Anaplastic Lymphoma Kinase (ALK) Receptor Tyrosine Kinase: A Catalytic Receptor with Many Faces


1
Department of Pediatric Oncology, Dana-Farber Cancer Institute, Boston, MA 02215, USA
2
Department of Pediatrics, Harvard Medical School, Boston, MA 02115, USA
Int. J. Mol. Sci. 2018, 19(11), 3448; https://0-doi-org.brum.beds.ac.uk/10.3390/ijms19113448
Submission received: 25 September 2018
/
Revised: 24 October 2018
/
Accepted: 30 October 2018
/
Published: 2 November 2018
(This article belongs to the Special Issue Advances in Molecular Oncology)
Abstract
:The anaplastic lymphoma kinase (ALK) receptor is a membrane-bound tyrosine kinase. The pathogenesis of several cancers is closely related to aberrant forms of ALK or aberrant ALK expression, including ALK fusion proteins, ALK-activated point mutations, and ALK amplification. Clinical applications of different ALK inhibitors represent significant progress in targeted therapy. Knowledge of different aspects of ALK biology can provide significant information to further the understanding of this receptor tyrosine kinase. In this mini-review, we briefly summarize different features of ALK. We also summarize some recent research advances on ALK fusion proteins in cancers.
1. Introduction
In 1994, anaplastic lymphoma kinase (ALK) was first found as a tyrosine kinase in anaplastic large-cell lymphoma (ALCL) cell lines [1,2]. In these cell lines, ALK fusion proteins (NPM–ALK) resulting from chromosomal translocation were found [1,2]. After the discovery of the first ALK fusion protein, researchers started to investigate the receptor tyrosine kinase itself. In 1997, several studies reported essential findings related to wild-type ALK [3,4].
The human ALK gene is located at chromosome region 2p23.2–p23.1. This gene, which contains 26 exons, encodes the full-length ALK protein with 1620 amino acids. ALK is an enzyme with tyrosine kinase activity, which catalyzes the transference of a gamma-phosphate group from adenosine triphosphate (ATP) to a tyrosine residue on a substrate protein. Therefore, it catalyzes a tyrosine residue phosphorylation reaction on its substrate proteins. The phosphorylation and dephosphorylation of proteins are critical reactions catalyzed by different enzymes (kinases and phosphatases), which play critical roles in various cellular functions.
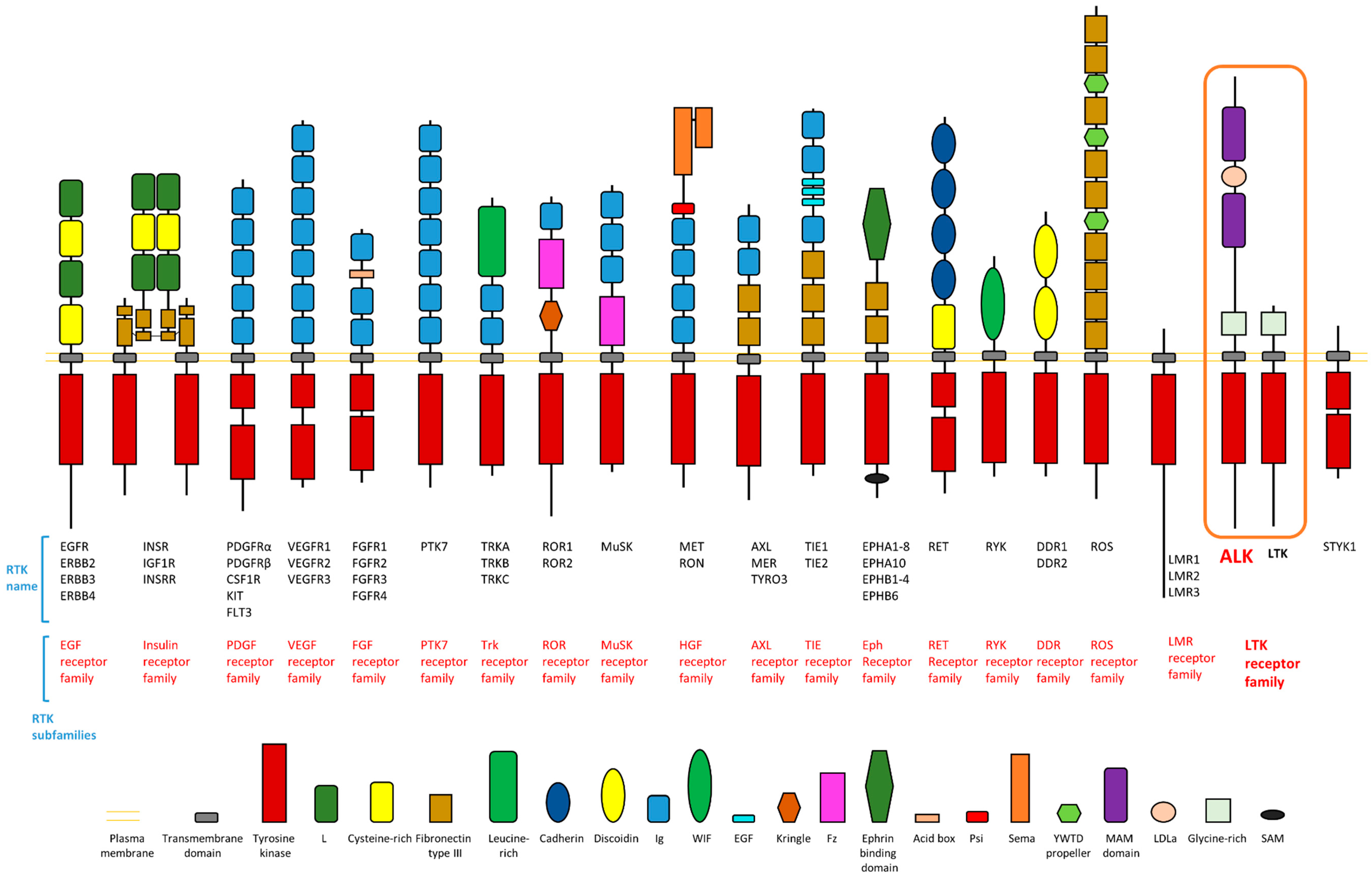
As one member of the receptor tyrosine kinase (RTK) family, ALK contains an extracellular domain (ECD), a transmembrane domain, and an intracellular domain (ICD) (Figure 1). There are more than 50 RTKs encoded in the human genome. These RTKs are grouped into 20 RTK subfamilies within the RTK family (Figure 1) [5]. All RTKs contain an extracellular region, a transmembrane domain, and intracellular domain (Figure 1). The tyrosine kinase domain of RTKs exists in the ICD (Figure 1). The ECD of RTKs usually varies in composition between the different RTK subfamilies (Figure 1). ALK belongs to the leukocyte tyrosine kinase (LTK) receptor subfamily (Figure 1), which includes two members: LTK and ALK. Based on the information on homology, the receptor LTK has the most similar features to ALK, although they differ in domain structure (Figure 1) [4,5]. Figure 1 shows the domain structure of human ALK and RTKs. ALK is a unique RTK member among the RTKs because the ALK ECD contains an extracellular domain structure, which does not exist in any other RTK member, including LTK (Figure 1). Detailed information is introduced in a subsequent section. RTKs are considered a large group of proteins called catalytic receptors, or enzyme-linked receptors [6]. Catalytic receptors are a large group of cell-surface proteins which bind to their ligands as cell-surface receptors in addition to carrying out their catalytic function [6]. Their roles, as both receptors and enzymes, are usually essential for the biological functions of RTKs. Numerous RTKs play an important role in transmembrane signaling and intercellular communication.
ALK is usually expressed during the development of the nervous system [4,7]. During mouse development, ALK expression was found in the central and peripheral nervous system, such as spinal cord motoneurons, sympathetic ganglia, and dorsal root ganglia [3,7]. A recent study showed that ALK was expressed by sympathetic neuroblasts during some stages (E12.5 and E13.5 stage) of mouse embryonic development [8]. After the birth of the mouse, the ALK expression level in the nervous system decreased. Additionally, during the development of chicks, ALK expression was found in the developing central and peripheral nervous system, including spinal cord motoneurons, sympathetic ganglia, and dorsal root ganglia [9]. In adult mammals, a relatively low level of ALK expression exists in certain regions of a few organs, such as the hippocampus within the brain [4,7,10,11]. Studies have shown that ALK is expressed in several regions of the hippocampus in the mouse brain, including the dentate gyrus, cornu ammonis (CA) 1 region, and CA3 region [10].
Although it is highly possible that the biological functions of mammalian ALK are related to the development and function of the nervous system, the direct biological roles of ALK are still not completely clarified. The study of Alk gene knockout mice indicates that ALK can affect the mouse brain functions [11,12,13,14]. Some behaviors closely related to brain functions were observed to differ between Alk gene knockout mice and wild-type mice [11,12,13,14]. For instance, several studies showed that Alk knockout mice displayed elevated ethanol consumption compared to wild-type mice [12,14].
This mini-review presents information on different aspects of ALK. Because several features of ALK biology are summarized and described in this review, a summarized illustration of these ALK features is presented (Figure 2).
2. ALK Domain Structure and 3-D Structure
Although ALK possesses characteristics that are common among RTKs, it also contains some unique features in its domain structure. The ECD of ALK is composed of 1038 amino acid residues (amino acids 1–1038) and has unique features (Figure 1 and Figure 2). In the ALK ECD, a low-density lipoprotein receptor class A domain (LDL, amino acids 453–471) is surrounded by two MAM domains (meprin/A5-protein/PTPmu; amino acids 264–427, 480–626) (Figure 2). In addition, the ALK ECD contains an N-terminal signal peptide (amino acids 1–18) and a glycine-rich region (amino acids 816–940) (Figure 2). ALK has a single transmembrane domain (amino acids 1039–1059). The ICD (amino acids 1060–1620) of ALK mainly comprises a tyrosine kinase domain (amino acids 1116–1392) and the juxtamembrane region (amino acids 1060–1115). In fact, ALK is the only RTK member that contains two MAM domains within its ECD. Among the RTKs, the combination of two MAM domains and one LDL domain is also unique to ALK. The biological roles of the LDL domain and the MAM domain are not yet clarified. The MAM domain consists of about 170 amino acid residues. Studies of the MAM domain in other cell surface proteins have shown that MAM domains usually participate in cell–cell interactions through homophilic binding [15,16,17]. The glycine-rich region of ALK contains consecutive glycine residues, but the function of the glycine-rich region within human ALK is still not clear. Previous studies using a Drosophila melanogaster model have shown that point mutations of glycine residues within the glycine-rich region of Drosophila Alk can lead to a loss of function [18].
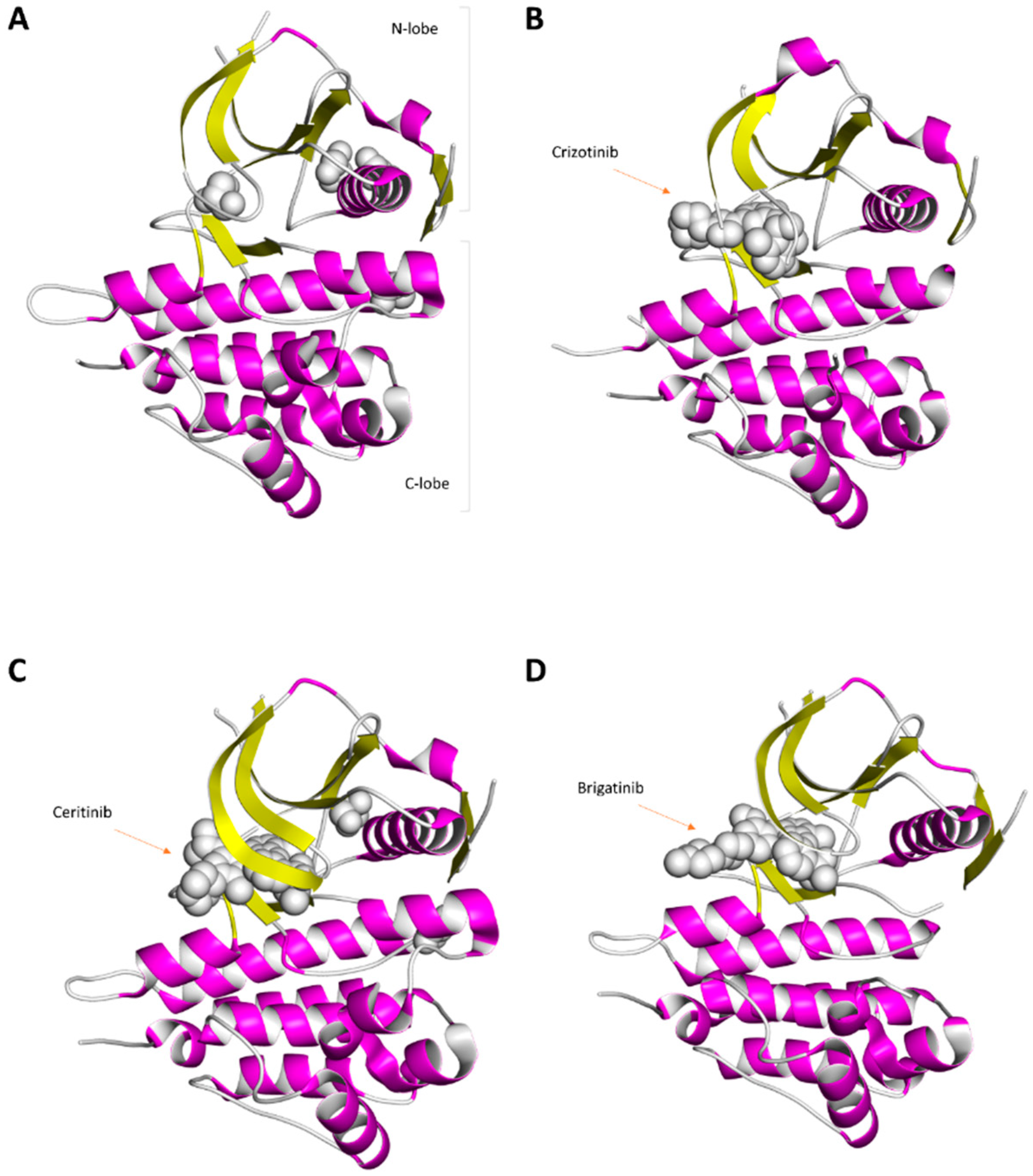
The complete structure of ALK is still not known, nor is the structure of the entire ALK ECD. Most structural research has focused on the ALK kinase domain, which directly conducts enzyme catalysis [19,20,21,22,23,24]. This kinase domain contains an amino-terminal lobe and a carboxy-terminal lobe [19,20,21,22,23,24] (Figure 3). The amino-terminal lobe of the ALK kinase domain contains several β-strands (forming antiparallel β-sheets), several loop regions (including a glycine-rich loop), and one helix (αC-helix) [19,20,21,22,23,24] (Figure 3). The carboxy-terminal lobe contains several α-helices, two short β-strands, and several loop regions (Figure 3). In this kinase domain, the catalytic activity of ALK in the kinase structure is affected by several essential segments, which include a catalytic loop, activation loop, αC-helix, glycine-rich loop, etc. [19,20,21,22,23,24]. The crucial residues in the human ALK kinase domain include E1167 within the αC-helix, HRD residues (H1247, R1248, D1249) within the catalytic loop, K1150 within the N-lobe, DFG residues (D1270, F1271, G1272) within the activation segment, the K1267 residue, etc. [19,20,21,22,23,24]. In Figure 3, the structure of the ALK kinase domain is displayed. Structures of the ALK kinase domain bound with three ALK inhibitors are also shown in this figure (Figure 3).
3. ALK Activation and Signaling Pathway
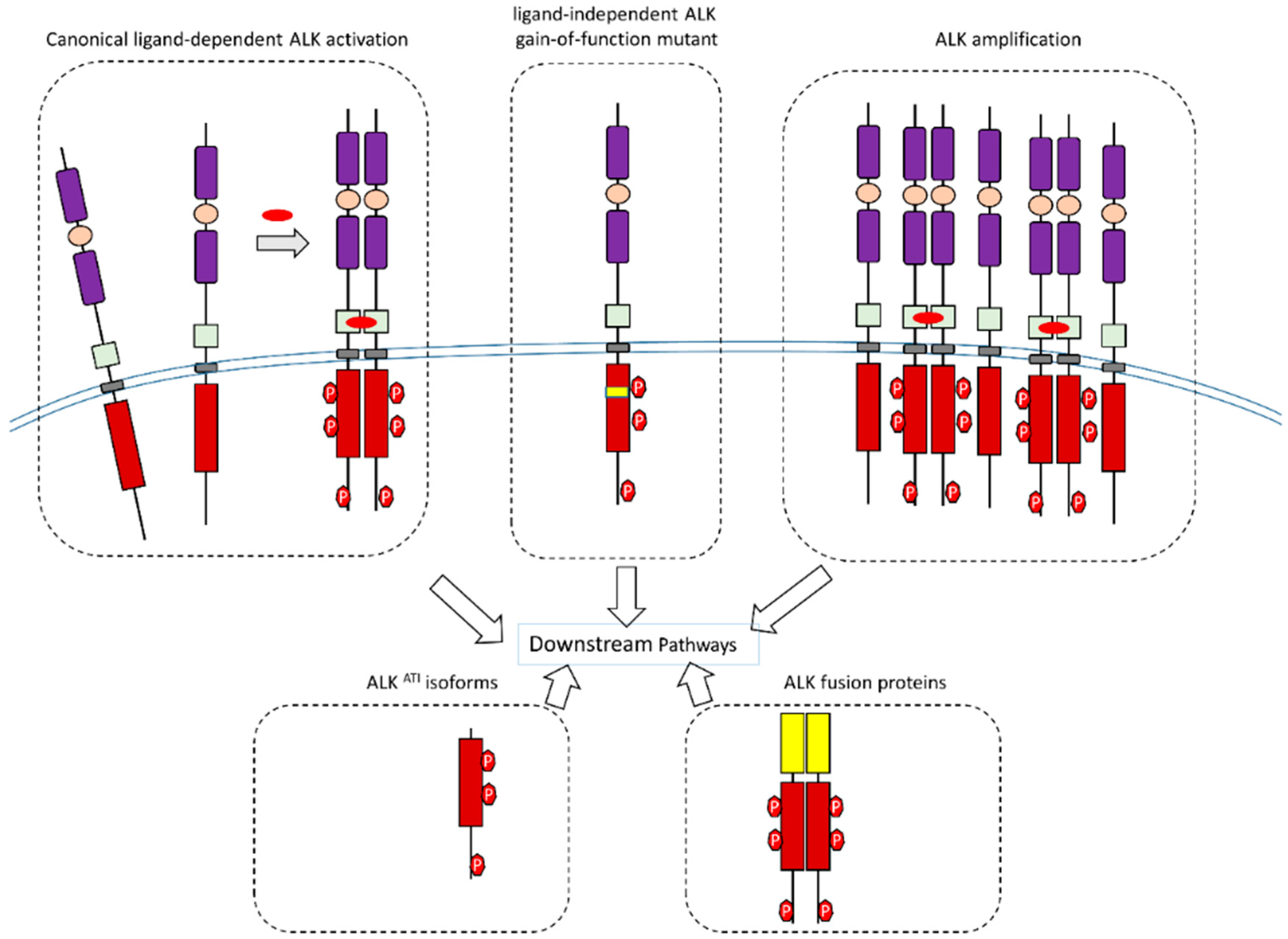
One property of an RTK is its mediation of downstream signaling pathways after it has been activated. The exact activation mechanism of ALK is still not completely understood, but may be achieved through the canonical RTK activation mechanism. The canonical model of RTK activation is through ligand-induced activation. After the extracellular region of one RTK binds to its ligand present in the extracellular space, homo-dimerization or hetero-dimerization induced by ligand binding occurs. This dimerization results in the trans-phosphorylation of specific tyrosine residues within the cytoplasmic domain of the RTK, which may lead to more tyrosine residues being phosphorylated on the same RTK. This phosphorylation can then activate the catalytic capability of the RTK. Activated RTK can then phosphorylate tyrosine residues on its substrate proteins, which can transmit the signals of the RTK. Several mechanisms can terminate an activated RTK once it has been activated, including dephosphorylation by tyrosine phosphatases and degradation after endocytosis. Dimerization of ALK may support the trans-phosphorylation of some tyrosine residues (probable sites are Y1278, Y1282, and Y1283) in the activation loop [24,25,26,27,28]. Then, other tyrosine residues can be phosphorylated after dimerization to activate ALK kinase activity [24,25,26,27,28]. Like other protein kinases, activated ALK can activate downstream pathways. The NPM–ALK fusion proteins were found to activate several downstream signal pathways. These pathways include the RAS/MAPK pathway, the JAK/STAT pathway, the PI3K/Akt pathway, and the PLC (phospholipase C)-γ pathway [24,25,26,27,28]. Activation of these pathways by NPM–ALK is completed through the phosphorylation of specific tyrosine residues of ALK. These residues include, but are not limited to, the tyrosine residues corresponding to Y1358, Y1507, and Y1604 of the full-length ALK (Figure 2). Phosphorylation of these ALK residues can transmit ALK-mediated signals to downstream signaling pathways [24,25,26,27,28].
In addition to canonical wild-type ALK activation, aberrant forms of ALK and ALK isoforms also can transmit signals to their downstream pathways (Figure 4).
4. ALK Ligands
To date, several potential human ALK ligands have been discovered (Figure 2), but more research is required to clarify all information of human ALK ligands. Recent research has discovered that ALKALs are ALK ligands [29,30,31,32,33,34]. These studies have shown that ALKALs (FAM150A and FAM150B), which were also found to be ligands of LTK, bind to the ALK ECD to activate ALK [29,30,31,32,33,34]. In vitro studies have shown that ALKALs activate ALK kinase activity. Conditioned medium containing ALKALs activated ALK in several ALK-expressing cell lines [30,31]. Expression of ALKALs also led to the activation of wild-type ALK in a Drosophila model. Additionally, in vivo studies using zebrafish as a model also supported ALKALs as ligands of the ALK/LTK receptor family [32,33]. Heparin was also found to be a putative ligand of mammalian ALK in a study [35]. A putative heparin-binding motif was found in the N-terminal region of the ALK ECD. Additionally, an in vitro study using canine ALK showed that the wild-type ALK ECD, but not a mutant ALK ECD with its N-terminal region deleted, could be purified using heparin–Sepharose chromatography. Heparins with a relatively long chain, such as heparins whose chains have a degree of polymerization (DP) of 25, can physically bind to the ALK ECD and activate ALK [35].
In Drosophila melanogaster, jelly belly (Jeb) has already been discovered as a biological ligand of Alk, which can activate it to promote visceral founder cell specification during the visceral musculature development of the gut [18,36,37]. During fruit fly embryogenesis, dAlk appears to function in gut development in Drosophila by activating its downstream signaling, such as the ERK signaling pathway [18,36,37]. Without a functional Alk gene in the fruit fly, the development of the gut is disrupted [18,36,37]. In addition, dAlk and its ligand Jeb play a critical role in the development of the visual system [38].
5. ALK Is a Dependence Receptor
One crucial characteristic of ALK is that it is a so-called dependence receptor [39,40,41] (Figure 2). Without ligand binding to activate its kinase activity, ALK can be cleaved by caspase-3 during apoptosis [39,40,41]. In the juxtamembrane region of ALK, there is a caspase-3 cleavage site (amino acids 1160–1163: DELD) (Figure 2). Elevated caspase-3 activity can cleave this ALK at this cleavage site, which releases an intracellular ALK fragment (about 60 kDa) into the cytoplasm. This caspase-dependent cleavage of ALK enhances apoptosis through the exposure of a pro-apoptotic segment (addiction/dependence domain, ADD) within the ALK juxtamembrane region [39,40,41]. ALK mutant D1160N abolishes the caspase-3 cleavage at this cleavage position, which also abrogates the ALK-mediated enhancement of apoptosis [39,40,41]. Moreover, people have found that synthesized peptides mimicking the proapoptotic domain of ALK caused cytotoxicity to ALK-positive ALCL and neuroblastoma (NB) cell lines [41]. This cytotoxic effect was found to be due to caspase-dependent apoptosis [41]. ALK is not the only RTK member belonging to the dependence receptor group [42,43,44,45,46]. Several other RTKs, such as MET, RET, and TrkC, are also dependence receptors that can be cleaved by caspases to enhance apoptosis [42,43,44,45,46,47]. Because this is a significant characteristic of ALK, studies on this topic could be a critical ALK research area.
6. ALK Extracellular and Intracellular Cleavage
Human ALK exists as a 220 kDa full-length ALK and a 140 kDa truncated ALK [48]. The full-length wild-type ALK can be cleaved in the ALK ECD (Figure 2). The extracellular cleavage of ALK results in a 140 kDa truncated ALK and a fragment shed into the extracellular space. The ALK ECD cleavage phenomenon can be detected in the developing brain of rats in vivo [49]. Also, this cleavage phenomenon has been frequently found in NB cancer cell lines and NB cancer tissues [48,49,50,51,52]. Previous research indicates that, during the development of rat dorsal root ganglion (DRG), ALK cleavage is regulated by Schwann cells [49].
7. ALK Glycosylation
One critical post-translational modification of ALK is N-glycosylation (Figure 2). There are 16 N-glycosylation sites within the ECD of ALK (Figure 2). As a result of ALK glycosylation, the molecular weight of the full-length wild-type ALK dramatically increases. The full-length ALK is about 180 kDa without glycosylation. After glycosylation, the molecular weight of the full-length ALK displayed on an SDS-PAGE gel is about 220 kDa. With respect to membrane glycoproteins, N-glycosylation typically contributes to glycoprotein folding, protein quality control, and membrane trafficking [53,54,55,56]. N-glycosylation of ALK may be involved in its function in protein folding, protein quality control, and membrane anchoring. One study indicated that inhibition of N-glycosylation of ALK negatively affected ALK phosphorylation and its downstream signaling [57].
8. ALK Isoforms
A recent study discovered an alternative transcription initiation (ATI) site in intron 19 of the ALK gene [58]. This ATI transcript results in the existence of three ALK isoforms (ALKATI) with molecular weights of 61.1, 60.8, and 58.7 kDa [58] (Figure 2). These three isoforms may result from the existence of three predicted in-frame translation start codons (ATGs) in the ALKATI transcript [58].
ALKATI isoforms are expressed in about 2% to 3% of melanomas. It is also sporadically expressed in several other human cancers, such as lung adenocarcinoma and kidney renal clear cell carcinoma [58]. The ALKATI isoforms, which are kinase active, contain oncogenic capacity [58]. An in vitro study showed that ALKATI could drive growth-factor-independent cell proliferation. Also, research using a mouse model showed that ALKATI promoted tumorigenesis [58]. The ALK inhibitor crizotinib efficiently inhibited both ALKATI kinase activity and ALKATI tumorigenesis ability [58]. An in vitro study using Ba/F3 cells stably expressing ALK isoforms, showed that ALK inhibitors ceritinib and TAE-684 also inhibited the IL-3-independent growth of the transformed Ba/F3 cells.
The positions of three translational start codons (ATG 1069, 1071, and 1089) in ALK alternative transcription initiation are not far from the codon that encodes D1160. Therefore, the molecular weights of these ALK isoforms are a little higher than that of the ALK fragment that is released into the cytoplasm after caspase-3 cleavage. ALKATI isoforms exist in the cytoplasm and the nucleus. Within the chromatin ATI region of ALK, there are two transposable elements, which include a long-terminal repeat (LTR) and a long interspersed nuclear element (LINE).
The existence of ALK isoforms indicates that ALK may not only function as a membrane binding RTK but also as a cytoplasmic tyrosine kinase when these isoforms of ALK are expressed. It is still not clear what the biological roles of these ALK isoforms are, nor the mechanisms which determine this alternative transcription.
9. Aberrant Forms of ALK and Aberrant ALK Expression in Cancers
Aberrant forms of ALK have been found in various cancers. Aberrant forms of ALK and aberrant ALK expression are generally caused by at least one of three primary mechanisms: ALK fusion mutations, ALK gain-of-function mutations, or ALK amplification.
ALK fusion proteins have been found be critical oncogenic drivers in some cancers, such as non-small cell lung cancer (NSCLC) [59,60], ALCL [1,2], and inflammatory myofibroblastic tumor (IMT) [61,62]. ALK fusion variants are usually caused by chromosomal translocation, which can lead to the creation of fusion proteins consisting of an ALK fragment and a fusion partner, such as NPM–ALK in ALCL and EML4–ALK in NSCLC [1,2,59,60]. Even in the same type of cancer, different ALK fusion proteins have been discovered. For instance, there are at least nine different ALK fusion proteins identified in ALCL [24,28]. Additionally, in some types of cancers, such as ALCL, the frequency of ALK rearrangements in patients is high [1,2,24,28,63,64]. ALK fusion proteins exist in more than 50% of ALCL cases [1,2,24,28,63,64]. NPM–ALK is the most frequently detected form of ALK fusion proteins in ALCL [1,2,24,28,63,64]. ALK fusion proteins are usually found to activate downstream signaling pathways that contribute to related cancer pathogenesis. Moreover, an increasing number of novel ALK fusion proteins are being identified in various types of cancers.
ALK activation mutation and ALK amplification were reported in pediatric cancer neuroblastoma many years ago [51,65,66,67,68]. Multiple ALK activation mutations were found in this cancer, which include, but are not limited to: ALK F1174I, ALK F1174L, ALK F1245C, ALK F1245V, ALK R1275Q, ALK R1275L, ALK D1091N, ALK G1128A, ALK M1166R, ALK I1171N, ALK R1192P, and ALK I1250T [51,65,66,67,68]. There are three major ALK mutated positions within the kinase domain: R1275, F1174, and F1245 [51,65,66,67,68]. These are three hotspot residues for ALK-activating point mutations. Both germline and somatic activating mutations have been found in neuroblastoma [51,65,66,67,68,69,70,71]. In addition to ALK-activated point mutations, truncated activated ALK mutants, including ALK Δ2–3, ALK Δ1–5, and ALK Δ4–11, have been found in several neuroblastoma-derived cell lines and tumor samples [72,73,74]. Additionally, one novel truncated form of an ALK variant (ALK Δ2–17) was identified recently in a ALK-positive anaplastic large cell lymphoma and one synovial sarcoma cell line [75,76]. The lack of several exons in ALK genes caused by genomic rearrangements leads to the generation of truncated ALK mutants [72,73,74]. Aberrant activation of ALK activity plays a crucial oncogenic role in neuroblastoma [51,65,66,67,68,77,78,79,80]. For instance, studies in multiple models have shown that aberrantly-activated ALK can potentiate the effect of another protein, MYCN, to drive neuroblastoma pathogenesis [52,77,78,79,80]. In addition to neuroblastoma, ALK amplification and ALK copy number gain have been found in other cancers, such as rhabdomyosarcomas [81,82]. Because several previous reviews also provide excellent summaries of aberrant ALK forms in cancers, this review does not present additional detailed information on this topic [24,28,64,71,83].
ALK Fusion Proteins in Cancers
In ALK fusion proteins, the ALK fusion partner may cause dimerization (or oligomerization) of the ALK fusion protein independent of ligand binding, causing oncogenic ALK activation. This is one canonical mechanism that can explain why ALK fusion proteins cause ALK activation. Moreover, the ALK fusion partner may also affect the subcellular location of the ALK fusion proteins. Because ALK fusion partners provide the N-terminal region of the fusion proteins, the transcription of a fusion protein is usually regulated by the promoter of ALK’s partner protein. The breakpoints for the translocations of ALK genes are typically located at exons 19–20 or exons 20–21. ALK fusion proteins usually contain the complete ALK kinase domain. The kinase activity of ALK fusion proteins leads to the activation of downstream signaling pathways, such as the RAS/MAPK pathway and the JAK/STAT pathway.
Some excellent reviews already summarize the ALK fusion proteins found in various types of cancers [24,28,64,83]. Because an increasing number of ALK fusion proteins are being identified in different types of cancers, we summarize the novel ALK fusion proteins that were found recently (from 2016 to 2018) (Table 1). The existence of numerous diverse ALK fusion proteins suggests that the establishment of ALK fusion proteins through translocation is an important molecular mechanism of oncogenesis in multiple cancer types.
10. ALK Tyrosine Kinase Inhibitors
Because oncogenic activation of ALK kinase activity is crucial to ALK fusion proteins and ALK gain-of-function point mutants, inhibition of ALK kinase activity is the key to targeting ALK in various cancers. To date, multiple generations of ALK tyrosine kinase inhibitors (TKIs) have been generated and evaluated, most of which are small molecular inhibitors.
Inhibition of ALK kinase activity using ALK–TKIs has been found to have potent antitumor efficacy in various research [105,106,107,108,109,110,111,112]. Furthermore, researchers have generated several highly potent selective ALK–TKIs, which can inhibit multiple aberrant forms of ALK, including ALK fusion proteins and ALK activated mutants [108,112,113,114,115,116,117,118,119,120,121,122,123,124,125]. Several ALK–TKIs have already been approved for use in the clinical treatment of specific cancers in some countries. ALK inhibitors that were approved by the U.S. Food and Drug Administration (FDA) for specific cancers include ceritinib, crizotinib, alectinib, and brigatinib. Some information related to these ALK inhibitors is listed in Table 2.
For ALK-directed therapy using ALK-TKIs, acquired drug resistance always arises in some patients, even though these patients may initially experience partial response or complete response. Drug resistance mechanisms after a specific ALK-TKI treatment are investigated widely. One mechanism of resistance is caused by acquired secondary point mutations in the ALK kinase domain. This type of ALK mutations found in previous studies include, but are not limited to: ALK G1202R, ALK F1174 L, ALK F1174C, ALK L1196 M, ALK I1171T, ALK G1269S, ALK V1180L, and ALK G1269A [108,112,113,114,115,116,117,118,119,120,121,122,123,124,125]. Other mechanisms include the activation of alternative survival pathways, ALK gene amplification, etc. In specific situations, resistance mutations may emerge during ALK–TKI treatment, or resistance mutations may already exist before ALK–TKI treatment. Overcoming acquired resistance is one critical challenge to ALK-targeted therapies, and thus many researchers are exploring methods to overcome drug resistance during ALK-targeted therapies. Developing new generations of ALK–TKIs and novel targeting strategies with optimization (such as drug combinations) are two critical approaches to defending against ALK–TKI drug resistance.
Several years ago, crizotinib was established as the standard first-line therapy for advanced ALK-positive NSCLC, because it was demonstrated to be superior to standard chemotherapy (both progression-free survival and objective response rates) in patients with ALK-positive NSCLC [127,128,129,130]. However, crizotinib is far from perfect as a first-line therapy for advanced ALK-positive NSCLC, because of its poor penetration of the central nervous system and the inevitable development of crizotinib resistance during therapy. A newer generation of ALK-TKIs, such as ceritinib and alectinib, have demonstrated efficacy in the treatment of crizotinib-resistant ALK-positive NSCLC [112,121,126,131,132,133,134]. Also, researchers are investigating, in clinical studies, the potential of newer generations of ALK-TKIs as first-line therapies of advanced ALK-positive NSCLC [135,136,137,138]. Several second-generation ALK-TKIs may become first-line therapies of this cancer in future.
Investigators can compare candidate ALK-TKIs with chemotherapy in clinical studies to evaluate whether a particular ALK-TKI can be developed as a first-line therapy. Ceritinib is one of the most widely investigated ALK-TKIs that has the potential to be used as a first-line therapy [137,139,140]. One recent phase 3 clinical trial compared ceritinib with chemotherapy in previously untreated ALK-rearranged NSCLC [137]. This open-labeled, randomized study in untreated patients with stage IIIB/IV ALK-positive non-squamous NSCLC showed that ceritinib displayed superiority, over platinum-based chemotherapy, as the first-line treatment in these patients [137]. The median progression-free survival for patients in the ceritinib group was 16.6 months (95% CI 12.6–27.2) compared to 8.1 months (5.8–11.1) in the chemotherapy group (hazard ratio 0.55 (95% CI 0.42–0.73); p < 0.00001) [137]. Additionally, the overall intracranial response rates of the ceritinib group were higher compared to the chemotherapy group.
Furthermore, researchers are investigating whether other ALK-TKIs can become the first-line therapy for ALK-positive NSCLC, by comparing other ALK-TKIs with crizotinib in clinical studies [135,136,138]. Phase 3 clinical studies, with a head-to-head comparison of alectinib and crizotinib, have suggested that alectinib has the potential to become the first-line treatment of ALK-positive NSCLC [135,136]. In these studies, alectinib showed superior efficacy and lower toxicity compared to crizotinib in the primary treatment of patients with ALK-positive NSCLC [135,136]. In a recent randomized, open-label, phase 3 clinical study, researchers compared the efficacy and safety of brigatinib with those of crizotinib, in patients with advanced ALK-positive NSCLC who had not previously received ALK inhibitor treatment [138]. In this study, brigatinib, as compared to crizotinib, displayed superior efficacy against this cancer. The rate of progression-free survival was significantly higher that among patients in the brigatinib group than among those in the crizotinib group [138].
11. Future Research of ALK
Several vital questions in the research area of ALK include, but are not limited to: (1) What is the biological role of human ALK in embryonic development? (2) What is the biological role of extracellular cleavage and intracellular cleavage of ALK? (3) What is the biological role of ALKATI isoforms? (4) What is the biological role of ALK as a dependence receptor? (5) What is the 3-D structure of whole ALK or ALK ECD? And (6) how does drug resistance (such as ALK acquired resistant mutations) become established with the stress of ALK tyrosine kinase inhibition? Investigations related to these questions can help us to further understand the biological role of ALK. Moreover, this knowledge can also provide vital information to improve the strategy of ALK-targeted therapy. For instance, this information may help to develop novel drug combination strategies.
12. Summary
ALK is an RTK with many characteristics to explore in the future. Different aspects of ALK biology are summarized in this review. Because aberrant forms of ALK are related to multiple cancers, understanding different aspects of this protein provides essential information for us to understand the role of ALK in diseases. This knowledge is also crucial for exploring novel ALK-related therapies.
Funding
This research received no external funding.
Acknowledgments
The author gratefully acknowledges Rani E. George for her support. The author gratefully acknowledges the support from Team Path to the Cure grant. The author apologizes to the authors whose work was not cited as a result of space and topic limitations.
Conflicts of Interest
The author declares no conflicts of interest.
References
- Shiota, M.; Fujimoto, J.; Semba, T.; Satoh, H.; Yamamoto, T.; Mori, S. Hyperphosphorylation of a novel 80 kDa protein-tyrosine kinase similar to Ltk in a human Ki-1 lymphoma cell line, AMS3. Oncogene 1994, 9, 1567–1574. [Google Scholar] [PubMed]
- Morris, S.W.; Kirstein, M.N.; Valentine, M.B.; Dittmer, K.; Shapiro, D.N.; Look, A.T.; Saltman, D.L. Fusion of a kinase gene, ALK, to a nucleolar protein gene, NPM, in non-Hodgkin’s lymphoma. Science 1995, 267, 316–317. [Google Scholar] [CrossRef] [PubMed]
- Morris, S.W.; Naeve, C.; Mathew, P.; James, P.L.; Kirstein, M.N.; Cui, X.; Witte, D.P. ALK, the chromosome 2 gene locus altered by the t(2;5) in non-Hodgkin’s lymphoma, encodes a novel neural receptor tyrosine kinase that is highly related to leukocyte tyrosine kinase (LTK). Oncogene 1997, 14, 2175–2188. [Google Scholar] [CrossRef] [PubMed]
- Iwahara, T.; Fujimoto, J.; Wen, D.; Cupples, R.; Bucay, N.; Arakawa, T.; Mori, S.; Ratzkin, B.; Yamamoto, T. Molecular characterization of ALK, a receptor tyrosine kinase expressed specifically in the nervous system. Oncogene 1997, 14, 439–449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lemmon, M.A.; Schlessinger, J. Cell signaling by receptor tyrosine kinases. Cell 2010, 141, 1117–1134. [Google Scholar] [CrossRef] [PubMed]
- Alexander, S.P.; Mathie, A.; Peters, J.A. Guide to Receptors and Channels (GRAC), 5th edition. Br. J. Pharmacol. 2011, 164, S13–S24. [Google Scholar] [CrossRef] [PubMed]
- Vernersson, E.; Khoo, N.K.; Henriksson, M.L.; Roos, G.; Palmer, R.H.; Hallberg, B. Characterization of the expression of the ALK receptor tyrosine kinase in mice. Gene Expr. Patterns GEP 2006, 6, 448–461. [Google Scholar] [CrossRef] [PubMed]
- Furlan, A.; Dyachuk, V.; Kastriti, M.E.; Calvo-Enrique, L.; Abdo, H.; Hadjab, S.; Chontorotzea, T.; Akkuratova, N.; Usoskin, D.; Kamenev, D.; et al. Multipotent peripheral glial cells generate neuroendocrine cells of the adrenal medulla. Science 2017, 357, eaal3753. [Google Scholar] [CrossRef] [PubMed]
- Hurley, S.P.; Clary, D.O.; Copie, V.; Lefcort, F. Anaplastic lymphoma kinase is dynamically expressed on subsets of motor neurons and in the peripheral nervous system. J. Comp. Neurol. 2006, 495, 202–212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weiss, J.B.; Xue, C.; Benice, T.; Xue, L.; Morris, S.W.; Raber, J. Anaplastic lymphoma kinase and leukocyte tyrosine kinase: Functions and genetic interactions in learning, memory and adult neurogenesis. Pharmacol. Biochem. Behav. 2012, 100, 566–574. [Google Scholar] [CrossRef] [PubMed]
- Bilsland, J.G.; Wheeldon, A.; Mead, A.; Znamenskiy, P.; Almond, S.; Waters, K.A.; Thakur, M.; Beaumont, V.; Bonnert, T.P.; Heavens, R.; et al. Behavioral and neurochemical alterations in mice deficient in anaplastic lymphoma kinase suggest therapeutic potential for psychiatric indications. Neuropsychopharmacol. Off. Publ. Am. Coll. Neuropsychopharmacol. 2008, 33, 685–700. [Google Scholar] [CrossRef] [PubMed]
- Lasek, A.W.; Lim, J.; Kliethermes, C.L.; Berger, K.H.; Joslyn, G.; Brush, G.; Xue, L.; Robertson, M.; Moore, M.S.; Vranizan, K.; et al. An evolutionary conserved role for anaplastic lymphoma kinase in behavioral responses to ethanol. PLoS ONE 2011, 6, e22636. [Google Scholar] [CrossRef] [PubMed]
- Witek, B.; El Wakil, A.; Nord, C.; Ahlgren, U.; Eriksson, M.; Vernersson-Lindahl, E.; Helland, A.; Alexeyev, O.A.; Hallberg, B.; Palmer, R.H. Targeted Disruption of ALK Reveals a Potential Role in Hypogonadotropic Hypogonadism. PLoS ONE 2015, 10, e0123542. [Google Scholar] [CrossRef] [PubMed]
- Mangieri, R.A.; Maier, E.Y.; Buske, T.R.; Lasek, A.W.; Morrisett, R.A. Anaplastic Lymphoma Kinase is a Regulator of Alcohol Consumption and Excitatory Synaptic Plasticity in the Nucleus Accumbens Shell. Front. Pharmacol. 2017, 8, 533. [Google Scholar] [CrossRef] [PubMed]
- Beckmann, G.; Bork, P. An adhesive domain detected in functionally diverse receptors. Trends Biochem. Sci. 1993, 18, 40–41. [Google Scholar] [CrossRef]
- Zondag, G.C.; Koningstein, G.M.; Jiang, Y.P.; Sap, J.; Moolenaar, W.H.; Gebbink, M.F. Homophilic interactions mediated by receptor tyrosine phosphatases mu and kappa. A critical role for the novel extracellular MAM domain. J. Boil. Chem. 1995, 270, 14247–14250. [Google Scholar] [CrossRef]
- Cismasiu, V.B.; Denes, S.A.; Reilander, H.; Michel, H.; Szedlacsek, S.E. The MAM (meprin/a5-protein/PTPmu) domain is a homophilic binding site promoting the lateral dimerization of receptor-like protein-tyrosine phosphatase mu. J. Boil. Chem. 2004, 279, 26922–26931. [Google Scholar] [CrossRef] [PubMed]
- Loren, C.E.; Englund, C.; Grabbe, C.; Hallberg, B.; Hunter, T.; Palmer, R.H. A crucial role for the Anaplastic lymphoma kinase receptor tyrosine kinase in gut development in Drosophila melanogaster. EMBO Rep. 2003, 4, 781–786. [Google Scholar] [CrossRef] [PubMed]
- Knighton, D.R.; Zheng, J.H.; Ten Eyck, L.F.; Ashford, V.A.; Xuong, N.H.; Taylor, S.S.; Sowadski, J.M. Crystal structure of the catalytic subunit of cyclic adenosine monophosphate-dependent protein kinase. Science 1991, 253, 407–414. [Google Scholar] [CrossRef] [PubMed]
- Taylor, S.S.; Kornev, A.P. Protein kinases: Evolution of dynamic regulatory proteins. Trends Biochem. Sci. 2011, 36, 65–77. [Google Scholar] [CrossRef] [PubMed]
- Bossi, R.T.; Saccardo, M.B.; Ardini, E.; Menichincheri, M.; Rusconi, L.; Magnaghi, P.; Orsini, P.; Avanzi, N.; Borgia, A.L.; Nesi, M.; et al. Crystal structures of anaplastic lymphoma kinase in complex with ATP competitive inhibitors. Biochemistry 2010, 49, 6813–6825. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.C.; Jia, Y.; Li, N.; Sun, X.; Ng, K.; Ambing, E.; Gao, M.Y.; Hua, S.; Chen, C.; Kim, S.; et al. Crystal structure of the ALK (anaplastic lymphoma kinase) catalytic domain. Biochem. J. 2010, 430, 425–437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Epstein, L.F.; Chen, H.; Emkey, R.; Whittington, D.A. The R1275Q neuroblastoma mutant and certain ATP-competitive inhibitors stabilize alternative activation loop conformations of anaplastic lymphoma kinase. J. Boil. Chem. 2012, 287, 37447–37457. [Google Scholar] [CrossRef] [PubMed]
- Roskoski, R., Jr. Anaplastic lymphoma kinase (ALK): Structure, oncogenic activation, and pharmacological inhibition. Pharmacol. Res. 2013, 68, 68–94. [Google Scholar] [CrossRef] [PubMed]
- Donella-Deana, A.; Marin, O.; Cesaro, L.; Gunby, R.H.; Ferrarese, A.; Coluccia, A.M.; Tartari, C.J.; Mologni, L.; Scapozza, L.; Gambacorti-Passerini, C.; et al. Unique substrate specificity of anaplastic lymphoma kinase (ALK): Development of phosphoacceptor peptides for the assay of ALK activity. Biochemistry 2005, 44, 8533–8542. [Google Scholar] [CrossRef] [PubMed]
- Tartari, C.J.; Gunby, R.H.; Coluccia, A.M.; Sottocornola, R.; Cimbro, B.; Scapozza, L.; Donella-Deana, A.; Pinna, L.A.; Gambacorti-Passerini, C. Characterization of some molecular mechanisms governing autoactivation of the catalytic domain of the anaplastic lymphoma kinase. J. Boil. Chem. 2008, 283, 3743–3750. [Google Scholar] [CrossRef] [PubMed]
- Chiarle, R.; Voena, C.; Ambrogio, C.; Piva, R.; Inghirami, G. The anaplastic lymphoma kinase in the pathogenesis of cancer. Nat. Rev. Cancer 2008, 8, 11–23. [Google Scholar] [CrossRef] [PubMed]
- Palmer, R.H.; Vernersson, E.; Grabbe, C.; Hallberg, B. Anaplastic lymphoma kinase: Signalling in development and disease. Biochem. J. 2009, 420, 345–361. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Pao, L.I.; Zhou, A.; Brace, A.D.; Halenbeck, R.; Hsu, A.W.; Bray, T.L.; Hestir, K.; Bosch, E.; Lee, E.; et al. Deorphanization of the human leukocyte tyrosine kinase (LTK) receptor by a signaling screen of the extracellular proteome. Proc. Natl. Acad. Sci. USA 2014, 111, 15741–15745. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guan, J.; Umapathy, G.; Yamazaki, Y.; Wolfstetter, G.; Mendoza, P.; Pfeifer, K.; Mohammed, A.; Hugosson, F.; Zhang, H.; Hsu, A.W.; et al. FAM150A and FAM150B are activating ligands for anaplastic lymphoma kinase. eLife 2015, 4, e09811. [Google Scholar] [CrossRef] [PubMed]
- Reshetnyak, A.V.; Murray, P.B.; Shi, X.; Mo, E.S.; Mohanty, J.; Tome, F.; Bai, H.; Gunel, M.; Lax, I.; Schlessinger, J. Augmentor α and β (FAM150) are ligands of the receptor tyrosine kinases ALK and LTK: Hierarchy and specificity of ligand-receptor interactions. Proc. Natl. Acad. Sci. USA 2015, 112, 15862–15867. [Google Scholar] [CrossRef] [PubMed]
- Mo, E.S.; Cheng, Q.; Reshetnyak, A.V.; Schlessinger, J.; Nicoli, S. Alk and Ltk ligands are essential for iridophore development in zebrafish mediated by the receptor tyrosine kinase Ltk. Proc. Natl. Acad. Sci. USA 2017, 114, 12027–12032. [Google Scholar] [CrossRef] [PubMed]
- Fadeev, A.; Mendoza-Garcia, P.; Irion, U.; Guan, J.; Pfeifer, K.; Wiessner, S.; Serluca, F.; Singh, A.P.; Nusslein-Volhard, C.; Palmer, R.H. ALKALs are in vivo ligands for ALK family receptor tyrosine kinases in the neural crest and derived cells. Proc. Natl. Acad. Sci. USA 2018, 115, E630–E638. [Google Scholar] [CrossRef] [PubMed]
- Reshetnyak, A.V.; Mohanty, J.; Tome, F.; Puleo, D.E.; Plotnikov, A.N.; Ahmed, M.; Kaur, N.; Poliakov, A.; Cinnaiyan, A.M.; Lax, I.; et al. Identification of a biologically active fragment of ALK and LTK-ligand 2 (augmentor-α). Proc. Natl. Acad. Sci. USA 2018, 115, 8340–8345. [Google Scholar] [CrossRef] [PubMed]
- Murray, P.B.; Lax, I.; Reshetnyak, A.; Ligon, G.F.; Lillquist, J.S.; Natoli, E.J., Jr.; Shi, X.; Folta-Stogniew, E.; Gunel, M.; Alvarado, D.; et al. Heparin is an activating ligand of the orphan receptor tyrosine kinase ALK. Sci. Signal. 2015, 8, ra6. [Google Scholar] [CrossRef] [PubMed]
- Englund, C.; Loren, C.E.; Grabbe, C.; Varshney, G.K.; Deleuil, F.; Hallberg, B.; Palmer, R.H. Jeb signals through the Alk receptor tyrosine kinase to drive visceral muscle fusion. Nature 2003, 425, 512–516. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.H.; Norris, A.; Weiss, J.B.; Frasch, M. Jelly belly protein activates the receptor tyrosine kinase Alk to specify visceral muscle pioneers. Nature 2003, 425, 507–512. [Google Scholar] [CrossRef] [PubMed]
- Bazigou, E.; Apitz, H.; Johansson, J.; Loren, C.E.; Hirst, E.M.; Chen, P.L.; Palmer, R.H.; Salecker, I. Anterograde Jelly belly and Alk receptor tyrosine kinase signaling mediates retinal axon targeting in Drosophila. Cell 2007, 128, 961–975. [Google Scholar] [CrossRef] [PubMed]
- Mourali, J.; Benard, A.; Lourenco, F.C.; Monnet, C.; Greenland, C.; Moog-Lutz, C.; Racaud-Sultan, C.; Gonzalez-Dunia, D.; Vigny, M.; Mehlen, P.; et al. Anaplastic lymphoma kinase is a dependence receptor whose proapoptotic functions are activated by caspase cleavage. Mol. Cell. Boil. 2006, 26, 6209–6222. [Google Scholar] [CrossRef] [PubMed]
- Allouche, M. ALK is a novel dependence receptor: Potential implications in development and cancer. Cell Cycle 2007, 6, 1533–1538. [Google Scholar] [CrossRef] [PubMed]
- Aubry, A.; Galiacy, S.; Ceccato, L.; Marchand, C.; Tricoire, C.; Lopez, F.; Bremner, R.; Racaud-Sultan, C.; Monsarrat, B.; Malecaze, F.; et al. Peptides derived from the dependence receptor ALK are proapoptotic for ALK-positive tumors. Cell Death Dis. 2015, 6, e1736. [Google Scholar] [CrossRef] [PubMed]
- Tulasne, D.; Deheuninck, J.; Lourenco, F.C.; Lamballe, F.; Ji, Z.; Leroy, C.; Puchois, E.; Moumen, A.; Maina, F.; Mehlen, P.; et al. Proapoptotic function of the MET tyrosine kinase receptor through caspase cleavage. Mol. Cell. Boil. 2004, 24, 10328–10339. [Google Scholar] [CrossRef] [PubMed]
- Tauszig-Delamasure, S.; Yu, L.Y.; Cabrera, J.R.; Bouzas-Rodriguez, J.; Mermet-Bouvier, C.; Guix, C.; Bordeaux, M.C.; Arumae, U.; Mehlen, P. The TrkC receptor induces apoptosis when the dependence receptor notion meets the neurotrophin paradigm. Proc. Natl. Acad. Sci. USA 2007, 104, 13361–13366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia-Lavandeira, M.; Diaz-Rodriguez, E.; Garcia-Rendueles, M.E.; Rodrigues, J.S.; Perez-Romero, S.; Bravo, S.B.; Alvarez, C.V. Functional role of the RET dependence receptor, GFRa co-receptors and ligands in the pituitary. Front. Horm. Res. 2010, 38, 127–138. [Google Scholar] [PubMed]
- Lefebvre, J.; Muharram, G.; Leroy, C.; Kherrouche, Z.; Montagne, R.; Ichim, G.; Tauszig-Delamasure, S.; Chotteau-Lelievre, A.; Brenner, C.; Mehlen, P.; et al. Caspase-generated fragment of the Met receptor favors apoptosis via the intrinsic pathway independently of its tyrosine kinase activity. Cell Death Dis. 2013, 4, e871. [Google Scholar] [CrossRef] [PubMed]
- Bordeaux, M.C.; Forcet, C.; Granger, L.; Corset, V.; Bidaud, C.; Billaud, M.; Bredesen, D.E.; Edery, P.; Mehlen, P. The RET proto-oncogene induces apoptosis: A novel mechanism for Hirschsprung disease. EMBO J. 2000, 19, 4056–4063. [Google Scholar] [CrossRef] [PubMed]
- Ancot, F.; Foveau, B.; Lefebvre, J.; Leroy, C.; Tulasne, D. Proteolytic cleavages give receptor tyrosine kinases the gift of ubiquity. Oncogene 2009, 28, 2185–2195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moog-Lutz, C.; Degoutin, J.; Gouzi, J.Y.; Frobert, Y.; Brunet-de Carvalho, N.; Bureau, J.; Creminon, C.; Vigny, M. Activation and inhibition of anaplastic lymphoma kinase receptor tyrosine kinase by monoclonal antibodies and absence of agonist activity of pleiotrophin. J. Boil. Chem. 2005, 280, 26039–26048. [Google Scholar] [CrossRef] [PubMed]
- Degoutin, J.; Brunet-de Carvalho, N.; Cifuentes-Diaz, C.; Vigny, M. ALK (Anaplastic Lymphoma Kinase) expression in DRG neurons and its involvement in neuron-Schwann cells interaction. Eur. J. Neurosci. 2009, 29, 275–286. [Google Scholar] [CrossRef] [PubMed]
- Osajima-Hakomori, Y.; Miyake, I.; Ohira, M.; Nakagawara, A.; Nakagawa, A.; Sakai, R. Biological role of anaplastic lymphoma kinase in neuroblastoma. Am. J. Pathol. 2005, 167, 213–222. [Google Scholar] [CrossRef]
- George, R.E.; Sanda, T.; Hanna, M.; Frohling, S.; Luther, W., 2nd; Zhang, J.; Ahn, Y.; Zhou, W.; London, W.B.; McGrady, P.; et al. Activating mutations in ALK provide a therapeutic target in neuroblastoma. Nature 2008, 455, 975–978. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schonherr, C.; Ruuth, K.; Kamaraj, S.; Wang, C.L.; Yang, H.L.; Combaret, V.; Djos, A.; Martinsson, T.; Christensen, J.G.; Palmer, R.H.; et al. Anaplastic Lymphoma Kinase (ALK) regulates initiation of transcription of MYCN in neuroblastoma cells. Oncogene 2012, 31, 5193–5200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roth, J. Protein N-glycosylation along the secretory pathway: Relationship to organelle topography and function, protein quality control, and cell interactions. Chem. Rev. 2002, 102, 285–303. [Google Scholar] [CrossRef] [PubMed]
- Zhou, F.; Xu, W.; Hong, M.; Pan, Z.; Sinko, P.J.; Ma, J.; You, G. The role of N-linked glycosylation in protein folding, membrane targeting, and substrate binding of human organic anion transporter hOAT4. Mol. Pharmacol. 2005, 67, 868–876. [Google Scholar] [CrossRef] [PubMed]
- Vagin, O.; Kraut, J.A.; Sachs, G. Role of N-glycosylation in trafficking of apical membrane proteins in epithelia. Am. J. Physiol. Renal Physiol. 2009, 296, F459–F469. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.; Ng, D.T. Glycosylation-directed quality control of protein folding. Nat. Rev. Mol. Cell Boil. 2015, 16, 742–752. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Del Grosso, F.; De Mariano, M.; Passoni, L.; Luksch, R.; Tonini, G.P.; Longo, L. Inhibition of N-linked glycosylation impairs ALK phosphorylation and disrupts pro-survival signaling in neuroblastoma cell lines. BMC Cancer 2011, 11, 525. [Google Scholar] [CrossRef] [PubMed]
- Wiesner, T.; Lee, W.; Obenauf, A.C.; Ran, L.; Murali, R.; Zhang, Q.F.; Wong, E.W.; Hu, W.; Scott, S.N.; Shah, R.H.; et al. Alternative transcription initiation leads to expression of a novel ALK isoform in cancer. Nature 2015, 526, 453–457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soda, M.; Choi, Y.L.; Enomoto, M.; Takada, S.; Yamashita, Y.; Ishikawa, S.; Fujiwara, S.; Watanabe, H.; Kurashina, K.; Hatanaka, H.; et al. Identification of the transforming EML4-ALK fusion gene in non-small-cell lung cancer. Nature 2007, 448, 561–566. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.L.; Takeuchi, K.; Soda, M.; Inamura, K.; Togashi, Y.; Hatano, S.; Enomoto, M.; Hamada, T.; Haruta, H.; Watanabe, H.; et al. Identification of novel isoforms of the EML4–ALK transforming gene in non-small cell lung cancer. Cancer Res. 2008, 68, 4971–4976. [Google Scholar] [CrossRef] [PubMed]
- Griffin, C.A.; Hawkins, A.L.; Dvorak, C.; Henkle, C.; Ellingham, T.; Perlman, E.J. Recurrent involvement of 2p23 in inflammatory myofibroblastic tumors. Cancer Res. 1999, 59, 2776–2780. [Google Scholar] [PubMed]
- Yamada, S.; Nomura, T.; Takano, K.; Fujita, S.; Miyake, M.; Miyake, J. Expression of a chimeric CSF1R-LTK mediates ligand-dependent neurite outgrowth. Neuroreport 2008, 19, 1733–1738. [Google Scholar] [CrossRef] [PubMed]
- Hallberg, B.; Palmer, R.H. The role of the ALK receptor in cancer biology. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2016, 27 (Suppl. S3), iii4–iii15. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.J.; Riely, G.J.; Shaw, A.T. Targeting ALK: Precision Medicine Takes on Drug Resistance. Cancer Discov. 2017, 7, 137–155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caren, H.; Abel, F.; Kogner, P.; Martinsson, T. High incidence of DNA mutations and gene amplifications of the ALK gene in advanced sporadic neuroblastoma tumours. Biochem. J. 2008, 416, 153–159. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Takita, J.; Choi, Y.L.; Kato, M.; Ohira, M.; Sanada, M.; Wang, L.; Soda, M.; Kikuchi, A.; Igarashi, T.; et al. Oncogenic mutations of ALK kinase in neuroblastoma. Nature 2008, 455, 971–974. [Google Scholar] [CrossRef] [PubMed]
- Janoueix-Lerosey, I.; Lequin, D.; Brugieres, L.; Ribeiro, A.; de Pontual, L.; Combaret, V.; Raynal, V.; Puisieux, A.; Schleiermacher, G.; Pierron, G.; et al. Somatic and germline activating mutations of the ALK kinase receptor in neuroblastoma. Nature 2008, 455, 967–970. [Google Scholar] [CrossRef] [PubMed]
- Mosse, Y.P.; Laudenslager, M.; Longo, L.; Cole, K.A.; Wood, A.; Attiyeh, E.F.; Laquaglia, M.J.; Sennett, R.; Lynch, J.E.; Perri, P.; et al. Identification of ALK as a major familial neuroblastoma predisposition gene. Nature 2008, 455, 930–935. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Devoto, M.; Specchia, C.; Laudenslager, M.; Longo, L.; Hakonarson, H.; Maris, J.; Mosse, Y. Genome-wide linkage analysis to identify genetic modifiers of ALK mutation penetrance in familial neuroblastoma. Hum. Hered. 2011, 71, 135–139. [Google Scholar] [CrossRef] [PubMed]
- Bresler, S.C.; Weiser, D.A.; Huwe, P.J.; Park, J.H.; Krytska, K.; Ryles, H.; Laudenslager, M.; Rappaport, E.F.; Wood, A.C.; McGrady, P.W.; et al. ALK mutations confer differential oncogenic activation and sensitivity to ALK inhibition therapy in neuroblastoma. Cancer Cell 2014, 26, 682–694. [Google Scholar] [CrossRef] [PubMed]
- Azarova, A.M.; Gautam, G.; George, R.E. Emerging importance of ALK in neuroblastoma. Semin. Cancer Boil. 2011, 21, 267–275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okubo, J.; Takita, J.; Chen, Y.; Oki, K.; Nishimura, R.; Kato, M.; Sanada, M.; Hiwatari, M.; Hayashi, Y.; Igarashi, T.; et al. Aberrant activation of ALK kinase by a novel truncated form ALK protein in neuroblastoma. Oncogene 2012, 31, 4667–4676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cazes, A.; Louis-Brennetot, C.; Mazot, P.; Dingli, F.; Lombard, B.; Boeva, V.; Daveau, R.; Cappo, J.; Combaret, V.; Schleiermacher, G.; et al. Characterization of rearrangements involving the ALK gene reveals a novel truncated form associated with tumor aggressiveness in neuroblastoma. Cancer Res. 2013, 73, 195–204. [Google Scholar] [CrossRef] [PubMed]
- Fransson, S.; Hansson, M.; Ruuth, K.; Djos, A.; Berbegall, A.; Javanmardi, N.; Abrahamsson, J.; Palmer, R.H.; Noguera, R.; Hallberg, B.; et al. Intragenic anaplastic lymphoma kinase (ALK) rearrangements: Translocations as a novel mechanism of ALK activation in neuroblastoma tumors. Genes Chromosom. Cancer 2015, 54, 99–109. [Google Scholar] [CrossRef] [PubMed]
- Fleuren, E.D.G.; Vlenterie, M.; van der Graaf, W.T.A.; Hillebrandt-Roeffen, M.H.S.; Blackburn, J.; Ma, X.; Chan, H.; Magias, M.C.; van Erp, A.; van Houdt, L.; et al. Phosphoproteomic Profiling Reveals ALK and MET as Novel Actionable Targets across Synovial Sarcoma Subtypes. Cancer Res. 2017, 77, 4279–4292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fukuhara, S.; Nomoto, J.; Kim, S.W.; Taniguchi, H.; Miyagi Maeshima, A.; Tobinai, K.; Kobayashi, Y. Partial deletion of the ALK gene in ALK-positive anaplastic large cell lymphoma. Hematol. Oncol. 2018, 36, 150–158. [Google Scholar] [CrossRef] [PubMed]
- Berry, T.; Luther, W.; Bhatnagar, N.; Jamin, Y.; Poon, E.; Sanda, T.; Pei, D.; Sharma, B.; Vetharoy, W.R.; Hallsworth, A.; et al. The ALK(F1174L) mutation potentiates the oncogenic activity of MYCN in neuroblastoma. Cancer Cell 2012, 22, 117–130. [Google Scholar] [CrossRef] [PubMed]
- Heukamp, L.C.; Thor, T.; Schramm, A.; De Preter, K.; Kumps, C.; De Wilde, B.; Odersky, A.; Peifer, M.; Lindner, S.; Spruessel, A.; et al. Targeted expression of mutated ALK induces neuroblastoma in transgenic mice. Sci. Transl. Med. 2012, 4, 141ra191. [Google Scholar] [CrossRef] [PubMed]
- Zhu, S.; Lee, J.S.; Guo, F.; Shin, J.; Perez-Atayde, A.R.; Kutok, J.L.; Rodig, S.J.; Neuberg, D.S.; Helman, D.; Feng, H.; et al. Activated ALK collaborates with MYCN in neuroblastoma pathogenesis. Cancer Cell 2012, 21, 362–373. [Google Scholar] [CrossRef] [PubMed]
- Ueda, T.; Nakata, Y.; Yamasaki, N.; Oda, H.; Sentani, K.; Kanai, A.; Onishi, N.; Ikeda, K.; Sera, Y.; Honda, Z.I.; et al. ALK(R1275Q) perturbs extracellular matrix, enhances cell invasion and leads to the development of neuroblastoma in cooperation with MYCN. Oncogene 2016, 35, 4447–4458. [Google Scholar] [CrossRef] [PubMed]
- Corao, D.A.; Biegel, J.A.; Coffin, C.M.; Barr, F.G.; Wainwright, L.M.; Ernst, L.M.; Choi, J.K.; Zhang, P.J.; Pawel, B.R. ALK expression in rhabdomyosarcomas: Correlation with histologic subtype and fusion status. Pediatr. Dev. Pathol. Off. J. Soc. Pediatr. Pathol. Paediatr. Pathol. Soc. 2009, 12, 275–283. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, R.; Takita, J.; Sato-Otsubo, A.; Kato, M.; Koh, K.; Hanada, R.; Tanaka, Y.; Kato, K.; Maeda, D.; Fukayama, M.; et al. Characterization of genetic lesions in rhabdomyosarcoma using a high-density single nucleotide polymorphism array. Cancer Sci. 2013, 104, 856–864. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hallberg, B.; Palmer, R.H. Mechanistic insight into ALK receptor tyrosine kinase in human cancer biology. Nat. Rev. Cancer 2013, 13, 685–700. [Google Scholar] [CrossRef] [PubMed]
- Hu, S.; Li, Q.; Peng, W.; Feng, C.; Zhang, S.; Li, C. VIT-ALK, a Novel Alectinib-Sensitive Fusion Gene in Lung Adenocarcinoma. J. Thorac. Oncol. Off. Publ. Int. Assoc. Study Lung Cancer 2018, 13, e72–e74. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.; Wu, X.; Tong, X.; Wei, W.; Chen, A.; Wang, X.; Shao, Y.W.; Huang, J. GCC2-ALK as a targetable fusion in lung adenocarcinoma and its enduring clinical responses to ALK inhibitors. Lung Cancer 2018, 115, 5–11. [Google Scholar] [CrossRef] [PubMed]
- Perron, E.; Pissaloux, D.; Charon Barra, C.; Karanian, M.; Lamant, L.; Parfait, S.; Alberti, L.; de la Fouchardiere, A. Melanocytic Myxoid Spindle Cell Tumor with ALK Rearrangement (MMySTAR): Report of 4 Cases of a Nevus Variant with Potential Diagnostic Challenge. Am. J. Surg. Pathol. 2018, 42, 595–603. [Google Scholar] [CrossRef] [PubMed]
- Fujimoto, M.; Togashi, Y.; Matsuzaki, I.; Baba, S.; Takeuchi, K.; Inaba, Y.; Jinnin, M.; Murata, S.I. A case report of atypical Spitz tumor harboring a novel MLPH–ALK gene fusion with discordant ALK immunohistochemistry results. Hum. Pathol. 2018, 80, 99–103. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Wang, Q.; Ding, Y.; Wang, G.; Chu, Y.; He, X.; Wu, X.; Shao, Y.W.; Lu, K. CUX1-ALK, a Novel ALK Rearrangement That Responds to Crizotinib in Non-Small-Cell Lung Cancer. J. Thorac. Oncol. Off. Publ. Int. Assoc. Study Lung Cancer 2018, 13, 1792–1797. [Google Scholar]
- Tian, Q.; Deng, W.J.; Li, Z.W. Identification of a novel crizotinib-sensitive BCL11A–ALK gene fusion in a nonsmall cell lung cancer patient. Eur. Respir. J. 2017, 49, 1602149. [Google Scholar] [CrossRef] [PubMed]
- Nakanishi, Y.; Masuda, S.; Iida, Y.; Takahashi, N.; Hashimoto, S. Case Report of Non-Small Cell Lung Cancer with STRN–ALK Translocation: A Nonresponder to Alectinib. J. Thorac. Oncol. Off. Publ. Int. Assoc. Study Lung Cancer 2017, 12, e202–e204. [Google Scholar] [CrossRef] [PubMed]
- Du, X.; Shao, Y.; Gao, H.; Zhang, X.; Zhang, H.; Ban, Y.; Qin, H.; Tai, Y. CMTR1-ALK: An ALK fusion in a patient with no response to ALK inhibitor crizotinib. Cancer Boil. Ther. 2018. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, M.; Kohashi, K.; Kushitani, K.; Yoshida, M.; Kurihara, S.; Kawashima, M.; Ueda, Y.; Souzaki, R.; Kinoshita, Y.; Oda, Y.; et al. Inflammatory myofibroblastic tumors of the lung carrying a chimeric A2M–ALK gene: Report of 2 infantile cases and review of the differential diagnosis of infantile pulmonary lesions. Hum. Pathol. 2017, 66, 177–182. [Google Scholar] [CrossRef] [PubMed]
- Inamura, K.; Kobayashi, M.; Nagano, H.; Sugiura, Y.; Ogawa, M.; Masuda, H.; Yonese, J.; Ishikawa, Y. A novel fusion of HNRNPA1-ALK in inflammatory myofibroblastic tumor of urinary bladder. Hum. Pathol. 2017, 69, 96–100. [Google Scholar] [CrossRef] [PubMed]
- Haimes, J.D.; Stewart, C.J.R.; Kudlow, B.A.; Culver, B.P.; Meng, B.; Koay, E.; Whitehouse, A.; Cope, N.; Lee, J.C.; Ng, T.; et al. Uterine Inflammatory Myofibroblastic Tumors Frequently Harbor ALK Fusions with IGFBP5 and THBS1. Am. J. Surg. Pathol. 2017, 41, 773–780. [Google Scholar] [CrossRef] [PubMed]
- Rao, N.; Iwenofu, H.; Tang, B.; Woyach, J.; Liebner, D.A. Inflammatory Myofibroblastic Tumor Driven by Novel NUMA1–ALK Fusion Responds to ALK Inhibition. J. Natl. Compr. Cancer Netw. 2018, 16, 115–121. [Google Scholar] [CrossRef] [PubMed]
- Amatu, A.; Somaschini, A.; Cerea, G.; Bosotti, R.; Valtorta, E.; Buonandi, P.; Marrapese, G.; Veronese, S.; Luo, D.; Hornby, Z.; et al. Novel CAD–ALK gene rearrangement is drugable by entrectinib in colorectal cancer. Br. J. Cancer 2015, 113, 1730–1734. [Google Scholar] [CrossRef] [PubMed]
- Siravegna, G.; Sartore-Bianchi, A.; Mussolin, B.; Cassingena, A.; Amatu, A.; Novara, L.; Buscarino, M.; Corti, G.; Crisafulli, G.; Bartolini, A.; et al. Tracking a CAD–ALK gene rearrangement in urine and blood of a colorectal cancer patient treated with an ALK inhibitor. Ann. Oncol. Off. J. Eur. Soc. Med Oncol. 2017, 28, 1302–1308. [Google Scholar] [CrossRef] [PubMed]
- Aghajan, Y.; Levy, M.L.; Malicki, D.M.; Crawford, J.R. Novel PPP1CB–ALK fusion protein in a high-grade glioma of infancy. BMJ Case Rep. 2016, 2016. [Google Scholar] [CrossRef] [PubMed]
- Panagopoulos, I.; Gorunova, L.; Lund-Iversen, M.; Lobmaier, I.; Bjerkehagen, B.; Heim, S. Recurrent fusion of the genes FN1 and ALK in gastrointestinal leiomyomas. Mod. Pathol. 2016, 29, 1415–1423. [Google Scholar] [CrossRef] [PubMed]
- Cajaiba, M.M.; Jennings, L.J.; George, D.; Perlman, E.J. Expanding the spectrum of ALK-rearranged renal cell carcinomas in children: Identification of a novel HOOK1–ALK fusion transcript. Genes Chromosom. Cancer 2016, 55, 814–817. [Google Scholar] [CrossRef] [PubMed]
- Kusano, H.; Togashi, Y.; Akiba, J.; Moriya, F.; Baba, K.; Matsuzaki, N.; Yuba, Y.; Shiraishi, Y.; Kanamaru, H.; Kuroda, N.; et al. Two Cases of Renal Cell Carcinoma Harboring a Novel STRN-ALK Fusion Gene. Am. J. Surg. Pathol. 2016, 40, 761–769. [Google Scholar] [CrossRef] [PubMed]
- Kazakov, D.V.; Kyrpychova, L.; Martinek, P.; Grossmann, P.; Steiner, P.; Vanecek, T.; Pavlovsky, M.; Bencik, V.; Michal, M.; Michal, M. ALK Gene Fusions in Epithelioid Fibrous Histiocytoma: A Study of 14 Cases, With New Histopathological Findings. Am. J. Dermatopathol. 2018, 40, 805–814. [Google Scholar] [CrossRef] [PubMed]
- Craig, J.W.; Quade, B.J.; Muto, M.G.; MacConaill, L.E. Endometrial cancer with an EML4–ALK rearrangement. Cold Spring Harb. Mol. Case Stud. 2018, 4, a003020. [Google Scholar] [CrossRef] [PubMed]
- Ise, M.; Kageyama, H.; Araki, A.; Itami, M. Identification of a novel GORASP2–ALK fusion in an ALK-positive large B-cell lymphoma. Leuk. Lymphoma 2018. [Google Scholar] [CrossRef] [PubMed]
- Sakamoto, H.; Tsukaguchi, T.; Hiroshima, S.; Kodama, T.; Kobayashi, T.; Fukami, T.A.; Oikawa, N.; Tsukuda, T.; Ishii, N.; Aoki, Y. CH5424802, a selective ALK inhibitor capable of blocking the resistant gatekeeper mutant. Cancer Cell 2011, 19, 679–690. [Google Scholar] [CrossRef] [PubMed]
- Guan, J.; Tucker, E.R.; Wan, H.; Chand, D.; Danielson, L.S.; Ruuth, K.; El Wakil, A.; Witek, B.; Jamin, Y.; Umapathy, G.; et al. The ALK inhibitor PF-06463922 is effective as a single agent in neuroblastoma driven by expression of ALK and MYCN. Dis. Model. Mech. 2016, 9, 941–952. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Guan, S.; Zhao, Y.; Yu, Y.; Woodfield, S.E.; Zhang, H.; Yang, K.L.; Bieerkehazhi, S.; Qi, L.; Li, X.; et al. The second-generation ALK inhibitor alectinib effectively induces apoptosis in human neuroblastoma cells and inhibits tumor growth in a TH-MYCN transgenic neuroblastoma mouse model. Cancer Lett. 2017, 400, 61–68. [Google Scholar] [CrossRef] [PubMed]
- Gettinger, S.N.; Bazhenova, L.A.; Langer, C.J.; Salgia, R.; Gold, K.A.; Rosell, R.; Shaw, A.T.; Weiss, G.J.; Tugnait, M.; Narasimhan, N.I.; et al. Activity and safety of brigatinib in ALK-rearranged non-small-cell lung cancer and other malignancies: A single-arm, open-label, phase 1/2 trial. Lancet Oncol. 2016, 17, 1683–1696. [Google Scholar] [CrossRef]
- Infarinato, N.R.; Park, J.H.; Krytska, K.; Ryles, H.T.; Sano, R.; Szigety, K.M.; Li, Y.; Zou, H.Y.; Lee, N.V.; Smeal, T.; et al. The ALK/ROS1 Inhibitor PF-06463922 Overcomes Primary Resistance to Crizotinib in ALK-Driven Neuroblastoma. Cancer Discov. 2016, 6, 96–107. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Anjum, R.; Squillace, R.; Nadworny, S.; Zhou, T.; Keats, J.; Ning, Y.; Wardwell, S.D.; Miller, D.; Song, Y.; et al. The Potent ALK Inhibitor Brigatinib (AP26113) Overcomes Mechanisms of Resistance to First- and Second-Generation ALK Inhibitors in Preclinical Models. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2016, 22, 5527–5538. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carneiro, B.A.; Pamarthy, S.; Shah, A.N.; Sagar, V.; Unno, K.; Han, H.; Yang, X.J.; Costa, R.B.; Nagy, R.J.; Lanman, R.B.; et al. Anaplastic Lymphoma Kinase Mutation (ALK F1174C) in Small Cell Carcinoma of the Prostate and Molecular Response to Alectinib. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2018, 24, 2732–2739. [Google Scholar] [CrossRef] [PubMed]
- Friboulet, L.; Li, N.; Katayama, R.; Lee, C.C.; Gainor, J.F.; Crystal, A.S.; Michellys, P.Y.; Awad, M.M.; Yanagitani, N.; Kim, S.; et al. The ALK inhibitor ceritinib overcomes crizotinib resistance in non-small cell lung cancer. Cancer Discov. 2014, 4, 662–673. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.L.; Soda, M.; Yamashita, Y.; Ueno, T.; Takashima, J.; Nakajima, T.; Yatabe, Y.; Takeuchi, K.; Hamada, T.; Haruta, H.; et al. EML4–ALK mutations in lung cancer that confer resistance to ALK inhibitors. N. Engl. J. Med. 2010, 363, 1734–1739. [Google Scholar] [CrossRef] [PubMed]
- Heuckmann, J.M.; Holzel, M.; Sos, M.L.; Heynck, S.; Balke-Want, H.; Koker, M.; Peifer, M.; Weiss, J.; Lovly, C.M.; Grutter, C.; et al. ALK mutations conferring differential resistance to structurally diverse ALK inhibitors. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2011, 17, 7394–7401. [Google Scholar] [CrossRef] [PubMed]
- Doebele, R.C.; Pilling, A.B.; Aisner, D.L.; Kutateladze, T.G.; Le, A.T.; Weickhardt, A.J.; Kondo, K.L.; Linderman, D.J.; Heasley, L.E.; Franklin, W.A.; et al. Mechanisms of resistance to crizotinib in patients with ALK gene rearranged non-small cell lung cancer. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2012, 18, 1472–1482. [Google Scholar] [CrossRef] [PubMed]
- Katayama, R.; Shaw, A.T.; Khan, T.M.; Mino-Kenudson, M.; Solomon, B.J.; Halmos, B.; Jessop, N.A.; Wain, J.C.; Yeo, A.T.; Benes, C.; et al. Mechanisms of acquired crizotinib resistance in ALK-rearranged lung Cancers. Sci. Transl. Med. 2012, 4, 120ra117. [Google Scholar] [CrossRef] [PubMed]
- Shaw, A.T.; Engelman, J.A. Ceritinib in ALK-rearranged non-small-cell lung cancer. N. Engl. J. Med. 2014, 370, 2537–2539. [Google Scholar] [CrossRef] [PubMed]
- Katayama, R.; Friboulet, L.; Koike, S.; Lockerman, E.L.; Khan, T.M.; Gainor, J.F.; Iafrate, A.J.; Takeuchi, K.; Taiji, M.; Okuno, Y.; et al. Two novel ALK mutations mediate acquired resistance to the next-generation ALK inhibitor alectinib. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2014, 20, 5686–5696. [Google Scholar] [CrossRef] [PubMed]
- Kodama, T.; Tsukaguchi, T.; Yoshida, M.; Kondoh, O.; Sakamoto, H. Selective ALK inhibitor alectinib with potent antitumor activity in models of crizotinib resistance. Cancer Lett. 2014, 351, 215–221. [Google Scholar] [CrossRef] [PubMed]
- Shaw, A.T.; Kim, D.W.; Mehra, R.; Tan, D.S.; Felip, E.; Chow, L.Q.; Camidge, D.R.; Vansteenkiste, J.; Sharma, S.; de Pas, T.; et al. Ceritinib in ALK-rearranged non-small-cell lung cancer. N. Engl. J. Med. 2014, 370, 1189–1197. [Google Scholar] [CrossRef] [PubMed]
- Ou, S.H.; Ahn, J.S.; De Petris, L.; Govindan, R.; Yang, J.C.; Hughes, B.; Lena, H.; Moro-Sibilot, D.; Bearz, A.; Ramirez, S.V.; et al. Alectinib in Crizotinib-Refractory ALK-rearranged Non-Small-Cell Lung Cancer: A Phase II Global Study. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2016, 34, 661–668. [Google Scholar] [CrossRef] [PubMed]
- Rotow, J.; Bivona, T.G. Understanding and targeting resistance mechanisms in NSCLC. Nat. Rev. Cancer 2017, 17, 637–658. [Google Scholar] [CrossRef] [PubMed]
- Gainor, J.F.; Dardaei, L.; Yoda, S.; Friboulet, L.; Leshchiner, I.; Katayama, R.; Dagogo-Jack, I.; Gadgeel, S.; Schultz, K.; Singh, M.; et al. Molecular Mechanisms of Resistance to First- and Second-Generation ALK Inhibitors in ALK-Rearranged Lung Cancer. Cancer Discov. 2016, 6, 1118–1133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holla, V.R.; Elamin, Y.Y.; Bailey, A.M.; Johnson, A.M.; Litzenburger, B.C.; Khotskaya, Y.B.; Sanchez, N.S.; Zeng, J.; Shufean, M.A.; Shaw, K.R.; et al. ALK: A tyrosine kinase target for cancer therapy. Cold Spring Harb. Mol. Case Stud. 2017, 3, a001115. [Google Scholar] [CrossRef] [PubMed]
- Rashdan, S.; Gerber, D.E. A crowded, but still varied, space: Brigatinib in anaplastic lymphoma kinase-rearranged non-small cell lung cancer. Transl. Cancer Res. 2017, 6, S78–S82. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.W.; Tiseo, M.; Ahn, M.J.; Reckamp, K.L.; Hansen, K.H.; Kim, S.W.; Huber, R.M.; West, H.L.; Groen, H.J.M.; Hochmair, M.J.; et al. Brigatinib in Patients with Crizotinib-Refractory Anaplastic Lymphoma Kinase-Positive Non-Small-Cell Lung Cancer: A Randomized, Multicenter Phase II Trial. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2017, 35, 2490–2498. [Google Scholar] [CrossRef] [PubMed]
- Shaw, A.T.; Kim, D.W.; Nakagawa, K.; Seto, T.; Crino, L.; Ahn, M.J.; de Pas, T.; Besse, B.; Solomon, B.J.; Blackhall, F.; et al. Crizotinib versus chemotherapy in advanced ALK-positive lung cancer. N. Engl. J. Med. 2013, 368, 2385–2394. [Google Scholar] [CrossRef] [PubMed]
- Solomon, B.J.; Mok, T.; Kim, D.W.; Wu, Y.L.; Nakagawa, K.; Mekhail, T.; Felip, E.; Cappuzzo, F.; Paolini, J.; Usari, T.; et al. First-line crizotinib versus chemotherapy in ALK-positive lung cancer. N. Engl. J. Med. 2014, 371, 2167–2177. [Google Scholar] [CrossRef] [PubMed]
- Solomon, B.J.; Kim, D.W.; Wu, Y.L.; Nakagawa, K.; Mekhail, T.; Felip, E.; Cappuzzo, F.; Paolini, J.; Usari, T.; Tang, Y.; et al. Final Overall Survival Analysis from a Study Comparing First-Line Crizotinib Versus Chemotherapy in ALK-Mutation-Positive Non-Small-Cell Lung Cancer. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2018, 36, 2251–2258. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.L.; Lu, S.; Lu, Y.; Zhou, J.; Shi, Y.K.; Sriuranpong, V.; Ho, J.C.M.; Ong, C.K.; Tsai, C.M.; Chung, C.H.; et al. Results of PROFILE 1029, a Phase III Comparison of First-Line Crizotinib versus Chemotherapy in East Asian Patients with ALK-Positive Advanced Non-Small Cell Lung Cancer. J. Thorac. Oncol. Off. Publ. Int. Assoc. Study Lung Cancer 2018, 13, 1539–1548. [Google Scholar] [CrossRef] [PubMed]
- Crino, L.; Ahn, M.J.; de Marinis, F.; Groen, H.J.; Wakelee, H.; Hida, T.; Mok, T.; Spigel, D.; Felip, E.; Nishio, M.; et al. Multicenter Phase II study of Whole-Body and Intracranial Activity with Ceritinib in Patients with Alk-Rearranged Non-Small-Cell Lung Cancer Previously Treated with Chemotherapy and Crizotinib: Results From ASCEND-2. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2016, 34, 2866–2873. [Google Scholar] [CrossRef] [PubMed]
- Shaw, A.T.; Gandhi, L.; Gadgeel, S.; Riely, G.J.; Cetnar, J.; West, H.; Camidge, D.R.; Socinski, M.A.; Chiappori, A.; Mekhail, T.; et al. Alectinib in ALK-positive, crizotinib-resistant, non-small-cell lung cancer: A single-group, multicentre, phase 2 trial. Lancet Oncol. 2016, 17, 234–242. [Google Scholar] [CrossRef]
- Domenech, M.; Jove, M.; Aso, S.; Marin, M.; Nadal, E. Successful treatment with brigatinib in a patient with ALK-rearranged lung adenocarcinoma who developed crizotinib-induced interstitial lung disease. Lung Cancer 2018, 119, 99–102. [Google Scholar] [CrossRef] [PubMed]
- Mezquita, L.; Planchard, D. The role of brigatinib in crizotinib-resistant non-small cell lung cancer. Cancer Manag. Res. 2018, 10, 123–130. [Google Scholar] [CrossRef] [PubMed]
- Hida, T.; Nokihara, H.; Kondo, M.; Kim, Y.H.; Azuma, K.; Seto, T.; Takiguchi, Y.; Nishio, M.; Yoshioka, H.; Imamura, F.; et al. Alectinib versus crizotinib in patients with ALK-positive non-small-cell lung cancer (J-ALEX): An open-label, randomised phase 3 trial. Lancet 2017, 390, 29–39. [Google Scholar] [CrossRef]
- Peters, S.; Camidge, D.R.; Shaw, A.T.; Gadgeel, S.; Ahn, J.S.; Kim, D.W.; Ou, S.I.; Perol, M.; Dziadziuszko, R.; Rosell, R.; et al. Alectinib versus Crizotinib in Untreated ALK-Positive Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2017, 377, 829–838. [Google Scholar] [CrossRef] [PubMed]
- Soria, J.C.; Tan, D.S.W.; Chiari, R.; Wu, Y.L.; Paz-Ares, L.; Wolf, J.; Geater, S.L.; Orlov, S.; Cortinovis, D.; Yu, C.J.; et al. First-line ceritinib versus platinum-based chemotherapy in advanced ALK-rearranged non-small-cell lung cancer (ASCEND-4): A randomised, open-label, phase 3 study. Lancet 2017, 389, 917–929. [Google Scholar] [CrossRef]
- Camidge, D.R.; Kim, H.R.; Ahn, M.J.; Yang, J.C.; Han, J.Y.; Lee, J.S.; Hochmair, M.J.; Li, J.Y.; Chang, G.C.; Lee, K.H.; et al. Brigatinib versus Crizotinib in ALK-Positive Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2018. [Google Scholar] [CrossRef] [PubMed]
- Claxton, L.; O’Connor, J.; Woolacott, N.; Wright, K.; Hodgson, R. Ceritinib for Untreated Anaplastic Lymphoma Kinase-Positive Advanced Non-Small-Cell Lung: An Evidence Review Group Evaluation of a NICE Single Technology Appraisal. PharmacoEconomics 2018. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.W.; Mehra, R.; Tan, D.S.W.; Felip, E.; Chow, L.Q.M.; Camidge, D.R.; Vansteenkiste, J.; Sharma, S.; De Pas, T.; Riely, G.J.; et al. Activity and safety of ceritinib in patients with ALK-rearranged non-small-cell lung cancer (ASCEND-1): Updated results from the multicentre, open-label, phase 1 trial. Lancet Oncol. 2016, 17, 452–463. [Google Scholar] [CrossRef]
Figure 1.
Domain structure of receptor tyrosine kinase families with anaplastic lymphoma kinase (ALK) highlighted. Modified from reference [5] with permission from Elsevier.
Figure 1.
Domain structure of receptor tyrosine kinase families with anaplastic lymphoma kinase (ALK) highlighted. Modified from reference [5] with permission from Elsevier.

Figure 2.
Summary of several ALK features. SP: Signal peptide; TM: Transmembrane domain; PTK: Protein kinase domain; G-rich: Glycine-rich domain; MAM: MAM domain; LDL: LDLα domain; ADD: Addiction/dependence domain.
Figure 2.
Summary of several ALK features. SP: Signal peptide; TM: Transmembrane domain; PTK: Protein kinase domain; G-rich: Glycine-rich domain; MAM: MAM domain; LDL: LDLα domain; ADD: Addiction/dependence domain.

Figure 3.
Ribbon diagram of the human ALK catalytic domain structure with or without ALK inhibitors. (A) Crystal structures of the ALK catalytic domain (PDB ID: 3L9P). This structure contains glycerol molecules (gray small molecules). (B) Ribbon diagram depicting the crystal structure of the ALK catalytic domain in complex with crizotinib (PDB ID: 2XP2). (C) Ribbon diagram depicting the crystal structure of the ALK catalytic domain in complex with ceritinib (PDB ID: 4MKC). (D) Crystal structure of the ALK catalytic domain bound to brigatinib (PDB ID: 5J7H).
Figure 3.
Ribbon diagram of the human ALK catalytic domain structure with or without ALK inhibitors. (A) Crystal structures of the ALK catalytic domain (PDB ID: 3L9P). This structure contains glycerol molecules (gray small molecules). (B) Ribbon diagram depicting the crystal structure of the ALK catalytic domain in complex with crizotinib (PDB ID: 2XP2). (C) Ribbon diagram depicting the crystal structure of the ALK catalytic domain in complex with ceritinib (PDB ID: 4MKC). (D) Crystal structure of the ALK catalytic domain bound to brigatinib (PDB ID: 5J7H).

Figure 4.
Schematic illustration of wild-type ALK, aberrant forms of ALK, ALK amplification, and ALK isoforms during signal transduction.
Figure 4.
Schematic illustration of wild-type ALK, aberrant forms of ALK, ALK amplification, and ALK isoforms during signal transduction.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Summary of several novel ALK fusion proteins discovered recently.
| Disease | Fusion Protein | Original Locus of Fusion Partner | References |
|---|---|---|---|
| Lung adenocarcinoma | VIT–ALK | 2p22.2 | [84] |
| Lung adenocarcinoma | GCC2–ALK | 2q12.3 | [85] |
| Melanocytic myxoid spindle cell tumor | FBXO28–ALK | 1q42.11 | [86] |
| Melanocytic myxoid spindle cell tumor | NPAS2–ALK | 2q11.2 | [86] |
| Spitz tumor | MLPH–ALK | 2q37.3 | [87] |
| Non-small-cell lung cancer | CUX1–ALK | 7q22.1 | [88] |
| Non-small-cell lung cancer | BCL11A–ALK | 2p16.1 | [89] |
| Non-small-cell lung cancer | STRN–ALK | 2p22.2 | [90] |
| Non-small-cell lung cancer | CMTR1–ALK | 6p21.2 | [91] |
| Inflammatory myofibroblastic tumor | A2M–ALK | 12p13.31 | [92] |
| Inflammatory myofibroblastic tumor | HNRNPA1–ALK | 12q13.13 | [93] |
| Inflammatory myofibroblastic tumor | IGFBP5–ALK | 2q35 | [94] |
| Inflammatory myofibroblastic tumor | THBS1–ALK | 15q14 | [94] |
| Inflammatory myofibroblastic tumor | NUMA1–ALK | 11q13.4 | [95] |
| Colorectal cancer | CAD–ALK | 2p23.3 | [96,97] |
| Glioma | PPP1CB–ALK | 2p23.2 | [98] |
| Gastrointestinal leiomyomas | FN1–ALK | 2q35 | [99] |
| Renal cell carcinomas | HOOK1–ALK | 1p32.1 | [100] |
| Renal cell carcinomas | STRN–ALK | 2p22.2 | [101] |
| Epithelioid fibrous histiocytoma | PRKAR2A–ALK | 3p21.31 | [102] |
| Epithelioid fibrous histiocytoma | MLPH–ALK | 2q37.3 | [102] |
| Endometrial cancer | EML4–ALK | 2p21 | [103] |
| Large B-cell lymphoma | GORASP2–ALK | 2q31.1 | [104] |
Table 2.
Summary information of several ALK inhibitors already used in clinical application.
| ALK Tyrosine Kinase Inhibitors | Generation | Other Targets | Indicated Application | Some Mutations in ALK Kinase Domain with Resistance | References |
|---|---|---|---|---|---|
| Crizotinib | First generation | ROS1, MET, et al. | ALK+ or ROS+ metastatic non–small cell lung cancer (NSCLC) | EML4-ALK: L1196M; G1269A; G1202R; I1151T | [112,113,114,115,116,117,120,122] |
| Ceritinib | Second generation | ROS1, IGF-1R, InsR | ALK+ metastatic NSCLC after the failure of prior crizotinib therapy | EML4-ALK: G1202R; C1156Y; F1174C | [112,116,119,122] |
| Alectinib | Second generation | LTK, GAK | ALK+ metastatic NSCLC after the failure of prior crizotinib therapy | EML4-ALK: G1202R I1171T; V1180L | [118,119,121,122] |
| Brigatinib | Second generation | ROS1, EGFR | ALK+ metastatic NSCLC after the failure of prior crizotinib therapy | EML4-ALK: G1202R | [108,122,123,124,125,126] |
© 2018 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Huang, H. Anaplastic Lymphoma Kinase (ALK) Receptor Tyrosine Kinase: A Catalytic Receptor with Many Faces. Int. J. Mol. Sci. 2018, 19, 3448. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms19113448
AMA Style
Huang H. Anaplastic Lymphoma Kinase (ALK) Receptor Tyrosine Kinase: A Catalytic Receptor with Many Faces. International Journal of Molecular Sciences. 2018; 19(11):3448. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms19113448
Chicago/Turabian StyleHuang, Hao. 2018. "Anaplastic Lymphoma Kinase (ALK) Receptor Tyrosine Kinase: A Catalytic Receptor with Many Faces" International Journal of Molecular Sciences 19, no. 11: 3448. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms19113448
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.