A Promising Biocompatible Platform: Lipid-Based and Bio-Inspired Smart Drug Delivery Systems for Cancer Therapy



Abstract
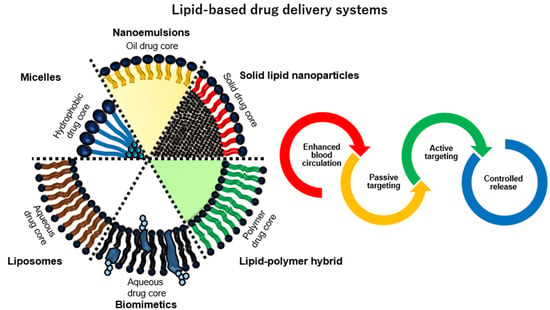
:
1. Introduction
2. Strategies for Prolonged Blood Circulation
2.1. Consideration of Physicochemical Characteristics
2.1.1. Particle Size
2.1.2. Particle Shape
2.1.3. Hydrophobicity
2.1.4. Surface Charge
2.2. Polyethylene Glycolylation
2.3. Biomimicry Inspired from Cells
3. Strategies for Targeting the Tumor Region
3.1. Passive Targeting
3.2. Direct Modification with Targeting Ligands
3.2.1. Antibodies
3.2.2. Aptamers
3.2.3. Other Ligands
3.3. Site-Specific Targeting
4. Strategies for Controlled Release
4.1. Typical Drug Release
4.2. Endogenous Stimuli-Induced Drug Release
4.3. Exogenous Stimuli-Induced Drug Release
5. Concluding Remarks and Future Perspectives
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| DDSs | Drug delivery systems |
| LBDDSs | Lipid-based drug delivery systems |
| TMEs | Tumor microenvironments |
| EPR | Enhanced permeability and retention |
| PEG | Polyethylene glycol |
| RBC | Red blood cell |
| mAb | Monoclonal antibody |
| SELEX | Systemic evolution of ligands using the exponential enrichment |
| HA | Hyaluronic acid |
| CTPs | Cell targeting peptides |
| SLNs | Solid lipid nanoparticles |
| Tm | Transition temperature |
| NIR | Near-infrared |
| EVs | Extracellular vesicles |
References
- Vera-Ramirez, L.; Ramirez-Tortosa, M.; Perez-Lopez, P.; Granados-Principal, S.; Battino, M.; Quiles, J.L. Long-term effects of systemic cancer treatment on DNA oxidative damage: The potential for targeted therapies. Cancer Lett. 2012, 327, 134–141. [Google Scholar] [CrossRef] [PubMed]
- Bakbak, B.; Gedik, S.; Koktekir, B.E.; Yavuzer, K.; Tulek, B.; Kanat, F.; Pancar, E. Assessment of ocular neurotoxicity in patients treated with systemic cancer chemotherapeutics. Cutan. Ocul. Toxicol. 2014, 33, 7–10. [Google Scholar] [CrossRef] [PubMed]
- Prinsloo, S.; Novy, D.; Driver, L.; Lyle, R.; Ramondetta, L.; Eng, C.; Lopez, G.; Li, Y.; Cohen, L. The Long-Term Impact of Neurofeedback on Symptom Burden and Interference in Patients With Chronic Chemotherapy-Induced Neuropathy: Analysis of a Randomized Controlled Trial. J. Pain Symptom Manag. 2018, 55, 1276–1285. [Google Scholar] [CrossRef] [PubMed]
- Curigliano, G.; Mayer, E.L.; Burstein, H.J.; Winer, E.P.; Goldhirsch, A. Cardiac toxicity from systemic cancer therapy: A comprehensive review. Prog. Cardiovasc. Dis. 2010, 53, 94–104. [Google Scholar] [CrossRef] [PubMed]
- Duffaud, F.; Therasse, P. New guidelines to evaluate the response to treatment in solid tumors. Bull. Cancer 2000, 87, 881–886. [Google Scholar] [PubMed]
- Bobo, D.; Robinson, K.J.; Islam, J.; Thurecht, K.J.; Corrie, S.R. Nanoparticle-Based Medicines: A Review of FDA-Approved Materials and Clinical Trials to Date. Pharm. Res. 2016, 33, 2373–2387. [Google Scholar] [CrossRef] [PubMed]
- Chaurasiya, B.; Mahanty, A.; Roy, D.; Shen, Y.; Tu, J.; Sun, C. Influence of Tumor Microenvironment on the Distribution and Elimination of Nano-formulations. Curr. Drug Metab. 2016, 17, 783–798. [Google Scholar] [CrossRef] [PubMed]
- Blanco, E.; Shen, H.; Ferrari, M. Principles of nanoparticle design for overcoming biological barriers to drug delivery. Nat. Biotechnol. 2015, 33, 941–951. [Google Scholar] [CrossRef] [Green Version]
- Cerqueira, B.B.; Lasham, A.; Shelling, A.N.; Al-Kassas, R. Nanoparticle therapeutics: Technologies and methods for overcoming cancer. Eur. J. Pharm. Biopharm. 2015, 97 Pt A, 140–151. [Google Scholar] [CrossRef]
- Zhang, H. Multifunctional nanomedicine platforms for cancer therapy. J. Nanosci. Nanotechnol. 2012, 12, 4012–4018. [Google Scholar] [CrossRef]
- Durymanov, M.O.; Rosenkranz, A.A.; Sobolev, A.S. Current Approaches for Improving Intratumoral Accumulation and Distribution of Nanomedicines. Theranostics 2015, 5, 1007–1020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaur, I.P.; Kakkar, V.; Deol, P.K.; Yadav, M.; Singh, M.; Sharma, I. Issues and concerns in nanotech product development and its commercialization. J. Control. Release 2014, 193, 51–62. [Google Scholar] [CrossRef] [PubMed]
- Tang, W.L.; Tang, W.H.; Li, S.D. Cancer theranostic applications of lipid-based nanoparticles. Drug Discov. Today 2018, 23, 1159–1166. [Google Scholar] [CrossRef] [PubMed]
- Xie, J.; Fan, Z.; Li, Y.; Zhang, Y.; Yu, F.; Su, G.; Xie, L.; Hou, Z. Design of pH-sensitive methotrexate prodrug-targeted curcumin nanoparticles for efficient dual-drug delivery and combination cancer therapy. Int. J. Nanomed. 2018, 13, 1381–1398. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; He, Y.; Zhang, S.; Qin, J.; Wang, J. Cell membrane-based nanoparticles: A new biomimetic platform for tumor diagnosis and treatment. Acta Pharm. Sin. B 2018, 8, 14–22. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.X.; Ahmed, T.; Li, L.Y.; Li, J.; Abbasi, A.Z.; Wu, X.Y. Design of nanocarriers for nanoscale drug delivery to enhance cancer treatment using hybrid polymer and lipid building blocks. Nanoscale 2017, 9, 1334–1355. [Google Scholar] [CrossRef]
- Stella, B.; Peira, E.; Dianzani, C.; Gallarate, M.; Battaglia, L.; Gigliotti, C.L.; Boggio, E.; Dianzani, U.; Dosio, F. Development and Characterization of Solid Lipid Nanoparticles Loaded with a Highly Active Doxorubicin Derivative. Nanomaterials (Basel) 2018, 8, 110. [Google Scholar] [CrossRef]
- Gilligan, K.E.; Dwyer, R.M. Engineering Exosomes for Cancer Therapy. Int. J. Mol. Sci. 2017, 18, 1122. [Google Scholar] [CrossRef]
- Bulbake, U.; Doppalapudi, S.; Kommineni, N.; Khan, W. Liposomal Formulations in Clinical Use: An Updated Review. Pharmaceutics 2017, 9, 12. [Google Scholar] [CrossRef]
- Jaiswal, M.; Dudhe, R.; Sharma, P.K. Nanoemulsion: An advanced mode of drug delivery system. 3 Biotech 2015, 5, 123–127. [Google Scholar] [CrossRef]
- Cerpnjak, K.; Zvonar, A.; Gasperlin, M.; Vrecer, F. Lipid-based systems as a promising approach for enhancing the bioavailability of poorly water-soluble drugs. Acta Pharm. 2013, 63, 427–445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanafy, N.A.N.; El-Kemary, M.; Leporatti, S. Micelles Structure Development as a Strategy to Improve Smart Cancer Therapy. Cancers (Basel) 2018, 10, 238. [Google Scholar] [CrossRef] [PubMed]
- Akbarzadeh, A.; Rezaei-Sadabady, R.; Davaran, S.; Joo, S.W.; Zarghami, N.; Hanifehpour, Y.; Samiei, M.; Kouhi, M.; Nejati-Koshki, K. Liposome: Classification, preparation, and applications. Nanoscale Res. Lett. 2013, 8, 102. [Google Scholar] [CrossRef] [PubMed]
- Gupta, A.; Eral, H.B.; Hatton, T.A.; Doyle, P.S. Nanoemulsions: Formation, properties and applications. Soft Matter 2016, 12, 2826–2841. [Google Scholar] [CrossRef] [PubMed]
- Geszke-Moritz, M.; Moritz, M. Solid lipid nanoparticles as attractive drug vehicles: Composition, properties and therapeutic strategies. Mater. Sci. Eng. C Mater. Biol. Appl. 2016, 68, 982–994. [Google Scholar] [CrossRef]
- Garg, N.K.; Tandel, N.; Jadon, R.S.; Tyagi, R.K.; Katare, O.P. Lipid-polymer hybrid nanocarrier-mediated cancer therapeutics: Current status and future directions. Drug Discov. Today 2018, 23, 1610–1621. [Google Scholar] [CrossRef]
- Ha, D.; Yang, N.; Nadithe, V. Exosomes as therapeutic drug carriers and delivery vehicles across biological membranes: Current perspectives and future challenges. Acta Pharm. Sin. B 2016, 6, 287–296. [Google Scholar] [CrossRef]
- Sun, Y.; Su, J.; Liu, G.; Chen, J.; Zhang, X.; Zhang, R.; Jiang, M.; Qiu, M. Advances of blood cell-based drug delivery systems. Eur. J. Pharm. Sci. 2017, 96, 115–128. [Google Scholar] [CrossRef]
- Fang, R.H.; Hu, C.M.; Luk, B.T.; Gao, W.; Copp, J.A.; Tai, Y.; O’Connor, D.E.; Zhang, L. Cancer cell membrane-coated nanoparticles for anticancer vaccination and drug delivery. Nano Lett. 2014, 14, 2181–2188. [Google Scholar] [CrossRef]
- Bottini, M.; Sacchetti, C.; Pietroiusti, A.; Bellucci, S.; Magrini, A.; Rosato, N.; Bottini, N. Targeted nanodrugs for cancer therapy: Prospects and challenges. J. Nanosci. Nanotechnol. 2014, 14, 98–114. [Google Scholar] [CrossRef]
- Shao, M.; Hussain, Z.; Thu, H.E.; Khan, S.; Katas, H.; Ahmed, T.A.; Tripathy, M.; Leng, J.; Qin, H.L.; Bukhari, S.N.A. Drug nanocarrier, the future of atopic diseases: Advanced drug delivery systems and smart management of disease. Colloids Surf. B Biointerfaces 2016, 147, 475–491. [Google Scholar] [CrossRef] [PubMed]
- Maeda, H.; Nakamura, H.; Fang, J. The EPR effect for macromolecular drug delivery to solid tumors: Improvement of tumor uptake, lowering of systemic toxicity, and distinct tumor imaging in vivo. Adv. Drug Deliv. Rev. 2013, 65, 71–79. [Google Scholar] [CrossRef] [PubMed]
- Khan, T.A.; Reddy, S.T. Immunological principles regulating immunomodulation with biomaterials. Acta Biomater. 2014, 10, 1720–1727. [Google Scholar] [CrossRef] [PubMed]
- Danaei, M.; Dehghankhold, M.; Ataei, S.; Hasanzadeh Davarani, F.; Javanmard, R.; Dokhani, A.; Khorasani, S.; Mozafari, M.R. Impact of Particle Size and Polydispersity Index on the Clinical Applications of Lipidic Nanocarrier Systems. Pharmaceutics 2018, 10, 57. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.S.; Liu, W.; Misra, P.; Tanaka, E.; Zimmer, J.P.; Itty Ipe, B.; Bawendi, M.G.; Frangioni, J.V. Renal clearance of quantum dots. Nat. Biotechnol. 2007, 25, 1165–1170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koide, H.; Asai, T.; Hatanaka, K.; Urakami, T.; Ishii, T.; Kenjo, E.; Nishihara, M.; Yokoyama, M.; Ishida, T.; Kiwada, H.; et al. Particle size-dependent triggering of accelerated blood clearance phenomenon. Int. J. Pharm. 2008, 362, 197–200. [Google Scholar] [CrossRef] [PubMed]
- Cho, N.J.; Hwang, L.Y.; Solandt, J.J.R.; Frank, C.W. Comparison of Extruded and Sonicated Vesicles for Planar Bilayer Self-Assembly. Materials (Basel) 2013, 6, 3294–3308. [Google Scholar] [CrossRef] [Green Version]
- Gentine, P.; Bourel-Bonnet, L.; Frisch, B. Modified and derived ethanol injection toward liposomes: Development of the process. J. Liposome Res. 2013, 23, 11–19. [Google Scholar] [CrossRef]
- Lebre, F.; Sridharan, R.; Sawkins, M.J.; Kelly, D.J.; O’Brien, F.J.; Lavelle, E.C. The shape and size of hydroxyapatite particles dictate inflammatory responses following implantation. Sci. Rep. 2017, 7, 2922. [Google Scholar] [CrossRef]
- Tan, J.; Shah, S.; Thomas, A.; Ou-Yang, H.D.; Liu, Y. The influence of size, shape and vessel geometry on nanoparticle distribution. Microfluid. Nanofluid. 2013, 14, 77–87. [Google Scholar] [CrossRef]
- Muller, K.; Fedosov, D.A.; Gompper, G. Margination of micro- and nano-particles in blood flow and its effect on drug delivery. Sci. Rep. 2014, 4, 4871. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geng, Y.; Dalhaimer, P.; Cai, S.; Tsai, R.; Tewari, M.; Minko, T.; Discher, D.E. Shape effects of filaments versus spherical particles in flow and drug delivery. Nat. Nanotechnol. 2007, 2, 249–255. [Google Scholar] [CrossRef] [PubMed]
- Owens, D.E., 3rd; Peppas, N.A. Opsonization, biodistribution, and pharmacokinetics of polymeric nanoparticles. Int. J. Pharm. 2006, 307, 93–102. [Google Scholar] [CrossRef] [PubMed]
- Chaudhari, K.R.; Ukawala, M.; Manjappa, A.S.; Kumar, A.; Mundada, P.K.; Mishra, A.K.; Mathur, R.; Monkkonen, J.; Murthy, R.S. Opsonization, biodistribution, cellular uptake and apoptosis study of PEGylated PBCA nanoparticle as potential drug delivery carrier. Pharm. Res. 2012, 29, 53–68. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.B.; Banerjee, A.; Onyuksel, H. Improvement of drug safety by the use of lipid-based nanocarriers. J. Control. Release 2012, 163, 34–45. [Google Scholar] [CrossRef]
- Aftab, S.; Shah, A.; Nadhman, A.; Kurbanoglu, S.; Aysil Ozkan, S.; Dionysiou, D.D.; Shukla, S.S.; Aminabhavi, T.M. Nanomedicine: An effective tool in cancer therapy. Int. J. Pharm. 2018, 540, 132–149. [Google Scholar] [CrossRef] [PubMed]
- Bradley, A.J.; Devine, D.V.; Ansell, S.M.; Janzen, J.; Brooks, D.E. Inhibition of liposome-induced complement activation by incorporated poly(ethylene glycol)-lipids. Arch. Biochem. Biophys. 1998, 357, 185–194. [Google Scholar] [CrossRef]
- Mare, R.; Paolino, D.; Celia, C.; Molinaro, R.; Fresta, M.; Cosco, D. Post-insertion parameters of PEG-derivatives in phosphocholine-liposomes. Int. J. Pharm. 2018, 552, 414–421. [Google Scholar] [CrossRef] [PubMed]
- Bernkop-Schnurch, A. Strategies to overcome the polycation dilemma in drug delivery. Adv. Drug Deliv. Rev. 2018, 136–137, 62–72. [Google Scholar] [CrossRef] [PubMed]
- Papageorgiou, F.; Pippa, N.; Naziris, N.; Demetzos, C. Physicochemical Study of the Protein-Liposome Interactions: Influence of Liposome Composition and Concentration on Protein Binding. J. Liposome Res. 2018, 1–29. [Google Scholar] [CrossRef] [PubMed]
- Sabin, J.; Vazquez-Vazquez, C.; Prieto, G.; Bordi, F.; Sarmiento, F. Double charge inversion in polyethylenimine-decorated liposomes. Langmuir 2012, 28, 10534–10542. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.W.; Jeong, H.Y.; Kang, S.J.; Choi, M.J.; You, Y.M.; Im, C.S.; Lee, T.S.; Song, I.H.; Lee, C.G.; Rhee, K.J.; et al. Cancer-targeted Nucleic Acid Delivery and Quantum Dot Imaging Using EGF Receptor Aptamer-conjugated Lipid Nanoparticles. Sci. Rep. 2017, 7, 9474. [Google Scholar] [CrossRef] [PubMed]
- Kang, J.H.; Jang, W.Y.; Ko, Y.T. The Effect of Surface Charges on the Cellular Uptake of Liposomes Investigated by Live Cell Imaging. Pharm. Res. 2017, 34, 704–717. [Google Scholar] [CrossRef] [PubMed]
- Barenholz, Y. Doxil(R)—The first FDA-approved nano-drug: Lessons learned. J. Control. Release 2012, 160, 117–134. [Google Scholar] [CrossRef] [PubMed]
- Riaz, M.K.; Riaz, M.A.; Zhang, X.; Lin, C.; Wong, K.H.; Chen, X.; Zhang, G.; Lu, A.; Yang, Z. Surface Functionalization and Targeting Strategies of Liposomes in Solid Tumor Therapy: A Review. Int. J. Mol. Sci. 2018, 19, 19. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, T.; Mima, Y.; Hashimoto, Y.; Ukawa, M.; Ando, H.; Kiwada, H.; Ishida, T. Anti-PEG IgM and complement system are required for the association of second doses of PEGylated liposomes with splenic marginal zone B cells. Immunobiology 2015, 220, 1151–1160. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, L.; Yi, Y.; Yu, Y. Effect of partial PEGylation on particle uptake by macrophages. Nanoscale 2017, 9, 288–297. [Google Scholar] [CrossRef]
- Fang, Y.; Xue, J.; Gao, S.; Lu, A.; Yang, D.; Jiang, H.; He, Y.; Shi, K. Cleavable PEGylation: A strategy for overcoming the “PEG dilemma” in efficient drug delivery. Drug Deliv. 2017, 24 (Suppl. 1), 22–32. [Google Scholar] [CrossRef]
- Muzykantov, V.R. Drug delivery by red blood cells: Vascular carriers designed by mother nature. Expert Opin. Drug Deliv. 2010, 7, 403–427. [Google Scholar] [CrossRef]
- Lizano, C.; Weissig, V.; Torchilin, V.P.; Sancho, P.; Garcia-Perez, A.I.; Pinilla, M. In vivo biodistribution of erythrocytes and polyethyleneglycol-phosphatidylethanolamine micelles carrying the antitumour agent dequalinium. Eur. J. Pharm. Biopharm. 2003, 56, 153–157. [Google Scholar] [CrossRef]
- Brenner, J.S.; Pan, D.C.; Myerson, J.W.; Marcos-Contreras, O.A.; Villa, C.H.; Patel, P.; Hekierski, H.; Chatterjee, S.; Tao, J.Q.; Parhiz, H.; et al. Red blood cell-hitchhiking boosts delivery of nanocarriers to chosen organs by orders of magnitude. Nat. Commun. 2018, 9, 2684. [Google Scholar] [CrossRef] [PubMed]
- Deak, R.; Mihaly, J.; Szigyarto, I.C.; Wacha, A.; Lelkes, G.; Bota, A. Physicochemical characterization of artificial nanoerythrosomes derived from erythrocyte ghost membranes. Colloids Surf. B Biointerfaces 2015, 135, 225–234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, C.M.; Zhang, L.; Aryal, S.; Cheung, C.; Fang, R.H.; Zhang, L. Erythrocyte membrane-camouflaged polymeric nanoparticles as a biomimetic delivery platform. Proc. Natl. Acad. Sci. USA 2011, 108, 10980–10985. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H. Erythrocytes in nanomedicine: An optimal blend of natural and synthetic materials. Biomater. Sci. 2016, 4, 1024–1031. [Google Scholar] [CrossRef] [PubMed]
- Fang, J.; Nakamura, H.; Maeda, H. The EPR effect: Unique features of tumor blood vessels for drug delivery, factors involved, and limitations and augmentation of the effect. Adv. Drug Deliv. Rev. 2011, 63, 136–151. [Google Scholar] [CrossRef] [PubMed]
- Golombek, S.K.; May, J.N.; Theek, B.; Appold, L.; Drude, N.; Kiessling, F.; Lammers, T. Tumor targeting via EPR: Strategies to enhance patient responses. Adv. Drug Deliv. Rev. 2018, 130, 17–38. [Google Scholar] [CrossRef] [PubMed]
- Stylianopoulos, T.; Jain, R.K. Design considerations for nanotherapeutics in oncology. Nanomedicine 2015, 11, 1893–1907. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Danhier, F. To exploit the tumor microenvironment: Since the EPR effect fails in the clinic, what is the future of nanomedicine? J. Control. Release 2016, 244 Pt A, 108–121. [Google Scholar] [CrossRef]
- Ojha, T.; Pathak, V.; Shi, Y.; Hennink, W.E.; Moonen, C.T.W.; Storm, G.; Kiessling, F.; Lammers, T. Pharmacological and physical vessel modulation strategies to improve EPR-mediated drug targeting to tumors. Adv. Drug Deliv. Rev. 2017, 119, 44–60. [Google Scholar] [CrossRef] [Green Version]
- Li, R.; Zheng, K.; Yuan, C.; Chen, Z.; Huang, M. Be Active or Not: The Relative Contribution of Active and Passive Tumor Targeting of Nanomaterials. Nanotheranostics 2017, 1, 346–357. [Google Scholar] [CrossRef]
- Lee, S.H.; Sato, Y.; Hyodo, M.; Harashima, H. Topology of Surface Ligands on Liposomes: Characterization Based on the Terms, Incorporation Ratio, Surface Anchor Density, and Reaction Yield. Biol. Pharm. Bull. 2016, 39, 1983–1994. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mishra, D.K.; Shandilya, R.; Mishra, P.K. Lipid based nanocarriers: A translational perspective. Nanomedicine 2018, 14, 2023–2050. [Google Scholar] [CrossRef]
- Chames, P.; Van Regenmortel, M.; Weiss, E.; Baty, D. Therapeutic antibodies: Successes, limitations and hopes for the future. Br. J. Pharmacol. 2009, 157, 220–233. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Rossi, J. Aptamers as targeted therapeutics: Current potential and challenges. Nat. Rev. Drug Discov. 2017, 16, 181–202. [Google Scholar] [CrossRef] [PubMed]
- Gilad, Y.; Firer, M.; Gellerman, G. Recent Innovations in Peptide Based Targeted Drug Delivery to Cancer Cells. Biomedicines 2016, 4, 11. [Google Scholar] [CrossRef] [PubMed]
- Kohler, G.; Milstein, C. Continuous cultures of fused cells secreting antibody of predefined specificity. Nature 1975, 256, 495–497. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Hu, W.; Li, L.; Ding, H.; Li, H. Therapeutic monoclonal antibodies and clinical laboratory tests: When, why, and what is expected? J. Clin. Lab. Anal. 2018, 32. [Google Scholar] [CrossRef]
- Muhamad, N.; Plengsuriyakarn, T.; Na-Bangchang, K. Application of active targeting nanoparticle delivery system for chemotherapeutic drugs and traditional/herbal medicines in cancer therapy: A systematic review. Int. J. Nanomed. 2018, 13, 3921–3935. [Google Scholar] [CrossRef]
- Bazak, R.; Houri, M.; El Achy, S.; Kamel, S.; Refaat, T. Cancer active targeting by nanoparticles: A comprehensive review of literature. J. Cancer Res. Clin. Oncol. 2015, 141, 769–784. [Google Scholar] [CrossRef]
- Singh, S.; Kumar, N.K.; Dwiwedi, P.; Charan, J.; Kaur, R.; Sidhu, P.; Chugh, V.K. Monoclonal Antibodies: A Review. Curr. Clin. Pharmacol. 2018, 13, 85–99. [Google Scholar] [CrossRef]
- Kontermann, R.E. Dual targeting strategies with bispecific antibodies. MAbs 2012, 4, 182–197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krishnamurthy, A.; Jimeno, A. Bispecific antibodies for cancer therapy: A review. Pharmacol. Ther. 2018, 185, 122–134. [Google Scholar] [CrossRef] [PubMed]
- Gotrik, M.R.; Feagin, T.A.; Csordas, A.T.; Nakamoto, M.A.; Soh, H.T. Advancements in Aptamer Discovery Technologies. Acc. Chem. Res. 2016, 49, 1903–1910. [Google Scholar] [CrossRef] [PubMed]
- Alshaer, W.; Hillaireau, H.; Fattal, E. Aptamer-guided nanomedicines for anticancer drug delivery. Adv. Drug Deliv. Rev. 2018, 134, 122–137. [Google Scholar] [CrossRef] [PubMed]
- Zhou, G.; Wilson, G.; Hebbard, L.; Duan, W.; Liddle, C.; George, J.; Qiao, L. Aptamers: A promising chemical antibody for cancer therapy. Oncotarget 2016, 7, 13446–13463. [Google Scholar] [CrossRef] [Green Version]
- Kang, S.J.; Jeong, H.Y.; Kim, M.W.; Jeong, I.H.; Choi, M.J.; You, Y.M.; Im, C.S.; Song, I.H.; Lee, T.S.; Park, Y.S. Anti-EGFR lipid micellar nanoparticles co-encapsulating quantum dots and paclitaxel for tumor-targeted theranosis. Nanoscale 2018, 10, 19338–19350. [Google Scholar] [CrossRef]
- Zhang, Y.; Chen, Y.; Han, D.; Ocsoy, I.; Tan, W. Aptamers selected by cell-SELEX for application in cancer studies. Bioanalysis 2010, 2, 907–918. [Google Scholar] [CrossRef] [Green Version]
- Hasegawa, H.; Savory, N.; Abe, K.; Ikebukuro, K. Methods for Improving Aptamer Binding Affinity. Molecules 2016, 21, 421. [Google Scholar] [CrossRef]
- Choudhury, H.; Pandey, M.; Chin, P.X.; Phang, Y.L.; Cheah, J.Y.; Ooi, S.C.; Mak, K.K.; Pichika, M.R.; Kesharwani, P.; Hussain, Z.; et al. Transferrin receptors-targeting nanocarriers for efficient targeted delivery and transcytosis of drugs into the brain tumors: A review of recent advancements and emerging trends. Drug Deliv. Transl. Res. 2018, 8, 1545–1563. [Google Scholar] [CrossRef]
- Heath, J.L.; Weiss, J.M.; Lavau, C.P.; Wechsler, D.S. Iron deprivation in cancer—Potential therapeutic implications. Nutrients 2013, 5, 2836–2859. [Google Scholar] [CrossRef]
- Chan, K.T.; Choi, M.Y.; Lai, K.K.; Tan, W.; Tung, L.N.; Lam, H.Y.; Tong, D.K.; Lee, N.P.; Law, S. Overexpression of transferrin receptor CD71 and its tumorigenic properties in esophageal squamous cell carcinoma. Oncol. Rep. 2014, 31, 1296–1304. [Google Scholar] [CrossRef] [PubMed]
- Tortorella, S.; Karagiannis, T.C. Transferrin receptor-mediated endocytosis: A useful target for cancer therapy. J. Membr. Biol. 2014, 247, 291–307. [Google Scholar] [CrossRef] [PubMed]
- Tran, T.H.; Nguyen, T.D.; Van Nguyen, H.; Nguyen, H.T.; Kim, J.O.; Yong, C.S.; Nguyen, C.N. Targeted and controlled drug delivery system loading artersunate for effective chemotherapy on CD44 overexpressing cancer cells. Arch. Pharm. Res. 2016, 39, 687–694. [Google Scholar] [CrossRef] [PubMed]
- Wickens, J.M.; Alsaab, H.O.; Kesharwani, P.; Bhise, K.; Amin, M.; Tekade, R.K.; Gupta, U.; Iyer, A.K. Recent advances in hyaluronic acid-decorated nanocarriers for targeted cancer therapy. Drug Discov. Today 2017, 22, 665–680. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rayahin, J.E.; Buhrman, J.S.; Zhang, Y.; Koh, T.J.; Gemeinhart, R.A. High and low molecular weight hyaluronic acid differentially influence macrophage activation. ACS Biomater. Sci. Eng. 2015, 1, 481–493. [Google Scholar] [CrossRef] [PubMed]
- Cyphert, J.M.; Trempus, C.S.; Garantziotis, S. Size Matters: Molecular Weight Specificity of Hyaluronan Effects in Cell Biology. Int. J. Cell Biol. 2015, 2015, 563818. [Google Scholar] [CrossRef] [PubMed]
- Zwicke, G.L.; Mansoori, G.A.; Jeffery, C.J. Utilizing the folate receptor for active targeting of cancer nanotherapeutics. Nano Rev. 2012, 3. [Google Scholar] [CrossRef]
- Ledermann, J.A.; Canevari, S.; Thigpen, T. Targeting the folate receptor: Diagnostic and therapeutic approaches to personalize cancer treatments. Ann. Oncol. 2015, 26, 2034–2043. [Google Scholar] [CrossRef]
- Gray, B.P.; Brown, K.C. Combinatorial peptide libraries: Mining for cell-binding peptides. Chem. Rev. 2014, 114, 1020–1081. [Google Scholar] [CrossRef]
- Mousavizadeh, A.; Jabbari, A.; Akrami, M.; Bardania, H. Cell targeting peptides as smart ligands for targeting of therapeutic or diagnostic agents: A systematic review. Colloids Surf. B Biointerfaces 2017, 158, 507–517. [Google Scholar] [CrossRef]
- Hirsjarvi, S.; Belloche, C.; Hindre, F.; Garcion, E.; Benoit, J.P. Tumour targeting of lipid nanocapsules grafted with cRGD peptides. Eur. J. Pharm. Biopharm. 2014, 87, 152–159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heckmann, D.; Kessler, H. Design and chemical synthesis of integrin ligands. Methods Enzymol. 2007, 426, 463–503. [Google Scholar] [PubMed]
- Ujula, T.; Huttunen, M.; Luoto, P.; Perakyla, H.; Simpura, I.; Wilson, I.; Bergman, M.; Roivainen, A. Matrix metalloproteinase 9 targeting peptides: Syntheses, 68Ga-labeling, and preliminary evaluation in a rat melanoma xenograft model. Bioconjug. Chem. 2010, 21, 1612–1621. [Google Scholar] [CrossRef] [PubMed]
- Gao, W.; Meng, T.; Shi, N.; Zhuang, H.; Yang, Z.; Qi, X. Targeting and microenvironment-responsive lipid nanocarrier for the enhancement of tumor cell recognition and therapeutic efficiency. Adv. Healthc. Mater. 2015, 4, 748–759. [Google Scholar] [CrossRef] [PubMed]
- Yanez-Mo, M.; Siljander, P.R.; Andreu, Z.; Zavec, A.B.; Borras, F.E.; Buzas, E.I.; Buzas, K.; Casal, E.; Cappello, F.; Carvalho, J.; et al. Biological properties of extracellular vesicles and their physiological functions. J. Extracell Vesicles 2015, 4, 27066. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garofalo, M.; Saari, H.; Somersalo, P.; Crescenti, D.; Kuryk, L.; Aksela, L.; Capasso, C.; Madetoja, M.; Koskinen, K.; Oksanen, T.; et al. Antitumor effect of oncolytic virus and paclitaxel encapsulated in extracellular vesicles for lung cancer treatment. J. Control. Release 2018, 283, 223–234. [Google Scholar] [CrossRef] [PubMed]
- Ohno, S.; Drummen, G.P.; Kuroda, M. Focus on Extracellular Vesicles: Development of Extracellular Vesicle-Based Therapeutic Systems. Int. J. Mol. Sci. 2016, 17, 172. [Google Scholar] [CrossRef]
- Wang, X.; Zhang, H.; Yang, H.; Bai, M.; Ning, T.; Li, S.; Li, J.; Deng, T.; Ying, G.; Ba, Y. Cell-derived Exosomes as Promising Carriers for Drug Delivery and Targeted Therapy. Curr. Cancer Drug Targets 2018, 18, 347–354. [Google Scholar] [CrossRef]
- Vader, P.; Mol, E.A.; Pasterkamp, G.; Schiffelers, R.M. Extracellular vesicles for drug delivery. Adv. Drug Deliv. Rev. 2016, 106 Pt A, 148–156. [Google Scholar] [CrossRef]
- Wiklander, O.P.; Nordin, J.Z.; O’Loughlin, A.; Gustafsson, Y.; Corso, G.; Mager, I.; Vader, P.; Lee, Y.; Sork, H.; Seow, Y.; et al. Extracellular vesicle in vivo biodistribution is determined by cell source, route of administration and targeting. J. Extracell Vesicles 2015, 4, 26316. [Google Scholar] [CrossRef] [Green Version]
- Tai, Y.L.; Chen, K.C.; Hsieh, J.T.; Shen, T.L. Exosomes in cancer development and clinical applications. Cancer Sci. 2018, 109, 2364–2374. [Google Scholar] [CrossRef] [PubMed]
- Sun, W.; Luo, J.D.; Jiang, H.; Duan, D.D. Tumor exosomes: A double-edged sword in cancer therapy. Acta Pharmacol. Sin. 2018, 39, 534–541. [Google Scholar] [CrossRef] [PubMed]
- Haney, M.J.; Klyachko, N.L.; Zhao, Y.; Gupta, R.; Plotnikova, E.G.; He, Z.; Patel, T.; Piroyan, A.; Sokolsky, M.; Kabanov, A.V.; et al. Exosomes as drug delivery vehicles for Parkinson’s disease therapy. J. Control. Release 2015, 207, 18–30. [Google Scholar] [CrossRef] [PubMed]
- Gomari, H.; Forouzandeh Moghadam, M.; Soleimani, M. Targeted cancer therapy using engineered exosome as a natural drug delivery vehicle. Onco Targets Ther. 2018, 11, 5753–5762. [Google Scholar] [CrossRef] [PubMed]
- Guerreiro, E.M.; Vestad, B.; Steffensen, L.A.; Aass, H.C.D.; Saeed, M.; Ovstebo, R.; Costea, D.E.; Galtung, H.K.; Soland, T.M. Efficient extracellular vesicle isolation by combining cell media modifications, ultrafiltration, and size-exclusion chromatography. PLoS ONE 2018, 13, e0204276. [Google Scholar] [CrossRef]
- Lin, Y.; Wu, J.; Gu, W.; Huang, Y.; Tong, Z.; Huang, L.; Tan, J. Exosome-Liposome Hybrid Nanoparticles Deliver CRISPR/Cas9 System in MSCs. Adv. Sci. (Weinh) 2018, 5, 1700611. [Google Scholar] [CrossRef] [PubMed]
- Sato, Y.T.; Umezaki, K.; Sawada, S.; Mukai, S.A.; Sasaki, Y.; Harada, N.; Shiku, H.; Akiyoshi, K. Engineering hybrid exosomes by membrane fusion with liposomes. Sci. Rep. 2016, 6, 21933. [Google Scholar] [CrossRef] [Green Version]
- Khaldoyanidi, S.K.; Glinsky, V.V.; Sikora, L.; Glinskii, A.B.; Mossine, V.V.; Quinn, T.P.; Glinsky, G.V.; Sriramarao, P. MDA-MB-435 human breast carcinoma cell homo- and heterotypic adhesion under flow conditions is mediated in part by Thomsen-Friedenreich antigen-galectin-3 interactions. J. Biol. Chem. 2003, 278, 4127–4134. [Google Scholar] [CrossRef]
- Gentile, P.; Chiono, V.; Carmagnola, I.; Hatton, P.V. An overview of poly(lactic-co-glycolic) acid (PLGA)-based biomaterials for bone tissue engineering. Int. J. Mol. Sci. 2014, 15, 3640–3659. [Google Scholar] [CrossRef]
- Pooja, D.; Tunki, L.; Kulhari, H.; Reddy, B.B.; Sistla, R. Optimization of solid lipid nanoparticles prepared by a single emulsification-solvent evaporation method. Data Brief 2016, 6, 15–19. [Google Scholar] [CrossRef]
- Winter, E.; Dal Pizzol, C.; Locatelli, C.; Crezkynski-Pasa, T.B. Development and Evaluation of Lipid Nanoparticles for Drug Delivery: Study of Toxicity In, Vitro and In Vivo. J. Nanosci. Nanotechnol. 2016, 16, 1321–1330. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Outschoorn, U.E.; Peiris-Pages, M.; Pestell, R.G.; Sotgia, F.; Lisanti, M.P. Cancer metabolism: A therapeutic perspective. Nat. Rev. Clin. Oncol. 2017, 14, 11–31. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.; Chen, C.; Huang, Y.; Zhang, F.; Lin, G. Study of the pH-sensitive mechanism of tumor-targeting liposomes. Colloids Surf. B Biointerfaces 2017, 151, 19–25. [Google Scholar] [CrossRef]
- Fritze, A.; Hens, F.; Kimpfler, A.; Schubert, R.; Peschka-Suss, R. Remote loading of doxorubicin into liposomes driven by a transmembrane phosphate gradient. Biochim. Biophys. Acta 2006, 1758, 1633–1640. [Google Scholar] [CrossRef]
- Gu, Z.; Chang, M.; Fan, Y.; Shi, Y.; Lin, G. NGR-modified pH-sensitive liposomes for controlled release and tumor target delivery of docetaxel. Colloids Surf. B Biointerfaces 2017, 160, 395–405. [Google Scholar] [CrossRef] [PubMed]
- Won, S.H.; Lee, J.U.; Sim, S.J. Fluorogenic pH-sensitive polydiacetylene (PDA) liposomes as a drug carrier. J. Nanosci. Nanotechnol. 2013, 13, 3792–3800. [Google Scholar] [CrossRef]
- Clawson, C.; Ton, L.; Aryal, S.; Fu, V.; Esener, S.; Zhang, L. Synthesis and characterization of lipid-polymer hybrid nanoparticles with pH-triggered poly(ethylene glycol) shedding. Langmuir 2011, 27, 10556–10561. [Google Scholar] [CrossRef] [PubMed]
- Qiao, Y.; Wan, J.; Zhou, L.; Ma, W.; Yang, Y.; Luo, W.; Yu, Z.; Wang, H. Stimuli-responsive nanotherapeutics for precision drug delivery and cancer therapy. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 2018, e1527. [Google Scholar] [CrossRef]
- Ta, T.; Porter, T.M. Thermosensitive liposomes for localized delivery and triggered release of chemotherapy. J. Control. Release 2013, 169, 112–125. [Google Scholar] [CrossRef] [Green Version]
- Alavizadeh, S.H.; Badiee, A.; Golmohammadzadeh, S.; Jaafari, M.R. The influence of phospholipid on the physicochemical properties and anti-tumor efficacy of liposomes encapsulating cisplatin in mice bearing C26 colon carcinoma. Int. J. Pharm. 2014, 473, 326–333. [Google Scholar] [CrossRef] [PubMed]
- Corso, C.R.; Acco, A. Glutathione system in animal model of solid tumors: From regulation to therapeutic target. Crit. Rev. Oncol. Hematol. 2018, 128, 43–57. [Google Scholar] [CrossRef] [PubMed]
- McCarley, R.L. Redox-responsive delivery systems. Annu. Rev. Anal. Chem. 2012, 5, 391–411. [Google Scholar] [CrossRef] [PubMed]
- Ren, G.; Jiang, M.; Guo, W.; Sun, B.; Lian, H.; Wang, Y.; He, Z. Construction and cellular uptake behavior of redox-sensitive docetaxel prodrug-loaded liposomes. Pharm. Dev. Technol. 2018, 23, 22–32. [Google Scholar] [CrossRef] [PubMed]
- Zhou, G.; Li, L.; Xing, J.; Jalde, S.; Li, Y.; Cai, J.; Chen, J.; Liu, P.; Gu, N.; Ji, M. Redox responsive liposomal nanohybrid cerasomes for intracellular drug delivery. Colloids Surf. B Biointerfaces 2016, 148, 518–525. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.H.; Kim, J.C. Redox-responsive solid lipid microparticles composed of octadecyl acrylate and allyl disulfide. J. Biomater. Sci. Polym. Ed. 2018, 29, 476–490. [Google Scholar] [CrossRef] [PubMed]
- Hu, Q.; Katti, P.S.; Gu, Z. Enzyme-responsive nanomaterials for controlled drug delivery. Nanoscale 2014, 6, 12273–12286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dai, Z.; Yao, Q.; Zhu, L. MMP2-Sensitive PEG-Lipid Copolymers: A New Type of Tumor-Targeted P-Glycoprotein Inhibitor. ACS Appl. Mater. Interfaces 2016, 8, 12661–12673. [Google Scholar] [CrossRef]
- Kessenbrock, K.; Plaks, V.; Werb, Z. Matrix metalloproteinases: Regulators of the tumor microenvironment. Cell 2010, 141, 52–67. [Google Scholar] [CrossRef]
- Zhu, L.; Kate, P.; Torchilin, V.P. Matrix metalloprotease 2-responsive multifunctional liposomal nanocarrier for enhanced tumor targeting. ACS Nano 2012, 6, 3491–3498. [Google Scholar] [CrossRef]
- Terada, T.; Iwai, M.; Kawakami, S.; Yamashita, F.; Hashida, M. Novel PEG-matrix metalloproteinase-2 cleavable peptide-lipid containing galactosylated liposomes for hepatocellular carcinoma-selective targeting. J. Control. Release 2006, 111, 333–342. [Google Scholar] [CrossRef]
- Fathi, M.; Sahandi Zangabad, P.; Majidi, S.; Barar, J.; Erfan-Niya, H.; Omidi, Y. Stimuli-responsive chitosan-based nanocarriers for cancer therapy. Bioimpacts 2017, 7, 269–277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, M.M.; Liu, Y.Y.; Su, G.H.; Song, F.F.; Liu, Y.; Zhang, Q.Q. NIR responsive liposomal system for rapid release of drugs in cancer therapy. Int. J. Nanomed. 2017, 12, 4225–4239. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Yang, X.; Zhou, Z.; Wang, K.; Li, C.; Qiao, H.; Oupicky, D.; Sun, M. Near-infrared light-triggered drug release from a multiple lipid carrier complex using an all-in-one strategy. J. Control. Release 2017, 261, 126–137. [Google Scholar] [CrossRef] [PubMed]
- Yao, C.; Wang, P.; Li, X.; Hu, X.; Hou, J.; Wang, L.; Zhang, F. Near-Infrared-Triggered Azobenzene-Liposome/Upconversion Nanoparticle Hybrid Vesicles for Remotely Controlled Drug Delivery to Overcome Cancer Multidrug Resistance. Adv. Mater. 2016, 28, 9341–9348. [Google Scholar] [CrossRef] [PubMed]
- Han, H.D.; Jeon, Y.W.; Kwon, H.J.; Jeon, H.N.; Byeon, Y.; Lee, C.O.; Cho, S.H.; Shin, B.C. Therapeutic efficacy of doxorubicin delivery by a CO2 generating liposomal platform in breast carcinoma. Acta Biomater. 2015, 24, 279–285. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, K.; Fuchigami, Y.; Hagimori, M.; Fumoto, S.; Miura, Y.; Kawakami, S. Efficient gene transfection to the brain with ultrasound irradiation in mice using stabilized bubble lipopolyplexes prepared by the surface charge regulation method. Int. J. Nanomed. 2018, 13, 2309–2320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chandan, R.; Banerjee, R. Pro-apoptotic liposomes-nanobubble conjugate synergistic with paclitaxel: A platform for ultrasound responsive image-guided drug delivery. Sci. Rep. 2018, 8, 2624. [Google Scholar] [CrossRef]
- Endo-Takahashi, Y.; Negishi, Y.; Suzuki, R.; Maruyama, K.; Aramaki, Y. MicroRNA Imaging in Combination with Diagnostic Ultrasound and Bubble Liposomes for MicroRNA Delivery. Methods Mol. Biol. 2016, 1372, 209–213. [Google Scholar]
- Zhang, N.; Li, J.; Hou, R.; Zhang, J.; Wang, P.; Liu, X.; Zhang, Z. Bubble-generating nano-lipid carriers for ultrasound/CT imaging-guided efficient tumor therapy. Int. J. Pharm. 2017, 534, 251–262. [Google Scholar] [CrossRef]
- Liang, J.; Zhang, X.; Miao, Y.; Li, J.; Gan, Y. Lipid-coated iron oxide nanoparticles for dual-modal imaging of hepatocellular carcinoma. Int. J. Nanomed. 2017, 12, 2033–2044. [Google Scholar] [CrossRef]
- Boucher, M.; Geffroy, F.; Preveral, S.; Bellanger, L.; Selingue, E.; Adryanczyk-Perrier, G.; Pean, M.; Lefevre, C.T.; Pignol, D.; Ginet, N.; et al. Genetically tailored magnetosomes used as MRI probe for molecular imaging of brain tumor. Biomaterials 2017, 121, 167–178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prevot, G.; Kauss, T.; Lorenzato, C.; Gaubert, A.; Lariviere, M.; Baillet, J.; Laroche-Traineau, J.; Jacobin-Valat, M.J.; Adumeau, L.; Mornet, S.; et al. Iron oxide core oil-in-water nanoemulsion as tracer for atherosclerosis MPI and MRI imaging. Int. J. Pharm. 2017, 532, 669–676. [Google Scholar] [CrossRef] [PubMed]
- Tian, J.; Yan, C.; Liu, K.; Tao, J.; Guo, Z.; Liu, J.; Zhang, Y.; Xiong, F.; Gu, N. Paclitaxel-Loaded Magnetic Nanoparticles: Synthesis, Characterization, and Application in Targeting. J. Pharm. Sci. 2017, 106, 2115–2122. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
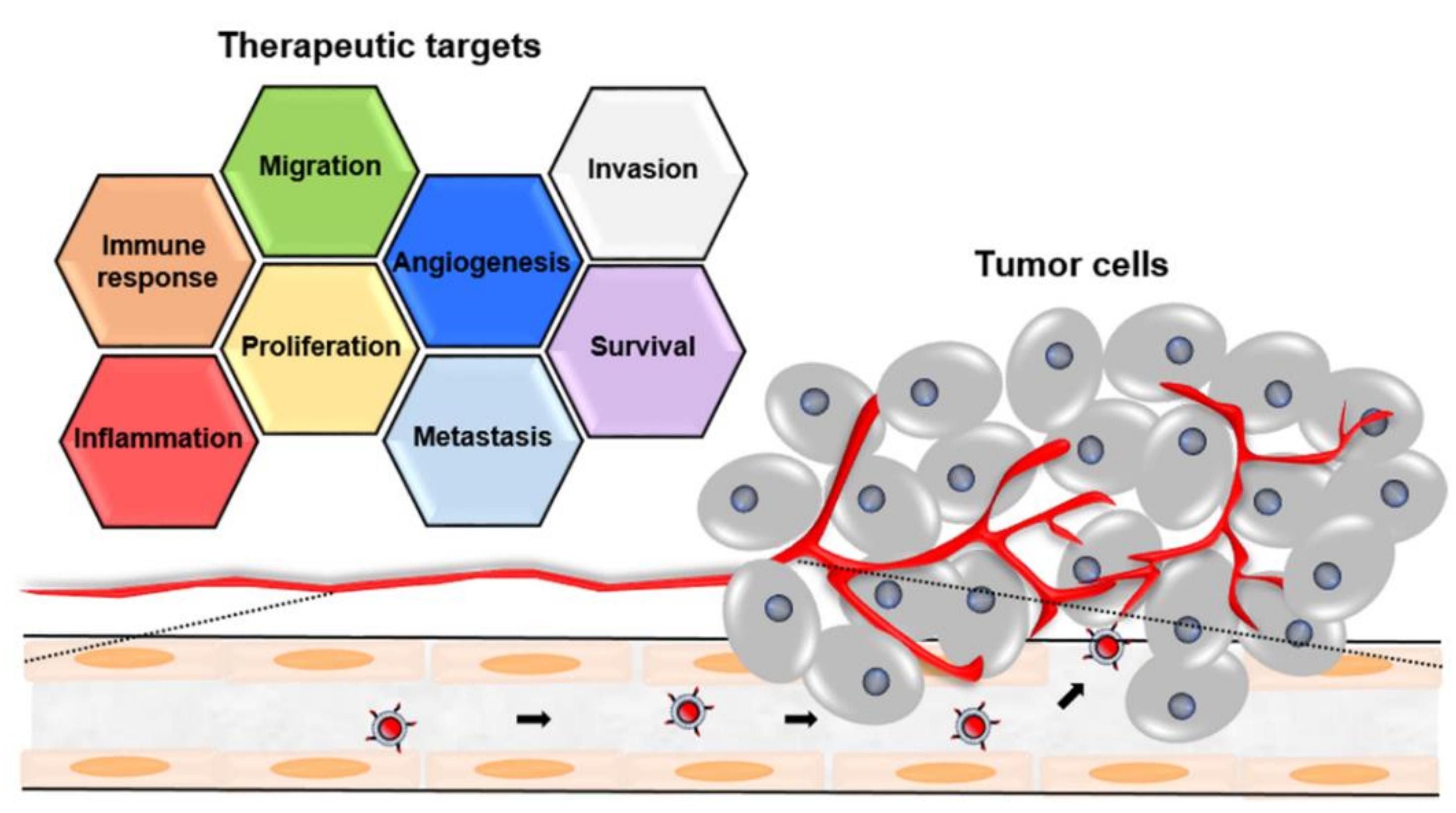{kind=link}
| Type | Core | Lipid lamellarity | Size | Characteristic | Reference | |
|---|---|---|---|---|---|---|
| Micelles | - Hydrophobic drug | - Monolayer | - 2 nm to 80 nm | - Lipid micelles are small-sized vesicles for solubilization of various poorly soluble pharmaceuticals. | [22] | |
| Liposomes | - Hydrophilic - Hydrophobic drug (inner membrane space) | - One to twenty bilayers | - 30 nm to 3000 nm | - Liposomes are synthetically constructed phospholipid vesicles can encapsulate both hydrophobic and hydrophilic drug. | [23] | |
| Nanoemulsions | - Hydrophobic drug (O/W) - Hydrophilic drug (W/O) | - Monolayer | - 50 nm to 500 nm | - Nanoemulsions are kinetically stable liquid-in-liquid dispersions with droplet sizes which has high surface area. | [24] | |
| Solid lipid nanoparticles | - Solid lipid core-drug matrix | - Monolayer | - 50 nm to 1000 nm | - Solid lipid core instead of liquid oils may provide stability and controlled drug release as the mobility of the drug in a solid lipid matrix. | [25] | |
| Polymer-lipid hybrids | - Polymeric core-drug (PLGA, gold, silica, iron oxide and etc.) | - Monolayer - Bilayer | - Polymer core (smaller than typically 300 nm) + bilayer (3 nm to 5 nm) | - Hybrid vesicles have an advanced vesicular structure to integrate best characteristics of liposomes and polymer in a new, single vesicle. | [26] | |
| Biomimetics | Exosomes | - Hydrophilic/hydrophilic drug | - Bilayer | - 40 nm to 100 nm | - Exosomes are small intracellular membrane-based vesicles with desirable features such as a long circulating half-life, the intrinsic ability to target tissue and biocompatibility | [27] |
| Blood cells (RBC, WBC and platelet) | - Polymeric core-drug - Hydrophilic/hydrophilic drug | - Monolayer - Bilayer | - 100 nm (nanovesicles) to 8 µm (whole cells) | - Blood cell-based vesicles have many unique advantages such as long life-span in circulation (especially erythrocytes), target release capacities (especially platelets), and natural adhesive properties (leukocytes and platelets). | [28] | |
| Cancer cells | - Polymeric core-drug - Hydrophilic/hydrophilic drug | - Monolayer - Bilayer | - Polymer core (smaller than typically 300 nm) + bilayer (3 nm to 5 nm) | - Cancer cell-derived vesicles carry the full array of cancer cell membrane antigens, and thus offer the inherent homotypic binding phenomenon frequently observed among tumor cells. | [29] | |
| Ligand | Targeted drug delivery | Reference | |
|---|---|---|---|
| Advantages | Disadvantages | ||
| Antibody | - Barely affected by nucleases in vivo - Different antibodies to bind to the same antigen which has multiple epitopes - High affinity to the target | - High immunogenicity - Easily loss of activity - Susceptible to high temperature and pH changes - Animal-based production | [73] |
| Aptamer | - Chemical synthesis - Smaller than antibodies (about ten times) - Low immunogenicity - Easy modification | - Susceptible to excess nucleases in a biological system - Low affinity compared to antibody - Limited diversity of possible secondary and tertiary structures | [74] |
| Peptide | - Peptide libraries for almost any desired target - Extremely high receptor affinity - Only around 10 amino acids - High stability | - More expensive and time-consuming - Limited production of peptides with a predetermined length - More potentially active sequences could be missed | [75] |
| Vehicle | Advantages | Disadvantages | Reference |
|---|---|---|---|
| Liposomes | - Various sizes with either single or multiple lipid bilayer - Diversity of lipid compositions - Mass production - Active clinical research | - Potential complement activation and low cellular uptake - Insufficient drug loading - Premature drug release | [107] [108] |
| Extracellular Vesicles | - Structurally similarity comparable to other membranous structures found in cells - Membrane-associated proteins, receptors, adhesion molecules, as well as a natural corona - Natural characteristics to deliver molecules to target cells - Non-immunogenic | - Limited clinical trials because of uncertainties regarding EVs - Necessity to establish methods for stable production and bulk preparation |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, M.W.; Kwon, S.-H.; Choi, J.H.; Lee, A. A Promising Biocompatible Platform: Lipid-Based and Bio-Inspired Smart Drug Delivery Systems for Cancer Therapy. Int. J. Mol. Sci. 2018, 19, 3859. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms19123859
Kim MW, Kwon S-H, Choi JH, Lee A. A Promising Biocompatible Platform: Lipid-Based and Bio-Inspired Smart Drug Delivery Systems for Cancer Therapy. International Journal of Molecular Sciences. 2018; 19(12):3859. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms19123859
Chicago/Turabian StyleKim, Min Woo, Seung-Hae Kwon, Jung Hoon Choi, and Aeju Lee. 2018. "A Promising Biocompatible Platform: Lipid-Based and Bio-Inspired Smart Drug Delivery Systems for Cancer Therapy" International Journal of Molecular Sciences 19, no. 12: 3859. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms19123859