Caspase-8: A Novel Target to Overcome Resistance to Chemotherapy in Glioblastoma

Abstract
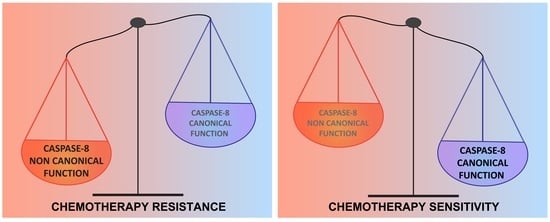
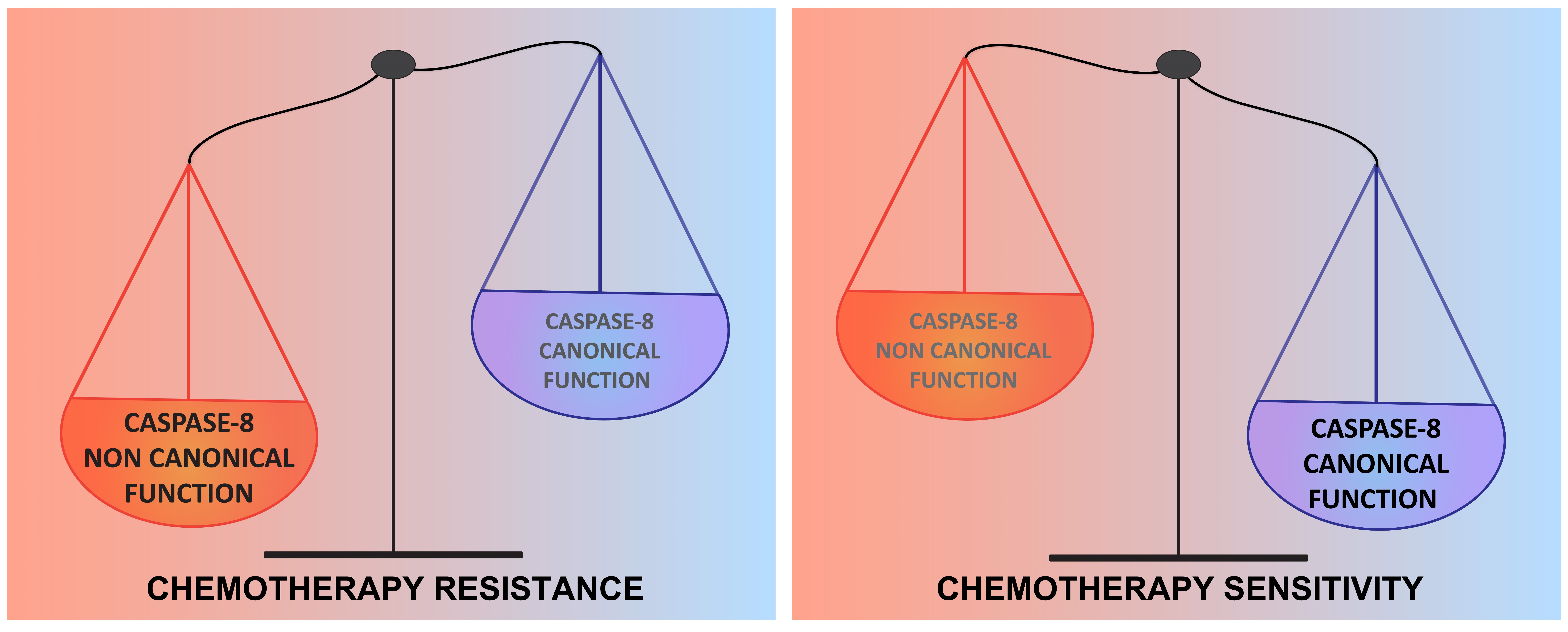
:
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Caspase-8 Structure and Enzymatic Function
3. Caspase-8 Cell Death Related Functions
3.1. Caspase-8 as a Central Transducer of Death Receptor-Induced Apoptosis
3.2. Caspase-8 in Other Programs of Death
4. Caspase-8 Cell Death Unrelated Functions
4.1. Caspase-8 Modulates Cell Adhesion and Migration
4.2. Caspase-8 Modulates the Immune System and NF-κB Activity
5. Molecular Mechanisms That Modulate Caspase-8 Enzymatic Activity and May Drive the Switch from Apoptotic to Non-Apoptotic Functions
5.1. Caspase-8 Modulation by FLIP Proteins and by Caspase-10
5.2. Caspase-8 Modulation by Post-Translational Modifications
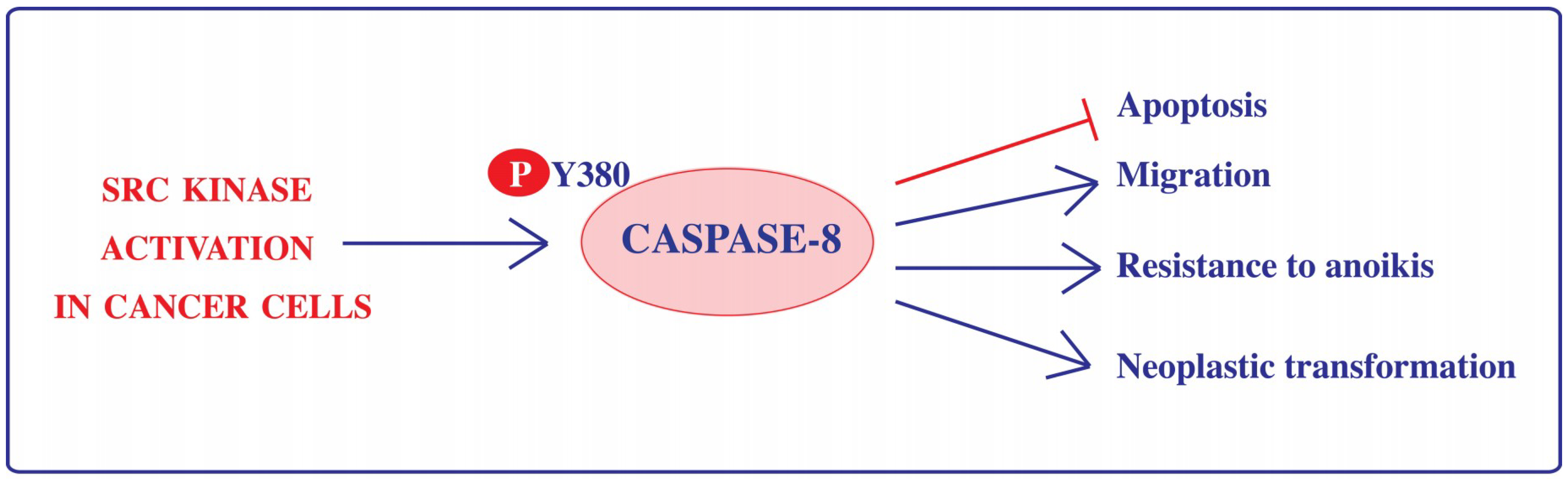
5.2.1. Phosphorylation
5.2.2. Ubiquitination
6. Caspase-8 in Glioblastoma
6.1. Caspase-8 Expression and Function
6.2. Caspase-8 and Glioblastoma Therapeutic Treatment
7. Conclusions
Funding
Conflicts of Interest
References
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Fulda, S. Targeting apoptosis for anticancer therapy. Semin. Cancer Biol. 2015, 31, 84–88. [Google Scholar] [CrossRef] [PubMed]
- Muzio, M.; Chinnaiyan, A.M.; Kischkel, F.C.; O’Rourke, K.; Shevchenko, A.; Ni, J.; Scaffidi, C.; Bretz, J.D.; Zhang, M.; Gentz, R.; et al. FLICE, a novel FADD-homologous ICE/CED-3-like protease, is recruited to the CD95 (Fas/APO-1) death-inducing signaling complex. Cell 1996, 85, 817–827. [Google Scholar] [CrossRef]
- Juo, P.; Kuo, C.J.; Yuan, J.; Blenis, J. Essential requirement for Caspase-8/FLICE in the initiation of the Fas-induced apoptotic cascade. Curr. Biol. 1998, 8, 1001–1008. [Google Scholar] [CrossRef]
- Stupack, D. Caspase-8 as a therapeutic target in cancer. Cancer Lett. 2013, 332, 133–140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Teitz, T.; Wei, T.; Valentine, M.B.; Vanin, E.F.; Grenet, J.; Valentine, V.A.; Behm, F.G.; Look, A.T.; Lahti, J.M.; Kidd, V.J. Caspase-8 is deleted or silenced preferentially in chidhood neuroblastomas with amplification of MYCN. Nat. Med. 2000, 6, 529–535. [Google Scholar] [CrossRef] [PubMed]
- Pingoud-Meier, C.; Lang, D.; Janss, A.J.; Rorke, L.B.; Phillips, P.C.; Shalaby, T.; Grotzer, M.A. Loss of Caspase-8 protein expression correlates with unfavorable survival outcome in childhood medulloblastoma. Clin. Cancer Res. 2003, 9, 6401–6409. [Google Scholar] [PubMed]
- Peter, M.E. The flip side of FLIP. Biochem. J. 2004, 382, e1–e3. [Google Scholar] [CrossRef] [PubMed]
- Humphreys, L.; Espona-Fiedler, M.; Longley, D.B. FLIP as a therapeutic target in cancer. FEBS J. 2018. [Google Scholar] [CrossRef] [PubMed]
- Cursi, S.; Rufini, A.; Stagni, V.; Condo, I.; Matafora, V.; Bachi, A.; Bonifazi, A.P.; Coppola, L.; Superti-Furga, G.; Testi, R.; et al. Src kinase phosphorylates Caspase-8 on Tyr380: A novel mechanism of apoptosis suppression. EMBO J. 2006, 25, 1895–1905. [Google Scholar] [CrossRef] [PubMed]
- Tummers, B.; Green, D.R. Caspase-8: Regulating life and death. Immunol. Rev. 2017, 277, 76–89. [Google Scholar] [CrossRef] [PubMed]
- Solier, S.; Fontenay, M.; Vainchenker, W.; Droin, N.; Solary, E. Non-apoptotic functions of Caspases in myeloid cell differentiation. Cell Death Differ. 2017, 24, 1337–1347. [Google Scholar] [CrossRef] [PubMed]
- Graf, R.P.; Keller, N.; Barbero, S.; Stupack, D. Caspase-8 as a regulator of tumor cell motility. Curr. Mol. Med. 2014, 14, 246–254. [Google Scholar] [CrossRef] [PubMed]
- Keller, N.; Ozmadenci, D.; Ichim, G.; Stupack, D. Caspase-8 function, and phosphorylation, in cell migration. Semin. Cell Dev. Biol. 2018, 82, 105–117. [Google Scholar] [CrossRef] [PubMed]
- Dunn, G.P.; Rinne, M.L.; Wykosky, J.; Genovese, G.; Quayle, S.N.; Dunn, I.F.; Agarwalla, P.K.; Chheda, M.G.; Campos, B.; Wang, A.; et al. Emerging insights into the molecular and cellular basis of glioblastoma. Genes Dev. 2012, 26, 756–784. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verhaak, R.G.; Hoadley, K.A.; Purdom, E.; Wang, V.; Qi, Y.; Wilkerson, M.D.; Miller, C.R.; Ding, L.; Golub, T.; Mesirov, J.P.; et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 2010, 17, 98–110. [Google Scholar] [CrossRef] [PubMed]
- Fianco, G.; Cenci, C.; Barilà, D. Caspase-8 expression and its Src-dependent phosphorylation on Tyr380 promote cancer cell neoplastic transformation and resistance to anoikis. Exp. Cell Res. 2016. [Google Scholar] [CrossRef] [PubMed]
- Fianco, G.; Mongiardi, M.P.; Levi, A.; De Luca, T.; Desideri, M.; Trisciuoglio, D.; del Bufalo, D.; Cinà, I.; di Benedetto, A.; Mottolese, A.; et al. Caspase-8 contributes to angiogenesis and chemotherapy resistance in glioblastoma. eLife 2017. [Google Scholar] [CrossRef] [PubMed]
- Pop, C.; Salvesen, G.S. Human caspases: Activation, specificity, and regulation. J. Biol. Chem. 2009, 284, 21777–21781. [Google Scholar] [CrossRef] [PubMed]
- Keller, N.; Grutter, M.; Zerbe, O. Studies of the molecular mechanism of Caspase-8 activation by solution NMR. Cell Death Differ. 2010, 17, 710–718. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kischkel, F.C.; Hellbardt, S.; Behrmann, I.; Germer, M.; Pawlita, M.; Krammer, P.H.; Peter, M.E. Cytotoxicity-dependent APO-1 (Fas/CD95)-associated proteins form a death-inducing signaling complex (DISC) with the receptor. EMBO J. 1995, 14, 5579–5588. [Google Scholar] [CrossRef] [PubMed]
- Chinnaiyan, A.M.; O’Rourke, K.; Tewari, M.; Dixit, V.M. FADD, a novel death domain-containing protein, interacts with the death domain of Fas and initiates apoptosis. Cell 1995, 81, 505–512. [Google Scholar] [CrossRef]
- Fu, T.; Li, Y.; Lu, A.; Li, Z.; Vajjhala, P.R.; Cruz, A.C.; Srivastava, D.B.; DiMaio, F.; Penczek, P.A.; Siegel, R.M.; et al. Cryo-EM structure of Caspase-8 tandem DED filament reveals assembly and regulation mechanisms of the death-inducing signaling complex. Mol. Cell 2016, 64, 236–250. [Google Scholar] [CrossRef] [PubMed]
- Salvesen, G.S.; Walsh, C.M. Functions of Caspase-8: The identified and the mysterious. Semin. Immunol. 2014, 26, 246–252. [Google Scholar] [CrossRef] [PubMed]
- Keller, N.; Mares, J.; Zerbe, O.; Grutter, M. Structural and biochemical studies on procaspase-8: New insights on initiator caspase activation. Structure 2009, 17, 438–448. [Google Scholar] [CrossRef] [PubMed]
- Pop, C.; Fitzgerald, P.; Green, D.R.; Salvesen, G.S. Role of proteolysis in Caspase-8 activation and stabilization. Biochemistry 2007, 46, 4398–4407. [Google Scholar] [CrossRef] [PubMed]
- Schug, Z.T.; Gonzalvez, F.; Houtkooper, R.H.; Vaz, F.M.; Gottlieb, E. BID is cleaved by Caspase-8 within a native complex on the mitochondrial membrane. Cell Death Differ. 2011, 18, 538–548. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; Alva, A.; Su, H.; Dutt, P.; Freundt, E.; Welsh, S.; Baehrecke, E.H.; Lenardo, M.J. Regulation of an ATG7-beclin 1 program of autophagic cell death by Caspase-8. Science 2004, 304, 1500–1502. [Google Scholar] [CrossRef] [PubMed]
- Tang, Z.; Takahashi, Y.; Chen, C.; Liu, Y.; He, H.; Tsotakos, N.; Serfass, J.M.; Gebru, M.T.; Chen, H.; Young, M.M.; et al. Atg2A/B deficiency switches cytoprotective autophagy to non-canonical Caspase-8 activation and apoptosis. Cell Death Differ. 2017, 24, 2127–2138. [Google Scholar] [CrossRef] [PubMed]
- Ye, C.P.; Qiu, C.Z.; Huang, Z.X.; Su, Q.C.; Zhuang, W.; Wu, R.L.; Li, X.F. Relationship between survivin expression and recurrence, and prognosis in hepatocellular carcinoma. World J. Gastroenterol. 2007, 13, 6264–6268. [Google Scholar] [CrossRef] [PubMed]
- Varfolomeev, E.E.; Schuchmann, M.; Luria, V.; Chiannilkulchai, N.; Beckmann, J.S.; Mett, I.L.; Rebrikov, D.; Brodianski, V.M.; Kemper, O.C.; Kollet, O.; et al. Targeted disruption of the mouse Caspase-8 gene ablates cell death induction by the TNF receptors, Fas/Apo1, and DR3 and is lethal prenatally. Immunity 1998, 9, 267–276. [Google Scholar] [CrossRef]
- Kaiser, W.J.; Upton, J.W.; Long, A.B.; Livingston-Rosanoff, D.; Daley-Bauer, L.P.; Hakem, R.; Caspary, T.; Mocarski, E.S. RIP3 mediates the embryonic lethality of Caspase-8-deficient mice. Nature 2011, 471, 368–372. [Google Scholar] [CrossRef] [PubMed]
- Oberst, A.; Dillon, C.P.; Weinlich, R.; McCormick, L.L.; Fitzgerald, P.; Pop, C.; Hakem, R.; Salvesen, G.S.; Green, D.R. Catalytic activity of the Caspase-8-FLIP(L) complex inhibits RIPK3-dependent necrosis. Nature 2011, 471, 363–367. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Zhou, X.; McQuade, T.; Li, J.; Chan, F.K.; Zhang, J. Functional complementation between FADD and RIP1 in embryos and lymphocytes. Nature 2011, 471, 373–376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheresh, D.A.; Stupack, D.G. Integrin-mediated death: An explanation of theintegrin-knockout phenotype? Nat. Med. 2002, 8, 193–194. [Google Scholar] [CrossRef] [PubMed]
- Stupack, D.G.; Teitz, T.; Potter, M.D.; Mikolon, D.; Houghton, P.J.; Kidd, V.J.; Lahti, J.M.; Cheresh, D.A. Potentiation of neuroblastoma metastasis by loss of Caspase-8. Nature 2006, 439, 95–99. [Google Scholar] [CrossRef] [PubMed]
- Helfer, B.; Boswell, B.C.; Finlay, D.; Cipres, A.; Vuori, K.; Bong Kang, T.; Wallach, D.; Dorfleutner, A.; Lahti, J.M.; Flynn, D.C.; et al. Caspase-8 promotes cell motility and calpain activity under nonapoptotic conditions. Cancer Res. 2006, 66, 4273–4278. [Google Scholar] [CrossRef] [PubMed]
- Barbero, S.; Mielgo, A.; Torres, V.; Teitz, T.; Shields, D.J.; Mikolon, D.; Bogyo, M.; Barilà, D.; Lahti, J.M.; Schlaepfer, D.; et al. Caspase-8 association with the focal adhesion complex promotes tumor cell migration and metastasis. Cancer Res. 2009, 69, 3755–3763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Torres, V.A.; Mielgo, A.; Barbero, S.; Hsiao, R.; Wilkins, J.A.; Stupack, D.G. Rab5 mediates Caspase-8-promoted cell motility and metastasis. Mol. Biol. Cell 2010, 21, 369–376. [Google Scholar] [CrossRef] [PubMed]
- Chun, H.; Zheng, L.; Ahmad, M.; Wang, J.; Speirs, C.K.; Siegel, R.M.; Dale, J.K.; Puck, J.; Davis, J.; Hall, C.G.; et al. Pleiotropic defects in lymphocyte activation caused by Caspase-8 mutations lead to human immunodeficiency. Nature 2002, 419, 395–399. [Google Scholar] [CrossRef] [PubMed]
- Salmena, L.; Lemmers, B.; Hakem, A.; Matysiak-Zablocki, E.; Murakami, K.; Au, P.Y.; Berry, D.M.; Tamblyn, L.; Shehabeldin, A.; Migon, E.; et al. Essential role for Caspase 8 in T-cell homeostasis and T-cell-mediated immunity. Genes Dev. 2003, 17, 883–895. [Google Scholar] [CrossRef] [PubMed]
- Chaudhary, P.M.; Eby, M.T.; Jasmin, A.; Kumar, A.; Liu, L.; Hood, L. Activation of the NF-κB pathway by Caspase 8 and its homologs. Oncogene 2000, 19, 4451–4460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, W.-H.; Johnson, H.; Sghu, H.-B. Activation of NF-κB by FADD, Casper, and Caspase-8. J. Biol. Chem. 2000, 275, 10838–10844. [Google Scholar] [CrossRef] [PubMed]
- Su, H.; Bidere, N.; Zheng, L.; Cubre, A.; Sakai, K.; Dale, J.; Salmena, L.; Hakem, R.; Straus, S.; Lenardo, M. Requirement for Caspase-8 in NF-κB activation by antigen receptor. Science 2005, 307, 1465–1468. [Google Scholar] [CrossRef] [PubMed]
- Bidère, N.; Snow, A.L.; Sakai, K.; Zheng, L.; Lenardo, M.J. Caspase-8 regulation by direct interaction with TRAF6 in T cell receptor-induced NF-κB activation. Curr. Biol. 2006, 16, 1666–1671. [Google Scholar] [CrossRef] [PubMed]
- Micheau, O.; Lens, S.; Gaide, O.; Alevizopoulos, K.; Tschopp, J. NF-κB signalsinduce the expression of c-FLIP. Mol. Cell Biol. 2001, 21, 5299–5305. [Google Scholar] [CrossRef] [PubMed]
- Kataoka, T.; Budd, R.C.; Holler, N.; Thome, M.; Martinon, F.; Irmler, M.; Burns, K.; Hahne, M.; Kennedy, N.; Kovacsovics, M.; et al. The Caspase-8 inhibitor FLIP promotes activation of NF-κB and Erk signaling pathways. Curr. Biol. 2000, 10, 640–648. [Google Scholar] [CrossRef]
- Dutton, A.; O’Neil, J.D.; Milner, A.E.; Reynolds, G.M.; Starczynski, J.; Crocker, J.; Young, L.S.; Murray, P.G. Expression of the cellular FLICE-inhibitory protein (c-FLIP) protects Hodgkin’s lymphoma cells from autonomous Fas-mediated death. Proc. Natl. Acad. Sci. USA 2004, 101, 6611–6616. [Google Scholar] [CrossRef] [PubMed]
- Mathas, S.; Lietz, A.; Anagnostopoulos, I.; Hummel, F.; Wiesner, B.; Janz, M.; Jundt, F.; Hirsch, B.; Johrens-Leder, K.; Vornlocher, H.P.; et al. c-FLIP mediates resistance of Hodgkin/Reed-Sternberg cells to death receptor-induced apoptosis. J. Exp. Med. 2004, 199, 1041–1052. [Google Scholar] [CrossRef] [PubMed]
- Safa, A.R.; Pollok, K.E. Targeting the anti-apoptotic protein c-FLIP for cancer therapy. Cancers 2011, 3, 1639–1671. [Google Scholar] [CrossRef] [PubMed]
- Eckhart, L.; Ballaun, C.; Hermann, M.; VandeBerg, J.L.; Sipos, W.; Uthman, A.; Fischer, H.; Tschachler, E. Identification of novel mammalian caspases reveals an important role of gene loss in shaping the human Caspase repertoire. Mol. Biol. Evol. 2008, 25, 831–841. [Google Scholar] [CrossRef] [PubMed]
- Sakamaki, K.; Imai, K.; Tomii, K.; Miller, D.J. Evolutionary analyses of Caspase-8 and its paralogs: Deep origins of the apoptotic signaling pathways. BioEssays 2015, 37, 767–776. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sprick, M.R.; Rieser, E.; Stah, L.H.; Grosse-Wilde, A.; Weigand, M.A.; Walczak, H. Caspase-10 is recruited to and activated at the native TRAIL and CD95 death-inducing signalling complexes in a FADD-dependent manner but can not functionally substitute Caspase-8. EMBO J. 2002, 21, 4520–4530. [Google Scholar] [CrossRef] [PubMed]
- Horn, S.; Hughes, M.A.; Schilling, R.; Sticht, C.; Tenev, T.; Ploesser, M.; Meier, P.; Sprick, M.R.; MacFarlane, M.; Leverkus, M. Caspase-10 negatively regulates Caspase-8-mediated cell death, switching the response to CD95L in favor of NF-κB activation and cell survival. Cell Rep. 2017, 19, 785–797. [Google Scholar] [CrossRef] [PubMed]
- Mohr, A.; Deedigan, L.; Jencz, S.; Mehrabadi, Y.; Houlden, L.; Albarenque, S.M.; Zwacka, R.M. Caspase-10: A molecular switch from cell-autonomous apoptosis to communal cell death in response to chemotherapeutic drug treatment. Cell Death Differ. 2018, 25, 340–352. [Google Scholar] [CrossRef] [PubMed]
- Powley, I.R.; Hughes, M.A.; Cain, K.; MacFarlane, M. Caspase-8 tyrosine-380 phosphorylation inhibits CD95 DISC function by preventing proCaspase-8 maturation and cycling within the complex. Oncogene 2016, 35, 5829–5840. [Google Scholar] [CrossRef] [PubMed]
- Frame, M.C. Src in cancer: Deregulation and consequences for cell behaviour. Biochim. Biophys. Acta 2002, 1602, 114–130. [Google Scholar] [CrossRef]
- Barbero, S.; Barilà, D.; Mielgo, A.; Stagni, V.; Clair, K.; Stupack, D. Identification of a critical tyrosine residue in Caspase-8 that promotes cell migration. J. Biol. Chem. 2008, 283, 13031–13034. [Google Scholar] [CrossRef] [PubMed]
- Finlay, D.; Vuori, K. Novel noncatalytic role for Caspase-8 in promoting SRC-mediated adhesion and Erk signaling in neuroblastoma cells. Cancer Res. 2007, 67, 11704–11711. [Google Scholar] [CrossRef] [PubMed]
- Senft, J.; Helfer, B.; Frisch, S.M. Caspase-8 interacts with the p85 subunit of phosphatidylinositol 3-kinase to regulate cell adhesion and motility. Cancer Res. 2007, 67, 11505–11509. [Google Scholar] [CrossRef] [PubMed]
- Graf, R.P.; Barbero, S.; Keller, N.; Chen, L.; Uryu, S.; Schlaepfer, D.; Stupack, D. Src-inducible association of CrkL with proCaspase-8 promotes cell migration. Cell Adh. Migr. 2013, 7, 362–369. [Google Scholar] [CrossRef] [PubMed]
- Jin, Z.; Li, Y.; Pitti, R.; Lawrence, D.; Pham, V.C.; Lill, J.R.; Ashkenazi, A. Cullin3-based polyubiquitination and p62-dependent aggregation of Caspase-8 mediate extrinsic apoptosis signaling. Cell 2009, 137, 721–735. [Google Scholar] [CrossRef] [PubMed]
- Gonzalvez, F.; Lawrence, D.; Yang, B.; Yee, S.; Pitti, R.; Marsters, S.; Pham, V.C.; Stephan, J.P.; Lill, J.; Ashkenazi, A. TRAF2 Sets a threshold for extrinsic apoptosis by tagging Caspase-8 with a ubiquitin shutoff timer. Mol. Cell 2012, 48, 888–899. [Google Scholar] [CrossRef] [PubMed]
- Nogueira, L.; Ruiz-Ontañon, P.; Vazquez-Barquero, A.; Moris, F.; Fernandez-Luna, J.L. The NF-κB pathway: A therapeutic target in glioblastoma. Oncotarget 2011, 2, 464–653. [Google Scholar] [CrossRef] [PubMed]
- Kargiotis, O.; Rao, J.S.; Kyritsis, A.P. Mechanisms of angiogenesis in gliomas. J. Neuro-Oncol. 2006, 78, 281–293. [Google Scholar] [CrossRef] [PubMed]
- Ashley, D.M.; Riffkin, C.D.; Muscat, A.M.; Knight, M.J.; Kaye, A.H.; Novak, U.; Hawkins, C.J. Caspase 8 is absent or low in many ex vivo gliomas. Cancer 2005, 104, 1487–1496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuijlen, J.M.A.; Bremer, E.; Mooij, J.J.A.; den Dunnen, W.F.A.; Helfrich, W. Review: On TRAIL for malignant glioma therapy? Neuropathol. Appl. Neurobiol. 2010, 36, 168–182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, J.; Bernasconi, P.; Clauser, K.R.; Mani, D.R.; Finn, S.P.; Beroukhim, R.; Burns, M.; Julian, B.; Peng, X.P.; Hieronymus, H.; et al. Bead-based profiling of tyrosine kinase phosphorylation identifies SRC as a potential target for glioblastoma therapy. Nat. Biotechnol. 2009, 27, 77–83. [Google Scholar] [CrossRef] [PubMed]
- Pfeffer, C.M.; Singh, A.T.K. Apoptosis: A target for anticancer therapy. Int. J. Mol. Sci. 2018, 19, 448. [Google Scholar] [CrossRef] [PubMed]
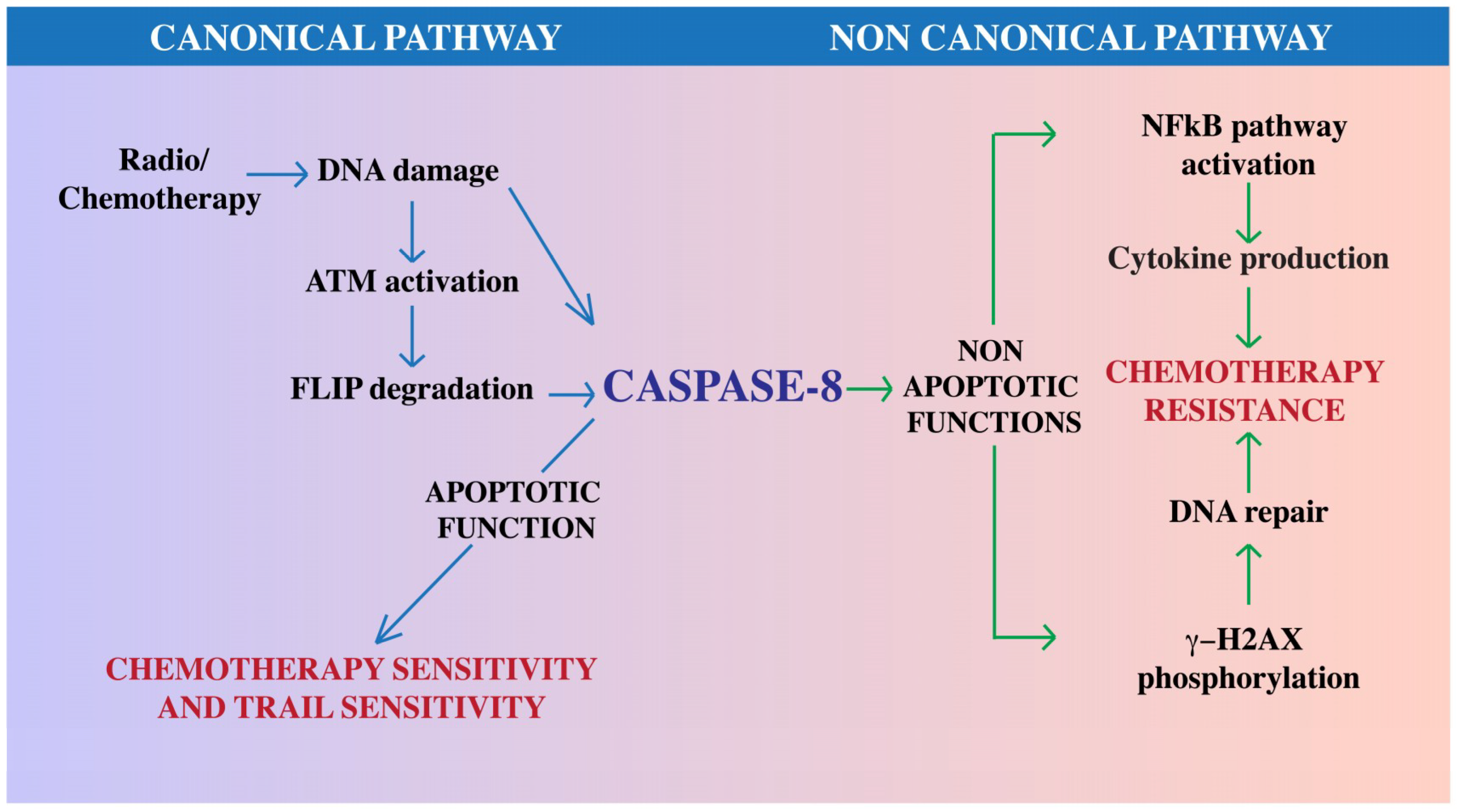
- Stagni, V.; di Bari, M.G.; Condò, I.; Cursi, S.; Testi, R.; Larenthal, Y.; Cundari, E.; Barilà, D. ATM kinase activity modulates Fas sensitivity through the regulation of FLIP in lymphoid cells. Blood 2008, 111, 829–837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stagni, V.; Mingardi, M.; Santini, S.; Giaccari, D.; Barilà, D. ATM kinase activity modulates cFLIP protein levels: Potential interplay between DNA damage signalling and TRAIL-induced apoptosis. Carcinogenesis 2010, 31, 1956–1963. [Google Scholar] [CrossRef] [PubMed]
- Kelly, S.K.; Ashkenazi, A. Targeting death receptors in cancer with Apo2L/TRAIL. Curr. Opin. Pharmacol. 2004, 4, 333–339. [Google Scholar] [CrossRef] [PubMed]
- Grotzer, M.A.; Eggert, A.; Zuzak, T.J.; Janss, A.J.; Marwaha, S.; Wiewrodt, B.R.; Ikegaki, N.; Brodeur, G.M.; Phillips, P.C. Resistance to TRAIL-induced apoptosis in primitive neuroectodermal brain tumor cells correlates with a loss of Caspase-8 expression. Oncogene 2000, 19, 4604–4610. [Google Scholar] [CrossRef] [PubMed]
- Stagni, V.; Santini, S.; Barilà, D. A New Player in the Development of TRAIL Based Therapies for Hepatocarcinoma Treatment: ATM Kinase. Cancers 2012, 4, 354–378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boege, Y.; Malehmir, M.; Healy, M.E.; Bettermann, K.; Lorentzen, A.; Vucur, M.; Ahuja, A.K.; Böhm, F.; Mertens, J.C.; Shimizu, Y.; et al. A dual role of Caspase-8 in triggering and sensing proliferation-associated DNA damage, a key determinant of liver cancer development. Cancer Cell 2017, 32, 342–359. [Google Scholar] [CrossRef] [PubMed]


© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fianco, G.; Contadini, C.; Ferri, A.; Cirotti, C.; Stagni, V.; Barilà, D. Caspase-8: A Novel Target to Overcome Resistance to Chemotherapy in Glioblastoma. Int. J. Mol. Sci. 2018, 19, 3798. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms19123798
Fianco G, Contadini C, Ferri A, Cirotti C, Stagni V, Barilà D. Caspase-8: A Novel Target to Overcome Resistance to Chemotherapy in Glioblastoma. International Journal of Molecular Sciences. 2018; 19(12):3798. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms19123798
Chicago/Turabian StyleFianco, Giulia, Claudia Contadini, Alessandra Ferri, Claudia Cirotti, Venturina Stagni, and Daniela Barilà. 2018. "Caspase-8: A Novel Target to Overcome Resistance to Chemotherapy in Glioblastoma" International Journal of Molecular Sciences 19, no. 12: 3798. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms19123798