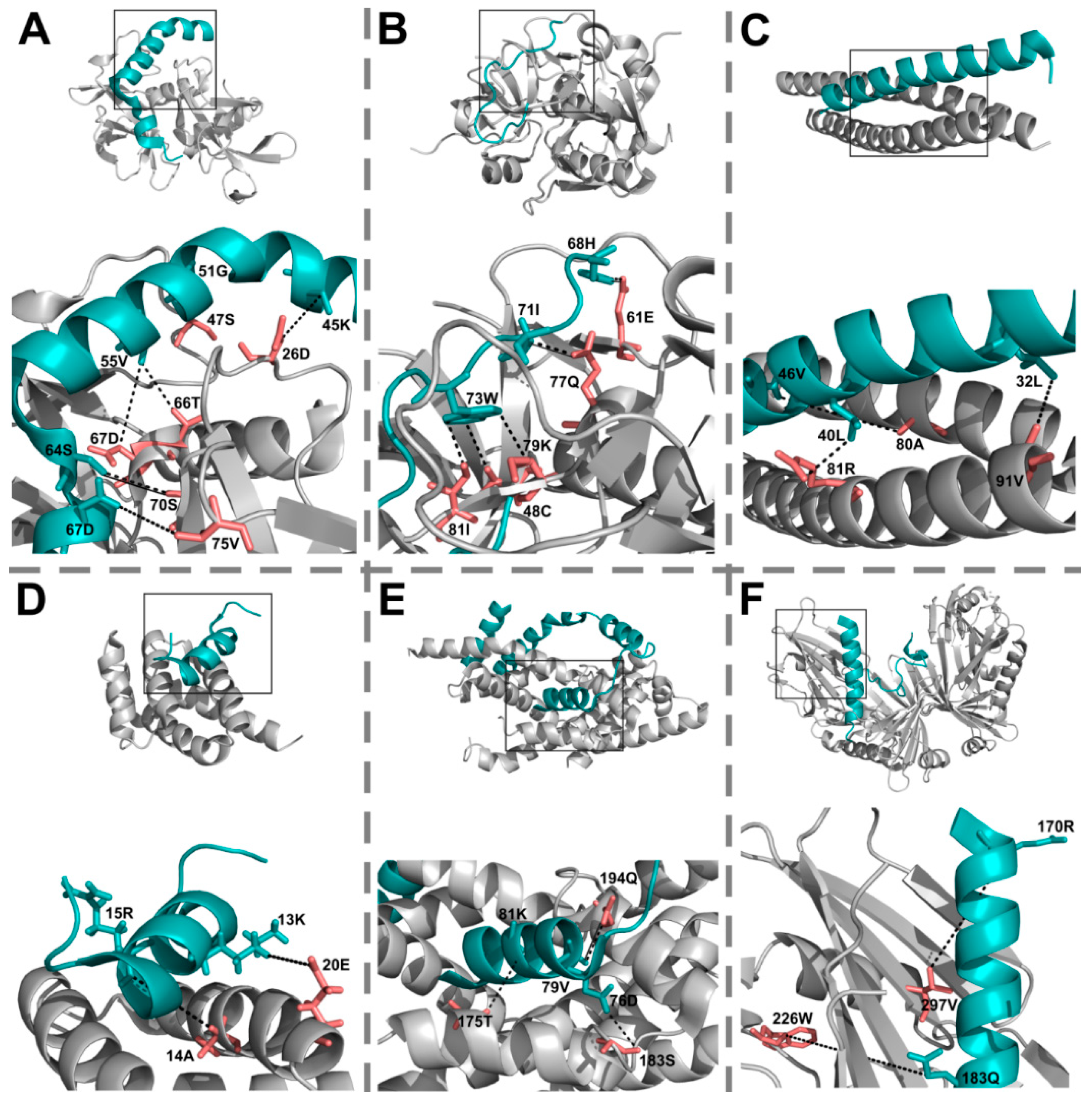
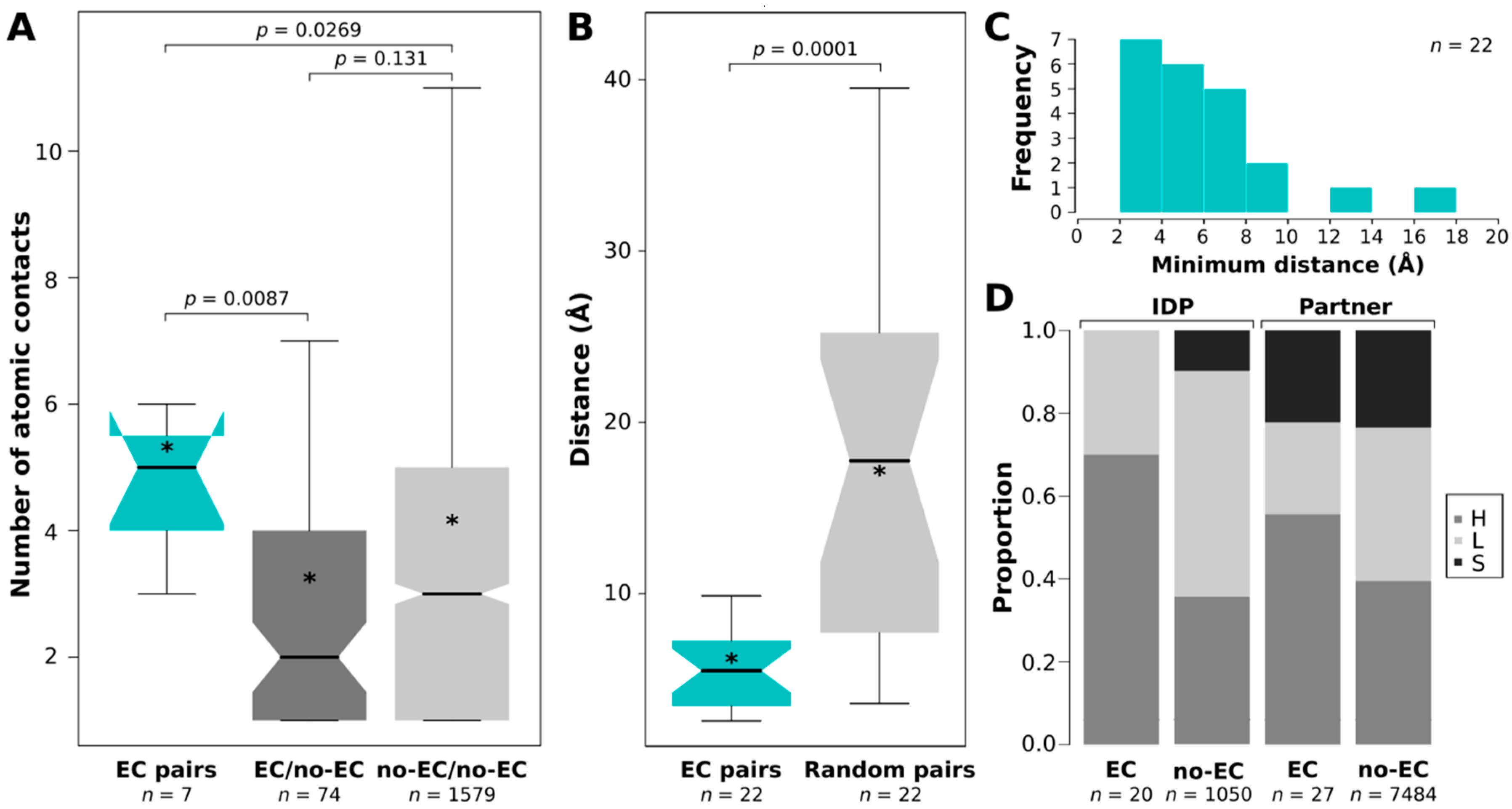
2.2. EC Pairs Have More Atomic Contacts between Them than Other Pairs
2.3. ECs Are Significantly Closer to Each Other than Randomly Paired IDP–Partner Interface Residues
2.4. EC Residues Preferentially Occur in Helices in Both IDPs and Partners
2.5. General Trends Observed for the Protein Complexes with High-Scoring Interprotein ECs
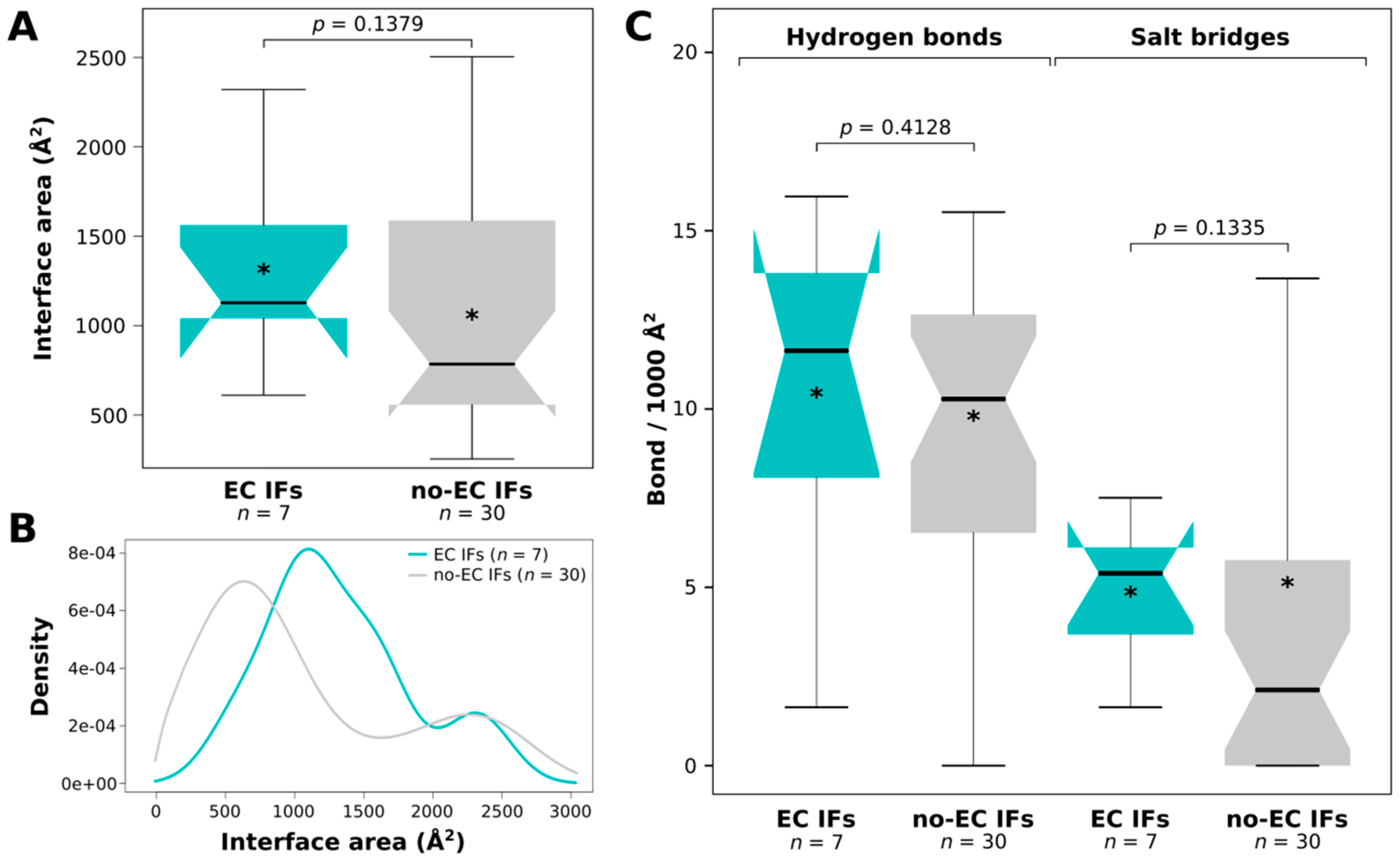
2.6. ECs Could Not Be Identified for Very Small Interfaces with no H-Bonds or Salt Bridges
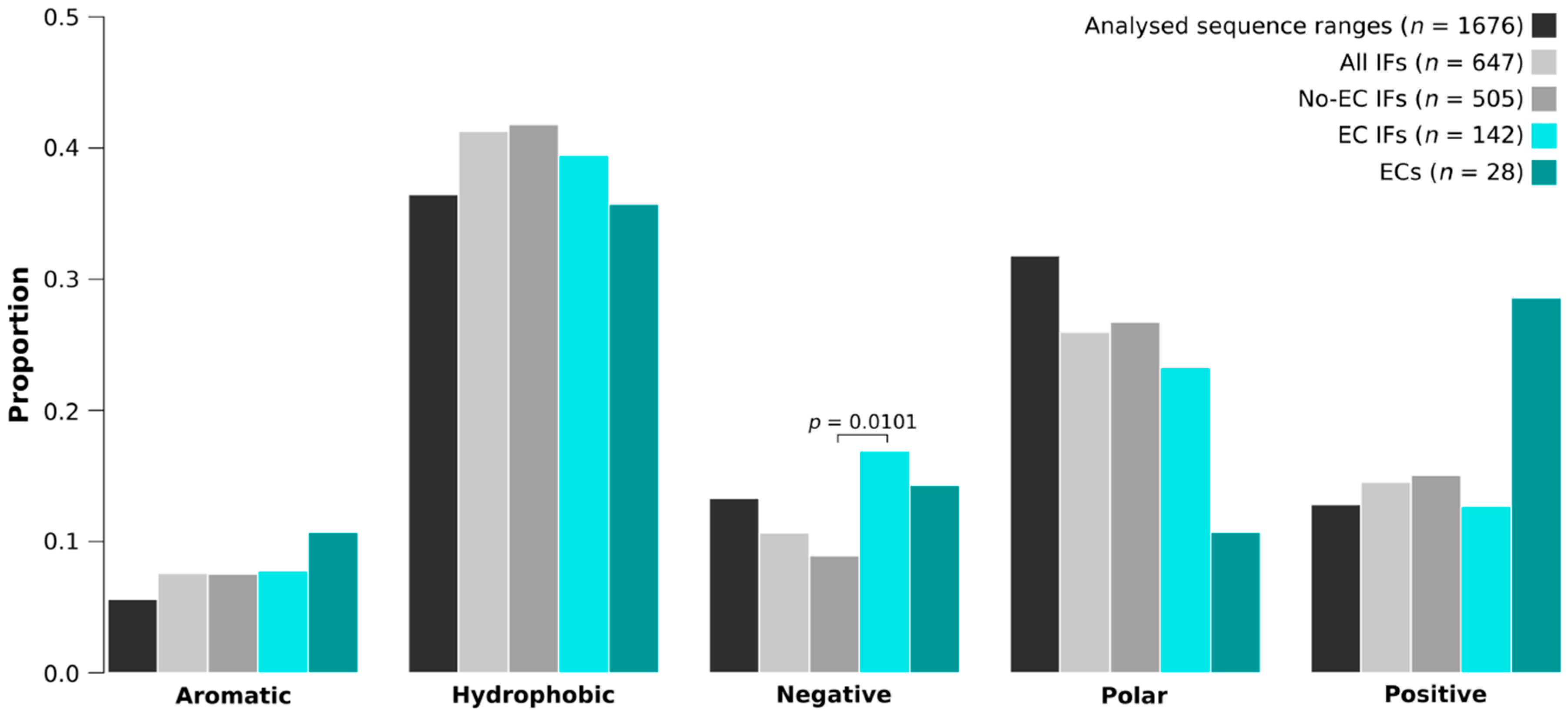
2.7. EC Interfaces Are Enriched in Negatively Charged Residues
3. Discussion
4. Materials and Methods
4.1. Dataset Preparation
4.2. Co-Evolution Analysis
4.3. Interface Properties
4.4. Amino Acid Compositions
4.5. Residue–Residue Contact Networks
4.6. Secondary Structure Assignments
4.7. Residue–Residue Distances
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Stein, R.R.; Marks, D.S.; Sander, C. Inferring Pairwise Interactions from Biological Data Using Maximum-Entropy Probability Models. PLoS Comput. Biol. 2015, 11, e1004182. [Google Scholar] [CrossRef] [PubMed]
- Marks, D.S.; Hopf, T.A.; Sander, C. Protein structure prediction from sequence variation. Nat. Biotechnol. 2012, 30, 1072–1080. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taylor, W.R.; Hamilton, R.S.; Sadowski, M.I. Prediction of contacts from correlated sequence substitutions. Curr. Opin. Struct. Biol. 2013, 23, 473–479. [Google Scholar] [CrossRef] [PubMed]
- Adhikari, B.; Bhattacharya, D.; Cao, R.; Cheng, J. CONFOLD: Residue-residue contact-guided ab initio protein folding. Proteins 2015, 83, 1436–1449. [Google Scholar] [CrossRef] [PubMed]
- Jones, D.T.; Singh, T.; Kosciolek, T.; Tetchner, S. MetaPSICOV: Combining coevolution methods for accurate prediction of contacts and long range hydrogen bonding in proteins. Bioinformatics 2015, 31, 999–1006. [Google Scholar] [CrossRef] [PubMed]
- Kosciolek, T.; Jones, D.T. De novo structure prediction of globular proteins aided by sequence variation-derived contacts. PLoS ONE 2014, 9, e92197. [Google Scholar] [CrossRef] [PubMed]
- Kosciolek, T.; Jones, D.T. Accurate contact predictions using covariation techniques and machine learning. Proteins 2015. [Google Scholar] [CrossRef] [PubMed]
- Ovchinnikov, S.; Kim, D.E.; Wang, R.Y.; Liu, Y.; DiMaio, F.; Baker, D. Improved de novo structure prediction in CASP11 by incorporating Co-evolution information into rosetta. Proteins 2015. [Google Scholar] [CrossRef]
- Ovchinnikov, S.; Kinch, L.; Park, H.; Liao, Y.; Pei, J.; Kim, D.E.; Kamisetty, H.; Grishin, N.V.; Baker, D. Large-scale determination of previously unsolved protein structures using evolutionary information. eLife 2015, 4, e09248. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Sun, S.; Li, Z.; Zhang, R.; Xu, J. Accurate De Novo Prediction of Protein Contact Map by Ultra-Deep Learning Model. PLoS Computat. Biol. 2017, 13, e1005324. [Google Scholar] [CrossRef] [PubMed]
- Hayat, S.; Sander, C.; Marks, D.S.; Elofsson, A. All-atom 3D structure prediction of transmembrane beta-barrel proteins from sequences. Proc. Natl. Acad. Sci. USA 2015, 112, 5413–5418. [Google Scholar] [CrossRef] [PubMed]
- Hopf, T.A.; Colwell, L.J.; Sheridan, R.; Rost, B.; Sander, C.; Marks, D.S. Three-dimensional structures of membrane proteins from genomic sequencing. Cell 2012, 149, 1607–1621. [Google Scholar] [CrossRef] [PubMed]
- Halabi, N.; Rivoire, O.; Leibler, S.; Ranganathan, R. Protein sectors: Evolutionary units of three-dimensional structure. Cell 2009, 138, 774–786. [Google Scholar] [CrossRef] [PubMed]
- Hopf, T.A.; Scharfe, C.P.; Rodrigues, J.P.; Green, A.G.; Kohlbacher, O.; Sander, C.; Bonvin, A.M.; Marks, D.S. Sequence co-evolution gives 3D contacts and structures of protein complexes. eLife 2014, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ovchinnikov, S.; Kamisetty, H.; Baker, D. Robust and accurate prediction of residue-residue interactions across protein interfaces using evolutionary information. eLife 2014, 3, e02030. [Google Scholar] [CrossRef] [PubMed]
- Kamisetty, H.; Ovchinnikov, S.; Baker, D. Assessing the utility of coevolution-based residue-residue contact predictions in a sequence- and structure-rich era. Proc. Natl. Acad. Sci. USA 2013, 110, 15674–15679. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dyson, H.J.; Wright, P.E. Intrinsically unstructured proteins and their functions. Nat. Rev. Mol. Cell Biol. 2005, 6, 197–208. [Google Scholar] [CrossRef] [PubMed]
- Habchi, J.; Tompa, P.; Longhi, S.; Uversky, V.N. Introducing protein intrinsic disorder. Chem. Rev. 2014, 114, 6561–6588. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van der Lee, R.; Buljan, M.; Lang, B.; Weatheritt, R.J.; Daughdrill, G.W.; Dunker, A.K.; Fuxreiter, M.; Gough, J.; Gsponer, J.; Jones, D.T.; et al. Classification of intrinsically disordered regions and proteins. Chem. Rev. 2014, 114, 6589–6631. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wright, P.E.; Dyson, H.J. Intrinsically unstructured proteins: Re-assessing the protein structure-function paradigm. J. Mol. Biol. 1999, 293, 321–331. [Google Scholar] [CrossRef] [PubMed]
- Pancsa, R.; Fuxreiter, M. Interactions via intrinsically disordered regions: What kind of motifs? IUBMB Life 2012, 64, 513–520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tompa, P.; Davey, N.E.; Gibson, T.J.; Babu, M.M. A Million Peptide Motifs for the Molecular Biologist. Mol. Cell 2014, 55, 161–169. [Google Scholar] [CrossRef] [PubMed]
- Iakoucheva, L.M.; Radivojac, P.; Brown, C.J.; O′Connor, T.R.; Sikes, J.G.; Obradovic, Z.; Dunker, A.K. The importance of intrinsic disorder for protein phosphorylation. Nucleic Acids Res. 2004, 32, 1037–1049. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wright, P.E.; Dyson, H.J. Intrinsically disordered proteins in cellular signalling and regulation. Nat. Rev. Mol. Cell Biol. 2014, 16, 18–29. [Google Scholar] [CrossRef] [PubMed]
- Hegyi, H.; Schad, E.; Tompa, P. Structural disorder promotes assembly of protein complexes. BMC Struct. Biol. 2007, 7, 65. [Google Scholar] [CrossRef] [PubMed]
- Balazs, A.; Csizmok, V.; Buday, L.; Rakacs, M.; Kiss, R.; Bokor, M.; Udupa, R.; Tompa, K.; Tompa, P. High levels of structural disorder in scaffold proteins as exemplified by a novel neuronal protein, CASK-interactive protein1. FEBS J. 2009, 276, 3744–3756. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mark, W.Y.; Liao, J.C.; Lu, Y.; Ayed, A.; Laister, R.; Szymczyna, B.; Chakrabartty, A.; Arrowsmith, C.H. Characterization of segments from the central region of BRCA1: An intrinsically disordered scaffold for multiple protein-protein and protein-DNA interactions? J. Mol. Biol. 2005, 345, 275–287. [Google Scholar] [CrossRef] [PubMed]
- Dosztanyi, Z.; Chen, J.; Dunker, A.K.; Simon, I.; Tompa, P. Disorder and sequence repeats in hub proteins and their implications for network evolution. J. Proteome Res. 2006, 5, 2985–2995. [Google Scholar] [CrossRef] [PubMed]
- Dunker, A.K.; Cortese, M.S.; Romero, P.; Iakoucheva, L.M.; Uversky, V.N. Flexible nets. The roles of intrinsic disorder in protein interaction networks. FEBS J. 2005, 272, 5129–5148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uversky, V.N.; Dave, V.; Iakoucheva, L.M.; Malaney, P.; Metallo, S.J.; Pathak, R.R.; Joerger, A.C. Pathological unfoldomics of uncontrolled chaos: Intrinsically disordered proteins and human diseases. Chem. Rev. 2014, 114, 6844–6879. [Google Scholar] [CrossRef] [PubMed]
- Davey, N.E.; Van Roey, K.; Weatheritt, R.J.; Toedt, G.; Uyar, B.; Altenberg, B.; Budd, A.; Diella, F.; Dinkel, H.; Gibson, T.J. Attributes of short linear motifs. Mol. BioSyst. 2012, 8, 268–281. [Google Scholar] [CrossRef] [PubMed]
- Van Roey, K.; Uyar, B.; Weatheritt, R.J.; Dinkel, H.; Seiler, M.; Budd, A.; Gibson, T.J.; Davey, N.E. Short linear motifs: Ubiquitous and functionally diverse protein interaction modules directing cell regulation. Chem. Rev. 2014, 114, 6733–6778. [Google Scholar] [CrossRef] [PubMed]
- Fuxreiter, M.; Tompa, P.; Simon, I. Local structural disorder imparts plasticity on linear motifs. Bioinformatics 2007, 23, 950–956. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buljan, M.; Chalancon, G.; Eustermann, S.; Wagner, G.P.; Fuxreiter, M.; Bateman, A.; Babu, M.M. Tissue-specific splicing of disordered segments that embed binding motifs rewires protein interaction networks. Mol. Cell 2012, 46, 871–883. [Google Scholar] [CrossRef] [PubMed]
- Weatheritt, R.J.; Davey, N.E.; Gibson, T.J. Linear motifs confer functional diversity onto splice variants. Nucleic Acids Res. 2012, 40, 7123–7131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weatheritt, R.J.; Gibson, T.J. Linear motifs: Lost in (pre)translation. Trends Biochem. Sci. 2012, 37, 333–341. [Google Scholar] [CrossRef] [PubMed]
- Brown, C.J.; Takayama, S.; Campen, A.M.; Vise, P.; Marshall, T.W.; Oldfield, C.J.; Williams, C.J.; Dunker, A.K. Evolutionary rate heterogeneity in proteins with long disordered regions. J. Mol. Evol. 2002, 55, 104–110. [Google Scholar] [CrossRef] [PubMed]
- Brown, C.J.; Johnson, A.K.; Daughdrill, G.W. Comparing models of evolution for ordered and disordered proteins. Mol. Biol. Evol. 2010, 27, 609–621. [Google Scholar] [CrossRef] [PubMed]
- Jeong, C.S.; Kim, D. Coevolved residues and the functional association for intrinsically disordered proteins. In Proceedings of the Pacific Symposium on Biocomputing, Kohala Coast, HI, USA, 3–7 January 2012; pp. 140–151. [Google Scholar] [CrossRef]
- Toth-Petroczy, A.; Palmedo, P.; Ingraham, J.; Hopf, T.A.; Berger, B.; Sander, C.; Marks, D.S. Structured States of Disordered Proteins from Genomic Sequences. Cell 2016, 167, 158–170. [Google Scholar] [CrossRef] [PubMed]
- Tompa, P.; Schad, E.; Tantos, A.; Kalmar, L. Intrinsically disordered proteins: Emerging interaction specialists. Curr. Opin. Struct. Biol. 2015, 35, 49–59. [Google Scholar] [CrossRef] [PubMed]
- Dunker, A.K.; Obradovic, Z.; Romero, P.; Garner, E.C.; Brown, C.J. Intrinsic protein disorder in complete genomes. Genome Inform. 2000, 11, 161–171. [Google Scholar]
- Pancsa, R.; Tompa, P. Structural disorder in eukaryotes. PLoS ONE 2012, 7, e34687. [Google Scholar] [CrossRef] [PubMed]
- Ward, J.J.; Sodhi, J.S.; McGuffin, L.J.; Buxton, B.F.; Jones, D.T. Prediction and functional analysis of native disorder in proteins from the three kingdoms of life. J. Mol. Biol. 2004, 337, 635–645. [Google Scholar] [CrossRef] [PubMed]
- Schad, E.; Ficho, E.; Pancsa, R.; Simon, I.; Dosztanyi, Z.; Meszaros, B. DIBS: A repository of disordered binding sites mediating interactions with ordered proteins. Bioinformatics 2018, 34, 535–537. [Google Scholar] [CrossRef] [PubMed]
- Finn, R.D.; Coggill, P.; Eberhardt, R.Y.; Eddy, S.R.; Mistry, J.; Mitchell, A.L.; Potter, S.C.; Punta, M.; Qureshi, M.; Sangrador-Vegas, A.; et al. The Pfam protein families database: Towards a more sustainable future. Nucleic Acids Res. 2016, 44, D279–D285. [Google Scholar] [CrossRef] [PubMed]
- Krissinel, E.; Henrick, K. Inference of macromolecular assemblies from crystalline state. J. Mol. Biol. 2007, 372, 774–797. [Google Scholar] [CrossRef] [PubMed]
- Zor, T.; Mayr, B.M.; Dyson, H.J.; Montminy, M.R.; Wright, P.E. Roles of phosphorylation and helix propensity in the binding of the KIX domain of CREB-binding protein by constitutive (c-Myb) and inducible (CREB) activators. J. Biol. Chem. 2002, 277, 42241–42248. [Google Scholar] [CrossRef] [PubMed]
- Selenko, P.; Gregorovic, G.; Sprangers, R.; Stier, G.; Rhani, Z.; Kramer, A.; Sattler, M. Structural basis for the molecular recognition between human splicing factors U2AF65 and SF1/mBBP. Mol. Cell 2003, 11, 965–976. [Google Scholar] [CrossRef]
- Tompa, P.; Fuxreiter, M. Fuzzy complexes: Polymorphism and structural disorder in protein-protein interactions. Trends Biochem. Sci. 2008, 33, 2–8. [Google Scholar] [CrossRef] [PubMed]
- Kayikci, M.; Venkatakrishnan, A.J.; Scott-Brown, J.; Ravarani, C.N.J.; Flock, T.; Babu, M.M. Visualization and analysis of non-covalent contacts using the Protein Contacts Atlas. Nat. Struct. Mol. Biol. 2018, 25, 185–194. [Google Scholar] [CrossRef] [PubMed]
- Anishchenko, I.; Ovchinnikov, S.; Kamisetty, H.; Baker, D. Origins of coevolution between residues distant in protein 3D structures. Proc. Natl. Acad. Sci. USA 2017, 114, 9122–9127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ait-Bara, S.; Carpousis, A.J.; Quentin, Y. RNase E in the gamma-Proteobacteria: Conservation of intrinsically disordered noncatalytic region and molecular evolution of microdomains. Mol. Genet. Genom. 2015, 290, 847–862. [Google Scholar] [CrossRef] [PubMed]
- Savvides, S.N.; Raghunathan, S.; Futterer, K.; Kozlov, A.G.; Lohman, T.M.; Waksman, G. The C-terminal domain of full-length E. coli SSB is disordered even when bound to DNA. Protein Sci. 2004, 13, 1942–1947. [Google Scholar] [CrossRef] [PubMed]
- Shereda, R.D.; Kozlov, A.G.; Lohman, T.M.; Cox, M.M.; Keck, J.L. SSB as an organizer/mobilizer of genome maintenance complexes. Crit. Rev. Biochem. Mol. Biol. 2008, 43, 289–318. [Google Scholar] [CrossRef] [PubMed]
- Lu, D.; Keck, J.L. Structural basis of Escherichia coli single-stranded DNA-binding protein stimulation of exonuclease I. Proc. Natl. Acad. Sci. USA 2008, 105, 9169–9174. [Google Scholar] [CrossRef] [PubMed]
- Tompa, P.; Szasz, C.; Buday, L. Structural disorder throws new light on moonlighting. Trends Biochem. Sci. 2005, 30, 484–489. [Google Scholar] [CrossRef] [PubMed]
- Finn, R.D.; Clements, J.; Arndt, W.; Miller, B.L.; Wheeler, T.J.; Schreiber, F.; Bateman, A.; Eddy, S.R. HMMER web server: 2015 update. Nucleic Acids Res. 2015, 43, W30–W38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Apweiler, R.; Bairoch, A.; Wu, C.H.; Barker, W.C.; Boeckmann, B.; Ferro, S.; Gasteiger, E.; Huang, H.; Lopez, R.; Magrane, M.; et al. UniProt: The Universal Protein knowledgebase. Nucleic Acids Res. 2004, 32, D115–D119. [Google Scholar] [CrossRef] [PubMed]
- Szklarczyk, D.; Franceschini, A.; Wyder, S.; Forslund, K.; Heller, D.; Huerta-Cepas, J.; Simonovic, M.; Roth, A.; Santos, A.; Tsafou, K.P.; et al. STRING v10: Protein-protein interaction networks, integrated over the tree of life. Nucleic Acids Res. 2015, 43, D447–D452. [Google Scholar] [CrossRef] [PubMed]
- Kersey, P.J.; Allen, J.E.; Armean, I.; Boddu, S.; Bolt, B.J.; Carvalho-Silva, D.; Christensen, M.; Davis, P.; Falin, L.J.; Grabmueller, C.; et al. Ensembl Genomes 2016: More genomes, more complexity. Nucleic Acids Res. 2016, 44, D574–D580. [Google Scholar] [CrossRef] [PubMed]
- Newcombe, R.G. Interval estimation for the difference between independent proportions: Comparison of eleven methods. Stat. Med. 1998, 17, 873–890. [Google Scholar] [CrossRef]
- Touw, W.G.; Baakman, C.; Black, J.; te Beek, T.A.; Krieger, E.; Joosten, R.P.; Vriend, G. A series of PDB-related databanks for everyday needs. Nucleic Acids Res. 2015, 43, D364–D368. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Complex | Folded Partner | IDP | Gremlin Analysis Results | ||||||
|---|---|---|---|---|---|---|---|---|---|
| DIBS ID-PDB ID (Chains Partner(s)|IDP) | IF Area (Å2) | Gene Name | Uniprot AC_region | Gene Name | Uniprot AC_Region | Length | # of Seq. in PFAM 31 Full | Coverage (seq/res) | ECs by Gremlin/by EVComplex/Gremlin IDP ECs on IF/in Bonds a |
| DI4200001-3B1K (BG|D) | 1016 | gap2 | Q9R6W2_78-215 | cp12 | Q6BBK3_46-75 | 30 | 507 | 0.76 | 1/0/1/1 |
| DI2200001-3HPW (AB|C) | 1483 | ccdB | P62554_1-101 | ccdA | P62552_37-72 | 36 | 395 | 4.06 | 7/0/6(1 inv b)/2 |
| DI2200002-5CQX (AB|C) | 1128 | mazF | P0AE70_1-111 | mazE | P0AE72_53-82 | 30 | 4349 | 3.00 | 9/1/5(4 inv)/5 |
| DI2200004-3M91 (AC|B) | 1065 | mpa | P9WQN5_46-96 | pup | P9WHN5_21-64 | 44 | 511 | 2.19 | 5/0/2(2 inv)/1 |
| DI2200006-3M4W (AC|E) | 2321 | rseB | P0AFX9_220-318 | rseA | P0AFX7_125-195 | 71 | 253 | 1.67 | 2/0/2/1 |
| DI1200004-1SC5 (A|B) | 1641 | fliA | O67268_1-236 | flgM | O66683_1-88 | 88 | 1450 | 3.43 | 3/10/3/1 |
| DI1210003-2A7U (B|A) | 611 | atpH | P0ABA5_1-134 | atpA | P0ABB3_1-30 | 30 | 14,854 | 8.34 | 4/3/2(2 inv)/1 |
| DI2200005-1SUY (AB|C) | 1588 | kaiA | Q79V62_177-283 | kaiC | Q79V60_485-518 | 34 | 2739 | 0.85 | 0/0/0/0 |
| DI1200003-1QFN (A|B) | 732 | grxA | P68688_1-85 | nrdA | P00452_732-761 | 30 | 7563 | 1.59 | 0/0/0/0 |
| DI1200001-1R1R (A|D) | 851 | nrdA | P00452_335-729 | nrdB | P69924_347-376 | 30 | 4425 | 0.78 | 0/1/0/0 |
| DI1200006-5D0O (D|C) | 2072 | bamD | P0AC02_1-245 | bamC | P0A903_30-85 | 56 | 726 | 0.38 | 0/0/0/0 |
| DI1210006-4Z0U (B|E) | 358 | rnhA | A7ZHV1_1-155 | ssb | P0AGE0_149-178 | 30 | 8735 | 0.31 | 0/0/0/0 |
| DI1210004-3C94 (A|B) | 320 | sbcB | P04995_13-355 | ssb | A0A0H3GL04_145-174 | 30 | 8735 | 0.11 | 0/0/0/0 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pancsa, R.; Zsolyomi, F.; Tompa, P. Co-Evolution of Intrinsically Disordered Proteins with Folded Partners Witnessed by Evolutionary Couplings. Int. J. Mol. Sci. 2018, 19, 3315. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms19113315
Pancsa R, Zsolyomi F, Tompa P. Co-Evolution of Intrinsically Disordered Proteins with Folded Partners Witnessed by Evolutionary Couplings. International Journal of Molecular Sciences. 2018; 19(11):3315. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms19113315
Chicago/Turabian StylePancsa, Rita, Fruzsina Zsolyomi, and Peter Tompa. 2018. "Co-Evolution of Intrinsically Disordered Proteins with Folded Partners Witnessed by Evolutionary Couplings" International Journal of Molecular Sciences 19, no. 11: 3315. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms19113315