Melatonin Improves Parthenogenetic Development of Vitrified–Warmed Mouse Oocytes Potentially by Promoting G1/S Cell Cycle Progression


 ,
, 
Abstract
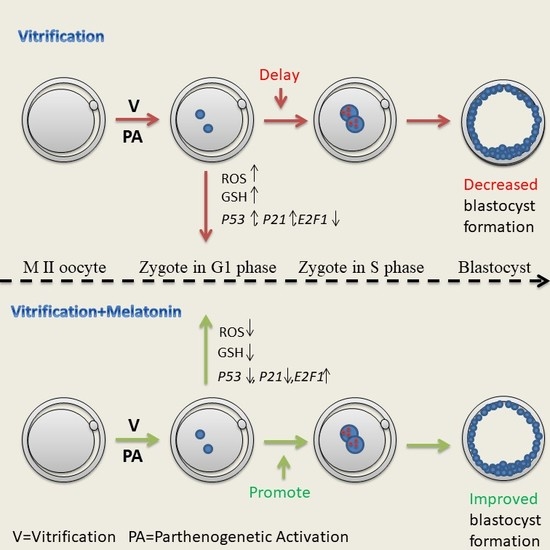
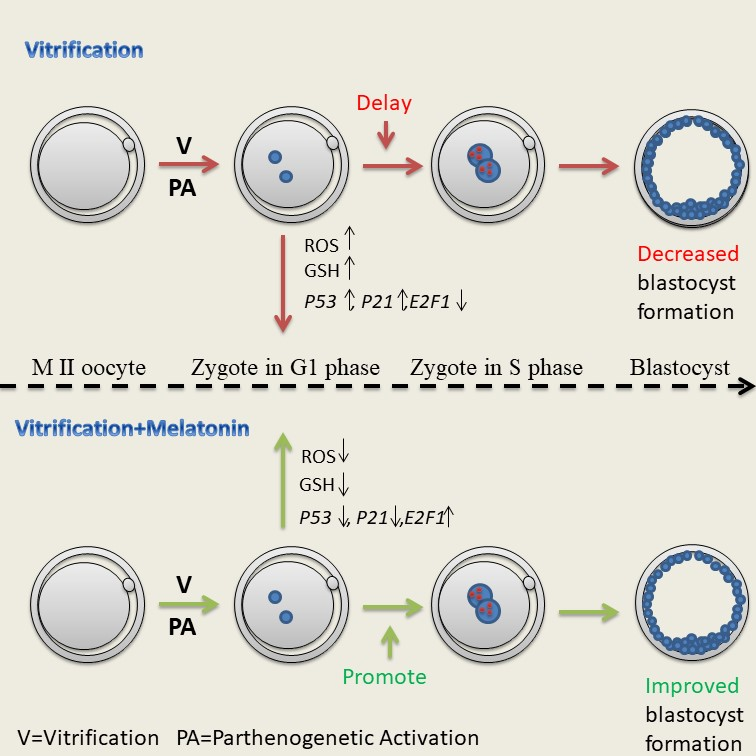
:
1. Introduction
2. Results
2.1. Melatonin Promotes the G1/S Transition of Parthenogenetic Zygotes Derived from Vitrified Mouse Oocytes
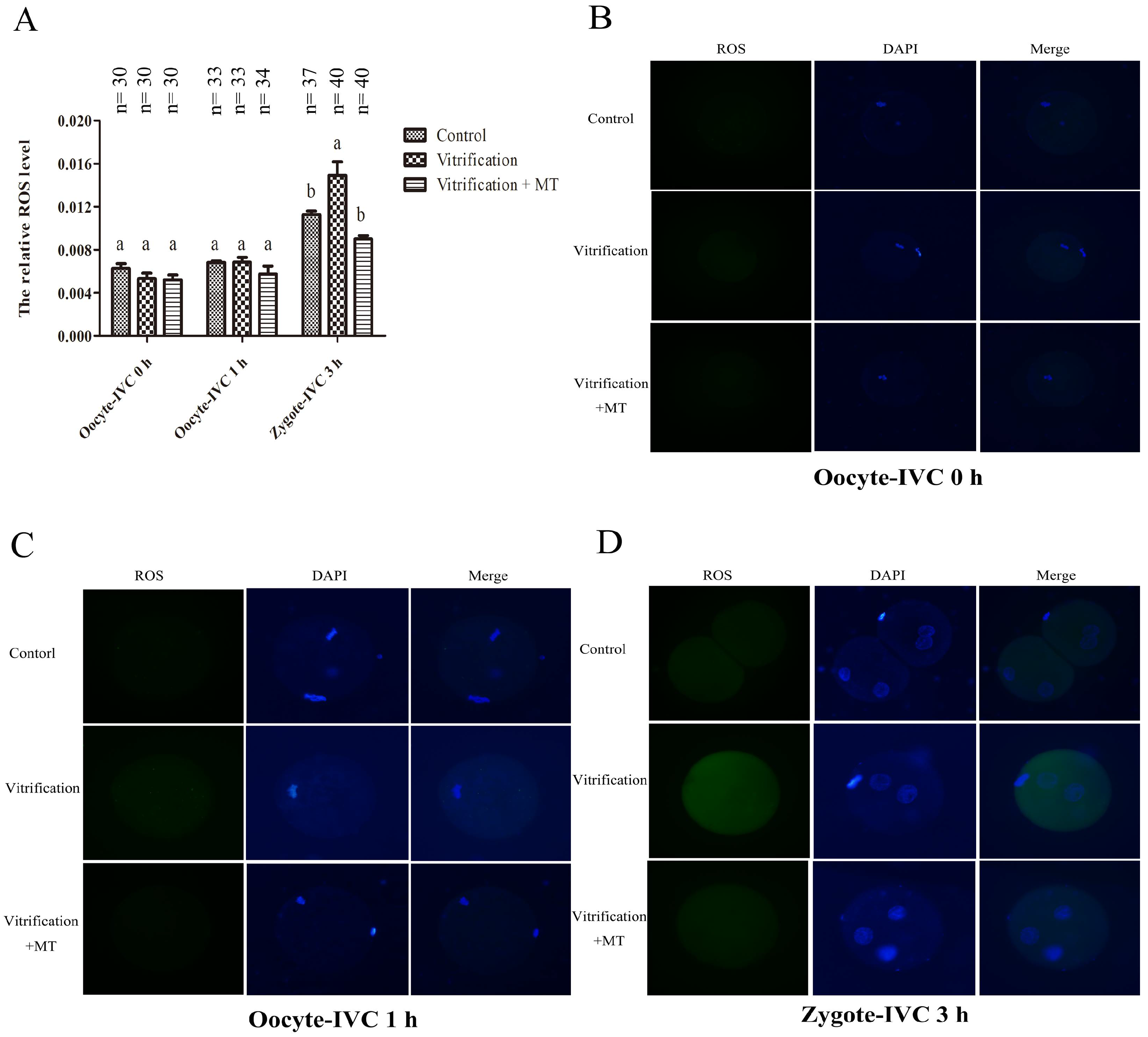
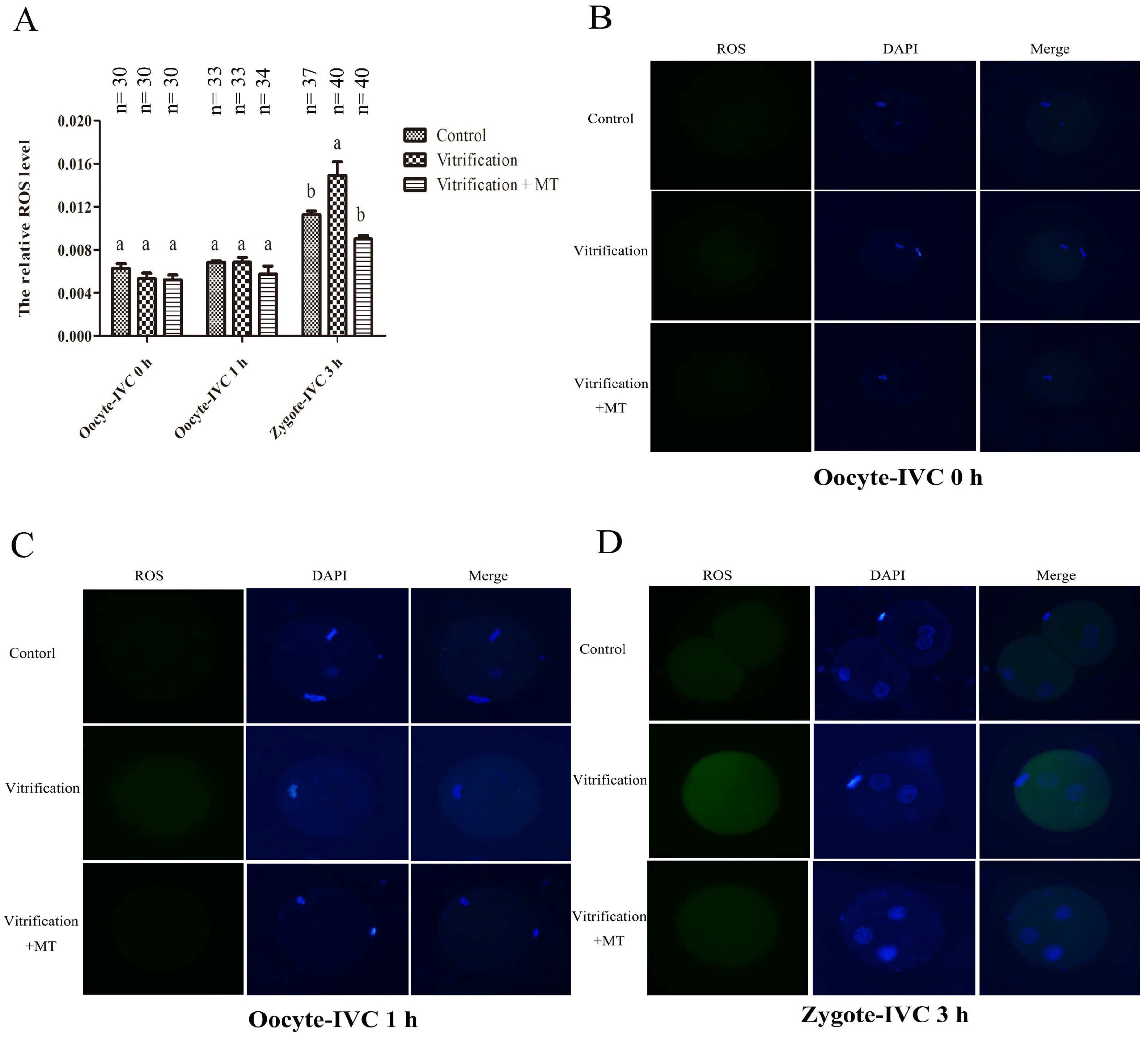
2.2. Melatonin Decreased ROS Levels in Parthenogenetic Zygotes from Vitrified Oocytes
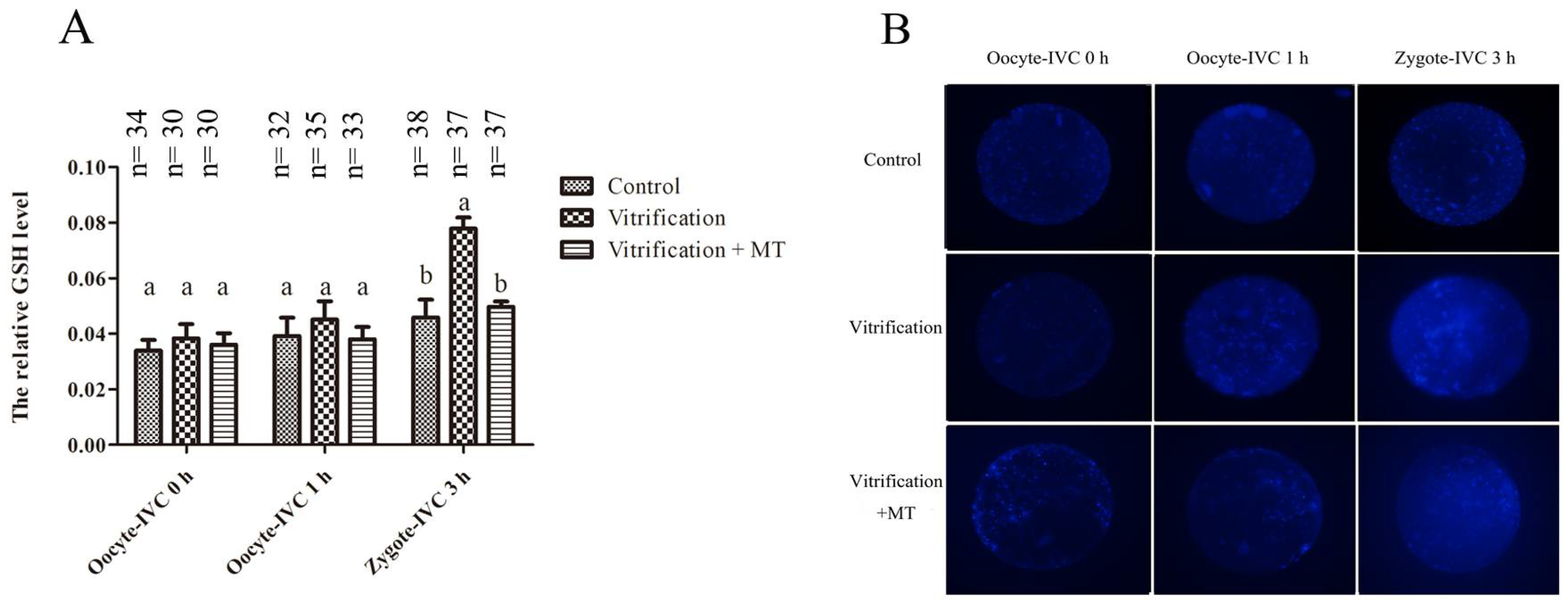
2.3. Melatonin Decreased GSH Levels in Parthenogenetic Zygotes from Vitrified Oocytes
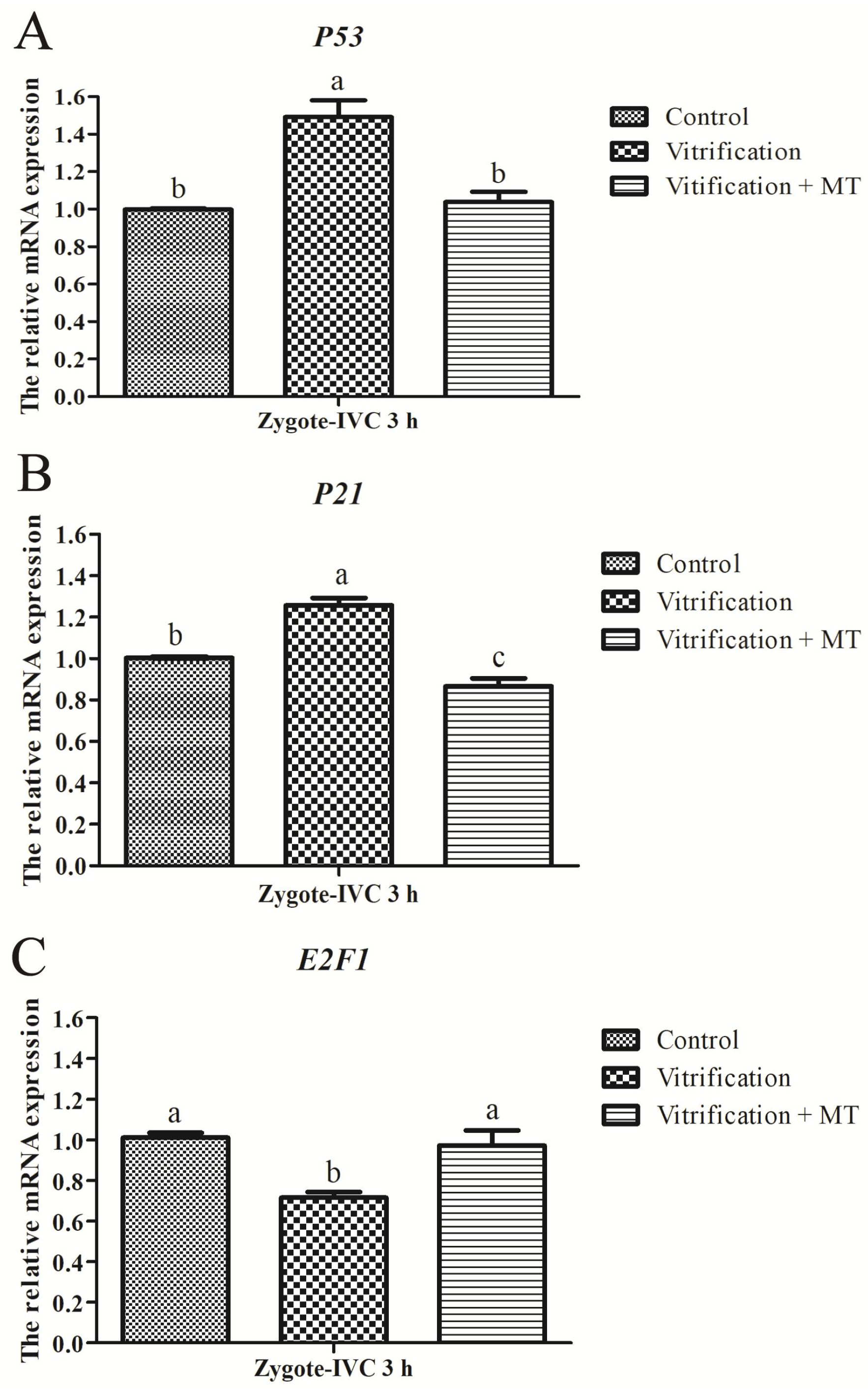
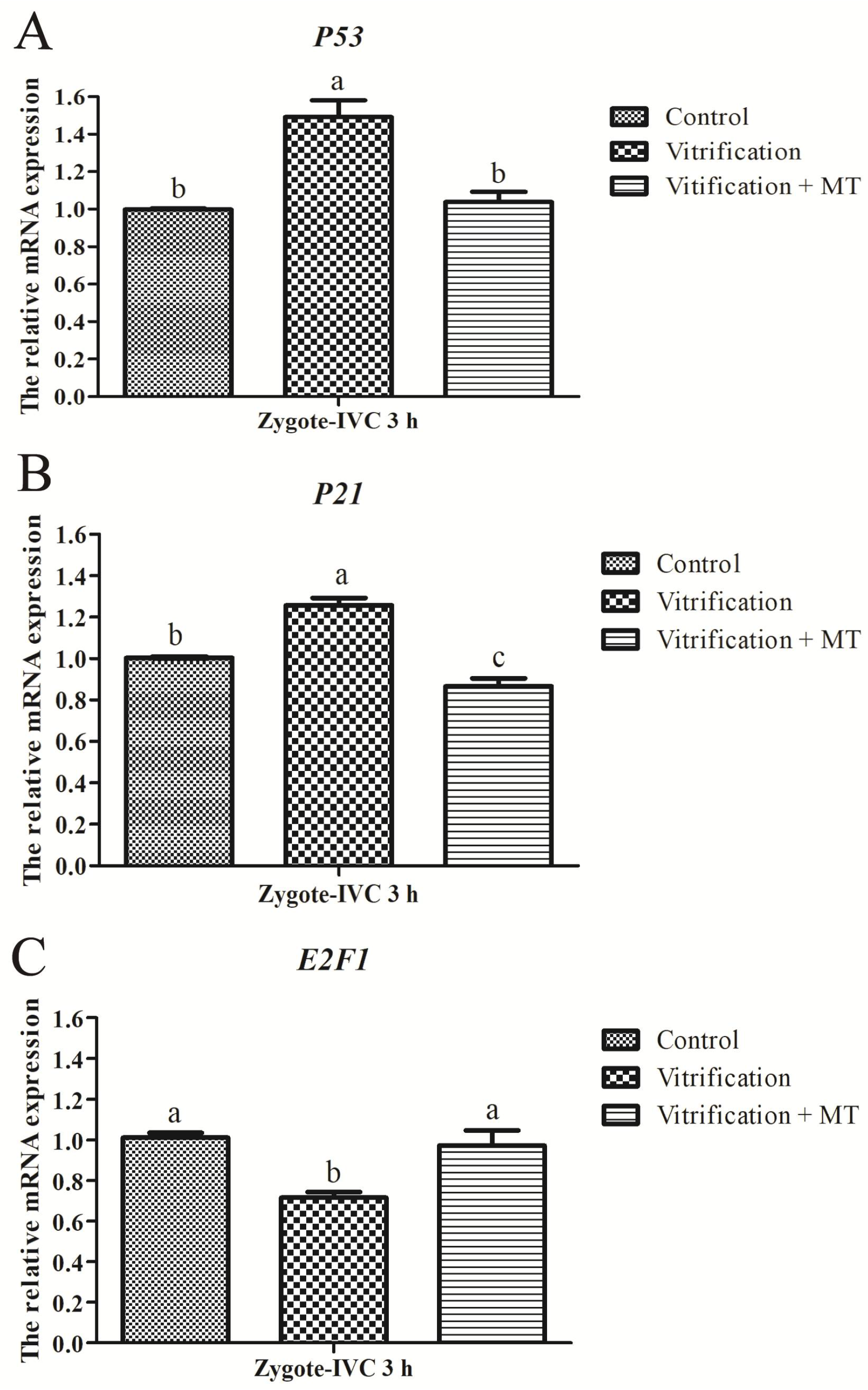
2.4. Melatonin Altered mRNA Expression of G1 Checkpoint Related Genes in Parthenogenetic Zygotes from Vitrified Oocytes
2.5. Melatonin Improved Parthenogenetic Development of Vitrified-Warmed Mouse Oocytes
3. Discussion
4. Materials and Methods
4.1. Oocyte Collection
4.2. Oocyte Vitrification and Warming
4.3. Oocyte Parthenogenetic Activation and Embryo Culture
4.4. Detection of Cell Cycle Progression
4.5. Measurement of Intracellular ROS and GSH
4.6. Quantitative Polymerase Chain Reaction (Q-PCR)
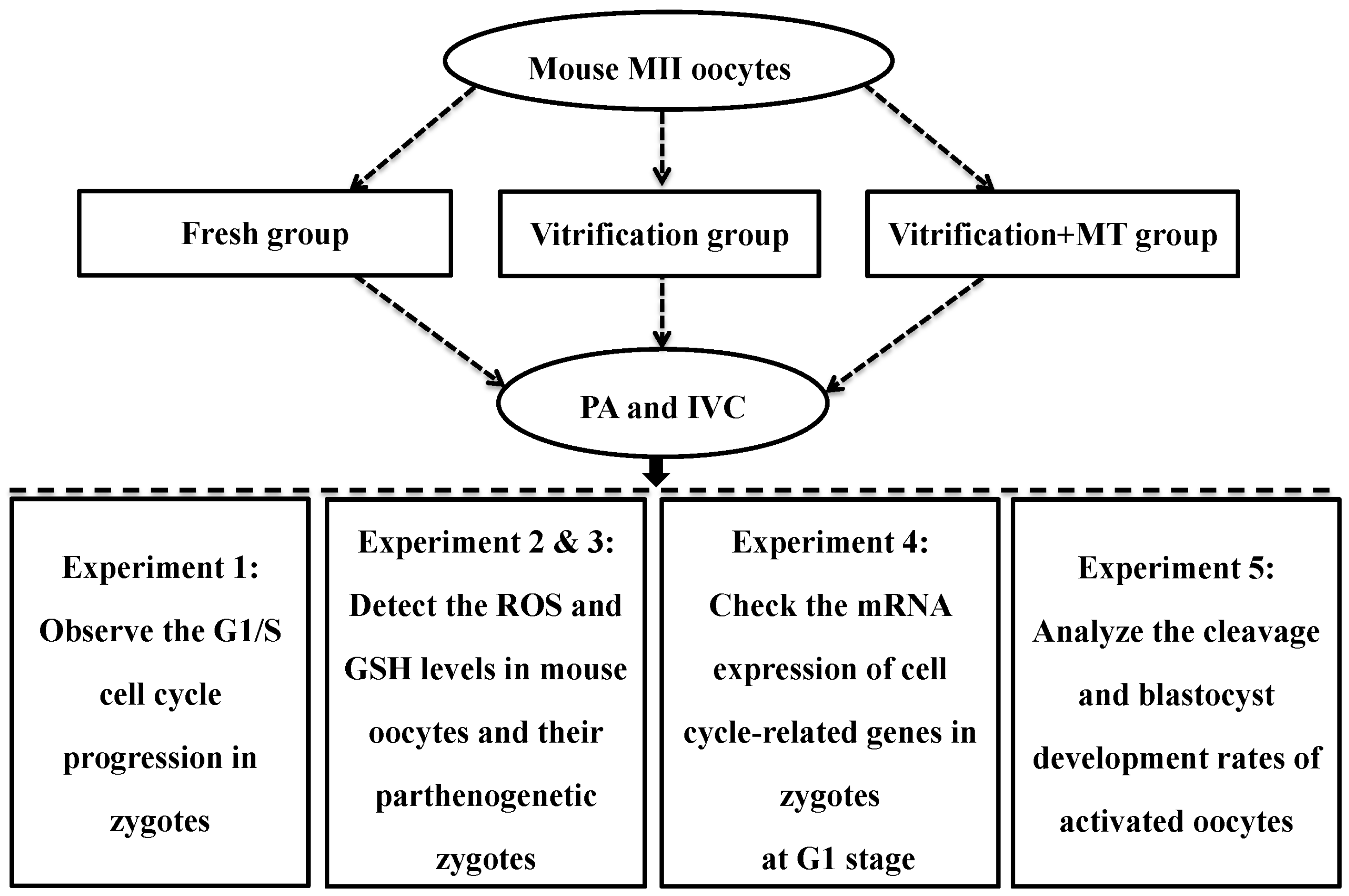
4.7. Experimental Design
4.8. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| DMSO | dimethyl sulfoxide |
| DPBS | dulbecco’s phosphate buffered saline |
| DAPI | 4′,6-diamidino-2-phenylindole |
| EG | ethylene glycol |
| FBS | fetal bovine serum |
| GAPDH | glyceraldehyde 3-phosphate dehydrogenase |
| GSH | glutathione |
| IVC | in vitro culture |
| IVM | in vitro maturation |
| LDs | lipid droplets |
| mtDNA | mitochondrial DNA |
| MII | metaphase II |
| MT | melatonin |
| OPS | open-pulled straws |
| PA | parthenogenetic activation |
| Q-PCR | quantitative polymerase chain reaction |
| ROS | reactive oxygen species |
| ZP | zona pellucida |
References
- Fabbri, R.; Porcu, E.; Marsella, T.; Rocchetta, G.; Venturoli, S.; Flamigni, C. Human oocyte cryopreservation: New perspectives regarding oocyte survival. Hum. Reprod. 2001, 16, 411–416. [Google Scholar] [CrossRef] [PubMed]
- Niemann, H. Cryopreservation of ova and embryos from livestock: Current status and research needs. Theriogenology 1991, 35, 109–124. [Google Scholar] [CrossRef]
- Kuwayama, M. Highly efficient vitrification for cryopreservation of human oocytes and embryos: The Cryotop method. Theriogenology 2007, 67, 73–80. [Google Scholar] [CrossRef] [PubMed]
- Singh, B.; Mal, G.; Singla, S.K. Chapter 18 Vitrification: A Reliable Method for Cryopreservation of Animal Embryos. Methods Mol. Biol. 2017, 1568, 243–249. [Google Scholar] [CrossRef] [PubMed]
- Porcu, E.; Venturoli, S.; Damiano, G.; Ciotti, P.M.; Notarangelo, L.; Paradisi, R.; Moscarini, M.; Ambrosini, G. Healthy twins delivered after oocyte cryopreservation and bilateral ovariectomy for ovarian cancer. Reprod. Biomed. Online 2008, 17, 265–267. [Google Scholar] [CrossRef]
- Oktay, K.; Bedoschi, G. Oocyte cryopreservation for fertility preservation in postpubertal female children at risk for premature ovarian failure due to accelerated follicle loss in turner syndrome or cancer treatments. J. Pediatr. Adolesc. Gynecol. 2014, 27, 342–346. [Google Scholar] [CrossRef] [PubMed]
- Kotera, T.; Shibata, T.; Kato, H.; Watanabe, H.; Nakago, S. Twin pregnancy in a 51-year-old woman who underwent autologous cryopreservation at the age of 36 years: Case report. Reprod. Med. Biol. 2016, 15, 187–189. [Google Scholar] [CrossRef]
- Turner, S.R.; Senaratna, T.; Bunn, E.; Tan, B.; Dixon, K.W.; Touchell, D.H. Cryopreservation of Shoot Tips from Six Endangered Australian Species using a Modified Vitrification Protocol. Ann. Bot. 2001, 87, 371–378. [Google Scholar] [CrossRef] [Green Version]
- Taylor-Robinson, A.W.; Walton, S.; Swain, D.L.; Walsh, K.B.; Vajta, G. The potential for modification in cloning and vitrification technology to enhance genetic progress in beef cattle in Northern Australia. Anim. Reprod. Sci. 2014, 148, 91–96. [Google Scholar] [CrossRef]
- Matsas, D.; Huntress, V.; Levine, H.; Saperstein, G.; Overstrom, E.W. 108 cryopreservation of germplasm from heritage breeds of domestic livestock: Seasonal effects on superovulation response and embryo production. Reprod. Fertil. Dev. 2003, 16, 176. [Google Scholar] [CrossRef]
- Punyawai, K.; Anakkul, N.; Srirattana, K.; Aikawa, Y.; Sangsritavong, S.; Nagai, T.; Imai, K.; Parnpai, R. Comparison of Cryotop and micro volume air cooling methods for cryopreservation of bovine matured oocytes and blastocysts. J. Reprod. Dev. 2015, 61, 431–437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Somfai, T.; Yoshioka, K.; Tanihara, F.; Kaneko, H.; Noguchi, J.; Kashiwazaki, N.; Nagai, T.; Kikuchi, K. Generation of live piglets from cryopreserved oocytes for the first time using a defined system for in vitro embryo production. PLoS ONE 2014, 9, e97731. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Li, W.; Ma, Y.; Wang, D.; Zhao, X.; Zeng, C.; Zhang, M.; Zeng, X.; Meng, Q.; Zhou, G. Improved development by melatonin treatment after vitrification of mouse metaphase II oocytes. Cryobiology 2016, 73, 335–342. [Google Scholar] [CrossRef] [PubMed]
- Ren, L.; Fu, B.; Ma, H.; Liu, D. Effects of mechanical delipation in porcine oocytes on mitochondrial distribution, ROS activity and viability after vitrification. Cryo. Lett. 2015, 36, 30–36. [Google Scholar]
- Cao, X.; Li, J.; Xue, H.; Wang, S.; Zhao, W.; Du, Z.; Yang, Y.; Yue, Z. Effect of vitrification on meiotic maturation, mitochondrial distribution and glutathione synthesis in immature silver fox cumulus oocyte complexes. Theriogenology 2017, 91, 104–111. [Google Scholar] [CrossRef] [PubMed]
- Monzo, C.; Haouzi, D.; Roman, K.; Assou, S.; Dechaud, H.; Hamamah, S. Slow freezing and vitrification differentially modify the gene expression profile of human metaphase II oocytes. Hum. Reprod. 2012, 27, 2160–2168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, J.; Ma, Y.; Wei, S.; Pan, B.; Qi, Y.; Hou, Y.; Meng, Q.; Zhou, G.; Han, H. Dynamic changes in the global transcriptome of bovine germinal vesicle oocytes after vitrification followed by in vitro maturation. Reprod. Fertil. Dev. 2018. [Google Scholar] [CrossRef]
- Ma, Y.; Pan, B.; Yang, H.; Qazi, I.H.; Wu, Z.; Zeng, C.; Zhang, M.; Meng, Q.; Zhou, G. Expression of CD9 and CD81 in bovine germinal vesicle oocytes after vitrification followed by in vitro maturation. Cryobiology 2018, 81, 206–209. [Google Scholar] [CrossRef]
- Forman, H.J.; Maiorino, M.; Ursini, F. Signaling functions of reactive oxygen species. Biochemistry 2010, 49, 835–842. [Google Scholar] [CrossRef]
- Paulsen, C.E.; Carroll, K.S. Orchestrating Redox Signaling Networks through Regulatory Cysteine Switches. ACS Chem. Biol. 2010, 5, 47–62. [Google Scholar] [CrossRef] [Green Version]
- Cabiscol, E.; Tamarit, J.; Ros, J. Oxidative stress in bacteria and protein damage by reactive oxygen species. Int. Microbiol. 2000, 3, 3–8. [Google Scholar] [PubMed]
- Lipinski, B. Hydroxyl radical and its scavengers in health and disease. Oxid. Med. Cell Longev. 2011, 2011, 809696. [Google Scholar] [CrossRef] [PubMed]
- Potts, R.J.; Notarianni, L.J.; Jefferies, T.M. Seminal plasma reduces exogenous oxidative damage to human sperm, determined by the measurement of DNA strand breaks and lipid peroxidation. Mutat. Res. 2000, 447, 249–256. [Google Scholar] [CrossRef]
- Munday, R.; Winterbourn, C.C. Reduced glutathione in combination with superoxide dismutase as an important biological antioxidant defence mechanism. Biochem. Pharmacol. 1989, 38, 4349–4352. [Google Scholar] [CrossRef]
- Handy, D.E.; Loscalzo, J. Redox regulation of mitochondrial function. Antioxid. Redox. Signal. 2012, 16, 1323–1367. [Google Scholar] [CrossRef] [PubMed]
- Fang, C.; Wei, X.; Zhang, Z.; Xianmei, L.I.; Zhang, X.; University, G. Effects of Vitrified Cryopreservation on GSH Content and Mitochondrial ATPase Activity in Oocytes of Zebrafish Danio rerio. Fish. Sci. 2017, 36, 773–777. [Google Scholar]
- Somfai, T.; Ozawa, M.; Noguchi, J.; Kaneko, H.; Kuriani Karja, N.W.; Farhudin, M.; Dinnyés, A.; Nagai, T.; Kikuchi, K. Developmental competence of in vitro-fertilized porcine oocytes after in vitro maturation and solid surface vitrification: Effect of cryopreservation on oocyte antioxidative system and cell cycle stage. Cryobiology 2007, 55, 115–126. [Google Scholar] [CrossRef]
- Nohales-Corcoles, M.; Sevillano-Almerich, G.; Di Emidio, G.; Tatone, C.; Cobo, A.C.; Dumollard, R.; De Los Santos Molina, M.J. Impact of vitrification on the mitochondrial activity and redox homeostasis of human oocyte. Hum. Reprod. 2016, 31, 1850–1858. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, M. Oxidative stress and redox regulation on in vitro development of mammalian embryos. J. Reprod. Dev. 2012, 58, 1–9. [Google Scholar] [CrossRef]
- Zhang, C.; Liu, C.; Li, D.; Yao, N.; Yuan, X.; Yu, A.; Lu, C.; Ma, X. Intracellular redox imbalance and extracellular amino acid metabolic abnormality contribute to arsenic-induced developmental retardation in mouse preimplantation embryos. J. Cell Physiol. 2010, 222, 444–455. [Google Scholar] [CrossRef]
- Spricigo, J.F.; Morato, R.; Arcarons, N.; Yeste, M.; Dode, M.A.; Lopez-Bejar, M.; Mogas, T. Assessment of the effect of adding L-carnitine and/or resveratrol to maturation medium before vitrification on in vitro-matured calf oocytes. Theriogenology 2017, 89, 47–57. [Google Scholar] [CrossRef]
- Castillo-Martin, M.; Yeste, M.; Soler, A.; Morato, R.; Bonet, S. Addition of L-ascorbic acid to culture and vitrification media of IVF porcine blastocysts improves survival and reduces HSPA1A levels of vitrified embryos. Reprod. Fertil. Dev. 2015, 27, 1115–1123. [Google Scholar] [CrossRef] [PubMed]
- Boonkusol, D.; Gal, A.B.; Bodo, S.; Gorhony, B.; Kitiyanant, Y.; Dinnyes, A. Gene expression profiles and in vitro development following vitrification of pronuclear and 8-cell stage mouse embryos. Mol. Reprod. Dev. 2006, 73, 700–708. [Google Scholar] [CrossRef]
- Majidi Gharenaz, N.; Movahedin, M.; Mazaheri, Z. Effects of Re-Vitrification of Mouse Morula and Early Blastocyst Stages on Apoptotic Gene Expression and Developmental Potential. Cell J. 2018, 19, 614–619. [Google Scholar] [CrossRef] [PubMed]
- Zhou, G.; Zeng, Y.; Guo, J.; Meng, Q.; Meng, Q.; Jia, G.; Cheng, K.; Zeng, C.; Zhang, M.; Liu, G.; et al. Vitrification transiently alters Oct-4, Bcl2 and P53 expression in mouse morulae but does not affect embryo development in vitro. Cryobiology 2016, 73, 120–125. [Google Scholar] [CrossRef]
- Miyauchi, H.; Minamino, T.; Tateno, K.; Kunieda, T.; Toko, H.; Komuro, I. Akt negatively regulates the in vitro lifespan of human endothelial cells via a p53/p21-dependent pathway. EMBO J. 2004, 23, 212–220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mikule, K.; Delaval, B.; Kaldis, P.; Jurcyzk, A.; Hergert, P.; Doxsey, S. Loss of centrosome integrity induces p38-p53-p21-dependent G1-S arrest. Nat. Cell Biol. 2007, 9, 160–170. [Google Scholar] [CrossRef]
- Waldman, T.; Kinzler, K.W.; Vogelstein, B. p21 Is Necessary for the p53-mediated G1 Arrest in Human Cancer Cells. Cancer Res. 1995, 55, 5187–5190. [Google Scholar] [PubMed]
- Kagawa, S.; Fujiwara, T.; Hizuta, A.; Yasuda, T.; Zhang, W.W.; Roth, J.A.; Tanaka, N. p53 expression overcomes p21WAF1/CIP1-mediated G1 arrest and induces apoptosis in human cancer cells. Oncogene 1997, 15, 1903–1909. [Google Scholar] [CrossRef]
- DeGregori, J.; Kowalik, T.; Nevins, J.R. Cellular targets for activation by the E2F1 transcription factor include DNA synthesis- and G1/S-regulatory genes. Mol. Cell. Biol. 1995, 15, 4215–4224. [Google Scholar] [CrossRef]
- Madan, E.; Gogna, R.; Kuppusamy, P.; Bhatt, M.; Pati, U.; Mahdi, A.A. TIGAR induces p53-mediated cell-cycle arrest by regulation of RB-E2F1 complex. Br. J. Cancer 2012, 107, 516–526. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Dai, L.; Zhang, B.; Xu, X.; Shi, J.; Fu, L.; Chen, X.; Li, J.; Bai, Y. miR-203 is a direct transcriptional target of E2F1 and causes G1 arrest in esophageal cancer cells. J. Cell. Physiol. 2015, 230, 903–910. [Google Scholar] [CrossRef] [PubMed]
- Berlinguer, F.; Leoni, G.G.; Succu, S.; Spezzigu, A.; Madeddu, M.; Satta, V.; Bebbere, D.; Contreras-Solis, I.; Gonzalez-Bulnes, A.; Naitana, S. Exogenous melatonin positively influences follicular dynamics, oocyte developmental competence and blastocyst output in a goat model. J. Pineal Res. 2009, 46, 383–391. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Liu, B.; Liu, W.; Xiao, Y.; Zhang, H.; Yang, L. The effects of melatonin on bovine uniparental embryos development in vitro and the hormone secretion of COCs. PeerJ 2017, 5, e3485. [Google Scholar] [CrossRef] [PubMed]
- Soto-Heras, S.; Roura, M.; Catala, M.G.; Menendez-Blanco, I.; Izquierdo, D.; Fouladi-Nashta, A.A.; Paramio, M.T. Beneficial effects of melatonin on in vitro embryo production from juvenile goat oocytes. Reprod. Fertil. Dev. 2018, 30, 253–261. [Google Scholar] [CrossRef] [PubMed]
- Mediavilla, M.D.; Sanchez-Barcelo, E.J.; Tan, D.X.; Manchester, L.; Reiter, R.J. Basic mechanisms involved in the anti-cancer effects of melatonin. Curr. Med. Chem. 2010, 17, 4462–4481. [Google Scholar] [CrossRef] [PubMed]
- Mandawala, A.; Harvey, S.; Roy, T.; Fowler, K. Cryopreservation of animal oocytes and embryos: Current progress and future prospects. Theriogenology 2016, 86, 1637–1644. [Google Scholar] [CrossRef] [Green Version]
- Vieira, A.D.; Mezzalira, A.; Barbieri, D.P.; Lehmkuhl, R.C.; Rubin, M.I.; Vajta, G. Calves born after open pulled straw vitrification of immature bovine oocytes. Cryobiology 2002, 45, 91–94. [Google Scholar] [CrossRef]
- Fujiwara, K.; Kamoshita, M.; Kato, T.; Ito, J.; Kashiwazaki, N. Generation of rats from vitrified oocytes with surrounding cumulus cells via in vitro fertilization with cryopreserved sperm. Anim. Sci. J. 2017, 88, 180–184. [Google Scholar] [CrossRef]
- Jimenez-Trigos, E.; Vicente, J.S.; Marco-Jimenez, F. First pregnancy and live birth from vitrified rabbit oocytes after intraoviductal transfer and in vivo fertilization. Theriogenology 2014, 82, 599–604. [Google Scholar] [CrossRef]
- Kim, M.K.; Lee, D.R.; Han, J.E.; Kim, Y.S.; Lee, W.S.; Won, H.J.; Kim, J.W.; Yoon, T.K. Live birth with vitrified-warmed oocytes of a chronic myeloid leukemia patient nine years after allogenic bone marrow transplantation. J. Assist. Reprod. Genet. 2011, 28, 1167–1170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kono, T.; Kwon, O.Y.; Nakahara, T. Development of vitrified mouse oocytes after in vitro fertilization. Cryobiology 1991, 28, 50–54. [Google Scholar] [CrossRef]
- Hou, Y.P.; Dai, Y.P.; Zhu, S.E.; Zhu, H.B.; Wu, T.Y.; Gong, G.C.; Wang, H.P.; Wang, L.L.; Liu, Y.; Li, R.; et al. Bovine oocytes vitrified by the open pulled straw method and used for somatic cell cloning supported development to term. Theriogenology 2005, 64, 1381–1391. [Google Scholar] [CrossRef] [PubMed]
- Yang, B.C.; Im, G.S.; Kim, D.H.; Yang, B.S.; Oh, H.J.; Park, H.S.; Seong, H.H.; Kim, S.W.; Ka, H.H.; Lee, C.K. Development of vitrified-thawed bovine oocytes after in vitro fertilization and somatic cell nuclear transfer. Anim. Reprod. Sci. 2008, 103, 25–37. [Google Scholar] [CrossRef] [PubMed]
- Lei, T.; Guo, N.; Liu, J.Q.; Tan, M.H.; Li, Y.F. Vitrification of in vitro matured oocytes: Effects on meiotic spindle configuration and mitochondrial function. Int. J. Clin. Exp. Pathol. 2014, 7, 1159–1165. [Google Scholar] [PubMed]
- Martinez-Burgos, M.; Herrero, L.; Megias, D.; Salvanes, R.; Montoya, M.C.; Cobo, A.C.; Garcia-Velasco, J.A. Vitrification versus slow freezing of oocytes: Effects on morphologic appearance, meiotic spindle configuration, and DNA damage. Fertil. Steril. 2011, 95, 374–377. [Google Scholar] [CrossRef] [PubMed]
- Dominguez, F.; Castello, D.; Remohi, J.; Simon, C.; Cobo, A. Effect of vitrification on human oocytes: A metabolic profiling study. Fertil. Steril. 2013, 99, 565–572. [Google Scholar] [CrossRef]
- Ohnishi, N.; Kodania, H.; Ando, S.; Komamine, A. Synthesis of protein and mRNA is necessary for transition of suspension-cultured Catharanthus roseus cells from the G1 to the S phase of the cell cycle. Physiol. Plant. 1990, 80, 95–101. [Google Scholar] [CrossRef]
- Deckbar, D.; Stiff, T.; Koch, B.; Reis, C.; Löbrich, M.; Jeggo, P.A. The limitations of the G1-S checkpoint. Cancer Res. 2010, 70, 4412–4421. [Google Scholar] [CrossRef]
- Khan, F.; Khan, I.; Farooqui, A.; Ansari, I.A. Carvacrol Induces Reactive Oxygen Species (ROS)-mediated Apoptosis Along with Cell Cycle Arrest at G0/G1 in Human Prostate Cancer Cells. Nutr. Cancer 2017, 69, 1075–1087. [Google Scholar] [CrossRef]
- Chung, Y.M.; Sun, M.C.; Kim, Y.H.; Kim, J.S.; Yoo, Y.D. A novel gene, Chemp-1, produces ROS and induces cell-cycle arrest at G1. Cancer Res. 2004, 64. [Google Scholar]
- Cos, S.; Blask, D.E.; Lemus-Wilson, A.; Hill, A.B. Effects of melatonin on the cell cycle kinetics and “estrogen-rescue” of MCF-7 human breast cancer cells in culture. J. Pineal Res. 1991, 10, 36–42. [Google Scholar] [CrossRef] [PubMed]
- Ookawa, K.; Tsuchida, S.; Kohno, T.; Yokota, J. Alterations in expression of E2F-1 and E2F-responsive genes by RB, p53 and p21(Sid1/WAF1/Cip1) expression. FEBS Lett. 2001, 500, 25–30. [Google Scholar] [CrossRef]
- Radhakrishnan, S.K.; Feliciano, C.S.; Najmabadi, F.; Haegebarth, A.; Kandel, E.S.; Tyner, A.L.; Gartel, A.L. Constitutive expression of E2F-1 leads to p21-dependent cell cycle arrest in S|[thinsp]|phase of the cell cycle. Oncogene 2004, 23, 4173–4176. [Google Scholar] [CrossRef] [PubMed]
- Qin, X.Q.; Livingston, D.M.; Ewen, M.; Sellers, W.R.; Arany, Z.; Kaelin, W.G., Jr. The transcription factor E2F-1 is a downstream target of RB action. Mol. Cell. Biol. 1995, 15, 742–755. [Google Scholar] [CrossRef] [PubMed]
- Amoushahi, M.; Salehnia, M.; Mowla, S.J. Vitrification of Mouse MII Oocyte Decreases the Mitochondrial DNA Copy Number, TFAM Gene Expression and Mitochondrial Enzyme Activity. J. Reprod. Infertil. 2017, 18, 343–351. [Google Scholar] [PubMed]
- Voehringer, D.W. BCL-2 and glutathione: Alterations in cellular redox state that regulate apoptosis sensitivity. Free Radic. Biol. Med. 1999, 27, 945–950. [Google Scholar] [CrossRef]
- Jiang, S.; Moriarty-Craige, S.E.; Orr, M.; Cai, J.; Sternberg, P., Jr.; Jones, D.P. Oxidant-induced apoptosis in human retinal pigment epithelial cells: Dependence on extracellular redox state. Investig. Ophthalmol. Vis. Sci. 2005, 46, 1054–1061. [Google Scholar] [CrossRef]
- Lee, E.K.; Kim, J.A.; Kim, J.S.; Park, S.J.; Heo, K.; Yang, K.M.; Son, T.G. Activation of de novo GSH synthesis pathway in mouse spleen after long term low-dose gamma-ray irradiation. Free Radic. Res. 2013, 47, 89–94. [Google Scholar] [CrossRef]
- He, B.; Yin, C.; Gong, Y.; Liu, J.; Guo, H.; Zhao, R. Melatonin-induced increase of lipid droplets accumulation and in vitro maturation in porcine oocytes is mediated by mitochondrial quiescence. J. Cell. Physiol. 2018, 233, 302–312. [Google Scholar] [CrossRef]
- Fair, T.; Hulshof, S.C.; Hyttel, P.; Greve, T.; Boland, M. Oocyte ultrastructure in bovine primordial to early tertiary follicles. Anat. Embryol. 1997, 195, 327–336. [Google Scholar] [CrossRef] [PubMed]
- Favetta, L.A.; St John, E.J.; King, W.A.; Betts, D.H. High levels of p66shc and intracellular ROS in permanently arrested early embryos. Free Radic. Biol. Med. 2007, 42, 1201–1210. [Google Scholar] [CrossRef] [PubMed]
- Gao, C.; Han, H.B.; Tian, X.Z.; Tan, D.X.; Wang, L.; Zhou, G.B.; Zhu, S.E.; Liu, G.S. Melatonin promotes embryonic development and reduces reactive oxygen species in vitrified mouse 2-cell embryos. J. Pineal Res. 2012, 52, 305–311. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; Hickman, J.H.; Wang, S.J.; Gu, W. Dynamic roles of p53-mediated metabolic activities in ROS-induced stress responses. Cell Cycle 2015, 14, 2881–2885. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Whittingham, D.G. Culture of mouse ova. J. Reprod. Fertil. Suppl. 1971, 14, 7–21. [Google Scholar] [PubMed]
- Vajta, G.; Holm, P.; Kuwayama, M.; Booth, P.J.; Jacobsen, H.; Greve, T.; Callesen, H. Open Pulled Straw (OPS) vitrification: A new way to reduce cryoinjuries of bovine ova and embryos. Mol. Reprod. Dev. 1998, 51, 53–58. [Google Scholar] [CrossRef]
- Yan, C.L.; Fu, X.W.; Zhou, G.B.; Zhao, X.M.; Suo, L.; Zhu, S.E. Mitochondrial behaviors in the vitrified mouse oocyte and its parthenogenetic embryo: Effect of Taxol pretreatment and relationship to competence. Fertil. Steril. 2010, 93, 959–966. [Google Scholar] [CrossRef]
- Quinn, P.; Kerin, J.F.; Warnes, G.M. Improved pregnancy rate in human in vitro fertilization with the use of a medium based on the composition of human tubal fluid. Fertil. Steril. 1985, 44, 493–498. [Google Scholar] [CrossRef]
- Carbone, M.C.; Tatone, C. Alterations in the protein kinase C signaling activated by a parthenogenetic agent in oocytes from reproductively old mice. Mol. Reprod. Dev. 2009, 76, 122–131. [Google Scholar] [CrossRef]
- Ho, Y.; Wigglesworth, K.; Eppig, J.J.; Schultz, R.M. Preimplantation development of mouse embryos in KSOM: Augmentation by amino acids and analysis of gene expression. Mol. Reprod. Dev. 1995, 41, 232–238. [Google Scholar] [CrossRef]
- Xiao, J.Y.; Liu, C.; Sun, X.H.; Yu, B.Z. Cdc25b subcellular localization influences one-cell stage mouse embryos. Chinese J. Biochem. Mol. Biol. 2013, 29, 33–41. [Google Scholar] [CrossRef]
- Wu, G.Q.; Jia, B.Y.; Li, J.J.; Fu, X.W.; Zhou, G.B.; Hou, Y.P.; Zhu, S.E. L-carnitine enhances oocyte maturation and development of parthenogenetic embryos in pigs. Theriogenology 2011, 76, 785–793. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Groups | No. of Oocytes Vitrified | No. of Oocytes Recovered | No. of Oocytes with Normal Morphology (%) | No. of Oocytes Activated | No. of Activated Oocytes Developed to | |
|---|---|---|---|---|---|---|
| Zygotes in G1 Phase (%) | Zygotes in S Phase (%) | |||||
| Control | - | 126 | 126 (100 ± 0) a | 118 | 60 (50.85 ± 18.78) a | 58 (49.15 ± 18.78) a |
| Vitrification | 183 | 171 | 158 (87.68 ± 8.22) b | 155 | 113 (72.91 ± 10.89) b | 42 (27.09 ± 10.89) b |
| Vitrification + MT | 174 | 163 | 153 (89.59 ± 5.71) b | 141 | 84 (59.58 ± 8.74) a | 57 (40.42 ± 8.74) a |
| Groups | No. of Oocytes Activated | No. of Activated Oocytes Developed to | ||||
|---|---|---|---|---|---|---|
| 2-Cell Embryos (%) | 4-Cell Embryos (%) | Morula (%) | Blastocysts (%) | Hatched Blastocysts (%) | ||
| Control | 150 | 141 (94.00 ± 2.55) a | 140 (93.33 ± 0.87) a | 129 (86.00 ± 1.94) a | 100 (66.67 ± 1.32) a | 50 (33.33 ± 15.35) a |
| Vitrfication | 122 | 90 (73.77 ± 11.96) b | 97 (79.51 ± 11.96) b | 80 (65.57 ± 12.32) b | 41 (33.61 ± 6.54) b | 8 (6.56 ± 4.78) c |
| Vitrification + MT | 175 | 160 (91.43 ± 9.62) a | 165 (94.29 ± 10.06) a | 147 (84.00 ± 7.72) a | 100 (57.14 ± 16.17) a | 39 (22.29 ± 3.82) b |
| Gene | Assay ID | Primer seq (5′-3′) | Product Length | Tm (°C) |
|---|---|---|---|---|
| P53 | NM_001127233.1 | F: AGGATTGTGGCCTTCTTTGA | 126 | 62 |
| R: CAGATGCCGGTTCAGGTACT | ||||
| P21 | NM_001111099.2 | F: TGGAGATGAACTGGACAGCA | 84 | 62 |
| R: TGAAGTTGCCATCAGCAAAC | ||||
| E2F1 | NM_001291105.1 | F: CGAGTCCTATGCCTTCAACA | 159 | 62 |
| R: GAGTCCAGCCAGGAGATGAC | ||||
| GAPDH | NM_001289726.1 | F: AGAACATCATCCCTGCATCC | 124 | 62 |
| R: AGATCCACGACGGACACATT |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pan, B.; Yang, H.; Wu, Z.; Qazi, I.H.; Liu, G.; Han, H.; Meng, Q.; Zhou, G. Melatonin Improves Parthenogenetic Development of Vitrified–Warmed Mouse Oocytes Potentially by Promoting G1/S Cell Cycle Progression. Int. J. Mol. Sci. 2018, 19, 4029. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms19124029
Pan B, Yang H, Wu Z, Qazi IH, Liu G, Han H, Meng Q, Zhou G. Melatonin Improves Parthenogenetic Development of Vitrified–Warmed Mouse Oocytes Potentially by Promoting G1/S Cell Cycle Progression. International Journal of Molecular Sciences. 2018; 19(12):4029. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms19124029
Chicago/Turabian StylePan, Bo, Haoxuan Yang, Zhenzheng Wu, Izhar Hyder Qazi, Guoshi Liu, Hongbing Han, Qingyong Meng, and Guangbin Zhou. 2018. "Melatonin Improves Parthenogenetic Development of Vitrified–Warmed Mouse Oocytes Potentially by Promoting G1/S Cell Cycle Progression" International Journal of Molecular Sciences 19, no. 12: 4029. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms19124029