Using Mouse and Drosophila Models to Investigate the Mechanistic Links between Diet, Obesity, Type II Diabetes, and Cancer

 and
and
Abstract
:
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Modeling Interactions between Diet and Cancer in Mice
2.1. Modeling Cancer in Mice
2.2. Investigating Interactions between Diet and Cancer in Mice
3. Investigating Interactions between Cancer, Type II Diabetes Mellitus, and Obesity in Mice
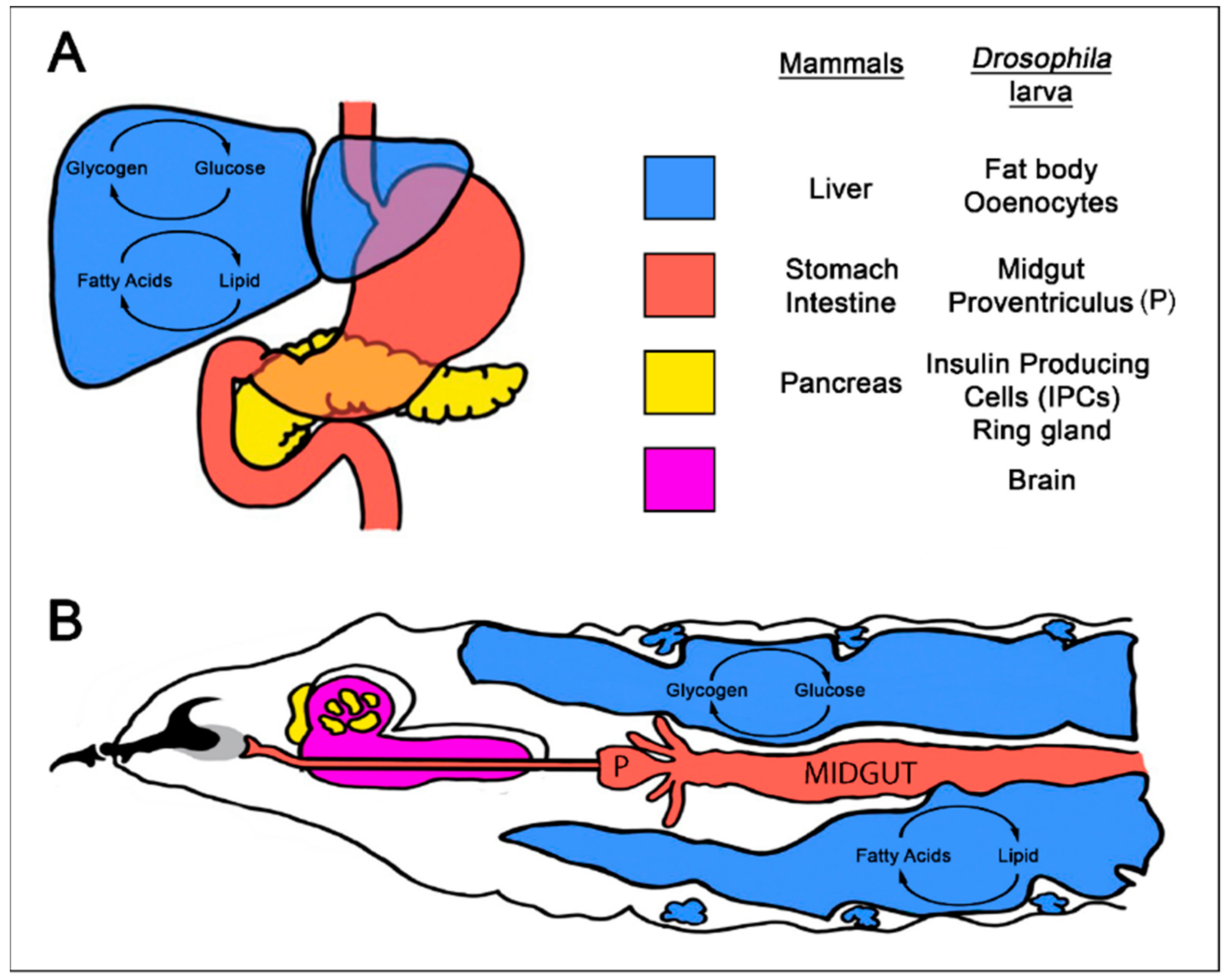
4. Using Drosophila to Model Interactions between Diet, Diet-Related Disease, and Cancer
4.1. Modeling Diet-Related Human Metabolic Diseases in Drosophila
4.2. Investigating Interactions between Diet and Cancer in Drosophila
4.3. Investigating Interactions between Dietary Related Metabolic Disorders and Cancer in Drosophila
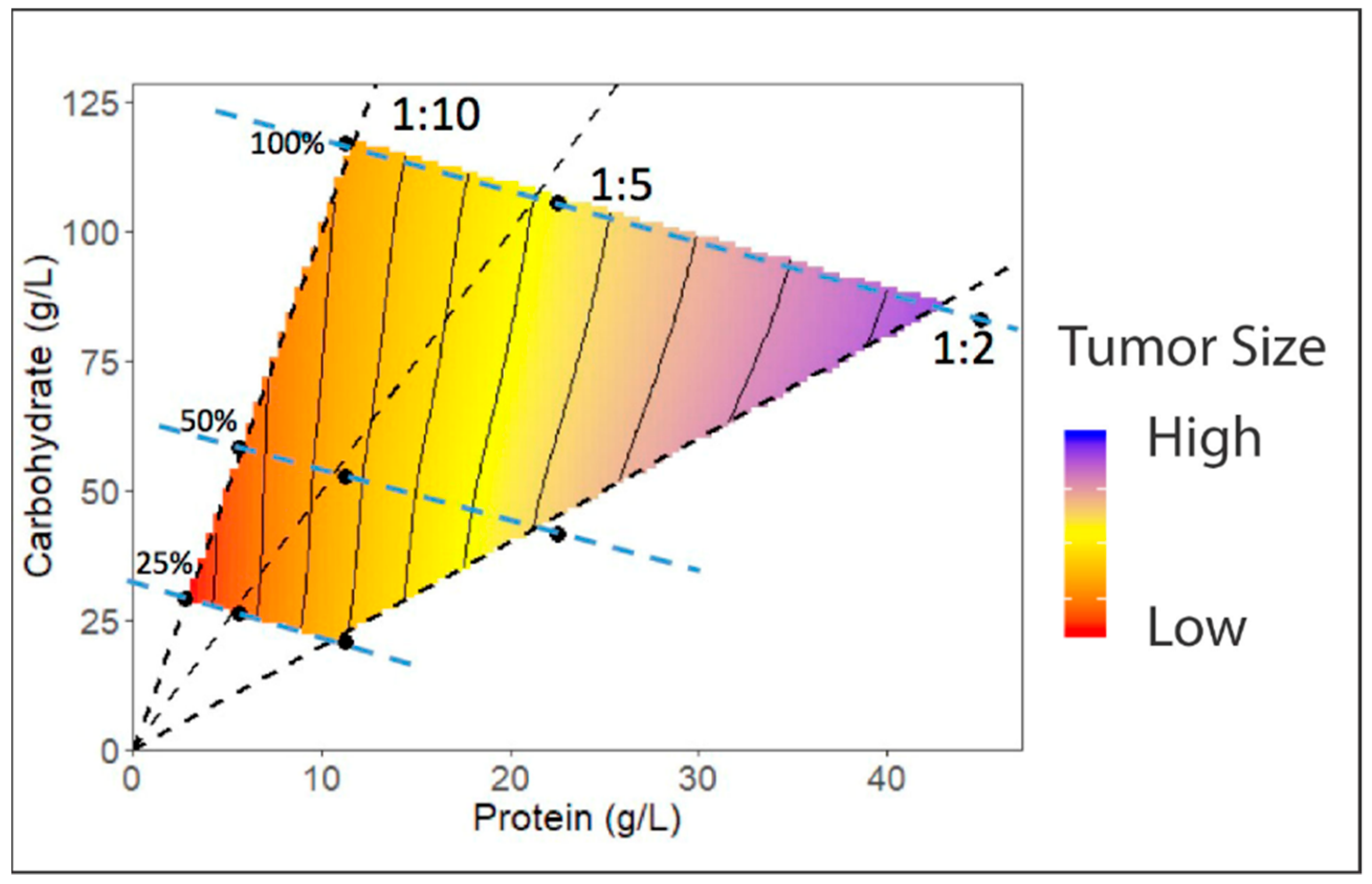
4.4. Development of Dietary Regimes in Drosophila to Study Interactions between Diet and Cancer
4.5. New Genetic Tools in Drosophila will Further Facilitate Studying Interactions between Diet-Related Metabolic Disorders and Cancer
5. Conclusions
Funding
Conflicts of Interest
References
- Wolin, K.Y.; Dart, H.; Colditz, G.A. Eight ways to stay healthy after cancer: An evidence-based message. Cancer Causes Control 2013, 24, 827–837. [Google Scholar] [CrossRef] [PubMed]
- Van Blarigan, E.L.; Fuchs, C.S.; Niedzwiecki, D.; Ye, X.; Zhang, S.; Song, M.; Saltz, L.B.; Mayer, R.J.; Mowat, R.B.; Whittom, R.; et al. Marine ω-3 Polyunsaturated Fatty Acid and Fish Intake after Colon Cancer Diagnosis and Survival: CALGB 89803 (Alliance). Cancer Epidemiol. Biomark. Prev. 2018, 27, 438–445. [Google Scholar] [CrossRef] [PubMed]
- Arends, J.; Bachmann, P.; Baracos, V.; Barthelemy, N.; Bertz, H.; Bozzetti, F.; Fearon, K.; Hütterer, E.; Isenring, E.; Kaasa, S.; et al. ESPEN guidelines on nutrition in cancer patients. Clin. Nutr. 2017, 36, 11–48. [Google Scholar] [CrossRef] [PubMed]
- Baena, R.; Salinas, P. Diet and colorectal cancer. Maturitas 2015, 80, 258–264. [Google Scholar] [CrossRef] [PubMed]
- Chlebowski, R.T. Nutrition and physical activity influence on breast cancer incidence and outcome. Breast 2013, 22 (Suppl. 2), S30–S37. [Google Scholar] [CrossRef] [PubMed]
- Sen, S.; He, Y.; Koya, D.; Kanasaki, K. Cancer biology in diabetes. J. Diabetes Investig. 2014, 5, 251–264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orgel, E.; Mittelman, S.D. The Links Between Insulin Resistance, Diabetes, and Cancer. Curr. Diabetes Rep. 2012, 13, 213–222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- García-Jiménez, C.; Gutiérrez-Salmerón, M.; Chocarro-Calvo, A.; García-Martinez, J.M.; Castaño, A.; De la Vieja, A. From obesity to diabetes and cancer: Epidemiological links and role of therapies. Br. J. Cancer 2016, 114, 716–722. [Google Scholar]
- Albanes, D. Total calories, body weight, and tumor incidence in mice. Cancer Res. 1987, 47, 1987–1992. [Google Scholar] [PubMed]
- Hirohata, T.; Nomura, A.M.; Hankin, J.H.; Kolonel, L.N.; Lee, J. An epidemiologic study on the association between diet and breast cancer. J. Natl. Cancer Inst. 1987, 78, 595–600. [Google Scholar] [PubMed]
- De Lorgeril, M.; Salen, P.; Martin, J.L.; Monjaud, I.; Boucher, P.; Mamelle, N. Mediterranean dietary pattern in a randomized trial: Prolonged survival and possible reduced cancer rate. Arch. Intern. Med. 1998, 158, 1181–1187. [Google Scholar] [CrossRef] [PubMed]
- Stepien, M.; Chajes, V.; Romieu, I. The role of diet in cancer: The epidemiologic link. Salud Publica Mex. 2016, 58, 261–273. [Google Scholar] [CrossRef] [PubMed]
- Norat, T.; Scoccianti, C.; Boutron-Ruault, M.-C.; Anderson, A.; Berrino, F.; Cecchini, M.; Espina, C.; Key, T.; Leitzmann, M.; Powers, H.; et al. European Code against Cancer 4th Edition: Diet and cancer. Cancer Epidemiol. 2015, 39 (Suppl. 1), S56–S66. [Google Scholar] [CrossRef] [Green Version]
- Baena Ruiz, R.; Salinas Hernández, P. Diet and cancer: Risk factors and epidemiological evidence. Maturitas 2014, 77, 202–208. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, B.B.; Shishodia, S. Molecular targets of dietary agents for prevention and therapy of cancer. Biochem. Pharmacol. 2006, 71, 1397–1421. [Google Scholar] [CrossRef] [PubMed]
- World Cancer Research Fund. Diet, Nutrition, Physical Activity and Cancer: A Global Perspective; The Third Expert Report; World Cancer Research Fund: London, UK; American Institute for Cancer: Arlington, VA, USA, 2018; pp. 1–116. [Google Scholar]
- Arnold, M.; Pandeya, N.; Byrnes, G.; Renehan, P.A.G.; Stevens, G.A.; Ezzati, P.M.; Ferlay, J.; Miranda, J.J.; Romieu, I.; Dikshit, R.; et al. Global burden of cancer attributable to high body-mass index in 2012: A population-based study. Lancet Oncol. 2015, 16, 36–46. [Google Scholar] [CrossRef]
- Niraula, S.; Ocana, A.; Ennis, M.; Goodwin, P.J. Body size and breast cancer prognosis in relation to hormone receptor and menopausal status: A meta-analysis. Breast Cancer Res. Treat. 2012, 134, 769–781. [Google Scholar] [CrossRef] [PubMed]
- Chan, D.S.M.; Vieira, A.R.; Aune, D.; Bandera, E.V.; Greenwood, D.C.; McTiernan, A.; Navarro Rosenblatt, D.; Thune, I.; Vieira, R.; Norat, T. Body mass index and survival in women with breast cancer-systematic literature review and meta-analysis of 82 follow-up studies. Ann. Oncol. 2014, 25, 1901–1914. [Google Scholar] [CrossRef] [PubMed]
- Osman, M.A.; Hennessy, B.T. Obesity Correlation with Metastases Development and Response to First-Line Metastatic Chemotherapy in Breast Cancer. Clin. Med. Insights Oncol. 2015, 9, 105–112. [Google Scholar] [CrossRef] [PubMed]
- Allott, E.H.; Masko, E.M.; Freedland, S.J. Obesity and prostate cancer: Weighing the evidence. Eur. Urol. 2013, 63, 800–809. [Google Scholar] [CrossRef] [PubMed]
- Vidal, A.C.; Freedland, S.J. Obesity and Prostate Cancer: A Focused Update on Active Surveillance, Race, and Molecular Subtyping. Eur. Urol. 2017, 72, 78–83. [Google Scholar] [CrossRef] [PubMed]
- Yongmei Xi, Y.Z. Fat Body Development and its Function in Energy Storage and Nutrient Sensing in Drosophila melanogaster. J. Tissue Sci. Eng. 2015, 6, 141. [Google Scholar] [CrossRef]
- Tammariello, A.E.; Milner, J.A. Mouse models for unraveling the importance of diet in colon cancer prevention. J. Nutr. Biochem. 2010, 21, 77–88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Day, C.-P.; Merlino, G.; Van Dyke, T. Preclinical Mouse Cancer Models: A Maze of Opportunities and Challenges. Cell 2015, 163, 39–53. [Google Scholar] [CrossRef] [PubMed]
- Cheng, L.Y.; Parsons, L.M.; Richardson, H.E. Modelling Cancer in Drosophila: The Next Generation. eLS 2013. [Google Scholar] [CrossRef]
- Lee, J.E.A.; Parsons, L.M.; Quinn, L.M. MYC function and regulation in flies: How Drosophila has enlightened MYC cancer biology. AIMS Genet. 2014, 1, 81–98. [Google Scholar]
- Sonoshita, M.; Cagan, R.L. Modeling Human Cancers in Drosophila. In Fly Models of Human Diseases; Current Topics in Developmental Biology; Elsevier: Cambridge, MA, USA, 2017; Volume 121, pp. 287–309. [Google Scholar]
- Thomas, R.M.; Van Dyke, T.; Merlino, G.; Day, C.-P. Concepts in Cancer Modeling: A Brief History. Cancer Res. 2016, 76, 5921–5925. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tentler, J.J.; Tan, A.C.; Weekes, C.D.; Jimeno, A.; Leong, S.; Pitts, T.M.; Arcaroli, J.J.; Messersmith, W.A.; Eckhardt, S.G. Patient-derived tumour xenografts as models for oncology drug development. Nat. Rev. Clin. Oncol. 2012, 9, 338–350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosfjord, E.; Lucas, J.; Li, G.; Gerber, H.-P. Advances in patient-derived tumor xenografts: From target identification to predicting clinical response rates in oncology. Biochem. Pharmacol. 2014, 91, 135–143. [Google Scholar] [CrossRef] [PubMed]
- Bult, C.J.; Krupke, D.M.; Begley, D.A.; Richardson, J.E.; Neuhauser, S.B.; Sundberg, J.P.; Eppig, J.T. Mouse Tumor Biology (MTB): A database of mouse models for human cancer. Nucleic Acids Res. 2014, 43, D818–D824. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Rideout, W.M.; Zi, T.; Bressel, A.; Reddypalli, S.; Rancourt, R.; Woo, J.-K.; Horner, J.W.; Chin, L.; Chiu, M.I.; et al. Chimeric mouse tumor models reveal differences in pathway activation between ERBB family- and KRAS-dependent lung adenocarcinomas. Nat. Biotechnol. 2010, 28, 71–78. [Google Scholar] [CrossRef] [PubMed]
- Meylan, E.; Dooley, A.L.; Feldser, D.M.; Shen, L.; Turk, E.; Ouyang, C.; Jacks, T. Requirement for NF-κB signalling in a mouse model of lung adenocarcinoma. Nature 2009, 462, 104–107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khasawneh, J.; Schulz, M.D.; Walch, A.; Rozman, J.; Hrabe de Angelis, M.; Klingenspor, M.; Buck, A.; Schwaiger, M.; Saur, D.; Schmid, R.M.; et al. Inflammation and mitochondrial fatty acid beta-oxidation link obesity to early tumor promotion. Proc. Natl. Acad. Sci. USA 2009, 106, 3354–3359. [Google Scholar] [CrossRef] [PubMed]
- Huang, M.; Narita, S.; Numakura, K.; Tsuruta, H.; Saito, M.; Inoue, T.; Horikawa, Y.; Tsuchiya, N.; Habuchi, T. A high-fat diet enhances proliferation of prostate cancer cells and activates MCP-1/CCR2 signaling. Prostate 2012, 72, 1779–1788. [Google Scholar] [CrossRef] [PubMed]
- Cho, H.; Kwon, G.; Park, H.; Song, H.; Lee, K.; Kim, J.-I.; Park, J. A High-Fat Diet Containing Lard Accelerates Prostate Cancer Progression and Reduces Survival Rate in Mice: Possible Contribution of Adipose Tissue-Derived Cytokines. Nutrients 2015, 7, 2539–2561. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Makowski, L.; Zhou, C.; Zhong, Y.; Kuan, P.F.; Fan, C.; Sampey, B.P.; Difurio, M.; Bae-Jump, V.L. Obesity increases tumor aggressiveness in a genetically engineered mouse model of serous ovarian cancer. Gynecol. Oncol. 2014, 133, 90–97. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cowen, S.; McLaughlin, S.; Hobbs, G.; Coad, J.; Martin, K.; Olfert, I.; Vona-Davis, L. High-Fat, High-Calorie Diet Enhances Mammary Carcinogenesis and Local Inflammation in MMTV-PyMT Mouse Model of Breast Cancer. Cancers 2015, 7, 1125–1142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fleet, J.C. Animal models of gastrointestinal and liver diseases. New mouse models for studying dietary prevention of colorectal cancer. Am. J. Physiol.-Gastrointest. Liver Physiol. 2014, 307, G249–G259. [Google Scholar] [CrossRef] [PubMed]
- Tang, F.-Y.; Pai, M.-H.; Chiang, E.-P.I. Consumption of high-fat diet induces tumor progression and epithelial–mesenchymal transition of colorectal cancer in a mouse xenograft model. J. Nutr. Biochem. 2012, 23, 1302–1313. [Google Scholar] [CrossRef] [PubMed]
- Van Kruijsdijk, R.C.M.; van der Wall, E.; Visseren, F.L.J. Obesity and Cancer: The Role of Dysfunctional Adipose Tissue. Cancer Epidemiol. Biomark. Prev. 2009, 18, 2569–2578. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kwong, L.N.; Dove, W.F. APC and its modifiers in colon cancer. Adv. Exp. Med. Biol. 2009, 656, 85–106. [Google Scholar] [PubMed]
- Day, S.D.; Enos, R.T.; McClellan, J.L.; Steiner, J.L.; Velázquez, K.T.; Murphy, E.A. Linking inflammation to tumorigenesis in a mouse model of high-fat-diet-enhanced colon cancer. Cytokine 2013, 64, 454–462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Norris, L.; Malkar, A.; Horner-Glister, E.; Hakimi, A.; Ng, L.L.; Gescher, A.J.; Creaser, C.; Sale, S.; Jones, D.J.L. Search for novel circulating cancer chemopreventive biomarkers of dietary rice bran intervention in Apc Minmice model of colorectal carcinogenesis, using proteomic and metabolic profiling strategies. Mol. Nutr. Food Res. 2015, 59, 1827–1836. [Google Scholar] [CrossRef] [PubMed]
- Berger, N.A. Murine Models, Energy Balance, and Cancer; Berger, N.A., Ed.; Springer International Publishing: Cham, Switzerland, 2015; Volume 10. [Google Scholar]
- Kleinert, M.; Clemmensen, C.; Hofmann, S.M.; Moore, M.C.; Renner, S.; Woods, S.C.; Huypens, P.; Beckers, J.; de Angelis, M.H.; Schürmann, A.; et al. Animal models of obesity and diabetes mellitus. Nat. Rev. Endocrinol. 2018, 14, 140–162. [Google Scholar] [CrossRef] [PubMed]
- Ford, N.A.; Nunez, N.P.; Holcomb, V.B.; Hursting, S.D. IGF1 dependence of dietary energy balance effects on murine Met1 mammary tumor progression, epithelial-to-mesenchymal transition, and chemokine expression. Endocr. Relat. Cancer 2013, 20, 39–51. [Google Scholar] [CrossRef] [PubMed]
- Novosyadlyy, R.; Lann, D.E.; Vijayakumar, A.; Rowzee, A.; Lazzarino, D.A.; Fierz, Y.; Carboni, J.M.; Gottardis, M.M.; Pennisi, P.A.; Molinolo, A.A.; et al. Insulin-Mediated Acceleration of Breast Cancer Development and Progression in a Nonobese Model of Type 2 Diabetes. Cancer Res. 2010, 70, 741–751. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferguson, R.D.; Novosyadlyy, R.; Fierz, Y.; Alikhani, N.; Sun, H.; Yakar, S.; LeRoith, D. Hyperinsulinemia enhances c-Myc-mediated mammary tumor development and advances metastatic progression to the lung in a mouse model of type 2 diabetes. Breast Cancer Res. 2012, 14, R8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olivo-Marston, S.E.; Hursting, S.D.; Lavigne, J.; Perkins, S.N.; Maarouf, R.S.; Yakar, S.; Harris, C.C. Genetic reduction of circulating insulin-like growth factor-1 inhibits azoxymethane-induced colon tumorigenesis in mice. Mol. Carcinog. 2009, 48, 1071–1076. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lashinger, L.M.; Harrison, L.M.; Rasmussen, A.J.; Logsdon, C.D.; Fischer, S.M.; McArthur, M.J.; Hursting, S.D. Dietary Energy Balance Modulation of Kras- and Ink4a/Arf+/−-Driven Pancreatic Cancer: The Role of Insulin-like Growth Factor-I. Cancer Prev. Res. 2013, 6, 1046–1055. [Google Scholar] [CrossRef] [PubMed]
- Lashinger, L.M.; Malone, L.M.; McArthur, M.J.; Goldberg, J.A.; Daniels, E.A.; Pavone, A.; Colby, J.K.; Smith, N.C.; Perkins, S.N.; Fischer, S.M.; et al. Genetic Reduction of Insulin-like Growth Factor-1 Mimics the Anticancer Effects of Calorie Restriction on Cyclooxygenase-2-Driven Pancreatic Neoplasia. Cancer Prev. Res. 2011, 4, 1030–1040. [Google Scholar] [CrossRef] [PubMed]
- Hao, J.; Zhang, Y.; Yan, X.; Yan, F.; Sun, Y.; Zeng, J.; Waigel, S.; Yin, Y.; Fraig, M.M.; Egilmez, N.K.; et al. Circulating Adipose Fatty Acid Binding Protein Is a New Link Underlying Obesity-Associated Breast/Mammary Tumor Development. Cell Metab. 2018, 28, 689–705.e5. [Google Scholar] [CrossRef] [PubMed]
- Nieman, K.M.; Kenny, H.A.; Penicka, C.V.; Ladanyi, A.; Buell-Gutbrod, R.; Zillhardt, M.R.; Romero, I.L.; Carey, M.S.; Mills, G.B.; Hotamisligil, G.S.; et al. Adipocytes promote ovarian cancer metastasis and provide energy for rapid tumor growth. Nat. Med. 2011, 17, 1498–1503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hancke, K.; Grubeck, D.; Hauser, N.; Kreienberg, R.; Weiss, J.M. Adipocyte fatty acid-binding protein as a novel prognostic factor in obese breast cancer patients. Breast Cancer Res. Treat. 2010, 119, 367. [Google Scholar] [CrossRef] [PubMed]
- Rudrapatna, V.A.; Cagan, R.L.; Das, T.K. Drosophila cancer models. Dev. Dyn. 2012, 241, 107–118. [Google Scholar] [CrossRef] [PubMed]
- Solon-Biet, S.M.; Mitchell, S.J.; Coogan, S.C.P.; Cogger, V.C.; Gokarn, R.; McMahon, A.C.; Raubenheimer, D.; de Cabo, R.; Simpson, S.J.; Le Couteur, D.G. Dietary Protein to Carbohydrate Ratio and Caloric Restriction: Comparing Metabolic Outcomes in Mice. Cell Rep. 2015, 11, 1529–1534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reiter, L.T.; Potocki, L.; Chien, S.; Gribskov, M.; Bier, E. A systematic analysis of human disease-associated gene sequences in Drosophila melanogaster. Genome Res. 2001, 11, 1114–1125. [Google Scholar] [CrossRef] [PubMed]
- Pandey, U.B.; Nichols, C.D. Human Disease Models in Drosophila melanogaster and the Role of the Fly in Therapeutic Drug Discovery. Pharmacol. Rev. 2011, 63, 411–436. [Google Scholar] [CrossRef] [PubMed]
- Gillooly, J.F.; Gomez, J.P.; Mavrodiev, E.V. A broad-scale comparison of aerobic activity levels in vertebrates: Endotherms versus ectotherms. Proc. R. Soc. B Biol. Sci. 2017, 284, 20162328. [Google Scholar] [CrossRef] [PubMed]
- Rand, M.D. Drosophotoxicology: The growing potential for Drosophila in neurotoxicology. Neurotoxicol. Teratol. 2010, 32, 74–83. [Google Scholar] [CrossRef] [PubMed]
- Padmanabha, D.; Baker, K.D. Drosophila gains traction as a repurposed tool to investigate metabolism. Trends Endocrinol. Metab. 2014, 25, 518–527. [Google Scholar] [CrossRef] [PubMed]
- Ugur, B.; Chen, K.; Bellen, H.J. Drosophila tools and assays for the study of human diseases. Dis. Model. Mech. 2016, 9, 235–244. [Google Scholar] [CrossRef] [PubMed]
- Lemaitre, B.; Miguel-Aliaga, I. The digestive tract of Drosophila melanogaster. Annu. Rev. Genet. 2013, 47, 377–404. [Google Scholar] [CrossRef] [PubMed]
- Bharucha, K.N. The epicurean fly: Using Drosophila melanogaster to study metabolism. Pediatr. Res. 2009, 65, 132–137. [Google Scholar] [CrossRef] [PubMed]
- Rajan, A.; Perrimon, N. Of flies and men: Insights on organismal metabolism from fruit flies. BMC Biol. 2013, 11, 38. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, D.; Katewa, S.D.; Qi, Y.; Jackson, S.A.; Kapahi, P.; Jasper, H. Control of metabolic adaptation to fasting by dILP6-induced insulin signaling in Drosophila oenocytes. Proc. Natl. Acad. Sci. USA 2014, 111, 17959–17964. [Google Scholar] [CrossRef] [PubMed]
- Gutierrez, E.; Wiggins, D.; Fielding, B.; Gould, A.P. Specialized hepatocyte-like cells regulate Drosophila lipid metabolism. Nature 2007, 445, 275–280. [Google Scholar] [CrossRef] [PubMed]
- Arrese, E.L.; Soulages, J.L. Insect Fat Body: Energy, Metabolism, and Regulation. Annu. Rev. Entomol. 2010, 55, 207–225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Das, R.; Dobens, L.L. Conservation of gene and tissue networks regulating insulin signalling in flies and vertebrates. Biochem. Soc. Trans. 2015, 43, 1057–1062. [Google Scholar] [CrossRef] [PubMed]
- Birsoy, K.; Festuccia, W.T.; Laplante, M. A comparative perspective on lipid storage in animals. J. Cell Sci. 2013, 126, 1541–1552. [Google Scholar] [CrossRef] [PubMed]
- Palm, W.; Sampaio, J.L.; Brankatschk, M.; Carvalho, M.; Mahmoud, A.; Shevchenko, A.; Eaton, S. Lipoproteins in Drosophila melanogaster—Assembly, Function, and Influence on Tissue Lipid Composition. PLoS Genet. 2012, 8, e1002828-18. [Google Scholar] [CrossRef] [PubMed]
- Zinke, I.; Schütz, C.S.; Katzenberger, J.D. Nutrient control of gene expression in Drosophila: Microarray analysis of starvation and sugar-dependent response. EMBO J. 2002, 21, 6162–6173. [Google Scholar] [CrossRef] [PubMed]
- Tripoli, G.; D’Elia, D.; Barsanti, P.; Caggese, C. Comparison of the oxidative phosphorylation (OXPHOS) nuclear genes in the genomes of Drosophila melanogaster, Drosophila pseudoobscura and Anopheles gambiae. Genome Biol. 2005, 6, R11. [Google Scholar] [CrossRef] [PubMed]
- Teleman, A.A. Molecular mechanisms of metabolic regulation by insulin in Drosophila. Biochem. J. 2009, 425, 13–26. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.K.; Rulifson, E.J. Conserved mechanisms of glucose sensing and regulation by Drosophila corpora cardiaca cells. Nature 2004, 431, 316–320. [Google Scholar] [CrossRef] [PubMed]
- Palanker Musselman, L.; Fink, J.L.; Narzinski, K.; Ramachandran, P.V.; Sukumar Hathiramani, S.; Cagan, R.L.; Baranski, T.J. A high-sugar diet produces obesity and insulin resistance in wild-type Drosophila. Dis. Model. Mech. 2011, 4, 842–849. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.-H.; Kang, M.; Lee, K.-S.; Yu, K. High fat diet-induced TGF-β/Gbb signaling provokes insulin resistance through the tribbles expression. Sci. Rep. 2016, 6, 30265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Birse, R.T.; Choi, J.; Reardon, K.; Rodriguez, J.; Graham, S.; Diop, S.; Ocorr, K.; Bodmer, R.; Oldham, S. High-fat-diet-induced obesity and heart dysfunction are regulated by the TOR pathway in Drosophila. Cell Metab. 2010, 12, 533–544. [Google Scholar] [CrossRef] [PubMed]
- Smith, W.W.; Thomas, J.; Liu, J.; Li, T.; Moran, T.H. From fat fruit fly to human obesity. Physiol. Behav. 2014, 136, 15–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gateff, E.; Schneiderman, H.A. Developmental Capacities of Benign and Malignant Neoplasms of Drosophila. Dev. Genes Evol. 1974, 176, 23–65. [Google Scholar]
- Lee, T.V.; Luo, L. Mosiac analysis with a repressible cell marker (MARCM) for Drosophila neural development. Trends Neurosci. 2001, 24, 251–254. [Google Scholar] [CrossRef]
- Cordero, J.B.; Macagno, J.P.; Stefanatos, R.K.; Strathdee, K.E.; Cagan, R.L.; Vidal, M. Oncogenic Ras Diverts a Host TNF Tumor Suppressor Activity into Tumor Promoter. Dev. Cell 2010, 18, 999–1011. [Google Scholar] [CrossRef] [PubMed]
- Nowak, K.; Seisenbacher, G.; Hafen, E.; Stocker, H. Nutrient restriction enhances the proliferative potential of cells lacking the tumor suppressor PTEN in mitotic tissues. eLife 2013, 2, e00380. [Google Scholar] [CrossRef] [PubMed]
- Hirabayashi, S.; Baranski, T.J.; Cagan, R.L. Transformed Drosophila Cells Evade Diet-Mediated Insulin Resistance through Wingless Signaling. Cell 2013, 154, 664–675. [Google Scholar] [CrossRef] [PubMed]
- Ishizawar, R.; Parsons, S.J. c-Src and cooperating partners in human cancer. Cancer Cell 2004, 6, 209–214. [Google Scholar] [CrossRef] [PubMed]
- Hirabayashi, S.; Cagan, R.L. Salt-Inducible Kinases Mediate Nutrient-Sensing to Link Dietary Sugar and Tumorigenesis in Drosophila. Available online: https://elifesciences.org/articles/08501 (accessed on 3 December 2018).
- Wang, B.; Moya, N.; Niessen, S.; Hoover, H.; Mihaylova, M.M.; Shaw, R.J.; Yates, J.R.; Fischer, W.H.; Thomas, J.B.; Montminy, M. A hormone-dependent module regulating energy balance. Cell 2011, 145, 596–606. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.; Lim, D.-S.; Chung, J. Feeding and Fasting Signals Converge on the LKB1-SIK3 Pathway to Regulate Lipid Metabolism in Drosophila. PLoS Genet. 2015, 11, e1005263. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.; Kim, W.; Chung, J. Drosophila salt-inducible kinase (SIK) regulates starvation resistance through cAMP-response element-binding protein (CREB)-regulated transcription coactivator (CRTC). J. Biol. Chem. 2011, 286, 2658–2664. [Google Scholar] [CrossRef] [PubMed]
- Henriksson, E.; Sall, J.; Gormand, A.; Wasserstrom, S.; Morrice, N.A.; Fritzen, A.M.; Foretz, M.; Campbell, D.G.; Sakamoto, K.; Ekelund, M.; et al. SIK2 regulates CRTCs, HDAC4 and glucose uptake in adipocytes. J Cell Sci. 2015, 128, 472–486. [Google Scholar] [CrossRef] [PubMed]
- Patel, K.; Marion, A.; Campbell, D.G.; Gourlay, R.; Boudaba, N.; Tournier, E.; Titchenell, P.; Peggie, M.; Deak, M.; Wan, M.; et al. The LKB1-salt-inducible kinase pathway functions as a key gluconeogenic suppressor in the liver. Nat. Commun. 2014, 5, 4535. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Teesalu, M.; Rovenko, B.M.; Hietakangas, V. Salt-Inducible Kinase 3 Provides Sugar Tolerance by Regulating NADPH/NADP(+) Redox Balance. Curr. Biol. 2017, 27, 458–464. [Google Scholar] [CrossRef] [PubMed]
- Bensinger, S.J.; Christofk, H.R. New aspects of the Warburg effect in cancer cell biology. Semin. Cell Dev. Biol. 2012, 23, 352–361. [Google Scholar] [CrossRef] [PubMed]
- Liberti, M.V.; Locasale, J.W. The Warburg Effect: How Does it Benefit Cancer Cells? Trends Biochem. Sci. 2016, 41, 211–218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, W.Q.; Zheng, J.N.; Pei, D.S. The diverse oncogenic and tumor suppressor roles of salt-inducible kinase (SIK) in cancer. Expert Opin. Ther. Targets 2016, 20, 477–485. [Google Scholar] [CrossRef] [PubMed]
- Charoenfuprasert, S.; Yang, Y.-Y.; Lee, Y.-C.; Chao, K.-C.; Chu, P.-Y.; Lai, C.-R.; Hsu, K.-F.; Chang, K.-C.; Chen, Y.-C.; Chen, L.-T.; et al. Identification of salt-inducible kinase 3 as a novel tumor antigen associated with tumorigenesis of ovarian cancer. Oncogene 2011, 30, 3570–3584. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmed, A.A.; Lu, Z.; Jennings, N.B.; Etemadmoghadam, D.; Capalbo, L.; Jacamo, R.O.; Barbosa-Morais, N.; Le, X.-F.; Vivas-Mejia, P.; Lopez-Berestein, G.; et al. SIK2 Is a Centrosome Kinase Required for Bipolar Mitotic Spindle Formation that Provides a Potential Target for Therapy in Ovarian Cancer. Cancer Cell 2010, 18, 109–121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amara, S.; Majors, C.; Roy, B.; Hill, S.; Rose, K.L.; Myles, E.L.; Tiriveedhi, V. Critical role of SIK3 in mediating high salt and IL-17 synergy leading to breast cancer cell proliferation. PLoS ONE 2017, 12, e0180097. [Google Scholar] [CrossRef] [PubMed]
- Miranda, F.; Mannion, D.; Liu, S.; Zheng, Y.; Mangala, L.S.; Redondo, C.; Herrero-Gonzalez, S.; Xu, R.; Taylor, C.; Chedom, D.F.; et al. Salt-Inducible Kinase 2 Couples Ovarian Cancer Cell Metabolism with Survival at the Adipocyte-Rich Metastatic Niche. Cancer Cell 2016, 30, 273–289. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, S.; Lagisz, M.; Hector, K.L.; Spencer, H.G. Comparative and meta-analytic insights into life extension via dietary restriction. Aging Cell 2012, 11, 401–409. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Speakman, J.R.; Mitchell, S.E.; Mazidi, M. Calories or protein? The effect of dietary restriction on lifespan in rodents is explained by calories alone. Exp. Gerontol. 2016, 86, 28–38. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.P.; Simpson, S.J.; Clissold, F.J.; Brooks, R.; Ballard, J.W.O.; Taylor, P.W.; Soran, N.; Raubenheimer, D. Lifespan and reproduction in Drosophila: New insights from nutritional geometry. Proc. Natl. Acad. Sci. USA 2008, 105, 2498–2503. [Google Scholar] [CrossRef] [PubMed]
- Bruce, K.D.; Hoxha, S.; Carvalho, G.B.; Yamada, R.; Wang, H.-D.; Karayan, P.; He, S.; Brummel, T.; Kapahi, P.; Ja, W.W. High carbohydrate-low protein consumption maximizes Drosophila lifespan. Exp. Gerontol. 2013, 48, 1129–1135. [Google Scholar] [CrossRef] [PubMed]
- Piper, M.D.W.; Soultoukis, G.A.; Blanc, E.; Mesaros, A.; Herbert, S.L.; Juricic, P.; He, X.; Atanassov, I.; Salmonowicz, H.; Yang, M.; et al. Matching Dietary Amino Acid Balance to the In Silico-Translated Exome Optimizes Growth and Reproduction without Cost to Lifespan. Cell Metab. 2017, 25, 610–621. [Google Scholar] [CrossRef] [PubMed]
- Stefana, M.I.; Driscoll, P.C.; Obata, F.; Pengelly, A.R.; Newell, C.L.; MacRae, J.I.; Gould, A.P. Developmental diet regulates Drosophila lifespan via lipid autotoxins. Nat. Commun. 2017, 8, 1384. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.P. Dietary protein:carbohydrate balance is a critical modulator of lifespan and reproduction in Drosophila melanogaster: A test using a chemically defined diet. J. Insect Physiol. 2015, 75, 12–19. [Google Scholar] [CrossRef] [PubMed]
- Simpson, S.J.; Raubenheimer, D. The Nature of Nutrition: A Unifying Framework from Animal Adaptation to Human Obesity; Princeton University Press: Princeton, NJ, USA; Oxfordshire, UK, 2012. [Google Scholar]
- Piper, M.D.W.; Blanc, E.; Leitão-Gonçalves, R.; Yang, M.; He, X.; Linford, N.J.; Hoddinott, M.P.; Hopfen, C.; Soultoukis, G.A.; Niemeyer, C.; et al. A holidic medium for Drosophila melanogaster. Nat. Methods 2014, 11, 100–105. [Google Scholar] [CrossRef] [PubMed]
- Simpson, S.J.; Le Couteur, D.G.; James, D.E.; George, J.; Gunton, J.E.; Solon-Biet, S.M.; Raubenheimer, D. The Geometric Framework for Nutrition as a tool in precision medicine. Nutr. Healthy Aging 2017, 4, 217–226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piper, M.D. Using artificial diets to understand the nutritional physiology of Drosophila melanogaster. Curr. Opin. Insect Sci. 2017, 23, 104–111. [Google Scholar] [CrossRef] [PubMed]
- Brand, A.H.; Perrimon, N. Targeted gene expression as a means of altering cell fates and generating dominant phenotypes. Development 1993, 118, 401–415. [Google Scholar] [PubMed]
- St Johnston, D. Using mutants, knockdowns, and transgenesis to investigate gene function in Drosophila. Wiley Interdiscip. Rev. Dev. Biol. 2012, 2, 587–613. [Google Scholar] [CrossRef] [PubMed]
- Lai, S.-L.; Lee, T. Genetic mosaic with dual binary transcriptional systems in Drosophila. Nat. Neurosci. 2006, 9, 703–709. [Google Scholar] [CrossRef] [PubMed]
- Potter, C.J.; Tasic, B.; Russler, E.V.; Liang, L.; Luo, L. The Q System: A Repressible Binary System for Transgene Expression, Lineage Tracing, and Mosaic Analysis. Cell 2010, 141, 536–548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riabinina, O.; Luginbuhl, D.; Marr, E.; Liu, S.; Wu, M.N.; Luo, L.; Potter, C.J. Improved and expanded Q-system reagents for genetic manipulations. Nat. Methods 2015, 12, 219–222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diao, F.; Ironfield, H.; Luan, H.; Diao, F.; Shropshire, W.C.; Ewer, J.; Marr, E.; Potter, C.J.; Landgraf, M.; White, B.H. Plug-and-play genetic access to Drosophila cell types using exchangeable exon cassettes. Cell Rep. 2015, 10, 1410–1421. [Google Scholar] [CrossRef] [PubMed]


© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Warr, C.G.; Shaw, K.H.; Azim, A.; Piper, M.D.W.; Parsons, L.M. Using Mouse and Drosophila Models to Investigate the Mechanistic Links between Diet, Obesity, Type II Diabetes, and Cancer. Int. J. Mol. Sci. 2018, 19, 4110. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms19124110
Warr CG, Shaw KH, Azim A, Piper MDW, Parsons LM. Using Mouse and Drosophila Models to Investigate the Mechanistic Links between Diet, Obesity, Type II Diabetes, and Cancer. International Journal of Molecular Sciences. 2018; 19(12):4110. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms19124110
Chicago/Turabian StyleWarr, Coral G., Katherine H. Shaw, Arani Azim, Matthew D. W. Piper, and Linda M. Parsons. 2018. "Using Mouse and Drosophila Models to Investigate the Mechanistic Links between Diet, Obesity, Type II Diabetes, and Cancer" International Journal of Molecular Sciences 19, no. 12: 4110. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms19124110